

Abstract
Chk2/hcds1, the human homolog of the Saccharomyces cerevisiae RAD53/SPK1 and Schizosaccharomyces pombe cds1 DNA damage checkpoint genes, encodes a protein kinase that is post-translationally modified after DNA damage. Like its yeast homologs, the Chk2/hCds1 protein phosphorylates Cdc25C in vitro, suggesting that it arrests cells in G2 in response to DNA damage. We expressed Chk2/hCds1 in human cells and analyzed their cell cycle profile. Wild-type, but not catalytically inactive, Chk2/hCds1 led to G1 arrest after DNA damage. The arrest was inhibited by cotransfection of a dominant-negative p53 mutant, indicating that Chk2/hCds1 acted upstream of p53. In vitro, Chk2/hCds1 phosphorylated p53 on Ser-20 and dissociated preformed complexes of p53 with Mdm2, a protein that targets p53 for degradation. In vivo, ectopic expression of wild-type Chk2/hCds1 led to increased p53 stabilization after DNA damage, whereas expression of a dominant-negative Chk2/hCds1 mutant abrogated both phosphorylation of p53 on Ser-20 and p53 stabilization. Thus, in response to DNA damage, Chk2/hCds1 stabilizes the p53 tumor suppressor protein leading to cell cycle arrest in G1.
Keywords: Chk2, hCds1, p53, ATM, DNA damage, checkpoints
In eukaryotic cells, DNA damage and stalled replication activate an evolutionarily conserved checkpoint (Elledge 1996; Weinert 1998). In Saccharomyces cerevisiae, the genes that comprise this checkpoint fall in three groups, those required only for the response to DNA damage, those required only for the response to stalled replication, and those required for both responses (Navas et al. 1996). Three genes that belong to the last group, MEC1, RAD53/SPK1, and CHK1, encode protein kinases, which become activated in response to DNA damage or stalled replication. The MEC1 kinase phosphorylates and activates the RAD53/SPK1 and CHK1 kinases, which then phosphorylate proteins that regulate progression through the cell cycle (Sanchez et al. 1996; Sun et al. 1996). Known targets of RAD53/SPK1 include SWI6, a transcriptional activator of G1 cyclins; DBF4, a regulatory kinase subunit involved in the firing of replication origins; and CDC5, a Polo kinase important for anaphase entry (Sidorova and Breeden 1997; Weinreich and Stillman 1999; Sanchez et al. 1999). CHK1 induces arrest in mitosis, but acts through a distinct mechanism other than RAD53/SPK1, as it targets the anaphase inhibitor PDS1 (Sanchez et al. 1999). CHK1 further differs from RAD53/SPK1 in that it responds to DNA damage but not to replication blocks (Cohen-Fix and Koshland 1997; Sanchez et al. 1999).
Homologs of MEC1, RAD53/SPK1, and CHK1, are present in S. pombe and higher eukaryotes. In S. pombe, the homolog of MEC1 is Rad3 (Bentley et al. 1996), whereas the functions of RAD53/SPK1 and CHK1 are mediated by Cds1 and Chk1, respectively. Cds1 is activated by both DNA damage and stalled replication, but only during S phase; Chk1 is activated in late S phase and G2 and responds primarily to DNA damage (Walworth and Bernards 1996; Lindsay et al. 1998; Brondello et al. 1999). Both Cds1 and Chk1 can phosphorylate and thereby inactivate Cdc25, a phosphatase needed for Cdc2 activation and entry into mitosis (Furnari et al. 1997; Zeng et al. 1998).
In humans, there are two known functional homologs of MEC1, ATM and ATR. ATM responds to DNA double stranded-breaks (DSBs) and its inactivation in patients with ataxia-telangiectasia (A-T) leads to checkpoint defects in G1, S, and G2 (Savitsky et al. 1995; Halazonetis and Shiloh 1999). ATR, an ATM-related kinase, may mediate the response to stalled replication or types of DNA damage other than DSBs (Bentley et al. 1996; Cimprich et al. 1996). The human homologs of S. pombe Cds1 and Chk1 are Chk2/hCds1 and hChk1, respectively. Chk2/hCds1 is phosphorylated in an ATM-dependent manner in response to ionizing radiation (IR), which induces DSBs, and in an ATM-independent manner in response to UV light and stalled replication (Matsuoka et al. 1998; Blasina et al. 1999; Brown et al. 1999). hChk1 is less well studied; the available evidence suggests that its response to DNA damage is ATM dependent (Flaggs et al. 1997).
The Chk2/hCds1 and hChk1 proteins have not yet been shown to induce cell cycle arrest when activated in human cells in response to DNA damage or stalled replication and their physiological substrates remain ill defined. The Cdc25C phosphatase is a potential in vivo substrate, as it is an in vitro substrate (Sanchez et al. 1997; Matsuoka et al. 1998; Blasina et al. 1999; Brown et al. 1999). Phosphorylation of Cdc25C in vivo could lead to arrest in G2, similar to the proposed mechanism for cell cycle arrest in S. pombe (Furnari et al. 1997; Zeng et al. 1998). Other targets of Chk2/hCds1 and hChk1 could regulate progression through the G1 and S phases of the cell cycle.
Arrest in the G1 phase of the cell cycle in mammalian cells exposed to DNA damage is mediated by the p53 tumor suppressor protein, a transcription factor for genes that induce cell cycle arrest or apoptosis (Kastan et al. 1991; Kuerbitz et al. 1992; Clarke et al. 1993; Lowe et al. 1993; Levine 1997). DNA damage leads to p53 stabilization and functional activation (Maltzman and Czyzyk 1984; Kastan et al. 1991; Fritsche et al. 1993; Lutzker and Levine 1996; Chernov and Stark 1997). Stabilization of p53 in cells exposed to IR or UV light is due to dissociation from Mdm2 (Shieh et al. 1997), a protein that targets p53 for degradation through the ubiquitin pathway (Haupt et al. 1997; Kubbutat et al. 1997; Midgley and Lane 1997). Functional activation is mediated by modifications that target the carboxy-terminal 30 residues of p53, a region that regulates the affinity of p53 for sequence-specific DNA (Kapoor and Lozano 1998; Lu et al. 1998; Sakaguchi et al. 1998; Waterman et al. 1998; Liu et al. 1999).
Stabilization of p53 in response to IR and UV light is dependent on the ATM and ATR kinases, respectively (Kastan et al. 1992; Khanna and Lavin 1993; Canman et al. 1994; Savitsky et al. 1995; Tibbetts et al. 1999). ATM and ATR directly phosphorylate p53 in vivo on Ser-15 and Ser-37 (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998; Tibbetts et al. 1999). Because p53 stabilization is dependent on phosphorylation of p53 on Ser-20 (Chehab et al. 1999), ATM and ATR must stabilize p53 by activating a kinase or kinases that phosphorylate(s) p53 on Ser-20. Chk2/hCds1 and hChk1 are obvious candidates, because they are downstream of ATM. In what follows, we examine whether Chk2/hCds1 has a role in p53 stabilization and induction of G1 arrest in response to DNA damage.
