

Abstract
Transcriptional silencing of the HM loci in yeast requires cis-acting elements, termed silencers, that function during S-phase passage to establish the silent state. To study the role of the regulatory elements in maintenance of repression, site-specific recombination was used to uncouple preassembled silent chromatin fragments from silencers. DNA rings excised from HMR were initially silent but ultimately reactivated, even in G1- or G2/M-arrested cells. In contrast, DNA rings bearing HML-derived sequence were stably repressed due to the presence of a protosilencing element. These data show that silencers (or protosilencers) are required continuously for maintenance of silent chromatin. Reactivation of unstably repressed rings was blocked by overexpression of silencing proteins Sir3p and Sir4p, and chromatin immunoprecipitation studies showed that overexpressed Sir3p was incorporated into silent chromatin. Importantly, the protein was incorporated even when expressed outside of S phase, during G1 arrest. That silencing factors can associate with and stabilize preassembled silent chromatin in non-S-phase cells demonstrates that heterochromatin in yeast is dynamic.
Keywords: Transcriptional repression, silencers, Saccharomyces cerevisiae, mating-type locus, Sir proteins, heterochromatin
Large regions of eukaryotic chromosomes are transcriptionally quiescent due to the packaging of DNA in repressive chromatin structures that are heritably propagated from one generation to the next. Heterochromatin, a constitutively condensed form of inactive chromatin, represents a primary example (Elgin 1996). Although heterochromatic regions are generally gene poor, heritable inactivation of either one of the two X chromosomes in female mammals involves formation of heterochromatin over the entire chromosome (Lee and Jaenisch 1997). In addition, chromosomal translocations that reposition active euchromatic genes near heterochromatin frequently result in a metastable form of repression that persists in subsequent generations.
Heritable inactivation of chromosomal domains in yeast Saccharomyces is typified by the HM loci (HMR and HML); in which endogenous copies of the mating- type genes are normally stored in a transcriptionally repressed state (Herskowitz et al. 1991). Inactivation of either HMR or HML involves a pair of flanking cis-acting regulatory sequences, referred to as the E and I silencers, that bind Rap1p, Abf1p, and the replication origin recognition complex (ORC) in various combinations (Loo and Rine 1995). An additional factor, Sir1p, is localized to silencers via protein–protein interactions (Triolo and Sternglanz 1996). Together, silencer-bound proteins recruit other Sir factors, Sir2p, Sir3p, and Sir4p, which associate with one another and histones throughout the repressed domain (Moretti et al. 1994; Hecht et al. 1995, 1996; Moazed and Johnson 1996; Strahl-Bolsinger et al. 1997). The resulting chromatin form, termed silent chromatin, bears many structural similarities to heterochromatin of higher eukaryotes. Both are assembled with hypoacetylated histones (Braunstein et al. 1993), both involve ORC as a DNA-binding component (Bell et al. 1993; Huang et al. 1998), and both are refractory to an array of DNA modification enzymes (Singh and Klar 1992; Loo and Rine 1994; Wallrath and Elgin 1995). This generalized chromatin inaccessibility, termed silencing in yeast, accounts for the block to transcription of both native and heterologous genes (Brand et al. 1985; Schnell and Rine 1986), as well as the diminished capacity for DNA repair (Terleth et al. 1989). Although heterochromatic structures are commonly assumed to be more or less static and inert, recent evidence suggests that cell cycle-dependent fluctuations occur. During mitotic chromosome condensation, much of the mouse heterochromatin protein HP1 is displaced from chromosomes (Murzina et al. 1999) and the accessibility of silenced yeast telomeric regions is increased in G2/M-arrested cells (Aparicio and Gottschling 1994).
A prevailing model for silencing has emerged in which repression consists of both establishment and maintenance phases. Accordingly, the establishment phase is one in which silent chromatin is reformed on nascent daughter duplexes following DNA replication. The maintenance phase, on the other hand, is one that sustains the silent state between successive establishment events. These concepts were first introduced by Miller and Nasmyth (1984) who used a conditional sir3 allele to show that de novo establishment of the silent state occurred during S-phase passage exclusively. Subsequent genetic studies isolated mutations in SIR1, RAP1, and silencers that impaired establishment but not maintenance of silencing (Pillus and Rine 1989; Mahoney et al. 1991; Sussel et al. 1993). In such mutants genotypically identical cells displayed variegated silencing phenotypes: In some cells the HM loci were “off”, whereas the loci were “on” in others. Switching between expression states occurred, albeit infrequently, indicating that the conditions that specify a particular state were reversible. The interpretation of this epigenetic behavior was that silencing could be maintained once established but that establishment in the mutants was an inefficient process. That all of these mutations were linked to silencers supported the notion that the elements were critical for establishment. Sir3p, on the other hand, was shown to be required continuously to maintain silencing; inactivation of the protein during any stage of the cell cycle led to immediate derepression (Miller and Nasmyth 1984). Therefore, Sir3p and other structural components that span the repressed domain (Sir2p and Sir4p) have come to be viewed as maintenance factors.
