

Abstract
Flap endonucleases (FENs) are divalent metal ion-dependent phosphodiesterases. Metallonucleases are often assigned a “two-metal ion mechanism” where both metals contact the scissile phosphate diester. The spacing of the two metal ions observed in T5FEN structures appears to preclude this mechanism. However, the overall reaction catalyzed by wild type (WT) T5FEN requires three Mg2+ ions, implying that a third ion is needed during catalysis, and so a two-metal ion mechanism remains possible. To investigate the positions of the ions required for chemistry, a mutant T5FEN was studied where metal 2 (M2) ligands are altered to eliminate this binding site. In contrast to WT T5FEN, the overall reaction catalyzed by D201I/D204S required two ions, but over the concentration range of Mg2+ tested, maximal rate data were fitted to a single binding isotherm. Calcium ions do not support FEN catalysis and inhibit the reactions supported by viable metal cofactors. To establish participation of ions in stabilization of enzyme-substrate complexes, dissociation constants of WT and D201I/D204S-substrate complexes were studied as a function of [Ca2+]. At pH 9.3 (maximal rate conditions), Ca2+ substantially stabilized both complexes. Inhibition of viable cofactor supported reactions of WT, and D201I/D204S T5FENs was biphasic with respect to Ca2+ and ultimately dependent on 1/[Ca2+]2. By varying the concentration of viable metal cofactor, Ca2+ ions were shown to inhibit competitively displacing two catalytic ions. Combined analyses imply that M2 is not involved in chemical catalysis but plays a role in substrate binding, and thus a two-metal ion mechanism is plausible.
Keywords: Biophysics, Enzyme Inhibitors, Enzyme Mechanisms, Metalloenzymes, Protein DNA-Interaction, Endonuclease, Nuclease
Introduction
Flap endonucleases (FENs)4, essential in all life forms, are members of the 5′-nuclease superfamily of structure-specific phosphate diesterases that carry out essential divalent metal ion-dependent nucleic acid hydrolysis (1). FENs act upon 5′- bifurcated structures known as flaps formed during lagging strand DNA replication and long patch base excision repair. Hydrolysis predominantly occurs one nucleotide into the 5′-duplex adjoining the site of bifurcation. An unusual feature of these enzymes is their high density of active site carboxylates that coordinate essential divalent metal ion cofactors. Bacterial and bacteriophage FENs possess eight active site acidic residues, seven of which are conserved in FENs from higher organisms and also in other members of the 5′-nuclease superfamily (1–4). In contrast, a typical metallonuclease active site consists of three or four carboxylates (5). However, a subclass of FEN paralogues, typified by Escherichia coli ExoIX, exists in eubacteria and appear to lack three of the metal-binding carboxylates (6).
Although the most common mechanism suggested for metal-dependent phosphoryl transferases involves two metal ions in contact with the scissile phosphate diester, this is not universally accepted (5, 7–10). Debate about the validity or otherwise of two-metal ion catalysis has focused on several enzymes including flap endonucleases (11–16). Crystallographic studies of substrate-free FENs have demonstrated two active site-bound metal ions (14, 17–19). Metal ion 1 (M1) occupies a similar position in all structures. However, there is some variation in the position of the site coordinating a second metal ion (M2), often observed in FEN crystal structures. The distance between the M1 and M2 sites varies, and the separations observed are generally much greater than the 4 Å required for both metals to contact the same phosphate diester. For example, in bacteriophage T5FEN,5 the M1-M2 separation of two Mn2+ ions is 8 Å (Fig. 1A) (17). However, in a substrate-free structure of hFEN1, two magnesium ions are bound with a separation of 3.4 Å in one of three proteins bound to proliferating cell nuclear antigen (Fig. 1B) (18). A very recent structure of hFEN1 bound to product DNA has two Sm3+ ions bound in equivalent positions, and these contact the cleaved phosphate monoester (4).
FIGURE 1.

The varying positions of metal ions in FEN structures. A, the active site structure of T5FEN (1UT5) illustrating the seven active site carboxylates present in similar positions in all FENs and the eighth carboxylate (Asp-201) present in the active sites of bacteriophage and bacterial enzymes. Two metal ions, M1 and M2, are bound with a separation of 8 Å. A metal site 2 mutant was created by alteration of the two aspartic acid residues (D210I/D204S). B, the active site structure of hFEN1 (1UL1x), illustrating the seven active site carboxylates present in similar positions in all FENs. Two metal ions, M1 and M2, are bound with a separation of 3.4 Å. Although all FENs conserve seven active site carboxylates, the position of M2 observed in each structure is variable. The N terminus (blue) occupies a similar position to M2 in the bacteriophage enzyme (A).
Despite the 8 Å distance between M1 and M2 in substrate-free T5FEN structures, magnesium ion stimulation data are consistent with a mechanism where the major rate acceleration is brought about by two metal ions but where overall (kcat/Km) has a three-divalent ion requirement (3). Thus an additional ion (M3) is bound with higher affinity to the enzyme-substrate complex. If two metal ions that participate in chemical catalysis are coordinated to the central carboxylate of the T5FEN active site (Asp-130), positioned as those observed in hFEN1 structures (Fig. 1) (2, 4), two-metal ion catalysis mechanisms where both metals coordinate the scissile phosphate diester could take place. As only M1 is observed in this position in substrate-free structures of bacteriophage FENs, in this scenario, the third ion implicated by functional studies would have to bind in this location, and the catalytic ions would therefore be M1 and M3 (Fig. 1A). However, alternative explanations of a two-ion requirement for the major rate acceleration are possible. For example, M2, as positioned in T5FEN substrate-free structures, would not be able to directly interact with the scissile phosphate diester but instead could catalyze chemical catalysis by more remote interactions. On the other hand, if catalytic M1 and M3 provide the main rate acceleration, M2 could play a role in enzyme-substrate complex stability.
Here we test earlier proposals for the roles of metal ions in reactions catalyzed by T5FEN. To investigate whether divalent metal ions alter the stability of enzyme-substrate complexes, dissociation constants were determined in the absence of catalysis at varying concentration of Ca2+ ions and pH. Previous studies of the T5FEN-catalyzed reactions demonstrated that catalytically inert Ca2+ ions inhibited reaction with a 1/[Ca2+]2 dependence, which is consistent with an absolute requirement for two viable metal cofactor ions that the Ca2+ ions displace. Potentially, this could be used to test for a requirement for two metal ions in mutated FENs and other metallonucleases. However, it is possible that decreases in the rate of reaction may involve Ca2+ binding non-competitively to the enzyme, substrate, or enzyme-substrate complex. To establish the competitive inhibition of FEN-catalyzed reactions by Ca2+ ions, the inhibitory characteristics were studied with different supporting metal cofactor ions at varying concentration. To probe the role of T5FEN M2, both the stimulation by viable metal cofactor ions and the inhibition by Ca2+ ions were evaluated for a mutated T5FEN where two of the carboxylates that bind M2 are altered. The combined analyses support a mechanism for T5FEN where the main rate acceleration is afforded by two ions, but not by M2, and imply a role for M2 in stabilization of enzyme-substrate interactions.