Results
As a first step to determine whether Chk2/hCds1 is involved in p53 stabilization, we examined whether it can phosphorylate p53 on Ser-20 in vitro and whether such phosphorylation disrupts the interaction of p53 with Mdm2. Purified recombinant His-tagged Chk2/hCds1 and full-length p53 proteins were incubated in the presence of [32P]ATP. Autoradiography of the reaction products indicated that p53 became phosphorylated, as did a proteolytic fragment of His-tagged Chk2/hCds1 and full-length His-tagged Chk2/hCds1 (Fig. 1A). A p53 mutant with substitution of Ser-20 with Ala (p53A20) was also phosphorylated by recombinant Chk2/hCds1 (data not shown), suggesting that either Chk2/hCds1 does not phosphorylate p53 on Ser-20 or that it phosphorylates multiple residues, including Ser-20. In favor of the latter possibility, full-length p53 incubated with Chk2/hCds1 reacted with AbS20p, an antibody specific for p53 phosphorylated on Ser-20 (Chehab et al. 1999); whereas p53A20, which serves as a negative control, did not react (Fig. 1B).
Figure 1.

Chk2/hCds1 phosphorylates p53 on Ser-20 in vitro and dissociates preformed p53/Mdm2 complexes. (A) Recombinant Chk2/hCds1 was incubated with [32P]ATP in the presence or absence of recombinant full-length p53. The reaction products were resolved by denaturing gel electrophoresis and visualized by autoradiography. (B) Full-length wild-type (wt) p53 or p53 with substitution of Ser-20 with Ala (A20) were incubated with Chk2/hCds1. Phosphorylation of p53 on Ser-20 (S20p) was monitored by immunoprecipitation of the reaction products with AbS20p, an antibody specific for p53 phosphorylated on Ser-20, followed by immunoblotting with antibody DO7, which recognizes p53 irrespective of its phosphorylation state. p53 protein levels were monitored by immunoblotting with DO7. (C) p53 phosphorylated by Chk2/hCds1 does not interact with Mdm2. Full-length p53 was incubated with Chk2/hCds1. One-half of the sample was stored on ice (input sample) and the other half was incubated with beads coated with GST–Mdm2(1–125). After washing the beads to remove unbound p53, the p53 protein bound to Mdm2 was eluted (Mdm2-bound sample). The fraction of p53 phosphorylated on Ser-20 in the input and Mdm2-bound samples was determined by comparing the amounts of p53 immunoprecipitated with antibodies AbS20p (p53 phosphorylated on Ser-20; S20p) and DO7 (p53 phosphorylated and nonphosphorylated on Ser-20; S20+S20p). (D) Chk2/hCds1 disrupts preformed p53/Mdm2 complexes. Complexes of GST–Mdm2 with wild-type p53 or p53A20 bound to glutathione beads were incubated with Chk2/hCds1. p53 that remained bound to Mdm2 after incubation of the beads with Chk2/hCds1 was resolved by denaturing gel electrophoresis and detected by immunoblotting with antibody DO7.
The interaction of p53 with Mdm2 is weakened when p53 is phosphorylated on Ser-20 (Chehab et al. 1999; Unger et al. 1999). To examine whether Chk2/hCds1 can regulate the binding of p53 to Mdm2 in vitro-purified recombinant full-length p53 was incubated with Chk2/hCds1 and then allowed to bind to glutathione beads coated with GST–Mdm2(1–125), a fusion protein consisting of GST and the amino-terminal 125 residues of Mdm2, which contain the entire domain of Mdm2 that interacts with p53 (Kussie et al. 1996). On the basis of its reactivity with AbS20p, about half of the total amount of p53 incubated with Chk2/hCds1 was phosphorylated on Ser-20; this fraction of p53, however, did not bind to the Mdm2-coated beads (Fig. 1C). In another experiment, purified full-length wild-type p53 or p53A20 was incubated with glutathione beads coated with GST–Mdm2(1–125). Excess p53 was washed off and the beads were incubated with Chk2/hCds1. The beads were washed again, and the fraction of p53 that still remained bound to Mdm2 was detected by immunoblotting. Chk2/hCds1 disrupted the wild-type p53/Mdm2 complex but did not affect the p53A20/Mdm2 complex (Fig. 1D). Thus, in vitro Chk2/hCds1 dissociated preformed p53/Mdm2 complexes, and this effect required phosphorylation of p53 on Ser-20.
The ability of Chk2/hCds1 to phosphorylate p53 on Ser-20 and dissociate p53 from Mdm2 was pursued in vivo. First, we examined whether Chk2/hCds1 activation and p53 stabilization exhibit similar kinetics in response to DNA damage. U2–OS osteosarcoma cells, which have wild-type endogenous p53, were stably transfected with a plasmid expressing Flag-tagged Chk2/hCds1. The cells were exposed to either IR or UV light and extracts were prepared at different time points after irradiation. Stabilization of endogenous p53 was monitored by immunoblotting; activation of Flag-tagged Chk2/hCds1 was monitored by changes in electrophoretic migration (Matsuoka et al. 1998; Blasina et al. 1999; Brown et al. 1999). Consistent with previous reports (Khanna and Lavin 1993; Lu and Lane 1993), IR induced an early and transient increase in p53 levels, whereas UV light induced a delayed and sustained increase. Activation of Chk2/hCds1 both in response to IR and UV light exhibited similar kinetics to p53 stabilization, supporting the hypothesis that Chk2/hCds1 activation leads to p53 stabilization (Fig. 2).
Figure 2.

Time course of p53 stabilization and Chk2/hCds1 activation in U2–OS cells exposed to IR or UV light. Extracts from cells exposed to DNA damage were resolved by denaturing gel electrophoresis and immunoblotted for p53 and Flag-tagged Chk2/hCds1. Chk2/hCds1 activated in response to DNA damage exhibits a mobility shift on denaturing gels.
A second prediction of the hypothesis that Chk2/hCds1 stabilizes p53 in vivo is that Chk2/hCds1 activation and p53 stabilization exhibit similar ATM dependence in response to DNA damage. Both Chk2/hCds1 activation and p53 stabilization are ATM dependent in response to IR, and ATM independent in response to UV light (Kastan et al. 1992; Khanna and Lavin 1993; Canman et al. 1994; Matsuoka et al. 1998; Blasina et al. 1999; Brown et al. 1999). To extend these observations, we examined the ATM dependence of p53 phosphorylation on Ser-20. Human nontransformed diploid fibroblasts from a normal individual and from two A-T patients, who have inactive ATM, were either untreated or exposed to IR or UV light. Extracts from these cells were prepared 2 and 16 hr after exposure to IR and UV light, respectively, and phosphorylation of p53 on Ser-20 was analyzed with antibody AbS20p. p53 from the normal fibroblasts became phosphorylated on Ser-20 after exposure to both IR and UV light; however, p53 from the A-T fibroblasts became phosphorylated on Ser-20 only after exposure to UV light (Fig. 3). Thus, phosphorylation of p53 on Ser-20 in response to IR and UV light matches the ATM dependence of Chk2/hCds1 activation.
Figure 3.