An unexpected role for silencers in maintenance of the silent state was first suggested by mutations in the ORC complex. Inactivation of conditional ORC subunits led to partial derepression of HMRa in G2/M-arrested cells, indicating that the silencer binding complex was required at times other than S phase for silencing (Fox et al. 1995). Complimentary data were obtained from more recent experiments in which preassembled silent chromatin domains were uncoupled from silencers by an inducible site-specific recombinase. Using this strategy, Holmes and Broach (1996) showed that removal of silencers from the chromosomal HMLα led to reactivation within a single cell cycle. In reciprocal studies by the Broach laboratory and our own, focus was placed on extrachromosomal rings that were excised from silent loci. Though initially silent, the nonreplicating rings were also not able to maintain silencing in the absence of silencers (Bi and Broach 1997; Cheng et al. 1998). In the case of HML-derived rings, cell cycle progression between G1 and G2 was required for reactivation (Bi and Broach 1997). A general view that emerges from this work is that derepression in the absence of silencers is triggered by a cell cycle specific event.
Here we capitalize on the DNA ring excision approach to investigate the relationship between silencers and silent chromatin stability. We show that the elements function continuously in cis to maintain silent chromatin, even in G1- and G2/M-arrested cells. Furthermore, we show that silencing proteins can be recruited to silent chromatin during G1 arrest. We propose that silencers act continuously throughout the cell cycle for this purpose. The experiments provide a unique and informative glimpse at the unexpected dynamic nature of silent chromatin.
Results
Maintenance of silencing at HMRa requires silencers
Previously, we found that DNA rings excised from HMR did not maintain silencing in logarithmically growing cells if the rings lacked silencers. Specifically, a ring-borne copy of the a1 gene was initially not expressed, yet it became fully activated within one to two doublings in cell density following excision. To determine whether reactivation required cell cycle progression, we measured the persistence of silencing in rings in non-cycling cells. To this end, chromosomal fragments containing the a mating-type genes, either with or without silencers (Fig. 1), were excised from HMR in cultures that had been treated with α-factor mating pheromone. Greater than 98% of cells arrested at G1 and remained there for the duration of the experiment, as confirmed by flow cytometry and a persistent unbudded shmoo morphology (data not shown). In the silencing-competent Δmat strains used here, no a1 transcript was detected prior to recombination (Cheng et al. 1998). However, in a strain containing a conditional sir3 allele, fully derepressed a1 levels were observed within 60 min of a shift to nonpermissive conditions (data not shown). These preliminary tests indicated that a1 gene expression could serve as a sensitive and rapid indicator of loss of silencing.
