

Abstract
Recruitment of p300/CBP by the hypoxia-inducible factor, HIF-1, is essential for the transcriptional response to hypoxia and requires an interaction between the p300/CBP CH1 region and HIF-1α. A new p300-CH1 interacting protein, p35srj, has been identified and cloned. p35srj is an alternatively spliced isoform of MRG1, a human protein of unknown function. Virtually all endogenous p35srj is bound to p300/CBP in vivo, and it inhibits HIF-1 transactivation by blocking the HIF-1α/p300 CH1 interaction. p35srj did not affect transactivation by transcription factors that bind p300/CBP outside the CH1 region. Endogenous p35srj is up-regulated markedly by the HIF-1 activators hypoxia or deferoxamine, suggesting that it could operate in a negative-feedback loop. In keeping with this notion, a p300 CH1 mutant domain, defective in HIF-1 but not p35srj binding, enhanced endogenous HIF-1 function. In hypoxic cells, p35srj may regulate HIF-1 transactivation by controlling access of HIF-1α to p300/CBP, and may keep a significant portion of p300/CBP available for interaction with other transcription factors by partially sequestering and functionally compartmentalizing cellular p300/CBP.
Keywords: Hypoxia, HIF-1, transcription, p35srj, p300, CBP, regulation
Localized tissue hypoxia is a major factor in the pathogenesis of tumor vascularization, myocardial ischemia, and stroke. An hypoxic signal is transduced through a hemoprotein oxygen sensor and results in the induction of the transcription factor, hypoxia inducible factor-1 (HIF-1). HIF-1 consists of HIF-1α and ARNT (aryl hydrocarbon nuclear translocator) subunits (Wang et al. 1995; for review, see Guillemin and Krasnow 1997). Hypoxia results in the stabilization of the HIF-1α protein, a marked increase in its transactivation potential, and heterodimerization of HIF-1α with ARNT to form active HIF-1 (Li et al. 1996; Jiang et al. 1997; Kallio et al. 1997; Pugh 1997; Huang et al. 1998). The active HIF-1 heterodimer binds to its cognate response element and activates transcription of several hypoxia-induced genes, including those encoding erythropoietin, vascular endothelial growth factor (VEGF), and key glycolytic enzymes. HIF-1α deficiency results in embryonic lethality, with severe neurological and cardiovascular developmental abnormalities and impaired expression of HIF-1 target genes. This suggests that HIF-1α is a global regulator of developmental and cellular O2 homeostasis (Iyer et al. 1998).
HIF-1α activates transcription by recruiting the transcriptional adapter/histone acetyltransferase proteins, p300 and CBP (CREB-binding protein), to a transcription complex. This is essential for the normal cellular response to hypoxia (Arany et al. 1996). p300 and CBP (Fig.1A) are homologous, ubiquitously expressed, nuclear phosphoproteins that contain three highly conserved cysteine–histidine-rich domains (CH1, -2, and -3). p300/CBP function as coactivators, linking signal-responsive DNA-bound transcriptional activators to the basal transcription machinery. It is likely that they also modify chromatin structure by virtue of intrinsic and associated (P/CAF, P/CIP-ACTR, src1) histone acetyltransferase activities (for review, see Shikama et al. 1997).
The CH1 region of p300 binds HIF-1α (Arany et al. 1996) and STAT2 directly (Bhattacharya et al. 1996), linking this region, at a minimum, to hypoxia and interferon-α signal transduction. The p300 CH1 domain is also important for the binding of the transcription factor ets-1 (Yang et al. 1998), the NFκB p65 subunit (Zhong et al. 1998), p53, and MDM2 (Grossman et al. 1998). A 35-kD cellular protein that binds p300-CH1 strongly was identified in our laboratory. We have cloned a human cDNA encoding this protein using an expression screen in which the p300-CH1 domain served as probe and named it p35srj for its unusual serine-glycine rich junction. p35srj is a novel p300/CBP binding protein, and is an alternatively spliced isoform of MRG1 (Shioda et al. 1996), a human protein of unknown function. Here we provide data indicating that p35srj is the product of a HIF-1-regulated gene, that functions, at least in part, to regulate access of HIF-1α and other CH1-binding proteins to p300/CBP and could serve to partition p300/CBP into functionally discrete subsets.
Results
Expression cloning of human p35srj, a highly conserved p300-CH1 binding protein
A HeLa λgt11 expression library was screened with a probe consisting of 32P labeled glutathione S-transferase fused to the CH1 domain of p300 (GST–CH1), as described previously (Bhattacharya et al. 1996). In addition to clones for STAT2 and HIF-1α (encoding amino acid residues 723-826), a single clone encoding a novel peptide was obtained. A full-length clone was obtained by screening a 293λZAP library with this cDNA. The open reading frame of this clone predicted the synthesis of a 270-amino acid polypeptide (Fig. 1B). In vitro-translated clonal p35srj comigrated with and had a similar peptide map to endogenous p35 present in anti-p300/CBP immunoprecipitates (data not shown). The murine p35srj peptide sequence (Fig. 1B) was deduced by cloning and sequencing murine genomic p35srj (S. Bhattacharya and D.M. Livingston, unpubl.). It and the human sequence are very similar (94.8% identity). Human and murine p35srj peptide sequences revealed an unusual, conserved 39-amino acid serine–glycine-rich junction (srj). p35srj does not contain a known DNA-binding domain or other known protein motifs.
Figure 1.

Cloning, expression, and stability of p35srj. (A) p300 and CBP contain multiple, conserved regions. These include three cysteine–histidine rich regions (CH1–3), the KIX domain (which binds CREB), a bromodomain, and a glutamine-rich region. The p300–CH1 domain, fused to GST bearing a protein kinase A phosphorylation site, was labeled with 32P and used as a probe to clone p35srj. (B) p35srj peptide sequence and homology. The serine–glycine rich junction is conserved in murine p35srj, and is boxed. The minimal p300-binding domain (see Fig. 3) is underlined. FG079M18bg6 is a fragment from the Fugu genome project, detected by database search. MSG1 is a homologous human gene also identified by database searches. (C) Expression of p35srj, as reflected the results of a multiple human tissue Northern blot (Clontech) probed with human p35srj cDNA. (D) The stability of p35srj was analyzed by pulse-chase analysis performed in exponentially growing C2C12 cells. 35S-labeled p35srj was immunoprecipitated at the indicated time points.
Database searches indicated that p35srj is similar to the products of two related human genes of unknown function, MRG1, and MSG1 (Shioda et al. 1996). MRG1 is an alternatively spliced isoform of p35srj that lacks the conserved serine–glycine rich junction. Comigration of in vitro translated, clonal p35srj with the endogenous immunoprecipitated protein indicates that the major endogenous isoform is p35srj (data not shown). As pointed out previously (Shioda et al. 1996), p35srj/MRG1 and MSG1 have similar carboxyl termini (76% identity over 45 amino acids). Because all data herein refer to the isoform containing the serine-rich junction, we will use the term p35srj. A search of the Fugu rubripes database also identified a DNA sequence encoding a protein fragment with strong (76.3% identity over 38 amino acids) homology to p35srj (Fig. 1B). p35srj homologs have not been found in the Saccharomyces cerevisiae, Caenorhabditis elegans, or Drosophila databases.
p35srj is an unstable nuclear protein that is almost entirely bound to p300/CBP
Northern blots of mRNA from multiple human tissues revealed that p35srj is ubiquitously expressed (Fig. 1C), like p300 and CBP (Eckner et al. 1994). Western blots probed with anti-p35srj polyclonal and monoclonal antibodies showed that the protein is present in all human and murine cell lines examined (data not shown). p35srj is unstable, with a half-life of ∼20 min (Fig. 1D). Immunostaining with anti-p35srj monoclonal antibody revealed that endogenous and ectopically expressed p35srj is nuclear in U2-OS and other cell lines studied (Fig. 2 and data not shown). Endogenous p35srj colocalized with p300 in dot-like structures that are detected when p300 is ectopically overproduced (Eckner et al. 1994; Fig. 2C).