EXPERIMENTAL PROCEDURES
Materials
Wild type (WT) and D201I/D204S T5FENs and 5′-overhanging hairpin substrate (5′-FAM-pd(CGCTGTCGAACACACGCTTGCGTGTGTTC)) (HP5F) were prepared and purified to homogeneity as described (3, 24, 25). Divalent metal ion contaminants were removed by treatment with Chelex resin.
Steady State Kinetic Analyses
Steady state kinetic parameters of WT and D201I/D204S T5FEN (E) were evaluated at 37 °C using HP5F substrate (S) in 25 mm CHES or potassium glycinate, pH 9.3, 0.1 mg/ml BSA, and 1 mm DTT as described (3). MgCl2 was added to the desired concentration, and the ionic strength (MgCl2 + KCl) was adjusted to 80 mm using KCl. Substrate concentrations were varied from 0.12 to 8 × Km, and reactions were sampled and quenched by the addition of an equal volume of 25 mm EDTA at the appropriate time intervals. Reactions were analyzed by denaturing HPLC equipped with a fluorescence detector, and initial rates (v) were calculated as described (26). Steady state catalytic parameters were determined at each Mg2+ concentration by curve fitting to the Michaelis-Menten equation. Plots of v/[E] versus [S], where [S] < Km, were used to determine kcat/Km.
Calcium Inhibition
For magnesium-supported T5FEN reactions, reaction mixtures containing 0.5 or 2 mm MgCl2, 1 μm HP5F, 25 mm potassium glycinate, pH 9.3, 0.1 mg/ml BSA, varying [CaCl2] with the appropriate amount of KCl to keep ionic strength constant as above, were initiated by the appropriate amounts of T5FEN (0.3–30 nm). For manganese-supported T5FEN reactions, reaction mixtures containing 0.1, 0.2, or 0.4 mm MnCl2, 0.2 μm HP5F, 25 mm MOPS, pH 7.5, 0.1 mg/ml BSA, CaCl2 with the appropriate amount of KCl were initiated by the addition of an appropriate amount of T5FEN (0.05–0.4 nm). For D201I/D204S-T5FEN reactions, reaction mixtures containing 0.5 or 2 mm MgCl2, 0.5 μm HP5F, 25 mm CHES, pH 9.3, 0.1 mg/ml BSA, CaCl2 with the appropriate amount of KCl were initiated by the addition of the appropriate amount of D201I/D204S T5FEN (final concentration 5–40 nm). All reactions were sampled and quenched as above and used to determine kobs (v/[E]) as a function of calcium ion concentration.
Fluorescence Anisotropy
T5FEN DNA interactions were evaluated by fluorescence anisotropy using a HORIBA Jobin Yvon FluoroMax-3® fluorometer with automatic polarizers. The excitation wavelength was 490 nm (excitation slit width 8 nm) with emission detected at 509–511 nm through an 8-nm-wide slit. Experiments contained 2 mm EDTA or varying concentrations of CaCl2 (0.5–10 mm), with the appropriate amount of KCl as for kinetic analyses, 100 nm HP5F (for WT T5FEN) or 10 nm HP5F (for D201I/D204S), 25 mm HEPES, pH 7.5, or 25 mm potassium glycinate, pH 9.3, 0.1 mg/ml BSA, and 1 mm DTT. All experiments were carried out at 37 °C. Anisotropy (r) was determined as described (27) initially prior to the addition of protein and then on the cumulative addition of enzyme.
Data were fitted, using KaleidaGraph, to the following equation with corrections made for dilution,
 |
where r is the measured anisotropy, [E] is the total protein concentration and [S] is the total HP5F concentration, rmin is the anisotropy of free DNA, rmax is the anisotropy of the DNA-protein complex, and KD is the dissociation constant (27).
Data Analyses
Curve fitting of Mg2+-dependent behavior and Ca2+-dependent inhibitions were carried out by non-linear regression fitting using KaleidaGraph software weighted according to individual error values when required, using Equations 2–7 (see “Results”).
RESULTS
The Effect of Divalent Metal Ions on WT FEN-Substrate Interactions
Studies on the magnesium ion dependence of the WT T5FEN-catalyzed reaction were carried out previously using the 5′-fluorescent overhanging hairpin substrate HP5F under pH-independent maximal rate conditions (pH = 9.3) (3). These earlier kinetic studies revealed a requirement for two Mg2+ ions to maximize 1/Km. However, the interpretation of this behavior was complicated by the nature of this parameter, which in FEN-catalyzed reactions that are partially rate-limited by the release of products reflects the stability of all forms of the enzyme-bound substrate and product. To examine binding of HP5F to T5FEN under similar conditions to catalytic experiments but in the absence of catalysis, fluorescence anisotropy was employed. In the presence of EDTA at pH 9.3, anisotropy (r) increased with the addition of protein up to 10 μm, but no maximal value was reached, implying KD > 5 μm (Fig. 2A). In contrast, in the presence of non-catalytically competent Ca2+ ions (1 mm), r reached a maximum value and could be fitted to a single binding isotherm (Equation 1) to yield a KD = 202 ± 27 nm (Fig. 2A). Thus divalent metal ions substantially stabilize the T5FEN-HP5F complex at pH 9.3.
FIGURE 2.

The effects of calcium ions on WT and D210I/D204S FEN-substrate dissociation constants evaluated by fluorescence anisotropy (r). Experimental details are contained under “Experimental Procedures,” and all experiments were carried out in triplicate. Constant ionic strength of potassium and calcium ions was maintained by varying the amount of KCl present. A, the addition of WT T5FEN to 100 nm HP5F at pH 9.3, in the presence of 2 mm EDTA (squares) and 1 mm Ca2+ ions (circles). Curve fitting to Equation 1 yields KD = 202 ± 27 nm (1 mm Ca2+). Without divalent metal ions present, the binding curve did not reach saturation. B, the addition of WT T5FEN to 100 nm HP5F at pH 7.5, in the presence of 2 mm EDTA (squares) and 1 mm Ca2+ ions (circles). Curve fitting to Equation 1 yields KD = 231 ± 13 nm (no divalent ion) and KD = 120 ± 13 nm (1 mm Ca2+). C, the addition of D210I/D204S T5FEN to 10 nm HP5F at pH 9.3, in the presence of 2 mm EDTA (squares) and 0.5 mm Ca2+ ions (circles). Curve fitting to Equation 1 yields KD = 2880 ± 160 nm (no divalent ion) and KD = 28 ± 3 nm (0.5 mm Ca2+). D, variation of log KD as a function of log [Ca2+] for WT T5FEN at pH 9.3 (circles), pH 7.5 (squares), and D210I/D204S T5FEN at pH 9.3 (diamonds). The slopes of the resultant plots are 0.15 ± 0.10, −0.06 ± 0.06, and 0.09 ± 0.04, respectively. Error bars indicate S.E.