Figure 1.
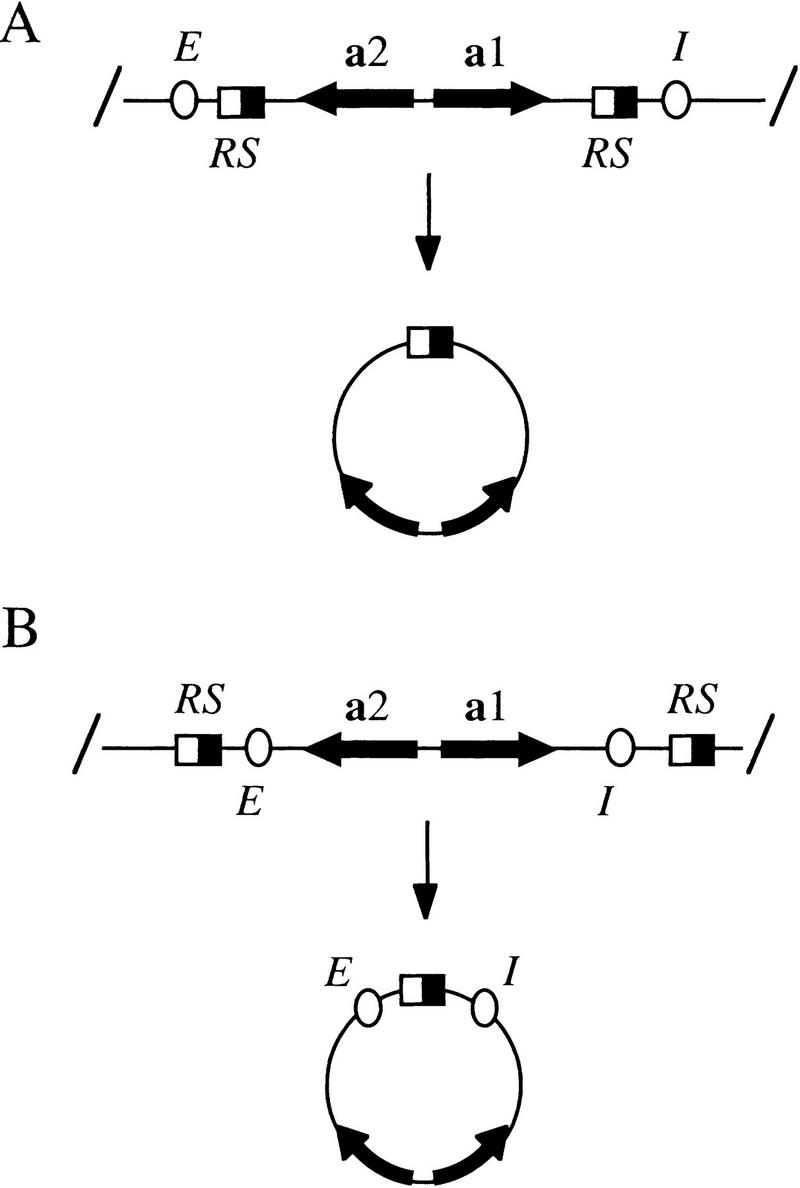
Excision of chromosomal fragments by site-specific recombination. RS target sites (half-filled boxes) for the Zygosaccharomyces rouxii R recombinase (Matsuzaki et al. 1990) were built into the HMR locus, either within the E and I silencers (A), or just beyond the E and I silencers (B) to produce rings that either lacked or contained the elements, respectively. In most laboratory strains, HMR contains the a1 and a2 mating-type genes (HMRa), whereas HML contains the α1 and α2 mating-type pair (HMLα). A third mating-type locus, MAT, contains an active copy of either pair of mating-type genes. In the various experiments described herein, either a mating type sequences, α mating type sequences, or a composite of the two were positioned at HMR for subsequent excision. All excision cassettes are named according to the rings they produce (e.g., locus hmr::rHMRa shown in A produces ring rHMRa).
Ring formation was initiated by galactose-induced expression of the R recombinase, and a1 mRNA levels were measured by Northern analysis at timed intervals thereafter. In a ring that lacked silencers, no a1 transcript was detected at the 60-min time point, indicating that the ring was initially repressed (Fig. 2A, lane 2). After 210 min, however, a1 mRNA began to accumulate, and by 360 min, the expression level approached that seen in a sir3 strain (lanes 1,3,4). In contrast, the a1 transcript was never observed if the ring contained both the E and I silencers (lanes 6–8). These data indicate that silencers are required for maintenance of repression in G1-arrested cells and that absence of the cis-acting elements results in deterioration of the repressed state in a time-dependent fashion.
Figure 2.
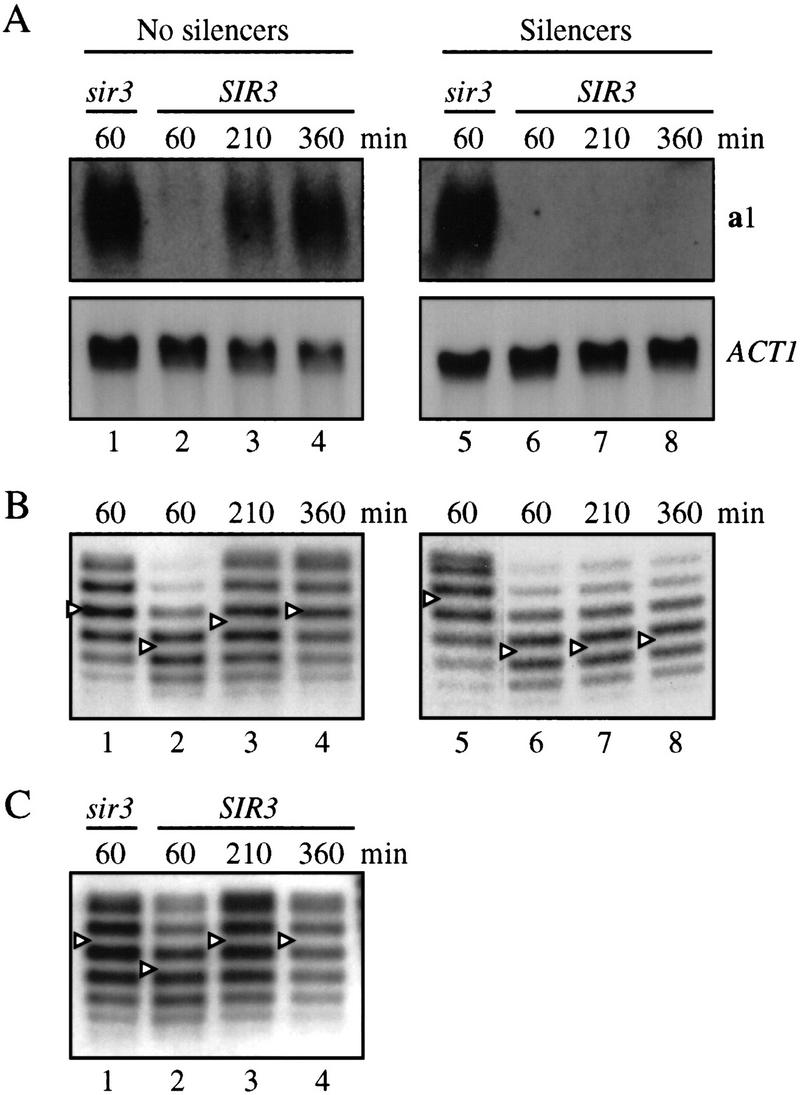
Maintenance of silencing requires silencers. DNA rings were excised from HMR by induction of recombination in cell cycle-arrested cultures. Strains THC78 hmr::rHMRa Δhml::kanMX Δsir3::HIS3 (lane 1) and THC43 hmr::rHMRa (lanes 2–4) produced rings that lacked silencers. Strains THC79 hmr::rHMRa+EI Δhml::kanMX Δsir3::HIS3 (lane 5) and THC37 hmr::rHMRa+EI (lanes 6–8) produced rings that contained silencers. HMLα was deleted in the sir3 strains; otherwise, expression of the locus would have prevented arrest by α-factor. MATa was deleted from all of the strains so that HMRa-derived rings provided the sole source of a1 mRNA. In cells arrested with α-factor or nocodazole, nucleic acids were harvested at timed intervals following galactose addition. (A) Northern analysis of a1 gene silencing in cells arrested by α factor in G1. Blots were hybridized sequentially with probes to a1 and the ACT1 gene. When normalized to ACT1, the levels of a1 mRNA in lanes 1 and 4 are comparable. (B) Chloroquine gel electrophoresis of DNA rings from α-factor-arrested cells. Centers of the topoisomer distributions were determined by the Gaussian method and marked with an arrowhead for clarity. Changes in DNA supercoiling were attributable to changes in chromatin structure and not due to the mechanics of transcription because rings that lacked promoters also bore SIR-dependent DNA topology changes (Cheng et al. 1998). (C) Analysis of a ring lacking silencers from cells arrested at G2/M with nocodazole.