Figure 2.
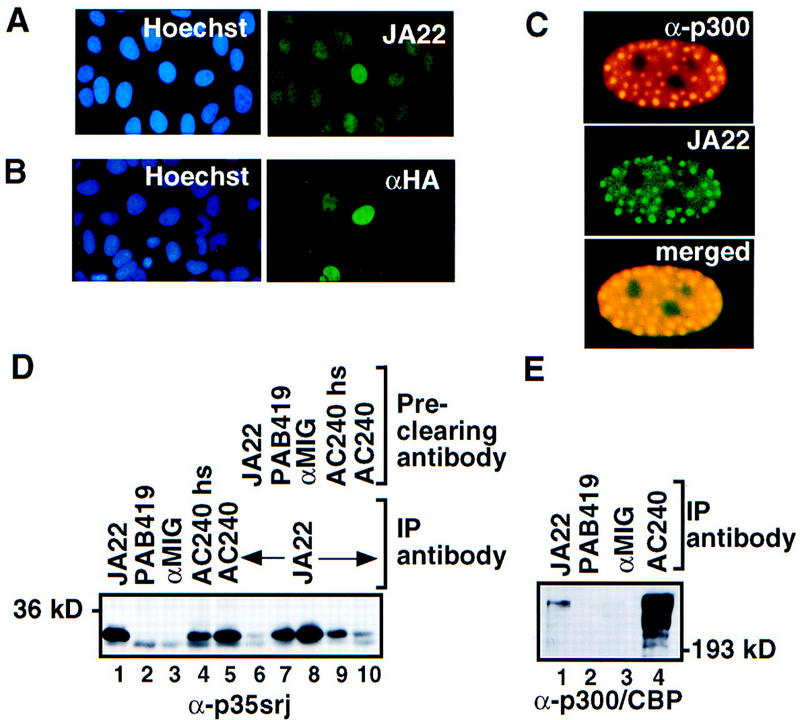
p35srj is a nuclear protein bound to p300/CBP. (A) Cellular localization of endogenous p35srj. U2-OS cells were stained with Hoechst to demonstrate cell nuclei, and with anti-p35srj monoclonal antibody JA22, to demonstrate endogenous p35srj. (B) U2-OS cells were transfected with HA–p35srj, cell nuclei stained with Hoechst, and immunostained with anti-HA monoclonal antibody. (C) Colocalization of endogenous p35srj with p300. U2-OS cells were transfected with HA–p300 and stained with anti-HA polyclonal antibody to reveal typical p300 dot-like structures characteristic of p300-overproducing cells. These cells were reacted simultaneously with anti-p35srj monoclonal antibody, JA22, to show the colocalization of endogenous p35srj in the p300 dots. The merged exposure confirms that the dots colocalize. (D) Efficiency of endogenous p35srj coimmunoprecipitation with anti-p300/CBP antibodies. Anti-p35srj Western blot of immunoprecipitates (IP) from U2-OS cell lysates. Immunoprecipitations were performed with monoclonal antibodies to p35srj (JA22, lane 1), control antibodies (PAB419 and rabbit anti-mouse IgG, αMIG, lanes 2,3), and antibody to p300/CBP (AC240) (lanes 4,5). The AC240hs immunoprecipitation (lane 4) was performed in 300 mm NaCl. The supernatants from the above immunoprecipitates were reprecipitated with JA22 (lanes 6–10) to assess the relative levels of free p35srj. (E) Anti-p300/CBP Western blot of immunoprecipitates (IP) from U2-OS cell lysates.
Monoclonal anti-p300/CBP (AC240) immunoprecipitates contained high levels of endogenous p35srj (Fig. 2D, lane 5). AC240 did not cross react with p35srj (data not shown), and p35srj was not detected in the control antibody immunoprecipitates. These data suggest that the two proteins interact in vivo. In this regard, AC240, which recognizes both p300 and CBP, coprecipitated p35srj just as well as an anti-p35srj monoclonal antibody (cf. Fig. 2D, lanes 1 and 5) implying that complex formation is an efficient process. In this regard, we found that AC240 immunoabsorption of p300/CBP depleted most of the p35srj in a U20S lysate at 150 mm NaCl (Fig. 2D, lane 10). The p35srj/p300 interaction was less stable in the presence of high (300 mm NaCl) salt-containing buffer (Fig. 2D, lane 4).Taken together, these results imply that nearly all steady-state p35srj is bound to p300/CBP.
Immunoprecipitation with the anti-p35srj mAb, JA22, also recovered significant amounts of endogenous p300/CBP, although the recovery of the latter was incomplete (Fig. 2E). This suggests that a significant fraction of the ambient p300/CBP is not complexed to p35srj.
Carboxy-terminal residues of p35srj are sufficient for binding to p300-CH1
To map the p35srj structures necessary for binding to p300-CH1, we constructed expression vectors encoding HA and VP16 activation domain-tagged wild-type and deletion mutant p35srj species. These vectors were used to synthesize 35S-labeled proteins by in vitro translation. Binding of these polypeptides to GST–p300 was then assayed (Fig. 3A). These experiments showed the carboxy-terminal region of p35srj, extending between residues 215 and 270, was necessary for p300–CH1 binding. To define the minimal binding region, discrete segments of the 215–270 region were fused to green fluorescent protein (as carrier) and assayed for GST–p300–CH1 binding (Fig. 3B). A region of 32 amino acids (224–255) was required for this reaction. When synthesized as a peptide, it efficiently competed with p35srj for binding to p300–CH1 (Fig. 3C), indicating that it is also sufficient for this interaction. Notably this peptide sequence is highly conserved in MSG1.
Figure 3.

p35srj carboxy-terminal residues are required for CH1 binding. (A) Binding of 35S-labeled, in vitro translated p35srj derivatives to GST or GST–p300CH1 fusion proteins (lanes 1–6). Twenty percent of the in vitro-translated input was loaded in lanes 7–9. (B) The indicated residues from the carboxyl terminus of p35srj, fused to green fluorescent protein (GFP), as carrier, were synthesized as 35S-labeled peptides by in vitro translation (lanes 1–4), and were assayed for ability to bind GST or GST–p300CH1 (lanes 5–12). (C) Effect of p35srj 224–255 peptide on in vitro binding of p35srj to p300–CH1. (Top) The binding of 35S-labeled p35srj, generated by in vitro translation, to GST–p300CH1 was tested in the presence of the p35srj peptide (20 μg, lane 4) and compared to either a control peptide (20 μg, lane 5) or no peptide (lane 3). Lane 1 shows 20% of the input in vitro translate. (Bottom) Coomassie stain of the gel demonstrating relative amounts of the GST and GST–p300CH1 proteins. (D) Mammalian two-hybrid assay in U2-OS cells transfected with a 3× E2–luciferase reporter (50 ng), E2–p300 (20 ng), expression vectors encoding the indicated p35srj derivatives fused to VP16 (25 ng), and CMV–lacZ (50 ng). The results of a representative experiment, performed in duplicate, are shown. E2–p300 contains the DNA-binding domain of BPV E2, fused to p300.