However, at pH 7.5 in the absence of divalent metal ions, KD = 231 ± 13 nm (Fig. 2B). At this pH, the presence of Ca2+ ions (1 mm) has a modest effect, with KD = 120 ± 13 nm. Thus either lowering the pH or adding divalent metal ions has a large effect on T5FEN-substrate interactions, but these effects are not additive. Similar stimulation of binding of DNA substrates at lowered pH or by Ca2+ ions has been observed with other metallonucleases and been assigned to protonation or binding of Ca2+ ions at the active site carboxylates (28). Here the protonation state of DNA-binding residues could also be a factor (29, 30).
The variation in KD as a function of [Ca2+] was determined at both pH 9.3 and pH 7.5. Calcium ion concentration was varied from 0.5 to 10 mm, maintaining the ionic strength by adjusting the concentration of KCl. Under these conditions, the slopes of log KD versus log [Ca2+] were 0.15 ± 0.10 (pH 9.3) and −0.06 ± 0.06 (pH 7.5), indicating that the changes in enzyme-substrate affinity were negligible (Fig. 2D). The relative insensitivity of KD to concentrations of divalent ions <20 mm has been noted in other systems where divalent ions are cofactors (31). Importantly, at both pH values, the changes in enzyme-substrate affinity on altering Ca2+ ion concentration are relatively small and so do not account for the inhibition characteristics of the enzyme-catalyzed reaction by calcium ions described below.
Inhibition of WT T5FEN-catalyzed Reactions with Ca2+ Ions
Earlier studies have revealed that Ca2+ ions inhibit the T5FEN-catalyzed reaction, and under some conditions, this is dependent on 1/[Ca2+]2 (3). Assuming competitive inhibition with viable metal cofactor ions, a two-Ca2+ ion inhibitory response could be a diagnostic test for a requirement for two viable metal ion cofactors. Previously, it was demonstrated that the concentration of Ca2+ ions required to reduce the rate of reaction by 50%, KI in Equation 2, was a function of both the concentration and the identity of the viable metal cofactor (3). This analysis assumes that replacement of a single viable cofactor ion with Ca2+ ion prevents reaction, where kobs is the observed normalized initial rate of reaction (v/[E]) and k0 is v/[E] in the absence of inhibitory ion. Thus at least one Ca2+ ion displaces one viable cofactor ion to inhibit reaction. However, the origins of the most informative feature of the inhibition, a two-Ca2+ ion inhibitory form, required further investigation.
 |
Assuming two inhibitory binding sites and competitive inhibition, where calcium ions compete for the same sites as viable metal cofactors (M2+), potentially eight enzyme mono or -dimetal species can exist when Ca2+ and/or M2+ are present, and so speciation could be very complex. This is particularly so when the inhibitory ions are bound with higher affinity by the protein than M2+ and especially in the context of reactions where M2+ is not supplied at saturating concentrations. The simplest responses are predicted to arise when M2+ is bound with higher affinity than the inhibitory ion, a criteria fulfilled by the Mn2+/Ca2+ pair. Hence to reveal whether Mn2+-supported FEN-catalyzed reactions are inhibited by displacement of two viable metal cofactor ions by calcium ions, reactions were studied at three concentrations of viable cofactor ions (0.1, 0.2, and 0.4 mm) with saturating HP5F substrate and varying concentrations of Ca2+. The reactions were studied at pH 7.5, where the Mn2+-supported FEN reaction is optimal but where formation of manganese hydroxides is avoided. These three concentrations of Mn2+ span subsaturating (0.1 mm) and maximal rate concentrations (0.2 and 0.4 mm) of viable metal cofactor ion. As the concentrations of Mn2+ and Ca2+ were varied, the ionic strength of reaction mixtures was kept constant by adjusting the amount of potassium chloride present. To avoid the concentration of ions being perturbed by binding to DNA, the substrate was supplied so that the total concentration of phosphate diesters in HP5F was equal to or less than 5% of the concentration of Mn2+.
As observed previously (3), but shown here with a more extensive data set, at lower concentrations of Ca2+ (up to 80% inhibition, kobs/k0 = 0.2), the data were fitted acceptably to the simple competitive inhibition scheme (Equation 2). As the concentration of Mn2+ was increased, KI was also increased, indicating that inhibition is competitive so that KI = 1.1 ± 0.1 mm at 0.1 mm Mn2+, KI = 2.2 ± 0.3 mm at 0.2 mm Mn2+, and KI = 3.8 ± 0.4 mm at 0.4 mm Mn2+ (Fig. 3A). The magnitude of KI confirms that in analogy to dissociation constants determined for the substrate-free enzyme, Mn2+ ions must bind to the protein with higher affinity than Ca2+ ions.
FIGURE 3.