ATM dependence of p53 phosphorylation on Ser-20 in response to IR, but not UV light. Primary fibroblasts from a normal individual (AG1522) or from A-T patients (AT1BR and AT5BI) were exposed to IR or UV light. Phosphorylation of p53 on Ser-20 (S20p) was monitored with the phosphate-specific antibody AbS20p. p53* indicates that to facilitate analysis of the p53 phosphorylation state, the amounts of extract used in each lane of the top and bottom panels were adjusted so that all lanes had equal levels of total p53 (bottom; immunoblotting with antibody DO7).
To further link Chk2/hCds1 and p53, we examined whether stabilization of p53 in response to DNA damage would be inhibited in cells transiently transfected with a dominant-negative Chk2/hCds1 mutant. For this experiment, a previously described kinase-inactive protein bearing an Asp347-to-Ala substitution (Chk2A347) served as a dominant-negative mutant (Matsuoka et al. 1998). In addition, we did not monitor the levels of endogenous p53, because p53 stabilization would be inhibited only in the subpopulation of transfected cells that actually expressed Chk2A347. Instead, the plasmid expressing Chk2A347 was cotransfected with a smaller amount of plasmid expressing an HA-tagged p53 protein to ensure that all cells that express HA-tagged p53 would also express Chk2A347. A third plasmid directing expression of green fluorescent protein (GFP) was cotransfected to monitor transfection efficiency. Stabilization of HA-tagged p53 was inhibited in cells expressing Chk2A347, but not in cells transfected with the empty vector, or in cells expressing Chk2Δ2-83 (Fig. 4A). Chk2Δ2-83 was used as a control; it should not act as a dominant-negative mutant, because it has a wild-type kinase domain, and it also lacks wild-type function, because its amino terminus is deleted (see below).
Figure 4.
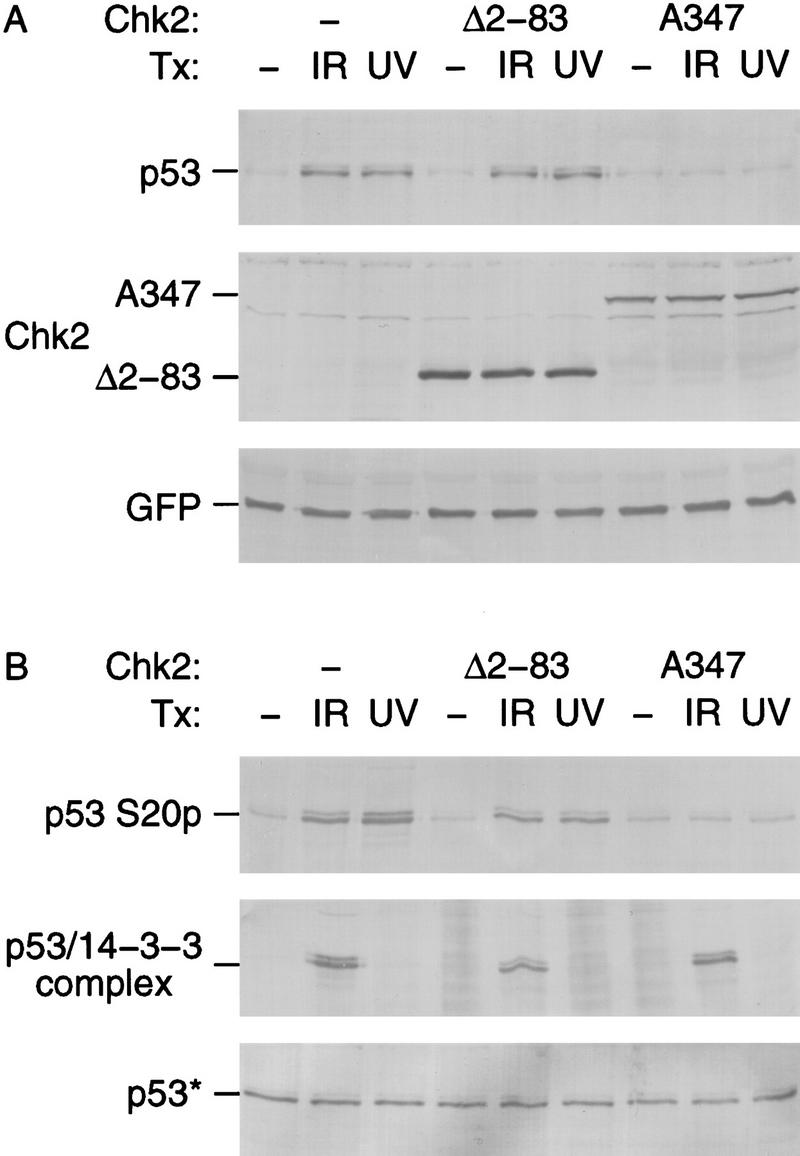
Dominant-negative Chk2/hCds1 mutants inhibit DNA damage-induced p53 stabilization (A) and Ser-20 phosphorylation (B). U2–OS cells were transiently transfected with plasmids expressing HA-tagged p53, the indicated Flag-tagged Chk2/hCds1 protein and GFP, as a marker of transfection efficiency. The cells were exposed to IR or UV light and cell extracts were prepared 2 and 16 hr later, respectively. (A) HA-tagged p53, Flag-tagged Chk2/hCds1, and GFP were resolved by denaturing gel electrophoresis and detected by immunoblotting. Equal levels of total protein were loaded in each lane to monitor the increase in p53 protein levels. (B) Phosphorylation of HA-tagged p53 on Ser-20 was monitored by immunoprecipitation with AbS20p, followed by immunoblotting with an antibody that recognizes the HA tag. The interaction of HA-tagged p53 with 14-3-3 proteins was monitored by immunoprecipitation with an antibody that recognizes 14-3-3 and detection of the coprecipitated HA-tagged p53 by immunoblotting. p53* indicates that to facilitate analysis of the p53 phosphorylation state and interaction with 14-3-3, the amounts of extract used in each lane of the top, middle, and bottom panels were adjusted so that all lanes had equal levels of total p53 (bottom; immunoblotting with an antibody that recognizes HA-tagged p53).
DNA damage induces multiple p53 post-translational modifications. These include phosphorylation of several amino-terminal serine residues, including Ser-20; an ATM-dependent dephosphorylation of phosphoserine 376, which creates a 14-3-3 binding site and leads to the association of p53 with 14-3-3 proteins; and other carboxy-terminal modifications (Knippschild et al. 1997; Shieh et al. 1997, 1999; Siliciano et al. 1997; Blades and Hupp 1998; Kapoor and Lozano 1998; Lu et al. 1998; Sakaguchi et al. 1998; Waterman et al. 1998; Liu et al. 1999). Chk2A347 inhibited the DNA damage-induced phosphorylation of p53 on Ser-20, but not the interaction of p53 with 14-3-3 proteins (Fig. 4B). These results support the model that Chk2/hCds1 regulates p53 protein levels through Ser-20 phosphorylation and also suggest that Chk2/hCds1 is involved only in a subset of the DNA damage signaling pathways that target p53.