The appearance of a ring-encoded a1 transcript suggested that changes in chromatin structure occur to relieve transcriptional repression following the removal of silencers. To test this idea more directly, we measured the level of supercoiling in excised DNA rings by electrophoresis in gels containing chloroquine. Previously, we showed that changes in the supercoiling of HMRa-derived rings correlated closely with transitions between silent and nonsilent states (Cheng et al. 1998). This is recapitulated in Figure 2B with rings from α-factor-arrested cells. At the earliest time point following excision (60 min), a ring lacking silencers was more negatively supercoiled by one to two turns when isolated from a SIR3 strain then when isolated from a sir3 strain (lanes 1,2). However, by 210 min the supercoiling shift of the ring from the SIR3 strain was slightly diminished, and by 360 min the supercoil density of the ring matched that of the ring from the sir3 strain (lanes 1,3,4). In contrast, the SIR-dependent supercoiling shift of the ring containing silencers did not change during the course of the experiment (lanes 6–8). These results indicate that silencers are required continuously in G1-arrested cells to maintain the alternate chromatin structure that is associated with transcriptional repression.
Cell cycle arrest in response to mating pheromones is mediated by a mitogen-activated protein (MAP) kinase pathway that triggers numerous physiological changes, including the hyperphosphorylation of Sir3 (Stone and Pillus 1996). Although activation of the pathway has been shown to strengthen telomeric silencing, repression of the extrachromosomal rings lacking silencers might be adversely affected. To test whether the persistence of silent chromatin structure was influenced by either α-factor treatment or G1 arrest, we examined the level of supercoiling of DNA rings in cells that had been arrested at G2/M with the microtubule destabilization agent nocodazole (Jacobs et al. 1988). Figure 2C shows that the supercoiling shift of a ring lacking silencers in nocodazole-arrested cells was similar to that in α-factor-arrested cells (cf. Fig. 2B). Sixty minutes after the induction of the recombinase, the ring was more negatively supercoiled when isolated from a SIR3 strain than when isolated from a sir3 mutant (lanes 1,2). At later time points, however, the altered supercoiling level of the ring reverted to that of the sir3 strain (lanes 3,4). Changes in the Sir-dependent supercoiling shift of rings that possessed silencers were not observed under these conditions (data not shown). Therefore, the role of silencers in maintaining silent chromatin is not restricted to cells arrested in G1 by α-factor. Rather, the results suggest that the cis-acting elements are required in a continuous manner throughout the cell cycle.