Inhibition of WT T5FEN-catalyzed reactions supported by Mn2+ and Mg2+ viable cofactors by the addition of calcium ions. All data points represent the average of three experiments, but error bars are omitted for clarity except for panel C. Data at 0.1 mm Mn2+ and 2 mm Mg2+ are from (3). A, inhibition of 0.1 mm (black circles), 0.2 mm (red squares), and 0.4 mm (blue diamonds) Mn2+-supported reactions by calcium ions. Experiments were carried out as described under “Experimental Procedures.” Data are fit to a simple single-site inhibition model (Equation 2) to yield KI = 1.1 ± 0.11, 2.2 ± 0.32, and 3.8 ± 0.41 mm, respectively. Although data fit well in the range kobs/k0 > 0.2, the deviation from this behavior at higher concentrations of Ca2+ ions is evident. B, the same data as in A fit to a more complex inhibition model where Ca2+ displaces either one or two Mn2+ ions (Equation 3) yielding KI1 = 5.3 mm (0.1 mm Mn2+) and 8.4 mm (0.2 mm Mn2+). In the case of data obtained at 0.4 mm Mn2+, where there is limited deviation from the single inhibition model (Equation 2), the value of KI is outside the data range. C, the dependence of log kobs/k0 (where kobs/k0 < 0.2) on log [Mn2+] at 20 mm Ca2+ (circles), 17.5 mm Ca2+ (squares), 15 mm Ca2+ (diamonds), 12.5 mm Ca2+ (crosses), and 10 mm Ca2+ (triangles). The slopes of the straight line fits are 1.8 ± 0.1 at 20 mm Ca2+, 1.8 ± 0.1 at 17.5 mm Ca2+, and 1.8 ± 0.1 at 15 mm Ca2+. Although there are only two available data points at 12.5–10 mm Ca2+, the same trend is observed in these data. The intercept in each case is a function of 1/[Ca2+]2. D, inhibition of 0.5 mm (black circles) and 2 mm (red squares) Mg2+-supported reactions by calcium ions. Experiments were carried out as described under “Experimental Procedures.” Data are fit to a simple single-site inhibition model (Equation 2) to yield KI = 0.1 ± 0.01 mm (0.5 mm Mg2+) and 0.2 ± 0.02 mm (2 mm Mg2+). Although data fit well in the range kobs/k0 > 0.2, the deviation from this behavior at higher concentrations of Ca2+ ions is evident. E, the same data as in D fit to a more complex inhibition model where Ca2+ displaces either one or two Mg2+ ions (Equation 3), yielding KI1 = 8.7 ± 0.1 mm (0.5 mm Mg2+) and 9.0 ± 1.3 mm (2 mm Mg2+).
In the case of the highest concentration of Mn2+, most of the data range fits acceptably to the simple inhibition model, although deviation is evident at the very highest concentrations of Ca2+. At lower concentrations of Mn2+, the data deviate from this simple inhibition scheme when inhibition is greater than 80% (kobs/k0 < 0.2). The slopes of log-log plots of normalized rates versus calcium ion concentration where kobs/k0 < 0.2 are −2.0 ± 0.1 (0.1 mm Mn2+) and −1.9 ± 0.1 (0.2 mm Mn2+) rather than the slope of −1 predicted by the simple inhibition scheme. This indicates that a two-Ca2+ form of the enzyme is also catalytically inactive. Although a complete data fit to the complex overall inhibition scenario requires a knowledge of all eight interrelated dissociation constants, it is possible to generate a relatively simple description that assumes two Ca2+ ions bind competitively and independently and that all forms of the enzyme that contain Ca2+ are inactive. Equation 3 models the expected variation of rate kobs/k0 with [Ca2+], defining two apparent KI values for Ca2+ ions, where KI1 is an apparent dissociation of dicalcium forms of the enzyme and KI2 is an apparent dissociation constant of monocalcium forms.
 |
Although good non-linear regression curve fits can be obtained using Equation 3, the similar magnitudes of the apparent inhibition constants result in the same value for each with large errors. Similar non-linear regression curve fitting problems have been noted previously (32). When KI2 is fixed according to the single-site inhibition model, resulting values of KI1 = 5.3 mm (0.1 mm Mn2+) and KI1 = 8.4 mm (0.2 mm Mn2+) are observed. At higher Mn2+, the data fit to a simple one-site inhibition model, implying that the value of KI1 is outside the data range (Fig. 3B). Hence the magnitude of this parameter is dependent on the concentration of viable metal cofactor ions present, implying that two Ca2+ ions compete for the same sites as the two Mn2+ ions.
Further evidence for a requirement for two catalytic metals can be derived by an alternative analysis of the data. Assuming that the only catalytically competent species has two viable metal ion cofactors bound, it follows that when the dominant species contains two inhibitory ions, the dependence of the normalized rate of reaction on viable cofactor metal ions will approach the square of its concentration. This will occur in situations where the viable metal cofactor ion has higher affinity than the inhibitory ion. Analyzing the data in the region kobs/k0 < 0.2, a plot of log kobs/k0 versus log [Mn2+] at fixed Ca2+ concentration does indeed display the expected slope close to 2 (Fig. 3C).
Similar analyses have also been conducted for Mg2+-supported T5FEN reactions, which also display a biphasic response to Ca2+ inhibition. These studies were carried out at pH 9.3, which is optimal for the Mg2+-supported reaction, and at saturating substrate. Two subsaturating concentrations of Mg2+ were studied (0.5 and 2 mm), a criteria imposed to keep total divalent ion concentration below a value that could potentially alter enzyme-substrate affinity. Analysis according to the one-site inhibition model (Equation 2) reveals KI = 0.11 ± 0.01 mm at 0.5 mm Mg2+ and KI = 0.22 ± 0.01 mm at 2 mm Mg2+ (Fig. 3D). The apparent KI values are smaller than the concentration of Mg2+, indicating that Mg2+ ions are bound more weakly than Ca2+. Applying the two-inhibitory ion model (Equation 3) yields apparent inhibition constants that are similar, KI1 = 8.7 ± 0.1 mm (0.5 mm Mg2+) and KI1 = 9.0 ± 1.3 mm (2 mm Mg2+), presumably reflecting the relative affinity of Ca2+ versus Mg2+ and the subsaturating conditions with respect to viable metal cofactor (Fig. 3E).
The Magnesium Ion Requirements of a Metal Site 2 Mutant (D201I/D204S)
Previous analyses, including the observation that D201I/D204S mutations do not drastically impair reaction, suggest that the T5FEN M2 may not play a direct role in catalysis (12, 24). In the designed D201I/D204S mutant, two of the carboxylates that bind M2 are altered (Fig. 1A) to abolish the binding site for this ion as observed in E. coli ExoIX. With HP5F as substrate, the D201I/D204S mutation slowed the maximal single turnover rate of the reaction by a factor of 40 at saturating Mg2+, a modest factor in the context of the entire T5FEN rate acceleration of 1016-1017 (33). To elucidate the role and minimal number of metal ions involved in the D201I/D204S mutant catalyzed reaction, kinetic parameters were determined as a function of magnesium ion concentration in an analogous fashion to experiments previously conducted on the WT enzyme using HP5F substrate (pH 9.3) and at constant ionic strength. Like WT T5FEN, the major site of reaction of D201I/D204S with HP5F is one nucleotide into the double-stranded region of the 5′-overhanging hairpin substrate, although slightly increased amounts of reaction elsewhere are observed with the mutant protein. Total concentrations of products were evaluated during all kinetic analyses (25).
The variations of the individual kinetic parameters of D201I/D204S mutant with magnesium ion concentration are shown in Fig. 4, A–C. The turnover number (kcat) is dependent upon increasing magnesium ion concentration until it reaches 10 mm Mg2+. Data could not be collected below 0.05 mm Mg2+ as the end points of the slowed reactions were <100% at these concentrations. A similar effect was previously noted for the WT enzyme, albeit at lower [Mg2+] (3).
FIGURE 4.