The ability of various VP16–p35srj fusion proteins to interact with E2–p300 (p300 fused to the BPV–E2 DNA-binding domain) in vivo was tested in a mammalian two-hybrid assay (Fig. 3D). In keeping with the above-noted results, the data show that the carboxy-terminal region of p35srj is also essential for an in vivo p35srj/p300 interaction. In return, the CH1 domain is required for efficient binding of this carboxy-terminal p35srj sequence to p300 in vivo (see Fig. 4D).
Figure 4.

p35srj competes with HIF-1α for binding to p300–CH1 in vitro and in vivo. (A) Effect of baculoviral p35srj on in vitro binding of HIF-1α to p300–CH1. (Top) The binding of 35S-labeled HIF-1α (lane 1) generated by in vitro translation to bacterially produced GST (lane 2) or GST–p300CH1 (lane 3) immobilized on glutathione–Sepharose beads was tested. The binding of HIF-1α to GST–p300CH1 was also tested in the presence of baculovirally expressed wild-type (lane 4) or mutant p35srj (expressing p35srj residues 1–160) as control (lane 5). (Bottom) Coomassie stain of the gel demonstrating relative amounts of the GST and GST–p300CH1 proteins. (B) Anti-p35srj Western blot of SF21 cell lysates used in A. (C) Effect of p35srj 224–255 peptide on in vitro binding of HIF-1α to p300–CH1. (Top) The binding of HIF-1α to GST–p300CH1 was tested in the presence of increasing concentrations of the wild-type peptide (lanes 5–8) and compared to either a control, irrelevant peptide (lane 4) or no peptide (lane 3).Amounts of peptide used are shown in micrograms. Twenty percent of the HIF-1α input was loaded in lane 1. (Bottom) Coomassie stain of the gel demonstrating relative amounts of the GST and GST–p300CH1 proteins. (D) Effect of DFO on a mammalian two-hybrid interaction between GAL4–CH1 and VP16–HIF1α 723–826 (left) or VP16–p35srj (right). Hep3B cells were cotransfected with the indicated GAL4 and VP16 fusion plasmids (40 ng each), 3× GAL4–luc reporter (100 ng), and CMV–lacZ (100 ng). GAL4–CH1 contains p300 residues 300–528. GAL4–CH1Δ lacks p300 residues 346–410, and served as a control. Results are presented as relative luciferase units (RLU, mean of three independent experiments ± s.e.m.). (E) Effect of p35srj on the two-hybrid interaction between GAL4–p300CH1 and VP16–HIF1α. Hep3B cells were cotransfected with VP16HIF1α and GAL4–CH1 (40 ng each), and either vector control or p35srj expression plasmids (80 ng each), and CMV–lacZ (100 ng). p35srjΔ lacks residues 215–270. Results (mean of three independent experiments ± s.e.m.) are presented as fold induction of luciferase activity by DFO. A fold induction of 1 represents absence of induction.
p35srj competes with HIF-1α for binding to p300-CH1 in vitro and in vivo
Because HIF-1α and p35srj both bind directly to the CH1 region, we asked whether they compete with one another for CH1 complex formation. As shown in Figure 4A (lane 4), baculovirus-encoded p35srj competed with HIF-1α efficiently for binding to p300–CH1 in vitro, whereas a mutant protein lacking its p300-binding domain failed in this regard. Similarly, the p35srj 32mer peptide competed with intact HIF-1α for CH1 binding (Fig. 4C, lanes 5–7).
HIF-1α has two transactivation domains. The amino-terminal one (TAD-N) overlaps an oxygen-dependent degradation domain and accounts for the instability of HIF-1α under normoxic conditions (Huang et al. 1998). The carboxy-terminal one (TAD-C) is only weakly active under normoxic conditions, but is induced markedly by hypoxia or the hypoxia mimic, deferoxamine (DFO), without there being any change in protein level (Li et al. 1996; Jiang et al. 1997; Kallio et al. 1997; Pugh 1997; Huang et al. 1998). The results of our expression cloning screen (see earlier discussion) and of in vitro binding assays indicated that HIF-1α residues 723–826, which overlap TAD-C, but not TAD-N, bind p300–CH1.
To test whether p35srj interferes with the binding of HIF-1α to p300–CH1 in vivo, we used a mammalian two-hybrid assay (Fig. 4D,E). First, GAL4–p300–CH1 was coexpressed with VP16–HIF1α TAD-C (residues 723–826) in Hep3B cells along with a GAL4-luciferase reporter. Under normoxic conditions, there was no evidence of an interaction between the two proteins relative to the control VP16 vector (Fig. 4D). Stimulation with deferoxamine led to a threefold activation over control, indicating the development of a specific interaction. This two-hybrid interaction depended on both the VP16 and HIF-1α components of the fusion protein, since a HIF-1α species lacking the VP16 domain did not have this effect (data not shown). No interaction was detected between VP16–HIF-1α and GAL4–CH11Δ, a mutant derivative lacking an intact CH1 domain.
The effect of cotransfected p35srj on the interaction between GAL4–p300–CH1, and VP16–HIF-1α was then assayed. As seen in Figure 4E, with cotransfection of a vector control or a p35srj1Δ expression vector (lacking residues 215–270), there was a 3.7-fold induction of the two-hybrid interaction by deferoxamine. Cotransfection of p35srj reduced this substantially (Fig. 4E). Notably, the apparent affinity of VP16–p35srj for p300–CH1 was not affected by deferoxamine stimulation and was at least 10-fold greater than that of VP16–HIF-1α (see Fig. 4D). Hence, the inhibitory effect of p35srj likely occurred independently of any increase in intrinsic ability of p35srj to bind to CH1.
p35srj inhibits HIF-1 transactivation
Fusion of HIF-1α residues 723–826 (TAD-C) to a GAL4–DNA-binding domain (GAL4–HIF-1α) permitted specific assays of its transcription function. Whereas GAL4–HIF-1α is a weak transcriptional activator under normoxic conditions, it was strongly activated by the hypoxia mimic, deferoxamine, without any change in protein level (Fig. 5A). Deletion of its carboxy-terminal 13 residues (GAL4–HIF1-αΔ) resulted in complete loss of transactivation. These results are consistent with those reported previously (Li et al. 1996; Jiang et al. 1997; Pugh 1997). Transactivation correlated with p300 binding, because deletion of the carboxy-terminal 13 residues of GAL4–HIF1α also resulted in complete loss of p300–CH1 binding (Fig. 5B).
Figure 5.