Maintenance of silencing at HMLα in the absence of E and I silencers
The observations described above are at apparent odds with those made by Holmes and Broach (1996), who showed that elimination of silencers from HMLα in G1-arrested cells did not disrupt silencing. The difference in persistence of transcriptional repression between HMLα and HMRa could be due to a host of factors, including long-range effects associated with either of the corresponding chromosomal domains or localized effects due to the specific sequences excised. To distinguish between these possibilities, we constructed a series of excision cassettes at HMR that contained sequences normally found at HMLα. The HML-based fragments were excised in G1-arrested cells, and persistence of the silent state was evaluated by measuring supercoil density of the resulting DNA rings. Figure 3 shows that the topoisomer distribution of a ring containing the α mating-type genes, but lacking silencers, remained constant during the 360-min experiment (lanes 2–4). At all times examined, the ring bore approximately one to two additional negative supercoils when isolated from a SIR3 strain than it did when isolated from a sir3 mutant. This result indicates that α genes, unlike a genes in Figure 2, maintain a silent chromatin structure upon excision and uncoupling from silencers at an HM locus.
Figure 3.
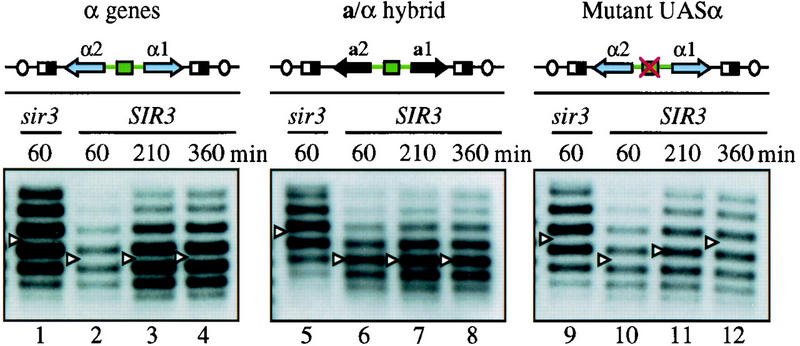
UASα maintains silencing in the absence of silencers. Following treatment with α-factor, DNA rings were formed in the following isogenic strain pairs: THC59 Δhmr::rHMRα Δsir3::HIS3 (lane 1), THC62 Δhmr::rHMRα (lanes 2–4); THC75 Δhmr::rHMRa/α Δsir3::HIS3 (lane 5), THC77 Δhmr::rHMRa/α (lanes 6–8); THC68 Δhmr::rHMRα(rap1pm) Δsir3::HIS3 (lane 9), THC69 Δhmr::rHMRα(rap1pm) (lanes 10–12). Diagrams of the excision cassettes are shown above each panel with UASα depicted as a green box. DNA rings were examined as in Fig. 2. Asynchronous cultures of THC59, THC75, and THC68 were used as nonsilent control because these sir3 strains contain HMLα and do not respond to α-factor. However, the supercoiling of rings in sir3 strains was influenced negligibly by α-factor arrest (data not shown).
Phenotypic evidence of persistent α gene repression was obtained to support the structural data. In MATa or Δmat haploid strains, α-factor treatment causes cells to arrest with an elongated shmoo morphology. If, however, the HMLα locus is derepressed (e.g., by inactivation of a conditional sir3 gene), cells adopt the a/α or α mating profile and rapidly escape α-factor-imposed arrest (Holmes and Broach 1996). Therefore, appearance of new buds in arrested cells can serve as a morphological marker for loss of silencing. A Δmat strain, THC62, was treated with α-factor, and a DNA ring containing a copy of the repressed α genes was excised from HMR. Greater than 98% of the cells adopted the shmoo morphology. Strikingly, no new buds emerged during the 6 hr incubation period following galactose-induced ring formation, indicating that expression of ring-borne α genes did not occur (data not shown). Together, these results show that silencing of the α genes, unlike the a genes, persists in G1-arrested cells in the absence of silencers.
A Rap1-binding site in the α gene UAS contributes to silencing at HML
The a and α mating-type loci are remarkably similar in primary sequence organization (Astell et al. 1981). The most notable difference is the Y region that spans the divergent a1/a2 promoters and the a1 gene at HMRa (Ya) versus the α1/α2 promoters and the α1 gene at HMLα (Yα). To identify the sequence determinants that facilitate maintenance of silencing at HML, an HMR/HML hybrid excision cassette was constructed in which the divergent a gene promoters were replaced with a fragment containing the α gene promoters (Fig. 3, middle). When this hybrid ring was excised from HMR in G1-arrested cells, the ring bore a SIR-dependent alteration in DNA supercoiling that did not change throughout the 360-min time course (lanes 6–8). This result indicates that a site within the α gene promoter region is sufficient for maintenance of preassembled silent chromatin.