Subsequently, we examined whether ectopic expression of wild-type Chk2/hCds1 would enhance p53 stabilization. U2–OS cells were selected for these experiments. Like many tumor cell lines with endogenous wild-type p53, these cells exhibit p53 stabilization in response to DNA damage, but its magnitude is modest and insufficient to lead to cell cycle arrest in G1 (Li et al. 1995; Nagasawa et al. 1998; Syljuasen et al. 1999; see below). Thus, there is potential for increased p53 stabilization and G1 arrest in response to Chk2/hCds1 expression. The cells were transiently transfected with a plasmid expressing wild-type Chk2/hCds1 or, as control, with a vector expressing Neo. A second plasmid directing expression of GFP was cotransfected to monitor transfection efficiency. The transfected cells were exposed to IR or were mock irradiated, and extracts were prepared 2 hr later. In nonirradiated cells, the levels of endogenous p53 were not affected by ectopic expression of Chk2/hCds1; however, in irradiated cells, p53 levels were higher in the cells transfected with the Chk2/hCds1 plasmid (Fig. 5A). The increase was modest, probably because only a small fraction of the transfected cells actually expressed Chk2/hCds1. We therefore repeated the experiment by monitoring the levels of p53 by immunofluorescence, which allows individual cells to be analyzed. After exposure to IR, cells transfected with the Neo vector showed a low uniform level of endogenous p53 expression. In contrast, within the cell population transfected with the Chk2/hCds1 plasmid, selected cells exhibited p53 staining almost as intense as cells transfected with a p53 vector (Fig. 5B). These cells also expressed GFP, marking them as cells that expressed proteins from the transfected plasmids (data not shown). Taken together, the immunoblotting and immunofluorescence experiments indicate that Chk2/hCds1 promotes stabilization of endogenous p53 in response to DNA damage.
Figure 5.
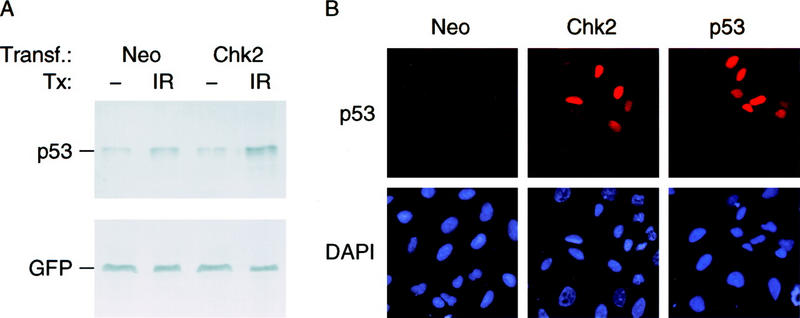
Ectopic expression of Chk2/hCds1 leads to p53 stabilization. (A) U2–OS cells transiently transfected with plasmids expressing Neo or Chk2/hCds1 and GFP, as a marker, were exposed to IR or were mock irradiated. Cell extracts were prepared 2 hr later, resolved by denaturing gel electrophoresis and immunoblotted for endogenous p53 with antibody DO7 and for GFP to monitor transfection efficiency. (B) U2–OS cells transiently transfected with plasmids expressing Neo, Chk2/hCds1, or wild-type p53 were exposed to IR. Two hours later, p53 levels were monitored by immunofluorescence and cell nuclei were visualized by DAPI staining.
To determine whether ectopic expression of Chk2/hCds1 restores the G1 checkpoint, U2–OS cells were transfected with plasmids expressing Chk2/hCds1 and GFP and examined for cell cycle arrest by flow cytometry analysis. The cell cycle profile was determined separately for the GFP-positive and GFP-negative cells; the former population should express Chk2/hCds1, whereas the latter serves as a negative control. In the absence of DNA damage, the cells were distributed in all phases of the cell cycle and expression of Chk2/hCds1 did not affect the cell cycle profile (data not shown). After exposure to IR, the nontransfected cells exhibited predominantly cell cycle arrest in G2 with practically no cells in S phase and very few cells arrested in G1, whereas cells transfected with a plasmid expressing Chk2/hCds1 had the G1 checkpoint restored almost as effectively as cells transfected with a wild-type p53 plasmid (Fig. 6C). G1 arrest induced by Chk2/hCds1 was dependent on endogenous wild-type p53, as p53W248, a dominant-negative p53 mutant, abrogated the ability of Chk2/hCds1 to induce G1 arrest (Fig. 6C) without affecting the levels of expressed Chk2/hCds1 protein (Fig. 6B). All three known protein domains of Chk2/hCds1 were required to restore the G1 checkpoint. Deletion of the region that is putatively phosphorylated by ATM (SQ), deletion of the FHA domain, or amino acid substitutions that inactivate the kinase domain (M249 and A347), abrogated the ability of Chk2/hCds1 to induce G1 arrest, even though there was no effect on the levels of expressed Chk2/hCds1 protein (Fig. 6; data not shown).
Figure 6.

Chk2/hCds1 functions as a G1 checkpoint in response to DNA damage. (A) Diagram of Chk2/hCds1 protein and deletion mutants. (SQ) Region that contains the SQ/TQ motifs that are putatively phosphorylated by ATM; (FHA) FHA domain; (KD) kinase domain. (B) Expression of Flag-tagged Chk2/hCds1 proteins in transiently transfected U2–OS cells exposed to IR. Extracts from transfected cells were resolved by denaturing gel electrophoresis and immunoblotted with an antibody that recognizes the Flag tag (p53W248, which does not have a Flag tag, is not recognized by this antibody). (C) Chk2/hCds1 induces cell cycle arrest in G1. U2–OS cells were either nontransfected (−) or transiently transfected with plasmids expressing the indicated Chk2/hCds1 and p53 checkpoint proteins and a plasmid expressing GFP (as a marker). Cell cycle profile was determined 12 hr after exposure of the cells to IR. GFP− cells do not express proteins from the transfected plasmids and serve as negative controls. 2N and 4N DNA contents correspond to cells in G1 and G2/M, respectively.
Discussion
An important component of the response of vertebrates to DNA damage is an increase in the levels of the p53 tumor suppressor protein, which is primarily due to decreased protein degradation (Maltzman and Czyzyk 1984; Kastan et al. 1991; Fritsche et al. 1993). Degradation of p53 involves Mdm2, an intracellular protein that binds to p53 (Momand et al. 1992) and exports it out of the nucleus (Roth et al. 1998; Lain et al. 1999). In the cytoplasm, Mdm2 targets p53 for ubiquitin-dependent proteolysis (Haupt et al. 1997; Kubbutat et al. 1997; Midgley and Lane 1997). There is consensus in the field that p53 stabilization in response to DNA damage is due to inhibition of Mdm2-dependent p53 degradation, because modified p53 proteins that fail to interact with Mdm2 are constitutively expressed at high levels (Ashcroft et al. 1999; Blattner et al. 1999; Chehab et al. 1999) and at least some DNA-damaging agents lead to dissociation of p53 from Mdm2 (Shieh et al. 1997).
The molecular basis for dissociation of p53 from Mdm2 in vivo was initially attributed to direct phosphorylation of p53 on Ser-15 by the DNA damage-inducible kinases ATM and ATR (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998; Tibbetts et al. 1999). However, substitution of Ser-15 with Ala does not abrogate stabilization of p53 after DNA damage (Ashcroft et al. 1999; Blattner et al. 1999; Chehab et al. 1999). Instead, the critical post-translational modification for p53 stabilization after DNA damage appears to be phosphorylation of Ser-20; phosphorylation of Ser-20 disrupts the interaction between p53 and Mdm2 in vitro and substitution of Ser-20 with Ala abolishes stabilization of p53 in response to IR and UV light in vivo (Chehab et al. 1999; Unger et al. 1999).