p35srj inhibits HIF-1 transactivation by specifically affecting CH1 function. (A) Effect of p35srj on the activation of a GAL4–luciferase reporter by GAL4–HIF-1α (723–826). (Top) Hep3B cells were transiently cotransfected with a 3× GAL4–luc reporter (0.2 μg), and the indicated GAL4–HIF1α (0.2 μg), and HA–p35srj expression plasmids (0.2 μg), and CMV–lacZ (0.1 μg). GAL4–HIF1αΔ lacks the carboxy-terminal 13 residues of HIF-1α. Results are presented as RLU. The differences noted were statistically significant in three independent experiments. (Middle; bottom) The levels of GAL4–HIF1α and HA-tagged p35srj proteins in the above cell lysates were assayed by Western blotting with anti-GAL4 and anti-HA antibodies, respectively. The p35srjΔ mutation deletes residues 215–270, i.e., the p300–CH1 binding domain. (B) Effect of mutating the HIF-1α carboxyl terminus on its binding to p300–CH1. 35S-labeled proteins encoded by the indicated GAL4–HIF-1α plasmids were generated by in vitro translation, and their ability to bind GST–p300CH1 (lanes 3,6) or GST (as control, lanes 2,5) was assayed. Twenty percent of the in vitro-translated input was loaded as control in each case (lanes 1,4). (C) Effect of p35srj on the activation of a VEGF promoter–luciferase reporter by DFO. Hep3B cells were transiently cotransfected with a VEGF–luc reporter (40 ng), the indicated p35srj expression plasmids (300 ng each) and CMV–lacZ (100 ng). Results are presented as RLU (mean of three independent experiments ± s.e.m.). (D) Effect of p35srj on HIF-1α, SREBP2, and src1 transactivation. The indicated GAL4 fusion proteins (2 ng), were cotransfected with p35srj expression plasmids (4 ng), GAL4–luc reporter (0.1 μg), and CMV–lacZ (0.2 μg) into Hep3B cells. Results are presented as RLU and are representative of several independent experiments. (E) Effect of p35srj on STAT2 and src1 transactivation. The indicated GAL4 fusion proteins (2 ng), were cotransfected with p35srj expression plasmid or vector control (10 ng), GAL4–luc reporter (0.1 μg), and CMV–lacZ (0.3 μg) into U2-OS cells. Results are presented as relative luciferase units (mean of four independent experiments ± s.e.m.).
Cotransfection of an HA-tagged p35srj wild-type expression plasmid led to marked inhibition of both basal normoxic and deferoxamine induced GAL4–HIF1α transcriptional activity, without affecting its abundance (Fig. 5A). A p35srj mutant lacking its p300–CH1 binding domain (HA–p35srjΔ) and present in similar amounts did not affect GAL4–HIF1α function. Induction of TAD-C transactivation by hypoxia was inhibited similarly by p35srj (data not shown). These results indicate that p35srj can interfere with HIF-1α TAD-C function. Taken together with the previous data, these results indicate that the affinity of HIF-1α for p300–CH1 increases with deferoxamine stimulation, and that p35srj inhibits HIF-1 transactivation function by interfering with the access of HIF-1α TAD-C to p300/CBP–CH1.
We next asked whether p35srj inhibits the activation of a natural HIF-1 response element by deferoxamine (Fig. 5C). The vascular endothelial cell growth factor promoter (pVEGF) fused to luciferase was used as a reporter of endogenous HIF-1 activity. This element contains a HIF-1 response element (Levy et al. 1995), is markedly activated by deferoxamine, and its activation was suppressed by p35srj, but not by HA–p35srjΔ. P35srj had no effect on basal activity of pVEGF–luciferase, indicating that p35srj effects are specific to deferoxamine activation of HIF-1. Induction of pVEGF–luciferase activity by hypoxia was also inhibited by p35srj (data not shown). Taken together, these results suggest that p35srj-mediated suppression of the interaction of p300/CBP with HIF-1α TAD-C resulted in inhibition of HIF-1 transactivation.
p35srj specifically inhibits p300–CH1 interacting transactivators
To determine whether p35srj is a general repressor of p300/CBP function, we tested its effect on three other p300/CBP interacting transcription factors: (1) STAT2, which binds CH1 (Bhattacharya et al. 1996); (2) SREBP2, which binds the KIX domain (Oliner et al. 1996); and (3) src-1, which interacts with the glutamine-rich domain (Yao et al. 1996). p35srj specifically inhibited transcription by GAL4–HIF-1α and GAL4–STAT2, but not by SREBP2 or src-1 (Fig. 5D,E). Moreover, p35srj competed with STAT2 for binding to p300–CH1 in vitro (data not shown). These results imply that p35srj specifically affects at least two p300–CH1-mediated events.
p35srj is induced by deferoxamine and hypoxia
We next asked whether p35srj is, itself, affected by HIF-1 activation. Surprisingly, we found that, in Hep3B cells, hypoxia or deferoxamine, both activators of HIF-1, strongly induced synthesis of p35srj mRNA and protein, which resulted in the formation of p300/CBP–p35srj complexes. The latter reached a plateau between 1 and 6 hr after initiating the stimulus (Fig. 6A), and could be detected by both anti-p300/CBP (Fig. 6A) as well as anti-p35srj monoclonal antibodies (Fig. 6B). We noted that p35srj protein level appeared to peak before the mRNA. Possible mechanisms for this include changes in synthesis and degradation rates at different time points. Pulse-chase experiments indicated that whereas p35srj protein synthesis was markedly upregulated by deferoxamine at 5 hr, there did not appear to be any significant effect on stability (data not shown). It is possible that at later time points either the protein synthesis rate falls, or that the degradation rate rises.
Figure 6.

p35srj is induced by deferoxamine and hypoxia. (A) Hep3B cells were grown to confluence and exposed to either 1 or 21% O2 for 6 hr or to 100 μm DFO for the indicated periods of time. Total RNA from these cells was Northern blotted and probed for p35srj (top). 28S and 18S rRNA are shown as loading controls. Whole-cell lysates (50 μg per lane) from these cells were Western blotted to detect p35srj, using JA22 mAb (α-p35srj). Cell lysates were also immunoprecipitated with anti-p300/CBP mAb, AC240 (α-p300/CBP IP), and Western blotted with anti-p300/CBP monoclonal antibodies (α-p300/CBP) and anti-p35srj monoclonal antibody JA22 (α-p35srj, bottom). (B) Hep3B cells were grown to confluence and exposed to 100 μm DFO for 5 hr. Cell lysates from control (C) cells or DFO treated (D) cells were immunoprecipitated with anti-p35srj monoclonal antibody JA22 (lanes 3 and 4, respectively), and Western blotted with anti-p300/CBP monoclonal antibodies. One-fortieth of the total cell lysate used for each immunoprecipitation was loaded in lanes 1 and 2, respectively. (C) Effect of DFO on the p35srj promoter. Hep3B cells were transfected with the indicated human p35srj promoter–luciferase construct (300 ng), and CMV–lacZ (100 ng). The −1202/−1159 wild-type reporter plasmid contains the sequence 5′-gtgtgcgcgtggtgccatacgggacgtgcagctacgtgcccacc, whereas the −1202/−1159 mutant contains the mutated sequence 5′-gtgtgcgaaaggtgccatacgggaaaagcagctaaaagcccacc, cloned upstream of TK–luciferase. The HIF-1 consensus sequences in the wild-type are underlined. Results are presented as RLU (mean of three independent experiments ± s.e.m.). (D) The p35srj promoter binds HIF-1 in vitro. A gel-shift assay using 32P-labeled double-stranded wild-type and mutant oligonucleotide probes, derived from the p35srj promoter (see B), was performed. The probes were incubated with HIF-1α, ARNT, or both proteins, which were generated by in vitro translation. Oligonucleotides, W18 and M18, are wild-type and mutant competitors for HIF-1 binding, and were introduced at 100 fold molar excess.