The variation of the catalytic parameters of D210I/D204S mutant T5FEN with concentration of magnesium ions and the inhibition of this reaction by calcium ions. Catalytic parameters and rates of reaction were determined at pH 9.3 with varying concentrations of Mg2+ ions, and when required, Ca2+ ions as described under “Experimental Procedures.” Experiments were carried out in triplicate, and in A–C, standard errors are shown. In D and E, standard errors are omitted for clarity. A, the variation in kcat as a function of magnesium ion concentration for D201I/D204S. Data have been fit to Equation 4 (black curve, KDMgES = 2.5 ± 0.3 mm and (kcat)max = 9.6 ± 0.3 min−1) and Equation 5 (red curve, KDMgES1 = 0.02 ± 0.004 mm, KDMgES2 = 2.7 ± 0.2 mm, and (kcat)max = 10.1 ± 0.5 min−1). B, the variation in 1/Km for D201I/D204S as a function of magnesium ion concentration was fitted to Equation 6 to yield KDMgE1 = 0.5 ± 0.2 mm and (1/Km)max = 0.03 ± 0.002 nm−1. C, the variation in kcat/Km for D201I/D204S as a function of magnesium ion concentration fit to Equation 7 with KDMgE1 = 0.5 mm to yield KDMgE2 = 3.3 ± 0.2 mm and (kcat/Km)max = 0.18 ± 0.01 min−1nm−1. D, inhibition of 0.5 mm (black circles) and 2 mm (red squares) Mg2+-supported reactions catalyzed by D210I/D204S by calcium ions. Experiments were carried out as described under “Experimental Procedures.” Data are fit to a simple single-site inhibition model (Equation 2) to yield KI = 1.1 ± 0.1 mm (0.5 mm Mg2+) and 2.2 ± 0.2 mm (2 mm Mg2+). Although data fit well in the range kobs/k0 > 0.2, the deviation from this behavior at higher concentrations of Ca2+ ions is evident. E, the same data as in D fit to a more complex inhibition model where Ca2+ displaces either one or two Mg2+ ions (Equation 3), yielding KI1 = 7.7 ± 0.9 mm (0.5 mm Mg2+) and 40 ± 18 mm (2 mm Mg2+).
A previous study of the kcat or kST response of WT T5FEN displayed a clear biphasic response to Mg2+ ions, where a second order dependence on Mg2+ at low concentrations of ions becomes first order at higher concentrations before approaching saturation (3). This situation can only be explained by the participation of two ions in chemical catalysis and was best fit to a model where reaction required the presence of two ions that were bound independently and with differing affinity (Equation 4).
 |
However, the kcat data for D201I/D204S fit acceptably to a single binding isotherm (Equation 5), where (kcat)obs is the observed turnover number at a given concentration of magnesium ions, KDMg2+ES is the magnesium ion dissociation constant from ES complex, and (kcat)max is the maximal turnover rate at infinite magnesium ions, giving KDMgES = 2.5 ± 0.3 mm. This value is similar to that obtained for the lower affinity magnesium ion associated with the kcat response of the WT protein (KD2MgWTES = 2.1 mm (3)).
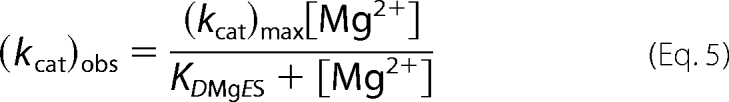 |
Applying a model where the reaction requires the presence of two ions that are bound independently, as used to fit WT data previously (Equation 4), places a higher affinity dissociation constant outside the data range, although this number can only be regarded as a potential upper limit (Fig. 4A).
Earlier studies of the WT protein revealed that both 1/Km and kcat/Km are Mg2+-dependent. Below 0.25 mm Mg2+, 1/Km has a [Mg2+]2 dependence (slope of 2 log-log plot). This required a two-ion model for the WT enzyme, where two ions were bound independently with similar affinity or cooperatively. The magnesium ion response of 1/Km for D201I/D204S differs from that of the WT protein and fits a single binding isotherm (Equation 6), yielding KDMgE = 0.5 ± 0.2 mm (Fig. 4B).
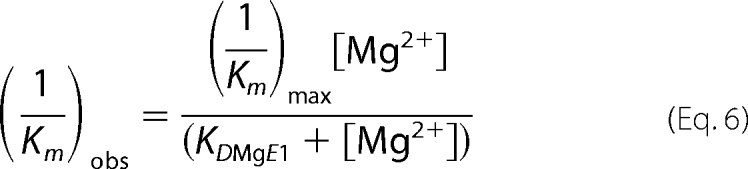 |
Below 0.25 mm, kcat/Km for the WT enzyme was dependent on [Mg2+]3, requiring a three-ion model. In contrast, a plot of log (kcat/Km) versus log [Mg2+] for D201I/D204S (Fig. 4C) reveals a slope of 1.8 ± 0.2, implying the overall involvement of two ions in the reaction catalyzed by the mutated protein, rather than the three-ion requirement of the WT enzyme. Fitting the data to Equation 7, assuming that the mutated FEN is only capable of reaction with two ions present that are bound independently, with KDMgE1 = 0.5 mm (as defined by the 1/Km data), yields KDMgE2 = 3.3 ± 0.2 mm.
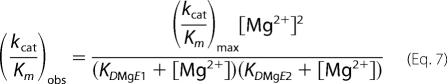 |
Thus combined magnesium stimulation data for D201I/D204S provide evidence that the overall reaction of the mutated protein requires at least two Mg2+ ions and differs from the WT protein, but the restricted data range makes it impossible to ascertain whether chemical catalysis (kcat) has a one- or two-ion requirement.
Substrate Binding and Calcium Ion Inhibition of D201I/D204S
Binding of HP5F and D201I/D204S was studied in an analogous fashion to the WT protein at pH 9.3 to ascertain whether divalent metal ions were required to bind substrate and whether binding of Ca2+ ions by DNA substrate could potentially be inhibitory. In contrast to the WT protein, in the absence of divalent metal ions, a dissociation constant could be determined (KD = 2300 ± 200 nm), presumably due to the substitution of two active site carboxylates with neutral residues (Fig. 2C). This was decreased by 2 orders of magnitude upon the addition of 0.5 mm Ca2+ ions (KD = 28 ± 3 nm). The effect of varying [Ca2+] on KD was modest; the slope of a log-log plot of KD versus [Ca2+] is 0.09 ± 0.04.