The observation that phosphorylation of Ser-20 is important for p53 stabilization after DNA damage implies the existence of one or more DNA damage-inducible kinases that can phosphorylate p53 on Ser-20, because the previously implicated kinases ATM and ATR target only serines 15 and 37 (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998; Tibbetts et al. 1999). The kinase(s) that phosphorylate p53 on Ser-20 would further be expected to be activated by ATM and ATR, as p53 stabilization in response to IR and UV light is ATM and ATR dependent, respectively (Kastan et al. 1992; Khanna and Lavin 1993; Canman et al. 1994; Savitsky et al. 1995; Tibbetts et al. 1999). We have identified Chk2/hCds1 as a kinase that can phosphorylate p53 on Ser-20 in response to DNA damage. Chk2/hCds1 fulfills several criteria that implicate it in regulation of p53 stabilization. First, Chk2/hCds1 phosphorylates p53 on Ser-20 and disrupts preformed p53/Mdm2 complexes in vitro. Second, Chk2/hCds1 activation, p53 phosphorylation on Ser-20 and p53 stabilization are all ATM dependent in response to IR and ATM independent in response to UV light. Third, a dominant-negative Chk2/hCds1 mutant suppresses p53 phosphorylation on Ser-20 and p53 stabilization after DNA damage. And fourth, ectopic expression of Chk2/hCds1 leads to increased p53 stabilization after DNA damage and to p53-dependent cell cycle arrest in G1. On the basis of these findings, we propose a revised model for the checkpoint-signaling pathways leading to p53 stabilization. The major difference from the previous models is that p53 stabilization is not attributed to direct phosphorylation of p53 by ATM or ATR (Banin et al. 1998; Canman et al. 1998; Khanna et al. 1998; Tibbetts et al. 1999) but to indirect phosphorylation by these kinases through Chk2/hCds1 (Fig. 7).
Figure 7.

Model for p53 stabilization in response to DNA damage and stalled replication. IR, induces DNA DSBs and activates Chk2/hCds1 in an ATM-dependent manner. Thymine dimers, which are induced by UV light and stalled replication, activate Chk2/hCds1 in an ATM-independent manner. Stabilization of p53 in response to UV light is dependent on ATR and requires DNA replication, suggesting that ATR signals the presence of thymine dimers and/or replication blocks to Chk2/hCds1 (? indicates that it is not known whether ATR responds to thymine dimers or stalled replication or both). In budding yeast, activation of RAD53/SPK1 (the Chk2/hCds1 homolog) is dependent on MEC1 (the ATM and ATR homolog) and leads to phosphorylation of the SWI6 transcription factor and G1 arrest.
In the revised model, the signaling pathways leading to p53 stabilization share similarities with the yeast DNA damage and stalled replication checkpoint-signaling pathways. Specifically, in budding yeast, the ATM homolog MEC1 induces G1 arrest by activating the downstream kinase RAD53/SPK1 (the homolog of Chk2/hCds1), which in turn phosphorylates SWI6, leading to decreased expression of G1 cyclins (Fig. 7). Thus, both in mammals and yeast, an evolutionarily conserved kinase cascade targets transcription factors (p53 and SWI6, respectively) that induce G1 arrest. Regulation of p53 and SWI6 through a kinase cascade, rather than by ATM or MEC1 directly, may allow amplification of the DNA damage signal.
Implicating Chk2/hCds1 in p53 stabilization allows us to consolidate some of the checkpoint-signaling pathways that target p53. It is well established that blocks in DNA replication induced by aphidicolin, hydroxyurea, or PALA lead to p53 stabilization (Linke et al. 1996; Agarwal et al. 1998; Chen et al. 1998). Some types of DNA damage, such as cross-linked pyrimidines induced by UV light, may also stabilize p53 by inhibiting DNA replication (Haapajarvi et al. 1997). The signaling pathway(s) leading to p53 stabilization in response to stalled replication have not been deciphered. As Figure 7 indicates, Chk2/hCds1 is activated in response to both DNA damage and replication blocks and is, in fact, a component of an evolutionarily conserved checkpoint that monitors both DNA damage and stalled replication (Matsuoka et al. 1998; Blasina et al. 1999; Brown et al. 1999). Because activated Chk2/hCds1 stabilizes p53, we propose that agents that block DNA replication stabilize p53 through Chk2/hCds1.
It is worth noting that Chk2/hCds1 is not involved in all aspects of p53 regulation in response to genotoxic stress. IR induces an ATM-dependent association of the carboxyl terminus of p53 with 14-3-3 proteins that is important for functional activation of p53 (Waterman et al. 1998). Chk2/hCds1 is not involved in regulating this association, as UV light, which activates Chk2/hCds1 as effectively as IR, did not promote the interaction between p53 and 14-3-3. Furthermore, a dominant-negative Chk2/hCds1 mutant inhibited p53 stabilization, but not the interaction of p53 with 14-3-3.
The budding yeast RAD53/SPK1 and fission yeast Cds1 checkpoint kinases induce cell cycle arrest when activated (Elledge 1996; Weinert 1998). Experimental evidence showing that human Chk2/hCds1 induces cell cycle arrest had been lacking. Our findings show a role of Chk2/hCds1 in promoting G1 arrest. We did not observe any effect of wild-type or dominant-negative Chk2/hCds1 on cell cycle arrest in S or G2. This does not preclude a role of Chk2/hCds1 in these phases of the cell cycle, but may indicate functional redundancy within the DNA damage and replication block checkpoint. In fact, we anticipate that Chk2/hCds1 functions as a DNA damage checkpoint in S and G2, as well as in G1, because IR induces post-translational modifications of Chk2/hCds1 in all phases of the cell cycle (Matsuoka et al. 1998). Functional redundancy in cell cycle checkpoints raises another issue; whether Chk2/hCds1 is the only kinase that phosphorylates p53 on Ser-20 after DNA damage and replication blocks. In fission yeast, there is redundancy between Cds1 and Chk1 (Boddy et al. 1998; Zeng et al. 1998). The human Chk2/hCds1 and hChk1 kinases may also exhibit functional redundancy, as they are both activated by DNA damage and have similar substrate specificities (Sanchez et al. 1997; Matsuoka et al. 1998; Blasina et al. 1999; Brown et al. 1999), which raises the possibility that not only Chk2/hCds1, but also hChk1 targets p53. There is now evidence that hChk1 phosphorylates p53 on Ser-20 and regulates p53 protein levels in vivo (Shieh et al. 2000).