These experiments suggested that p35srj may be transcriptionally regulated by HIF-1. To investigate this further, we cloned and sequenced ∼3.5 kb of the human p35srj promoter. Three consensus HIF-1 response elements (HREs, 5′-RCGTG) (Semenza et al. 1996) were identified in tandem within a 32 nucleotide sequence of the p35srj promoter (−1196/−1165, relative to the start of p35srj cDNA). A human p35srj promoter–luciferase reporter plasmid (−2186/+65), containing the 3× HRE, was constructed. Reporter activity from this plasmid was activated about fourfold by deferoxamine (Fig. 6C). A deletion mutant reporter (−973/+65), lacking the 3× HRE, was not activated. A short promoter segment within which the 3× HRE sequence is imbedded, (−1202/−1159), when cloned upstream of a HSV–TK minimal promoter–luciferase reporter, resulted in similar inducibility by deferoxamine.
Gel-shift analysis showed that a 32P-labeled, 3× HRE sequence from the p35srj promoter specifically formed a complex with HIF-1 (i.e., HIF-1α + ARNT, Fig. 6D, lane 5), but not with either protein individually (lanes 1 and 3). Formation of this complex was specifically competed by excess, cold wild-type HIF-1 binding oligonucleotide (W18, lane 7), but not by a mutant oligonucleotide (M18, lane 9) (Wang and Semenza 1995). A mutant 3× HRE probe from the p35srj promoter did not bind HIF-1 (lane 6).
These results show that p35srj is transcriptionally induced by hypoxia and deferoxamine, and that this induction could be mediated by HIF-1. Taken together with the previous results, they suggest that p35srj can function in a negative-feedback mechanism to regulate HIF-1 transactivation.
Sequestration of endogenous p35srj enhances endogenous HIF-1 function in vivo
To explore the function of endogenous p35srj, we attempted to specifically titrate endogenous p35srj (but not HIF-1α) away from endogenous p300/CBP. In preparation for such an experiment, a systematic linker scanning mutagenesis, replacing six consecutive amino acid residues of the p300–CH1 region with the peptide sequence, NAAIRS, was performed (A.L. Kung and D.M. Livingston, unpubl.). The NAAIRS linker can assume an α-helical or a β-sheet configuration, thereby minimally affecting protein folding when inserted at the site of a small deletion (Wilson et al. 1993; Sellers et al. 1998). A mutation replacing p300 amino acids 413–418 resulted in a mutant CH1 protein that bound tightly to in vitro translated p35srj, but was completely defective for binding to HIF-1α (Fig. 7A). A mutation that replaced p300 residues 371–376 bound very weakly to p35srj but, like CH1 413–418, appeared to be wholly defective for binding HIF-1α. We assayed the effects of overproducing these mutant p300–CH1 domains on the response of the VEGF promoter to DFO. Importantly, neither p35srj, nor HIF-1α, nor intact p300 were transfected in this experiment. As shown in Figure 7B, in the presence of both mutant p300–CH1 domains, there was clear DFO activation of the VEGF promoter. However, by comparison with the effect noted in cells cotransfected with the mutant CH1 species that bound p35srj poorly (CH1 371–376), a net increase in DFO-mediated pVEGF activation was observed repeatedly in the presence of the CH1 413–418 mutant, which strongly and specifically binds p35srj. These results imply that specific sequestration of endogenous p35srj by an ectopic CH1-containing polypeptide leads to enhanced activation of the VEGF promoter by endogenous HIF-1, which, in turn, was activated by deferoxamine. Hence, one can argue that p35srj participates in the modulation of an endogenous HIF-1 response to hypoxia or an equivalent, deferoxamine exposure.
Figure 7.

Sequestering p35srj enhances HIF-1 transactivation. (A) Effect of mutations in GST p300–CH1 on binding to HIF-1α and to p35srj. GST–CH1 wild type includes p300 amino acids 300–528. In GST–CH1–371–6, and CH1–413–8, the indicated residues were replaced with a NAAIRS sequence. The relative efficiency of binding of in vitro translated HIF-1α or p35srj to the wild-type and mutant GST–CH1 peptides was assayed. (B) Effect of mutant CH1 peptides on activation of VEGF promoter by deferoxamine. The mutants are the NAAIRS-substituted derivatives of CH1 (p300 residues 300–528) described in A. Hep3B cells were transiently cotransfected with a VEGF–luc reporter (40 ng), the indicated HA-tagged CH1 expression plasmids (300 ng each) and CMV–lacZ (100 ng). Results are presented as fold induction of the VEGF reporter by DFO relative to the uninduced activity. Two independent experiments are shown. The cells in the experiment represented at left were analyzed at a higher density than those at right. (C) Model: Putative roles of p35srj in HIF-1 transactivation.
Discussion
A novel, p300–CH1 interacting protein, p35srj, was cloned by an expression strategy. It is ubiquitously expressed, unstable, nuclear, and almost entirely bound directly to p300/CBP in vivo. However, most p300/CBP is apparently uncomplexed with p35srj, implying that variations in the p35srj protein level could, in principle, alter the level of free p300/CBP.
HIF-1α and p35srj bind directly to the p300/CBP-CH1 domain. p35srj blocked the HIF-1α–p300 interaction in vitro and in vivo, suggesting that the mechanisms by which p35srj and HIF-1α bind to p300–CH1 may be related. In keeping with this suggestion, both p35srj and HIF-1α have similar, negatively charged, leucine-rich p300–CH1 binding regions (data not shown). As a small peptide derived from the p35srj carboxy-terminal region also blocked the HIF-1α–p300 interaction, it is likely that the mechanism of the observed competition is direct occupancy of a CH1 binding site(s) rather than steric hindrance. The existence of p300 mutations that discriminate between p35srj and HIF-1α suggest that the two proteins do not occupy the same site on p300. They could however occupy overlapping sites. Alternatively, binding of p35srj might alter p300–CH1 structure, affecting its ability to bind to HIF-1α.
The data also suggest strongly that HIF-1α transactivation requires the efficient binding of its carboxyl terminus to p300/CBP–CH1, and that deferoxamine activation of HIF-1α appeared to be associated with marked enhancement of its affinity for p300–CH1. This could result from a conformational change in the HIF-1α carboxy-terminal region. p35srj inhibited basal and deferoxamine-activated HIF-1α carboxy-terminal region transactivation, as well as activation of the VEGF promoter by endogenous HIF-1. As the carboxy-terminal transactivating domain of HIF-1α is hypoxia/deferoxamine-activated, it appears that p35srj can interfere directly with the physiological association of p300/CBP with an important functional domain of HIF-1α TAD-C. Indeed, this increase in affinity and the marked increase in total HIF-1α level resulting from protein stabilization, would allow it to compete more efficiently with the constitutive, seemingly high-affinity CH1-interacting protein, p35srj, for access to limited quantities of p300/CBP. Such a mechanism would allow HIF-1α to recruit p300/CBP into a productive transcription complex, thereby increasing its transactivation potential.
Why there are two, independently activated HIF-1α domains is not clear. The amino-terminal transactivation domain (TAD-N) might function through p300/CBP, or it might contact the transcriptional machinery by another route. Conceivably, the two transactivation domains of HIF-1α may be involved in the fine-tuning of the hypoxia response through potential cooperative interactions with other vital components of the transcription machinery. The presence of TAD-N activity might explain why p35srj only partially inhibited HIF-1 transactivation (Fig. 5C), as opposed to its more marked inhibition of the carboxy-terminal transactivation domain (Fig. 5A,D). Indeed, we have observed that hypoxia, or deferoxamine activation of GAL4–HIF-1α (401–603) is not inhibited by p35srj (data not shown).