To determine the number of viable metal cofactor ions required by D201I/D204S, Ca2+ inhibition of the Mg2+-supported reactions was investigated in an identical manner to the WT protein. Applying the single-site inhibition model (Equation 2) yielded KI = 2.1 ± 0.2 mm (2 mm Mg2+) and KI = 1.1 ± 0.1 mm (0.5 mm Mg2+) (Fig. 4D). Importantly, data obtained at 0.5 mm Mg2+ clearly display 1/[Ca]2 dependence at Ca2+ ion concentrations above 6 mm, whereas increasing the concentration of Mg2+ leads to deviation from the single-site inhibition model only at the very highest [Ca2+] employed (>15 mm) (Fig. 4E). Respective values of KI1 = 7.7 ± 0.9 mm (0.5 mm Mg2+) and 40 ± 18 mm (2 mm Mg2+) were obtained by fitting the data to Equation 3. As the addition of Ca2+ ions at low concentration stimulates substrate binding and the stability of complexes is similar at inhibitory concentrations, the data imply that when a substantive part of the T5FEN M2-binding site is abolished, the FEN-catalyzed reaction still requires at least two catalytically viable ions.
DISCUSSION
The work described here supports the hypothesis that the major rate acceleration in the reactions catalyzed by flap endonucleases requires the presence of two metal ions. Stimulation of chemical catalysis (kcat or kST) by WT T5FEN has revealed previously that the reaction has a requirement for two magnesium ions that are bound independently with differing affinity (3). Confirmation of this requirement is demonstrated by the characteristics of inhibition of the WT reaction by catalytically inert Ca2+ ions, shown here to be competitive with viable metal cofactor ions. The response of the FEN reaction to added Ca2+ is biphasic and ultimately dependent on 1/[Ca]2, indicative of a requirement for two viable metal cofactors. The possibility that higher order inhibition is the result of altered T5FEN-substrate interactions as a result of added Ca2+ is ruled out. Moreover, stimulation of reactions under higher Ca2+ inhibitory conditions by Mn2+ is a second order process with respect to stimulatory metal cofactor ion. Thus the conclusion of more than one experimental approach is that the chemical process catalyzed by T5FEN involves at least two catalytic metal ions.
A designed metal site 2 mutant catalyzes phosphate diester hydrolysis with a decrease in the rate of reaction of only 40-fold, implying that the T5FEN M2 is not responsible for major rate accelerations. Previous experiments have indicated that the overall reaction of the WT enzyme (kcat/Km) requires at least three divalent ions. In contrast, magnesium ion stimulation data demonstrate that the overall reaction catalyzed by D201I/D204S requires at least two ions. However, within the experimental data range tested, chemical catalysis requirements for the mutated protein (kcat) can be assigned to a single-ion dependence. In theory, these data could indicate that a single metal ion is required for catalysis by the mutant. However, inhibition of the mutated protein by Ca2+ ions clearly demonstrates a requirement for two viable metal cofactor ions, in an analogous fashion to the WT protein. The combined data therefore imply that another ion (referred to here as M3), not present in substrate-free T5FEN structures, is bound by both the WT and the mutated enzymes. Together with M1, this provides the major rate enhancement of phosphate diester hydrolysis.
Although the data presented here implicate two ions in catalysis, they do not provide information on the location of the T5FEN M3. For a two-metal ion mechanism where both ions contact the scissile phosphate diester, M3 would have to be located within <4 Å of M1, where both could be bound by the central carboxylate of the T5FEN active site (Asp-130). As this is the arrangement of two Mg2+ ions shown in a substrate-free structure of hFEN (Fig. 1B) (18) and the position of two Sm3+ ions in an hFEN1 product complex (4), this positioning of ions for chemical catalysis appears highly plausible. Thus we suggest that the T5FEN M3, not visible in substrate-free structures of bacteriophage enzymes, is equivalent to the ion designated M2 in hFEN1 structures (Fig. 1, A and B).
However, despite the necessity of two ions for chemical catalysis, the WT T5FEN protein does have a three-metal ion requirement to maximize the rate of its overall (kcat/Km) reaction (3), which is reduced to two ions for D201I/D204S. In the absence of catalysis at pH 9.3, the dissociation constant of substrate HP5F is >5 μm, but the addition of Ca2+ ions substantially lowers this value. Enhancement of substrate affinity is also observed with the T5FEN M2 site mutant upon the addition of Ca2+, although overall dissociation constants in the presence and absence of divalent ions are lowered as the result of the neutralizing mutations. This, together with the 1/Km divalent metal dependences of both the WT and the D201I/D204S mutant proteins, implies a role for metal ions in FEN-substrate complex stability. Unlike the WT protein, which requires two ions to maximize 1/Km, data for the mutated FEN fit to a single binding isotherm. Therefore, the combined data suggest that the T5FEN M2 stabilizes protein-substrate interaction in WT T5FEN but is not an absolute requirement when neutralizing mutations are present in D201I/D204S.
Earlier consideration of functional and structural analyses led us to suggest that for FENs and related superfamily members to achieve a state where the scissile phosphate diester contacted metal ions involved in chemical catalysis, the terminus of the duplex where the reaction occurs would have to be unpaired (3). Structural analyses of mutant T4FEN bound to a pseudo-Y substrate in the absence of divalent ions (15) demonstrate that the scissile phosphate diester is located within a duplex bound in front of and parallel to the carboxylate-rich active site (Fig. 5, A and B). Modeling indicated that the unpairing of two nucleotides would be required to place the scissile phosphate in the vicinity of M1 (and presumably T5FEN M3) (Fig. 5C). Recent structural analyses of DNA complexes of hFEN1 and hEXO1 lend support to this unpairing mechanism (2, 4). When the 5′-phosphate monoester of products contacts active site metal ions, one nucleotide of the duplex is unpaired, implying that for the scissile bond to occupy the same site, two nucleotides must unpair.
FIGURE 5.

Proposed double nucleotide unpairing in T5FEN catalyzed reactions. A, model of T5FEN (1UT5) bound to a pseudo-Y DNA created by overlay with the structure of T4 RNase H (2IHN, T4FEN) bound to DNA (T4 protein not shown). Active site carboxylates are shown as sticks with red oxygens, and T5 metal ions (M2 foreground) are shown in gray. The duplex where the reaction occurs is bound in front of and parallel to the active site. The protein-DNA structure 2IHN was solved in the absence of metal ions; hence the substrate arches away and does not occupy the carboxylate-rich active site. The scissile phosphate diester bond positioned 7 Å from M1 is colored yellow, and the adjacent duplex phosphate −1 is colored lilac. The two 5′-terminal nucleobases of the duplex are depicted in blue. B, close up of the active site in this model with coloring as in A. C, proposed unpairing of the two terminal base pairs of the duplex to allow the scissile phosphate diester (yellow) to move toward M1. This relocation requires that the adjacent phosphate diester −1 moves farther toward the carboxylate active site and closer to M2. In other FENs and related superfamily members, the N terminus plays a similar role to M2 (2, 4).