Because p53 is a tumor suppressor and Chk2/hCds1 activates p53, one might anticipate that Chk2/hCds1 is inactivated in human cancer. There are already two reports pointing in this direction. First, Chk2/hCds1 is expressed at much lower levels in tumor cell lines that have endogenous wild-type p53 as compared with cell lines with mutant p53 (Tominaga et al. 1999). Using an antibody that recognizes endogenous protein, we also observed low levels of Chk2/hCds1 in U2–OS cells (data not shown). Low levels of Chk2/hCds1 might compromise the p53 checkpoint and can explain why ectopic expression of Chk2/hCds1 enhanced p53 stabilization and p53-dependent G1 arrest in response to genotoxic stress. The most dramatic observation linking Chk2/hCds1 to human cancer is the identification of mutations in the chk2/hcds1 gene in a subset of Li-Fraumeni syndrome families, who do not have p53 mutations (Bell et al. 1999). In what can be considered as genetic evidence for chk2/hcds1 and p53 being in the same pathway, mutations targeting either gene lead to a similar phenotype, the Li-Fraumeni syndrome.
Materials and methods
Recombinant plasmids and proteins
A clone within the EST database (GenBank accession no. AA285249) containing the entire coding sequence of Chk2/hCds1 was obtained from Robert Hawley (The Toronto Hospital, Canada). For expression in mammalian cells, the Chk2/hCds1-coding sequence was cloned in a modified pcDNA–zeocin vector (Invitrogen) that fuses a Flag tag (Sigma) to the Chk2/hCds1 amino terminus. For expression in Escherichia coli, the Chk2/hCds1-coding sequence was cloned in a modified pT5T vector (Eisenberg et al. 1990) that fuses a six-histidine tag to the Chk2/hCds1 amino terminus; His-tagged Chk2/hCds1 protein was purified from crude E. coli lysates on a Hi-Trap Ni-chelating column (Pharmacia, Piscataway, NJ). Plasmids expressing full-length p53 in mammalian cells and E. coli have been described (Muller-Tiemann et al. 1998; Chehab et al. 1999). Full-length p53 expressed in E. coli was purified by sequential chromatography on Sepharose SP ion exchange and Sephadex 200 gel filtration columns on a high resolution SMART protein purification system (Pharmacia). Mutants of Chk2/hCds1 and p53 were constructed by PCR-based mutagenesis. The domain of human Mdm2 that binds p53 (residues 1–125) was expressed in E. coli as a GST fusion protein with a pGEX4T1 plasmid (Pharmacia) GST–Mdm2(1–125) was purified with glutathione beads (Pharmacia).
Antibodies
AbS20p, a polyclonal antibody (clone 430) that recognizes p53 phosphorylated on Ser-20, was obtained from New England Biolabs (Beverly, MA). DO7, a monoclonal antibody that recognizes p53 irrespective of its phosphorylation state, was obtained from Calbiochem (San Diego, CA). Y11 and M5, monoclonal antibodies that recognize amino-terminal hemagglutinin (HA) and Flag tags, respectively, were obtained from Santa Cruz Biotech (Santa Cruz, CA) and Sigma (St. Louis, MO). Antibodies GFP–FL and K-19 that recognize GFP and 14-3-3 proteins, respectively, were obtained from Santa Cruz Biotech.
In vitro kinase assay
Purified recombinant full-length wild-type or mutant p53 proteins (150 ng) were incubated with or without purified His-tagged Chk2/hCds1 protein (200 ng) for 30 min in 1× kinase buffer [50 mm HEPES (pH 7.4), 10 mm MgCl2, 10 mm MnCl2, 10 mm DTT] supplemented with either 500 nm unlabeled ATP or 200 nm unlabeled and 200 nm [32P]ATP. The protein mixtures were either resolved by denaturing gel electrophoresis and subjected to autoradiography to monitor 32P incorporation or immunoprecipitated with AbS20p, resolved by denaturing gel electrophoresis and immunoblotted with antibody DO7 to monitor phosphorylation of p53 on Ser-20.
Binding of p53 to Mdm2
Purified full-length p53 (150 ng) was incubated with 200 ng of purified His-tagged Chk2/hCds1 for 30 min in 1× kinase buffer. Then, half of the sample (input sample) was stored on ice; the other half was incubated for 1 hr with glutathione beads coated with 200 ng of GST–Mdm2(1–125) in 1× IP buffer [25 mm HEPES (pH 7.4), 100 mm NaCl, 5 mm MgCl2, 100 mm EDTA, 200 ng/ml BSA, 0.1% Tween 20]. The beads were washed with 2×IP buffer and p53 that remained bound to Mdm2 was released from the beads by adding 20 μl of SDS sample buffer, and then diluted 50-fold with 1× IP buffer (Mdm2-bound sampled). The fraction of p53 phosphorylated on Ser-20 in the input and Mdm2-bound samples was determined by comparing the amounts of p53 immunoprecipitated with AbS20p (p53 phosphorylated on Ser-20) versus the amount of p53 immunoprecipitated with DO7 (p53 phosphorylated and nonphosphorylated on Ser-20). p53 immunoprecipitated with AbS20p or DO7 was detected by immunoblotting with DO7.
Disruption of preformed p53/Mdm2 complexes
Purified full-length p53 (150 ng) was incubated for 1 hr with glutathione beads coated with 200 ng of GST–Mdm2(1–125) in 1× IP buffer. Unbound p53 was removed by washing the beads three times with 2× IP buffer. The beads were then incubated with 200 ng of purified His-tagged Chk2/hCds1 for 30 min in 1× kinase buffer and washed again three times with 2× IP buffer to remove any p53 protein that had dissociated from Mdm2. The fraction of p53 that remained bound to the beads after washing was visualized by immunoblotting with antibody DO7.
Kinetics of Chk2/hCds1 activation and p53 stabilization
U2–OS osteosarcoma cells were transfected by calcium phosphate precipitation with 5 μg of Flag-tagged Chk2/hCds1 expression plasmid, 1 μg of plasmid pSV7neo, and 24 μg of pBC12/PLseap carrier plasmid (Wieczorek et al. 1996). Stably transfected cells were selected with G418 and pooled when visible colonies emerged. The pools of stably transfected cells were exposed to 9 Gy IR or 50 J/m2 UV light. Whole cell extracts were prepared 1–16 hr after exposure to IR or UV light by lysis in 1× extraction buffer [50 mm Tris (pH 8), 120 mm NaCl, 0.5% NP-40, 1 mm DTT, 0.4 μg/ml Pefabloc SC, 2 μg/ml pepstatin, 0.2 μm wortmannin, 0.1 μm staurosporine, 15 mm NaF, 1 mm sodium vanadate]. DNA damage-induced post-translational modifications of Flag-tagged Chk2/hCds1 and stabilization of endogenous p53 were monitored by immunoblotting with M5 and DO7 antibodies, respectively.
ATM dependence of p53 Ser-20 phosphorylation
Cultures of diploid nontransformed fibroblasts from a normal individual (AG1522) or from A-T patients (AT1BR and AT5BI) were exposed to 9 Gy IR or 50 J/m2 UV light. Whole cell extracts were prepared 2 hr after exposure to IR or 16 hr after exposure to UV light by lysis in 1× extraction buffer. Phosphorylation of p53 on Ser-20 was assayed by immunoprecipitation with AbS20p, followed by immunoblotting with DO7. The amounts of extracts used were adjusted to have equal p53 levels in all lanes, as shown by immunoblotting with DO7.