Taken together, the results presented in this report lead us to propose the following model (Fig. 7C). Hypoxia, and other activators such as deferoxamine, induce HIF-1α + ARNT complex formation and DNA-binding, and activate the carboxy-terminal transactivation domain of HIF-1α. This results in an increase in the affinity of the HIF-1α carboxyl terminus for p300/CBP–CH1. The increased affinity for CH1 and the increase in HIF-1α protein level allow it to compete more effectively with p35srj, and possibly other CH1-interacting proteins, for binding to CH1. Activated HIF-1α then signals through p300/CBP to the basal transcription complex, leading to the transcription of genes essential for the hypoxic response. These events also lead to increased transcription of p35srj, which is encoded by a gene bearing HIF-1 sites within its promoter. This, in turn, leads to accumulation of p35srj, which complexes with p300/CBP by binding to the CH1 region.
The increased binding of p35srj to p300/CBP following HIF-1 activation may have important functional consequences. p35srj could interfere with the HIF-1α p300/CBP interaction, by blocking functional p300/CBP–CH1 sites, resulting in a degree of negative feedback, and fine-tuning HIF-1 transactivation. In support of this hypothesis, we found that p300–CH1 species that can successfully titrate endogenous p35srj enhanced endogenous HIF-1 function reproducibly. Also, consistent with this model, the timing of hypoxia induction of erythropoietin and heme-oxygenase mRNAs is characterized by an increase followed by a decrease, indicating that the response becomes attenuated with time (Fandrey and Bunn 1993; Lee et al. 1997). Against this hypothesis is the fact that most of cellular p300/CBP is apparently not bound to p35srj (see Fig. 6B). A number of other factors, e.g., STAT2 (Bhattacharya et al. 1996), ets-1 (Yang et al. 1998), NFκB-p65 (Zhong et al. 1998), p53, and MDM2 (Grossman et al. 1998) bind to p300/CBP–CH1, and could occupy available binding sites. A recent study has in fact shown that STAT2 and NFκB compete for p300/CBP at the CH1 binding site (Hottiger et al. 1998). This indicates that functional p300/CBP–CH1 sites could be limited in vivo by occupying factors. Thus, in spite of there being a considerable excess of total p300/CBP over p35srj, variations in p35srj level could significantly affect the proportion of available functional CH1 sites in vivo, and thus modulate HIF-1 function. The transcriptional activation of a negative feedback regulator has been described in at least two other systems: NFκB and p53 transcriptionally activate the synthesis of their respective inhibitors, IκB-α and MDM2 (Le Bail et al. 1993; for review, see Ko and Prives 1996).
Our data indicate that p35srj is constitutively bound to p300–CH1. In the context of the aforementioned model, this may serve as a safety mechanism, restricting inappropriate access of HIF-1α molecules that are in the basal state, to p300/CBP. This may be important in preventing illegitimate transcription at promoters containing HIF-1 sites. Because p35srj can also inhibit the binding and function of at least one other CH1-dependent transcription factor (STAT2), it is possible that it serves to regulate transcription factor access to this region of p300/CBP more generally.
The specificity of the p35srj–CH1 interaction suggests that it may function to prevent titration of cellular p300/CBP by HIF-1. A number of studies have shown that transcription factors can compete for limiting amounts of p300/CBP, resulting in cross-talk between different signaling pathways (Kamei et al. 1996; Avantaggiati et al. 1997; Horvai et al. 1997). Moreover, studies of the Rubinstein-Taybi syndrome (Petrij et al. 1995), of heterozygous p300 and CBP knockout mice (Yao et al. 1998), indicate that partial loss of p300 or CBP function results in an abnormal phenotype. This, again, shows that p300/CBP proteins are functionally limiting in the cell.
Because p35srj does not affect the function of at least two, non-CH1 binding, p300-interacting transcription factors, it is conceivable that it reserves a subset of p300/CBP for use by these factors. Thus, p35srj may compartmentalize p300/CBP into certain discrete, functional subsets. This may be important for maintaining normal cellular function during hypoxia, possibly by limiting competition between HIF-1 and p300/CBP-dependent transcription factors binding at other sites than CH1 for use of the coactivation function of p300/CBP.
P35srj may also function to link certain transcription factors to p300/CBP, as is the case for src-1, which participates in the interactions between certain nuclear hormone receptors and p300/CBP (Hanstein et al. 1996; Yao et al. 1996). In keeping with this idea, the carboxyl terminus of p35srj/MRG1 contains a strong transactivation domain (Shioda et al. 1997), which can be inhibited by blocking p300/CBP function with adenoviral E1A (S. Bhattacharya and D.M. Livingston, unpubl.). Also supporting this idea is the observation that the related protein, MSG1, which contains a conserved, high-affinity p300/CBP-binding domain, has been shown recently to function as a coactivator for the transcription factor SMAD4 (Shioda et al. 1998), and could presumably link SMAD4 to p300/CBP-CH1. Thus HIF-1 activation may result in the generation of a p35srj–p300/CBP coactivator complex that could have an important role in cellular function.
In addition to a role in the HIF response, p35srj, and the related protein, MSG1, may have other, as yet unsuspected functions. Both p35srj and MSG1 likely play a significant role in development, as their mRNA levels are regulated during embryogenesis (Dunwoodie et al. 1998). Gene-targeting experiments will serve to distinguish the functions of these developmentally regulated p300/CBP-binding proteins and will further test the models of p35srj function, proposed above.
Materials and methods
Cloning
Expression cloning using GST–CH1 and a λgt11 HeLa cell library (Clontech) was performed as described previously (Bhattacharya et al. 1996). Standard cloning procedures (Ausubel et al. 1995) were used to obtain a full-length p35srj clone from a λZAP HEK293 cell library (gift of Bill Kaelin, Dana-Farber Cancer Institute). The HeLa and the longest 293 clone were sequenced completely. A human EST clone encoding MSG1 was obtained (EST accession no. 270311 N41476, Research Genetics) and was sequenced. The sequence of murine p35srj was deduced from the murine p35srj genomic sequence (S. Bhattacharya and D.M. Livingston, unpubl.). The p35srj promoter was cloned by screening a human λEMBL3 SP6/T7 genomic library (Clontech) with a 32P-labeled fragment of p35srj cDNA. Promoter fragments were subcloned into pBluescript (Stratagene) and sequenced.
Polyclonal and monoclonal antibodies
Anti-p35srj polyclonal and monoclonal antibodies (JA22) were raised by immunizing rabbits or mice with GST–p35srj (residues 66–270). JA22 is IgG1k, and its epitope maps between residues 66–124 of p35srj (S. Bhattacharya and D.M. Livingston, unpubl.). The antibodies do not cross-react with MSG1 (data not shown). Anti-p300/CBP antibodies (AC26, AC238, AC240, RW128, and RW105) have been described previously (Eckner et al. 1996). PAB419 is a monoclonal antibody to SV40 Tag and was a gift of Ed Harlow (MGH Cancer Center, Boston, MA). Anti-MIG polyclonal was obtained from Cappel.