To achieve contact between the scissile phosphate and the metal ions, the unpaired DNA has to traverse the active site. It is interesting to note that the phosphate diester adjacent to the scissile bond (−1) would also have to move farther toward the carboxylate-rich region, closer to the T5FEN M2, which would presumably act to stabilize this unpaired state. We suggest that in accord with the functional analyses of WT T5FEN, two metal ions are required to position the substrate in the reactive conformation: M1, which later plays a direct role in chemical catalysis, and “non-catalytic” M2. It is tempting to speculate that an M2 that plays a substrate-positioning role can be replaced with Ca2+, explaining why WT T5FEN has an overall three-metal ion requirement but is inhibited by replacement of only two viable metal cofactor ions.
In unpaired hFEN1 and hEXO1 DNA complexes, the −1 phosphate interacts with the protein N terminus (2, 4), which occupies a similar position to the T5FEN M2 (Fig. 1, A and B). The N-terminal region of the protein is disordered in bacterial and bacteriophage FEN structures and is thought to be a site of protein-protein interaction (17, 21, 22). In T5FEN, the first 19 residues can be removed while retaining full enzymatic activity, and so the N terminus is unlikely to play a substrate-positioning role (12, 23). Thus apparent discrepancies between metal ion-binding sites in FEN structures could be explained by a functional equivalence of a divalent metal ion and the N terminus.
The designed neutralization of Asp-201/Asp-204 to Ile-201/Ser-204, based on the sequence of another 5′-nuclease family member (E. coli ExoIX (6)), presumably facilitates passage of the substrate across the same region of the protein without a requirement for a M2, yet still with the participation of M3. Individual mutations of the equivalent carboxylates (Asp-185 and Asp-188) in the FEN domain of E. coli DNA polymerase to alanine, which could be viewed as more conservative than the D201I/D204S double mutation, were both reported to have a much larger deleterious effect on enzyme activity than the designed double mutation introduced here (34). This is presumably in part because a greater active site charge remains in the individual Ala mutants, but stabilizing substrate interactions with the side chains of Ile and Ser are also a possibility in D201I/D204S. A mutated T5FEN where the same two carboxylates are changed to arginine (D201R/D204R) retains activity in line with a non-catalytic substrate-positioning/stabilizing role for the T5FEN M2, perhaps compensating for the lack of a bound metal ion with the cationic arginine (12). The decrease in the reaction rate observed with our designed M2 mutant may reflect a change in the partitioning of the substrate structure between a bound duplex form and an unpaired, active site occupying form. Such a situation would be consistent with the properties of exonucleolytic reactions catalyzed by the mutated protein (24). Although not a feature of the T5FEN-catalyzed reactions of 5′-flap substrates,6 the exonucleolytic reactions of T5FEN are diffusion-controlled, indicating that forward reaction steps are faster than substrate dissociation. Despite more stable enzyme-DNA complexes with D201I/D204S, both in the presence and in the absence of divalent ions, reactions are no longer diffusion-controlled, implying a slowed later step in the reaction sequence. Furthermore this rate-limiting later step is insensitive to the nature of the leaving group, consistent with a physical rather than chemical process, such as substrate unpairing.
In contrast to the ExoIX subgroup, the carboxylate-rich active site is conserved in the majority of the DNA 5′-nuclease superfamily (1). These include enzymes that catalyze structure-specific reactions of an apparently disparate group of nucleic acid substrates including DNA bubbles (xeroderma pigmentosum complementation group G (XPG)) (35), four-way junctions (GEN-1 (xeroderma pigmentosum complementation group G-like gap endonuclease, a putative human Holliday junction resolvase)) (36), as well as flapped, nicked, and 3′-overhang DNAs (FEN and EXO1) (2, 20, 37, 38). Because all of these enzymes carry out the same chemical transformation, like T5FEN, they too will require two ions to effect phosphate diester hydrolysis. Notably, the reaction specificities of the superfamily do have a common feature; the major site of reaction occurs one nucleotide into a double-stranded region of the substrate, located at a junction. It seems highly likely that the unpairing mechanism is universally conserved in the seven/eight-carboxylate enzymes to give rise to this reaction specificity. If this is the case, FEN superfamily members may also require either a further active site metal or a functionality of the protein such as the N terminus for substrate unpairing in addition to two catalytic ions. Analyses of the nuclear RNA degradative 5′-exoribonuclease Rat1 (nuclear 5′-3′-exoribonuclease; also known as Xrn2) (41) demonstrate similar secondary structure to FENs (5). In addition, we also note that two recent structures of the corresponding cytosolic enzyme Xrn1 (cytosolic 5′-3′-exoribonuclease) (39, 40) demonstrate that these enzymes conserve the DNA 5′-nuclease superfamily active site and that RNA 5′-nucleases may in turn may also utilize a similar catalytic mechanism.
Acknowledgments
We thank L. David Finger, Nikesh Patel (Sheffield), and Susan Tsutakawa (Lawrence Berkeley National Laboratory) for insightful discussions and Elaine Frary for technical assistance.
This work was supported by Biotechnology and Biological Sciences Research Council Grant F0147321. This work was also supported by National Institutes of Health Grant CA081967 (to J. A. T.).
In early literature, T5FEN was referred to as T5 5′-3′-exonuclease and T5 5′-nuclease. In early literature, T4FEN was referred to as referred to also as T4 RNase H.
B. Sengerová and J. A. Grasby, unpublished observations.
- FEN
- flap endonuclease
- T5FEN
- bacteriophage T5 flap endonuclease
- T4FEN
- T4 flap endonuclease
- hFEN1
- human flap endonuclease
- ExoIX
- E. coli exonuclease IX
- EXO1
- exonuclease 1
- hEXO1
- human EXO1
- FAM
- 6-carboxyfluorescein
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- MOPS
- 3-(N-morpholino)propane-sulfonic acid.