We hypothesized that the α gene promoters recruited a protein that favored the persistence of silencing. A well-characterized binding site for the silencer binding protein Rap1p within the α gene UAS (UASα) represented a likely candidate. When the α genes are located at MAT, binding of the dual function protein to the site is required for expression (Giesman et al. 1991; Kurtz and Shore 1991). Footprinting studies have shown that a region encompassing the 15-bp UASα is not occluded by nucleosomes at HML, indicating that the site might be available to Rap1p, even within the silent chromatin (Weiss and Simpson 1998). To test whether the UASα contributes to silencing of the genes when they are located at an HM locus, we examined the supercoiling of a ring bearing a nonfunctional Rap1p-binding site. The central cytosine of the conserved CCC triplet of the Rap1 site was converted to an adenosine, a mutation that blocks transactivation of MATα in vivo and prevents the binding of Rap1p in vitro (Vignais and Sentenac 1989; Giesman et al. 1991). Sixty minutes after excision the mutated and nonmutated rings produced similar SIR-dependent supercoiling shifts (Fig. 3, lanes 2,10). At later time points, however, the ring with the mutant Rap1p-binding site reverted back to the nonsilent state (cf. lanes 11 and 12 to lanes 3 and 4). Together, these results indicate that the Rap1p-binding site in UASα is both necessary and sufficient for maintenance of the repressed state in rings that lack silencers.
A sensitive genetic assay was used to test whether UASα contributes to silencing at the chromosomal HML locus. A chimeric reporter gene in which the URA3 promoter was fused to the ADE2 ORF was integrated between α2 and the HMLE silencer (Fig. 4). Cells that express ADE2 give rise to white colonies on media containing low adenine, whereas cells that do not express the gene give rise to red colonies (Roman 1957). Cells containing URA3P–ADE2 at HML produced uniformly red colonies (Fig. 4, left), indicating that the reporter gene was silenced. When the gene was integrated at a derivatized HML locus that contained a point mutation in the UASα Rap1 site, a mixture of derepressed white colonies, partially repressed pink colonies, and fully repressed red colonies was observed (Fig. 4, right). Some colonies were either predominantly red or white but contained small sectors of the opposite color, indicating that a stable switch between expression states occurred during colony formation. The data reveal that the Rap1p-binding site in the α gene promoters is a significant contributor to repression of the genes when they are located at HML.
Figure 4.
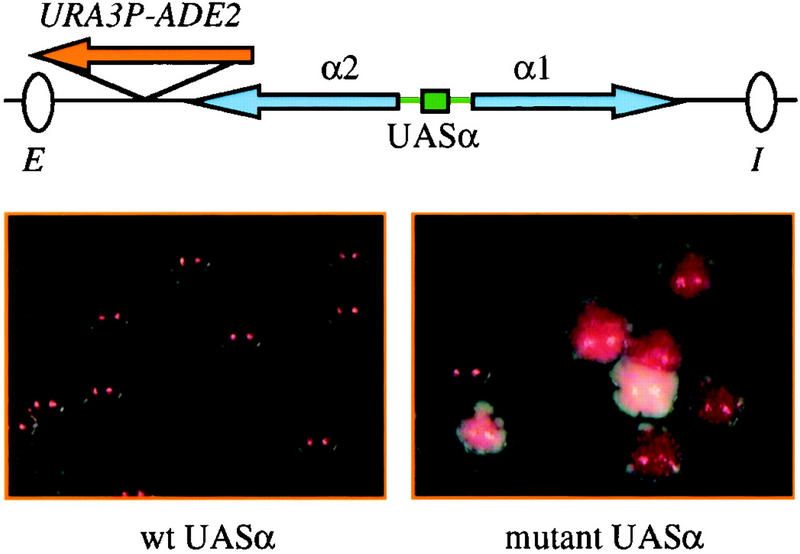
UASα contributes to silencing of the chromosomal HMLα locus. Strains THC74 hml::URA3P–ADE2 and THC76 hml(rap1pm)::URA3P–ADE2 were plated on SC media containing minimal adenine after overnight growth on nonselective media. Red or pink pigmentation corresponds to full or partial repression of ADE2, respectively. Unpigmented (white) colonies correspond to complete ADE2 derepression. Previously, the URA3P–ADE2 construct was used to monitor transcriptional repression at HMRa (Rivier et al. 1999). The URA3 promoter is up-regulated when uracil is omitted from the growth media. On SC plates lacking uracil, more frequent derepression was observed in the UASα mutant strain (data not shown).