Plasmids
Plasmids were generated using recombinant techniques (Ausubel et al. 1995). All mammalian expression plasmids used the CMV promoter. p35srj expression plasmids were constructed in the vector pcDNA3 (Invitrogen), and had a HA tag fused at the amino terminus. GAL4 and VP16 fusions were made in plasmid vectors, PCMXGAL4N, and PCMXVPN (gifts from Ron Evans, Salk Institute, San Diego, CA). Mutations were generated using a site-directed mutagenesis kit (Bio-Rad). p35srj promoter–reporter plasmids were constructed in pGL3-Basic (Promega). The p35srj 3× HRE–luc reporter (−1202/−1159) and mutant reporter plasmids were constructed in pTK–luc (gift of Tso-Pang Yao, Dana-Farber Cancer Institue). p300–CH1 linker-scanning mutants were generated by site directed mutagenesis, replacing six consecutive amino acid residues of the p300 CH1 region with an NAAIRS peptide linker. The mutant versions of CH1 were expressed in mammalian cells using the vector pCDNA3. Details of constructions are available upon request. E2–p300 and HA–p300 were described previously (Eckner et al. 1994; Arany et al. 1995). The E2–luc plasmid was a gift from Steve Grossman. The VEGF–luc reporter was a gift from Mark Goldberg (Arany et al. 1996). The GAL4–luc plasmid was a gift from Ron Evans. GAL4–SREBP2 (residues 1–330) was a gift from Jon Oliner and Robert Tjian (HHMI, Berkeley, CA). GAL4–src1 (residues 789–993) was a gift from Tso-Pang Yao. GAL4–STAT2 and GST–CH1 have been described previously (Bhattacharya et al. 1996). The HIF-1α and ARNT plasmids used to generate in vitro translates were gifts from Steve McKnight and Oliver Hankinson, respectively.
Immunoprecipitation, GST fusion-binding, Northern blotting, Western blotting, and gel-shift assays
All reagents were obtained from Sigma unless otherwise indicated. Northern blotting, Western blotting, and immunoprecipitations were performed using standard techniques (Ausubel et al. 1995) as described (Bhattacharya et al. 1996). The Northern blot in Figure 6A was a gift from Mark Goldberg (Brigham and Women’s Hospital, Boston, MA). p35srj Northern blots were probed with an EcoRI–EcoNI fragment encoding the amino terminus of the full-length human protein. GST fusion proteins were generated using pGEX vectors from Pharmacia, and in vitro binding reactions were performed as described previously, in buffer Hyb-75 + 1% milk (Bhattacharya et al. 1996). All immunoprecipitations were performed, unless otherwise stated, in buffer containing Tris at pH 8 50 mm, NaCl 150 mm, NP-40 0.5%, protease inhibitors (2 μg/ml leupeptin; 3.6 μg/ml aprotinin; 1 mm PMSF), and 0.5 mm DTT. Western blots for p35srj were transferred in 10 mm CAPS pH11/20% methanol buffer onto PVDF membranes (Immobilon), which were blocked in 5% milk in PBS at 37°C. Membranes were incubated with a 1:10 dilution of JA22 tissue culture supernatant in TBS-T (Tris-buffered saline 0.05% Tween 20, 1% BSA) overnight, washed with TBS, and probed with anti-mouse IgG–HRP conjugate (Amersham). For some immunoprecipitation–Western blot experiments, an anti-p35srj polyclonal antibody was used for Western blotting (1:1000 dilution) and probed using protein-A HRP (Amersham). p300/CBP were detected in Western blots using a cocktail of antibodies (AC238, AC26, RW128). GAL4 proteins were detected using mAb RK51 (Santa Cruz). Signals were detected by ECL (Amersham) following the instructions of the manufacturer. Gel-shift assays were performed as described (Wang and Semenza 1995) using 2.5 μl of programmed reticulocyte lysate as a source of HIF-1α or ARNT in a 20-μl reaction.
Immunostaining
p35srj immunostaining was performed using a method described previously (Eckner et al. 1994). JA22 tissue culture supernatent was routinely employed as a source of antibody at a 1:10 dilution. Alternatively, anti-HA polyclonal antibody (BabCO) in PBTB (PBS containing 0.1%Triton X-100, and 3% BSA) was used. Secondary antibodies were donkey anti-mouse IgG–FITC to detect the monoclonal antibody, and goat anti-rabbit rhodamine (both from Jackson Labs), to detect the rabbit polyclonal antibody. Nuclei were counterstained with Hoechst 33452 (2 μg/ml).
Pulse-chase experiments
Proliferating C2C12 cells (in 3-cm dishes) were washed twice using warm methionine-free medium and starved for 30 min in methionine-free medium containing 10% dialyzed bovine serum. They were pulsed with 1 mCi (15 μl)/ml of [35S]methionine for 10 min at 37°C, and washed three times with chase medium (20% FCS + 3 mg/ml cold excess methionine). Chased cells were collected at the indicated time points, lysed in RIPA buffer, and immunoprecipitated with anti-p35srj polyclonal antibody. Immunoprecipitates were fractionated by SDS-PAGE, and p35srj was visualized by autoradiography and quantitated using a Betascope.
In vitro peptide and protein competition/binding experiments
Baculoviruses were generated using pFastbacHTb (GIBCO BRL) following the recommendations of the manufacturer. Baculoviral proteins were obtained by infecting SF21 cells and then harvesting them in five-pellet volumes of immunoprecipitation buffer containing 180 mm NaCl. Cell lysates (protein content 3.5 mg/ml) were frozen after the addition of glycerol to 10%. SF21 lysates contained equal amounts of p35srj or p35srj mut (mutant) by Western blot (data not shown). All binding reactions were performed at 4°C. Competition experiments using baculovirally encoded proteins was performed using 20 μl of GST–CH1 or GST beads (50% by volume), and 100 μl of SF21 cell lysate in 300 μl of immunoprecipitation buffer. Beads were preincubated with SF21 lysate for 1 hr before adding in vitro-translated proteins (10 μl). Binding proceeded for a further 2 hr following which beads were washed three times in immunoprecipitation buffer, boiled in sample buffer, and fractionated by SDS-PAGE. The p35srj peptide (DEEVLMSLVIEMGLDRIKELPELWLGQNEFDF) and a control, irrelevant 25-mer peptide (ALTIQLIQNHFVDEYDPTIEDSYRK) were dissolved in DMSO, and used in the competition reactions as shown. For these experiments, GST–CH1 or GST, (1 μg of eluted protein), was preincubated with the indicated peptide (2–20 μg) for 5 hr in 300 μl Hyb-75 + 1% milk buffer. All reaction mixtures were constituted to contain equal amounts of the DMSO vehicle (final 1.76%), which did not interfere with binding. 35S-labeled, in vitro translated proteins were then added and binding reactions were carried out overnight, following which the relevant proteins were precipitated with glutathione–Sepharose beads, and analyzed as above.
Cells
Hep3B cells (gift of Eric Huang, Brigham and Women’s Hospital), were cultivated in α-MEM (GIBCO BRL) containing 10% fetal bovine serum in a 5% CO2-containing atmosphere, and passaged every three days. They were grown to confluence before addition of deferoxamine to 100 μm. U2OS cells (ATCC), and the Hep3B cells in Figure 6A, were cultivated in DMEM + 10% fetal clone (HYCLONE).