REFERENCES
- 1. Tomlinson C. G., Atack J. M., Chapados B., Tainer J. A., Grasby J. A. (2010) Biochem. Soc. Trans. 38, 433–437 [DOI] [PubMed] [Google Scholar]
- 2. Orans J., McSweeney E. A., Iyer R. R., Hast M. A., Hellinga H. W., Modrich P., Beese L. S. (2011) Cell 145, 212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Syson K., Tomlinson C., Chapados B. R., Sayers J. R., Tainer J. A., Williams N. H., Grasby J. A. (2008) J. Biol. Chem. 283, 28741–28746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsutakawa S. E., Classen S., Chapados B. R., Arvai A. S., Finger L. D., Guenther G., Tomlinson C. G., Thompson P., Sarker A. H., Shen B., Cooper P. K., Grasby J. A., Tainer J. A. (2011) Cell 145, 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang W. (2011) Q. Rev. Biophys. 44, 1–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Allen L. M., Hodskinson M. R., Sayers J. R. (2009) Biochem. J. 418, 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sigel R. K., Pyle A. M. (2007) Chem. Rev. 107, 97–113 [DOI] [PubMed] [Google Scholar]
- 8. Yang W., Lee J. Y., Nowotny M. (2006) Mol. Cell 22, 5–13 [DOI] [PubMed] [Google Scholar]
- 9. Pingoud A., Fuxreiter M., Pingoud V., Wende W. (2005) Cell. Mol. Life Sci. 62, 685–707 [DOI] [PubMed] [Google Scholar]
- 10. Dupureur C. M. (2010) Metallomics 2, 609–620 [DOI] [PubMed] [Google Scholar]
- 11. Zheng L., Li M., Shan J., Krishnamoorthi R., Shen B. (2002) Biochemistry 41, 10323–10331 [DOI] [PubMed] [Google Scholar]
- 12. Feng M., Patel D., Dervan J. J., Ceska T., Suck D., Haq I., Sayers J. R. (2004) Nat. Struct. Mol. Biol. 11, 450–456 [DOI] [PubMed] [Google Scholar]
- 13. Hosfield D. J., Mol C. D., Shen B., Tainer J. A. (1998) Cell 95, 135–146 [DOI] [PubMed] [Google Scholar]
- 14. Mueser T. C., Nossal N. G., Hyde C. C. (1996) Cell 85, 1101–1112 [DOI] [PubMed] [Google Scholar]
- 15. Devos J. M., Tomanicek S. J., Jones C. E., Nossal N. G., Mueser T. C. (2007) J. Biol. Chem. 282, 31713–31724 [DOI] [PubMed] [Google Scholar]
- 16. Tock M. R., Frary E., Sayers J. R., Grasby J. A. (2003) EMBO J. 22, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ceska T. A., Sayers J. R., Stier G., Suck D. (1996) Nature 382, 90–93 [DOI] [PubMed] [Google Scholar]
- 18. Sakurai S., Kitano K., Yamaguchi H., Hamada K., Okada K., Fukuda K., Uchida M., Ohtsuka E., Morioka H., Hakoshima T. (2005) EMBO J. 24, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hwang K. Y., Baek K., Kim H. Y., Cho Y. (1998) Nature Struct. Biol. 5, 707–713 [DOI] [PubMed] [Google Scholar]
- 20. Chapados B. R., Hosfield D. J., Han S., Qiu J., Yelent B., Shen B., Tainer J. A. (2004) Cell 116, 39–50 [DOI] [PubMed] [Google Scholar]
- 21. Bhagwat M., Meara D., Nossal N. G. (1997) J. Biol. Chem. 272, 28531–28538 [DOI] [PubMed] [Google Scholar]
- 22. Kim Y., Eom S. H., Wang J., Lee D. S., Suh S. W., Steitz T. A. (1995) Nature 376, 612–616 [DOI] [PubMed] [Google Scholar]
- 23. Gangisetty O., Jones C. E., Bhagwat M., Nossal N. G. (2005) J. Biol. Chem. 280, 12876–12887 [DOI] [PubMed] [Google Scholar]
- 24. Sengerová B., Tomlinson C., Atack J. M., Williams R., Sayers J. R., Williams N. H., Grasby J. A. (2010) Biochemistry 49, 8085–8093 [DOI] [PubMed] [Google Scholar]
- 25. Williams R., Sengerová B., Osborne S., Syson K., Ault S., Kilgour A., Chapados B. R., Tainer J. A., Sayers J. R., Grasby J. A. (2007) J. Mol. Biol. 371, 34–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patel D., Tock M. R., Frary E., Feng M., Pickering T. J., Grasby J. A., Sayers J. R. (2002) J. Mol. Biol. 320, 1025–1035 [DOI] [PubMed] [Google Scholar]
- 27. Reid S. L., Parry D., Liu H. H., Connolly B. A. (2001) Biochemistry 40, 2484–2494 [DOI] [PubMed] [Google Scholar]
- 28. Lagunavicius A., Grazulis S., Balciunaite E., Vainius D., Siksnys V. (1997) Biochemistry 36, 11093–11099 [DOI] [PubMed] [Google Scholar]
- 29. Dervan J. J., Feng M., Patel D., Grasby J. A., Artymiuk P. J., Ceska T. A., Sayers J. R. (2002) Proc. Natl. Acad. Sci. 99, 8542–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pickering T. J., Garforth S., Sayers J. R., Grasby J. A. (1999) J. Biol. Chem. 274, 17711–17717 [DOI] [PubMed] [Google Scholar]
- 31. Datta K., LiCata V. J. (2003) J. Biol. Chem. 278, 5694–5701 [DOI] [PubMed] [Google Scholar]
- 32. Bastock J. A., Webb M., Grasby J. A. (2007) J. Mol. Biol. 368, 421–433 [DOI] [PubMed] [Google Scholar]
- 33. Pickering T. J., Garforth S. J., Thorpe S. J., Sayers J. R., Grasby J. A. (1999) Nucleic Acids Res. 27, 730–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu Y., Derbyshire V., Ng K., Sun X. C., Grindley N. D., Joyce C. M. (1997) J. Mol. Biol. 268, 284–302 [DOI] [PubMed] [Google Scholar]
- 35. Hohl M., Thorel F., Clarkson S. G., Schärer O. D. (2003) J. Biol. Chem. 278, 19500–19508 [DOI] [PubMed] [Google Scholar]
- 36. Rass U., Compton S. A., Matos J., Singleton M. R., Ip S. C., Blanco M. G., Griffith J. D., West S. C. (2010) Genes Dev. 24, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Finger L. D., Blanchard M. S., Theimer C. A., Sengerová B., Singh P., Chavez V., Liu F., Grasby J. A., Shen B. (2009) J. Biol. Chem. 284, 22184–22194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee B. I., Wilson D. M., 3rd (1999) J. Biol. Chem. 274, 37763–37769 [DOI] [PubMed] [Google Scholar]
- 39. Chang J. H., Xiang S., Xiang K., Manley J. L., Tong L. (2011) Nat. Struct. Mol. Biol. 18, 270–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jinek M., Coyle S. M., Doudna J. A. (2011) Mol. Cell 41, 600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiang S., Cooper-Morgan A., Jiao X., Kiledjian M., Manley J. L., Tong L. (2009) Nature 458, 784–788 [DOI] [PMC free article] [PubMed] [Google Scholar]