Elevated levels of Sir3p and Sir4p stabilize rings of silent chromatin
Silencing of the HM loci is compromised when they are ectopically positioned far from telomeres where the Sir proteins are normally concentrated (Thompson et al. 1994; Maillet et al. 1996). The defect can be suppressed, however, by overexpressing a subset of the SIR genes. To test whether elevated SIR gene dosage would improve silencing in a ring that lacked silencers, a strain carrying the excision cassette shown in Figure 1A was transformed with CEN -based plasmids carrying SIR1, SIR2, SIR3, or SIR4. Maintenance of silencing was then measured in G1-arrested cells. Figure 5A shows that SIR-dependent supercoiling shift of the ring did not persist in cells containing empty vector or an extra copy of SIR1 or SIR2 (lanes 1–9). In all three cases, the rings reverted to the level of supercoiling associated with the derepressed state. This result indicates that neither Sir1p nor Sir2p is limiting for maintenance of silencing under these conditions. In contrast, the silent state was stabilized by an extra copy of either SIR3 or SIR4 (lanes 13–18). Particularly in the case of elevated SIR3 expression, supercoiling levels of the ring remained roughly constant throughout the duration of the experiment. Minor changes in the distribution of ring topoisomers probably reflect the presence of cells in the population that have lost the SIR expression vectors.
Figure 5.
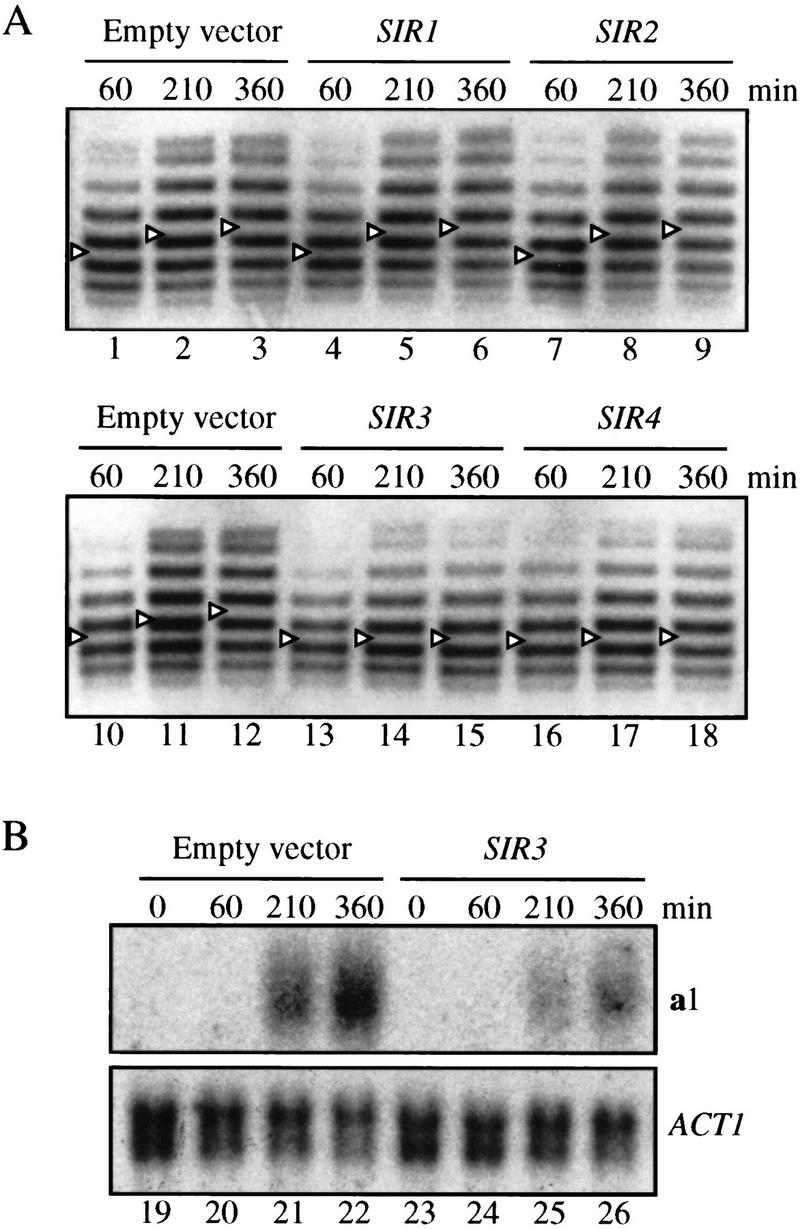
Stabilization of silent chromatin rings by elevated SIR gene dosage. Strain THC43 hmr::rHMRa was transformed with single copy, CEN-based vectors pJR910 (SIR1, lanes 4–6), pJR69 (SIR2, lanes 7–9), pJR273 (SIR3, lanes 13–15, 23–26), pJR368 (SIR4, lanes 16–18), or pRS416 (empty vector, lanes 1–3, 10–12, 19–22). In each case, the gene of interest was transcribed from its own promoter. Supercoiling of DNA rings (A) and RNA levels (B) were examined as in Fig. 2.