Transfections and luciferase assays
Hep3B cells were plated in 24-well plates at 2.5 × 104 cells per well, and were transfected the following day using Fugene 6 (Boehringer). Transfection mixtures contained 1 μl Fugene 6 and up to 0.5 μg of total plasmid per well, as recommended by the manufacturer. DFO (100 μm) was added the following day; cells were harvested 16 hr later; and luciferase and β-galactosidase activity (from a cotransfected CMV–lacZ reporter) analyzed as described previously (Bhattacharya et al. 1996). Luciferase activity was corrected for lacZ activity to normalize for transfection efficiency, and the data was presented as relative luciferase units (RLU). Amounts of DNA used per transfection refer to the amounts added per well of a 24-well plate. For immunostaining, U2OS cells were transfected on coverslips using the calcium-phosphate method, as described previously (Bhattacharya et al. 1996). For transfections U20S were plated as described previously (Bhattacharya et al. 1996).
Acknowledgments
We are indebted to N. Chandramouli (Novartis) for synthesizing the p35srj peptides and to Lidia Sambucetti and Dahlia Cohen for their help in obtaining these reagents and for their support and cooperation during the course of this work. We are very grateful to Frank Bunn and Mark Goldberg for discussions, and Bill Kaelin for the gift of the 293ZAP library. We also wish to thank Jim DeCaprio, Bill Sellers, Pang Yao, and Steve Grossman for their many helpful discussions, Jenny Gan for her expert help with generating monoclonal antibodies, Liz Oldread for superb technical assistance, and Richard Eckner for his help and advice during the initial part of this project and invaluable gifts of reagents. S.B. thanks Jane for her support. This work was funded by grants from the National Institutes of Health (NIH) to D.M.L., by an NIH Mentored Clinical Scientist Development Award and a Welkome Senior Fellowship Award to S.B., and by a grant from the Dana-Farber Cancer Institute/Novartis Drug Development Program to D.M.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
The GenBank accession number for the human p35srj sequence is AF109161.
Footnotes
E-MAIL David_Livingston@dfci.harvard.edu; FAX (617) 632-4381.
References
- Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel F, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short protocols in molecular biology. New York, NY: John Wiley & Sons; 1995. [Google Scholar]
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, Livingston DM. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-α. Nature. 1996;383:344–347. doi: 10.1038/383344a0. [DOI] [PubMed] [Google Scholar]
- Dunwoodie SL, Rodriguez TA, Beddington RSP. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech Dev. 1998;72:27–40. doi: 10.1016/s0925-4773(98)00011-2. [DOI] [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, Livingston DM. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes & Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- Eckner R, Ludlow JW, Lill NL, Oldread E, Arany Z, Modjtahedi N, DeCaprio JA, Livingston DM, Morgan JA. Association of p300 and CBP with simian virus 40 large T antigen. Mol Cell Biol. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandrey J, Bunn HF. In vivo and in vitro regulation of erythropoietin mRNA: Measurement by competitive polymerase chain reaction. Blood. 1993;81:617–623. [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- Guillemin K, Krasnow MA. The hypoxic response: Huffing and HIFing. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- Hanstein B, Eckner R, DiRenzo J, Halachmi S, Liu H, Searcy B, Kurokawa R, Brown M. p300 is a component of an estrogen receptor coactivator complex. Proc Natl Acad Sci. 1996;93:11540–11545. doi: 10.1073/pnas.93.21.11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen TM, Rose DW, Rosenfeld MG, Glass CK. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc Natl Acad Sci. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger MO, Felzien LK, Nabel GJ. Modulation of cytokine-induced HIV gene expression by competitive binding of transcription factors to the coactivator p300. EMBO J. 1998;17:3124–3134. doi: 10.1093/emboj/17.11.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 α. Genes & Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1α. Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- Kallio PJ, Pongratz I, Gradin K, McGuire J, Poellinger L. Activation of hypoxia-inducible factor 1α: Posttranscriptional regulation and conformational change by recruitment of the Arnt transcription factor. Proc Natl Acad Sci. 1997;94:5667–5672. doi: 10.1073/pnas.94.11.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Le Bail O, Schmidt-Ullrich R, Israel A. Promoter analysis of the gene encoding the I kappa B-alpha/MAD3 inhibitor of NF-κB: Positive regulation by members of the rel/NF-κB family. EMBO J. 1993;12:5043–5049. doi: 10.1002/j.1460-2075.1993.tb06197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. 1997;272:5375–5381. [PubMed] [Google Scholar]
- Levy AP, Levy NS, Wegner S, Goldberg MA. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem. 1995;270:13333–13340. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- Li H, Ko HP, Whitlock JP. Induction of phosphoglycerate kinase 1 gene expression by hypoxia. Roles of Arnt and HIF1α. J Biol Chem. 1996;271:21262–21267. doi: 10.1074/jbc.271.35.21262. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Andresen JM, Hansen SK, Zhou S, Tjian R. SREBP transcriptional activity is mediated through an interaction with the CREB-binding protein. Genes & Dev. 1996;10:2903–2911. doi: 10.1101/gad.10.22.2903. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, Tommerup N, Ommen GB, Goodman RH, Peters DJM, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Pugh CW. Oxygen and genes in health and disease [editorial] Quart J Med. 1997;90:307–310. doi: 10.1093/qjmed/90.5.307. [DOI] [PubMed] [Google Scholar]
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG., Jr Stable binding to E2F is not required for the retinoblastoma protein to activate transcription, promote differentiation, and suppress tumor cell growth. Genes & Dev. 1998;12:95–106. doi: 10.1101/gad.12.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Rue EA, Iyer NV, Pang MG, Kearns WG. Assignment of the hypoxia-inducible factor 1α gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics. 1996;34:437–439. doi: 10.1006/geno.1996.0311. [DOI] [PubMed] [Google Scholar]
- Shikama N, Lyon J, La Thangue NB. The p300/CBP family: Integrating signals with transcription factors and chromatin. Trends Cell Biol. 1997;7:230–236. doi: 10.1016/S0962-8924(97)01048-9. [DOI] [PubMed] [Google Scholar]
- Shioda T, Fenner MH, Isselbacher KJ. msg1, a novel melanocyte-specific gene, encodes a nuclear protein and is associated with pigmentation. Proc Natl Acad Sci. 1996;93:12298–12303. doi: 10.1073/pnas.93.22.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda T, Fenner MH, Isselbacher KJ. MSG1 and its related protein MRG1 share a transcription activating domain. Gene. 1997;204:235–241. doi: 10.1016/s0378-1119(97)00551-9. [DOI] [PubMed] [Google Scholar]
- Shioda T, Lechleider RJ, Dunwoodie SL, Li H, Yahata T, de Caestecker MP, Fenner MH, Roberts AB, Isselbacher KJ. Transcriptional activating activity of Smad4: Roles of SMAD hetero-oligomerization and enhancement by an associating transactivator. Proc Natl Acad Sci. 1998;95:9785–9790. doi: 10.1073/pnas.95.17.9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TE, Padgett KA, Johnston M, Milbrandt J. A genetic method for defining DNA-binding domains: Application to the nuclear receptor NGFI-B. Proc Natl Acad Sci. 1993;90:9186–9190. doi: 10.1073/pnas.90.19.9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Shapiro LH, Rivera M, Kumar A, Brindle PK. A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol Cell Biol. 1998;18:2218–2229. doi: 10.1128/mcb.18.4.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TP, Ku G, Zhou N, Scully R, Livingston DM. The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc Natl Acad Sci. 1996;93:10626–10631. doi: 10.1073/pnas.93.20.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T-P, Oh SP, Fuchs M, Zhou N-D, Ch’ng L-E, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]