Effects of dominant-negative Chk2/hCds1 mutants
U2–OS osteosarcoma cells were transfected by calcium phosphate precipitation with 0.5 μg of a plasmid expressing HA-tagged p53IND, 5 μg of a plasmid expressing mutant Chk2/hCds1, or 5 μg of the expression plasmid without insert, 1 μg of a plasmid expressing GFP (to monitor transfection efficiency), and 24 μg of pBC12/PLseap carrier plasmid (Wieczorek et al. 1996; Palm et al. 1997; Chehab et al. 1999). p53IND differs from wild-type p53 by seven amino acid substitutions in the oligomerization domain; it forms homotetramers and maintains the native p53 structure and function, but does not hetero-oligomerize with native p53 (Stavridi et al. 1999). The transfected cells were exposed to 50 J/m2 UV light or 9 Gy IR 36 and 48 hr after transfection, respectively. Whole cell extracts were prepared 16 hr after exposure to UV light or 2 hr after exposure to IR by lysis in 1× extraction buffer. The levels of HA-tagged p53IND, Flag-tagged Chk2/hCds1 and GFP were assayed by immunoblotting. Phosphorylation of HA-tagged p53IND on Ser-20 was assayed by immunoprecipitation with AbS20p, followed by immunoblotting with antibody Y11. The interaction of HA-tagged p53IND with 14-3-3 proteins was monitored by immunoprecipitation with antibody K-19, followed by immunoblotting with Y11.
Effects of ectopic expression of wild-type Chk2/hCds1 on p53 levels
U2–OS osteosarcoma cells were transfected by calcium phosphate precipitation with 5 μg of a plasmid expressing wild-type Chk2/hCds1 or 5 μg of a plasmid expressing Neo, 1 μg of a plasmid expressing GFP (as a marker), and 24 μg of pBC12/PLseap carrier plasmid (Wieczorek et al. 1996; Palm et al. 1997). Thirty-six hours after transfection, the cells were exposed to 5 Gy IR and analyzed for p53 levels 2 hr later by immunoblotting and immunofluorescence. For immunoblotting, the cells were lysed in 1× extraction buffer; the lysates were resolved by denaturing gel electrophoresis, and levels of endogenous p53 were assayed by immunoblotting with antibody DO7. For immunofluorescence, cells that were transfected on glass coverslips (Fisher, Pittsburgh, PA) were washed 2 hr after irradiation, once with PBS and three times with KM buffer [10 mm MES (pH 6.2), 10 mm NaCl, 10 mm MgCl2, and 2.5% glycerol] and were then fixed with 1% paraformaldehyde (in PBS) on ice for 15 min. After three washes with PBS, the cells were permeabilized by incubation with 0.2% Triton X-100/PBS for 20 min on ice, washed again three times with PBS, and incubated with antibody DO7 for 1 hr at room temperature. The cells were washed three times with PBS, incubated with Texas Red-conjugated secondary antibody (Vector Laboratories, Burlingame, CA) for 30 min at room temperature, washed again with PBS, and stained with 3.0 mg/ml DAPI (Sigma) for 5 min. The coverslips containing the cells were then washed three times with PBS and mounted onto glass slides with Fluoromount-G (Fisher). The cells were visualized with a fluorescence microscope (Leica, Deerfield, IL) and separate images were acquired with filters corresponding to the excitation maxima of DAPI (to visualize cell nuclei), GFP (which serves as a marker of transfected cells), and Texas Red (to visualize p53).
Effects of Chk2/hCds1 on cell cycle progression
U2–OS osteosarcoma cells were transfected by calcium phosphate precipitation with 2.5 μg of a plasmid expressing various Chk2/hCds1 and/or p53 proteins or 2.5 μg of the expression plasmid without insert, 1 μg of a plasmid expressing GFP (as a marker), and 24 μg of pBC12/PLseap carrier plasmid (Wieczorek et al. 1996). Twenty-four hours after transfection, the cells were exposed to 5 Gy IR or were mock irradiated. The cells were trypsinized 12 hr later, washed once with PBS supplemented with 1% FBS (GIBCO-BRL, Grand Island, NY), resuspended in 200 μl of 0.4% paraformaldehyde in PBS, and incubated for 12 min at 37°C and, subsequently, for 10 min on ice. The fixed cells were overlaid with 1800 μl of cold (−20°C) methanol with gentle vortexing. After a 10-min incubation on ice, the cells were washed in 1× PBS–TF (PBS with 0.1% Tween 20 and 2% FBS) and incubated in 1 ml of PBS–TF containing 20 μl of RNase (GIBCO-BRL) and 10 μl of propidium iodide (Boehringer Mannheim, Indianapolis, IN) for 1 hr at 37°C. Flow cytometry analysis was performed on a FACSscan flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Acknowledgments
We thank Carol Prives, Philip Leder, Jules Shafer, Frank Rauscher III, and Giovanni Rovera for support and discussions. We also thank Daniel Scolnick for identifying the Chk2/hCds1 clone in the EST database before it was published, Robert Hawley for supplying us with the Chk2/hCds1 EST clone, George Pavlakis for the plasmid expressing enhanced GFP, and Andreas Nelsbach for supplying us with antibody AbS20P prior to its commercial introduction. Financial support was provided by the National Cancer Institute (CA76367 and CA25874), the American Cancer Society, and the Wistar Institute NIH Training Grant (CA09171).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL halazonetis@wistar.upenn.edu; FAX (215) 898-3929.
References
- Agarwal ML, Agarwal A, Taylor WR, Chernova O, Sharma Y, Stark GR. A p53-dependent S-phase checkpoint helps to protect cells from DNA damage in response to starvation for pyrimidine nucleotides. Proc Natl Acad Sci. 1998;95:14775–14780. doi: 10.1073/pnas.95.25.14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 function and stability by phosphorylation. Mol Cell Biol. 1999;19:1751–1758. doi: 10.1128/mcb.19.3.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, et al. Heterozygous germline hChk2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Blades JP, Hupp T. DNA damage triggers DRB-resistant phosphorylation of p53 at the CKII site. Oncogene. 1998;17:1045–1052. doi: 10.1038/sj.onc.1202014. [DOI] [PubMed] [Google Scholar]
- Blasina A, Weyer de IV, Laus MC, Luyten WH, Parker AE, McGowan CH. A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol. 1999;9:1–10. doi: 10.1016/s0960-9822(99)80041-4. [DOI] [PubMed] [Google Scholar]
- Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P. DNA damage induced p53 stabilization: No indication for an involvement of p53 phosphorylation. Oncogene. 1999;18:1723–1732. doi: 10.1038/sj.onc.1202480. [DOI] [PubMed] [Google Scholar]
- Boddy MN, Furnari B, Mondesert O, Russell P. Replication checkpoint enforced by kinases Cds1 and Chk1. Science. 1998;280:909–912. doi: 10.1126/science.280.5365.909. [DOI] [PubMed] [Google Scholar]
- Brondello JM, Boddy MN, Furnari B, Russell P. Basis for the checkpoint signal specificity that regulates Chk1 and Cds1 protein kinases. Mol Cell Biol. 1999;19:4262–4269. doi: 10.1128/mcb.19.6.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Lee CH, Schwarz JK, Mitiku N, Piwnica-Worms H, Chung JH. A human Cds1-related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc Natl Acad Sci. 1999;96:3745–3750. doi: 10.1073/pnas.96.7.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Wolff AC, Chen CY, Fornace AJ, Jr, Kastan MB. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54:5054–5058. [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of p53 in response to DNA damage. Proc Natl Acad Sci. 1999;96:13777–13782. doi: 10.1073/pnas.96.24.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Hall I, Lansing TJ, Gilmer TM, Tlsty TD, Kastan MB. Separate pathways for p53 induction by ionizing radiation and N-(phosphonoacetyl)-L-aspartate. Cancer Res. 1996;56:3659–3662. [PubMed] [Google Scholar]
- Chernov MV, Stark GR. The p53 activation and apoptosis induced by DNA damage are reversibly inhibited by salicylate. Oncogene. 1997;14:2503–2510. doi: 10.1038/sj.onc.1201104. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci. 1997;94:14361–14366. doi: 10.1073/pnas.94.26.14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg SP, Evans RJ, Arend WP, Verderber E, Brewer MT, Hannum CH, Thompson RC. Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature. 1990;343:341–346. doi: 10.1038/343341a0. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: Preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Flaggs G, Plug AW, Dunks KM, Mundt KE, Ford JC, Quiggle MR, Taylor EM, Westphal CH, Ashley T, Hoekstra MF, et al. Atm-dependent interactions of a mammalian chk1 homolog with meiotic chromosomes. Curr Biol. 1997;7:977–986. doi: 10.1016/s0960-9822(06)00417-9. [DOI] [PubMed] [Google Scholar]
- Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene. 1993;8:307–318. [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- Haapajarvi T, Pitkanen K, Tsubari M, Laiho M. p53 transactivation and protein accumulation are independently regulated by UV light in different phases of the cell cycle. Mol Cell Biol. 1997;17:3074–3080. doi: 10.1128/mcb.17.6.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Shiloh Y. Many faces of ATM: Eighth international workshop on ataxia-telangiectasia. Bioch Bioph Acta. 1999;1424:R45–R55. doi: 10.1016/s0304-419x(99)00023-2. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Kapoor M, Lozano G. Functional activation of p53 via phosphorylation following DNA damage by UV but not gamma radiation. Proc Natl Acad Sci. 1998;95:2834–2837. doi: 10.1073/pnas.95.6.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Lavin MF. Ionizing radiation and UV induction of p53 protein by different pathways in ataxia-telangiectasia cells. Oncogene. 1993;8:3307–3312. [PubMed] [Google Scholar]
- Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller SP, et al. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Milne DM, Campbell LE, DeMaggio AJ, Christenson E, Hoekstra MF, Meek DW. p53 is phosphorylated in vitro and in vivo by the delta and epsilon isoforms of casein kinase 1 and enhances the level of casein kinase 1 delta in response to topoisomerase-directed drugs. Oncogene. 1997;15:1727–1736. doi: 10.1038/sj.onc.1201541. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Lain S, Midgley C, Sparks A, Lane EB, Lane DP. An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp Cell Res. 1999;248:457–472. doi: 10.1006/excr.1999.4433. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Li CY, Nagasawa H, Dahlberg WK, Little JB. Diminished capacity for p53 in mediating a radiation-induced G1 arrest in established human tumor cell lines. Oncogene. 1995;11:1885–1892. [PubMed] [Google Scholar]
- Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes & Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke SP, Clarkin KC, Leonardo Di A, Tsou A, Wahl GM. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes & Dev. 1996;10:934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Lu H, Taya Y, Ikeda M, Levine AJ. Ultraviolet radiation, but not gamma radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc Natl Acad Sci. 1998;95:6399–6402. doi: 10.1073/pnas.95.11.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Lane DP. Differential induction of transcriptionally active p53 following UV or ionizing radiation: Defects in chromosome instability syndromes? Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- Lutzker SG, Levine AJ. A functionally inactive p53 protein in teratocarcinoma cells is activated by either DNA damage or cellular differentiation. Nat Med. 1996;2:804–810. doi: 10.1038/nm0796-804. [DOI] [PubMed] [Google Scholar]
- Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–1694. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179–1189. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Muller-Tiemann BF, Halazonetis TD, Elting JJ. Identification of an additional negative regulatory region for p53 sequence-specific DNA binding. Proc Natl Acad Sci. 1998;95:6079–6084. doi: 10.1073/pnas.95.11.6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa H, Keng P, Maki C, Yu Y, Little JB. Absence of a radiation-induced first-cycle G1-S arrest in p53+ human tumor cells synchronized by mitotic selection. Cancer Res. 1998;58:2036–2041. [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase epsilon form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes & Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Palm GJ, Zdanov A, Gaitanaris GA, Stauber R, Pavlakis GN, Wlodawer A. The structural basis for spectral variations in green fluorescent protein. Nat Struct Biol. 1997;4:361–365. doi: 10.1038/nsb0597-361. [DOI] [PubMed] [Google Scholar]
- Roth J, Dobbelstein M, Freedman DA, Shenk T, Levine AJ. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998;17:554–564. doi: 10.1093/emboj/17.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes & Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by Chk1 and Rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Taya Y, Prives C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999;18:1815–1823. doi: 10.1093/emboj/18.7.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes & Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM, Breeden LL. Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae. Genes & Dev. 1997;11:3032–3045. doi: 10.1101/gad.11.22.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes & Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavridi ES, Chehab NH, Caruso LC, Halazonetis TD. Change in oligomerization specificity of the p53 tetramerization domain by hydrophobic amino acid substitutions. Protein Sci. 1999;8:1773–1779. doi: 10.1110/ps.8.9.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes & Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Krolewski B, Little JB. Loss of normal G1 checkpoint control is an early step in carcinogenesis, independent of p53 status. Cancer Res. 1999;59:1008–1014. [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga K, Morisaki H, Kaneko Y, Fujimoto A, Tanaka T, Ohtsubo M, Hirai M, Okayama H, Ikeda K, Nakanishi M. Role of human cds1 (Chk2) kinase in DNA damage checkpoint and its regulation by p53. J Biol Chem. 1999;274:31463–31467. doi: 10.1074/jbc.274.44.31463. [DOI] [PubMed] [Google Scholar]
- Unger T, Juven-Gershon T, Moallem E, Berger M, Sionov RV, Lozano G, Oren M, Haupt Y. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 1999;18:1805–1814. doi: 10.1093/emboj/18.7.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet. 1998;19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- Weinert T. DNA damage and checkpoint pathways: Molecular anatomy and interactions with repair. Cell. 1998;94:555–558. doi: 10.1016/s0092-8674(00)81597-4. [DOI] [PubMed] [Google Scholar]
- Weinreich M, Stillman B. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 1999;18:5334–5346. doi: 10.1093/emboj/18.19.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek AM, Waterman JL, Waterman MJ, Halazonetis TD. Structure-based rescue of common tumor-derived p53 mutants. Nat Med. 1996;2:1143–1146. doi: 10.1038/nm1096-1143. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Forbes KC, Wu Z, Moreno S, Piwnica-Worms H, Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507–510. doi: 10.1038/26766. [DOI] [PubMed] [Google Scholar]