Northern analysis of the a1 transcript confirmed the structural data. Appearance of a1 mRNA following excision in strains containing an extra copy of SIR3 was greatly reduced relative to strains that contained empty vector (Fig. 5B). These results indicate that elevated expression of the Sir3p and Sir4p structural components of the silent chromatin increases the persistence of the repressive structure, even in the absence of silencers.
Preassembled silent chromatin is stabilized by Sir3p overexpression in G1
Stabilization of repressed rings by elevated Sir3p or Sir4p levels could occur during the establishment or maintenance phases of silencing. Conceptually, extra Sir proteins could facilitate the establishment of a more stable repressive structure. Alternatively, extra Sir proteins could add to and stabilize a pre-existing repressive structure. To determine whether elevated Sir3p increases the stability of preassembled silent chromatin, we induced expression of the protein in α-factor-treated cultures. Arrest with the pheromone in G1 prevents cells from transiting through S phase, the period during which establishment of silencing is known to occur.
Induction of Sir3p was achieved with a chromosomally integrated SIR3 gene fused to the GAL10 promoter (GAL10P–SIR3). This tightly regulated construct provided functional Sir3p at nontoxic levels when induced (see Materials and Methods). Following uniform G1 arrest, Sir3p and recombinase production were induced simultaneously by the addition of galactose. Recombination yielded the ring shown in Figure 1A. In a SIR3 strain, production of additional Sir3p prevented loss of silencing of the ring (Fig. 6). The initial SIR-dependent DNA supercoiling level of the ring was maintained throughout the duration of the experiment (lanes 4–6). This result indicates that elevation of Sir3p levels can stabilize preassembled silent chromatin, even without passage through S phase.
Figure 6.

Stabilization of silent chromatin rings by elevated SIR3 expression in G1-arrested cells. Strains THC43 hmr::rHMRa (lanes 1–3), THC57 hmr::rHMRa ura3-1::GAL10P–SIR3::URA3 (lanes 4–6), and THC70 hmr::rHMRa Δsir3::HIS3 ura3-1::GAL10P–SIR3HA::URA3 (lanes 7–9) containing recombinase expression vector pHM153 were arrested with α-factor. Subsequently, expression of the R recombinase gene and SIR3 were induced simultaneously with galactose. Supercoiling of DNA rings was examined as in Fig. 2.
To test whether non-S-phase expression of Sir3p could silence a locus that was initially derepressed, we induced GAL10P–SIR3 in a sir3 mutant strain. As before, cells were uniformly arrested in G1 with α-factor and coproduction of recombinase and Sir3p was induced. Upon excision, the ring bore a nonsilent DNA topology (Fig. 6; cf. lanes 3 and 7) This condition was not changed by >6 hr of Sir3p expression (lanes 8,9). Therefore, Sir3p cannot impart silencing de novo to a derepressed locus in noncycling cells. Apparently, stabilization of silent chromatin by Sir3p in non-S-phase cells requires a pre-existing silent chromatin to be in place (lanes 4–6). The result is consistent with a requirement for passage through S phase to establish silencing (Miller and Nasmyth 1984; Fox et al. 1997). It seems likely that some specialized feature of chromatin assembly, such as histone deposition or modification, represents the critical S-phase event in the establishment of silent chromatin.
Association of Sir3p with silent chromatin does not require S-phase passage
Stabilization of the silent chromatin by Sir3p expression in non-S-phase cells could occur directly by incorporation of the protein into chromatin or by less direct means. To determine whether the protein was incorporated into preassembled silent chromatin structure, chromatin immunoprecipitation assays (ChImp assays) were performed with an induced, HA-tagged Sir3 derivative, Sir3HAp. Following uniform G1 arrest, the epitope-tagged protein was expressed from a GAL10P–SIR3HA gene fusion. Immunoprecipitations were performed with anti-HA antibody and sonicated extract from formaldehyde cross-linked cells. Coimmunoprecipitation of representative genomic loci, including the a1 gene at HMRa, as well as GAL1, ACT1, and PHO5, were evaluated by PCR using corresponding primer pairs. Figure 7 shows that a1 was selectively precipitated upon induction of SIR3HA in a SIR3 strain (lane 3). Precipitation of the gene did not occur if Sir3HAp was not induced or if the epitope was removed from the expression vector (lanes 1,2). Finally, Sir3HAp was not incorporated into chromatin at a1 in a sir3 strain (lane 4), in agreement with the DNA supercoiling data in Figure 6 (lanes 7–9). These data show that incorporation of Sir3p into the silent chromatin is not restricted to S phase. The results suggest that yeast heterochromatin is a dynamic structure that possess the ability to exchange chromatin-bound Sir3p for soluble unbound protein throughout the cell cycle.
Figure 7.

Incorporation of Sir3p into silent chromatin in G1-arrested cells. Strains THC57 ura3-1::GAL10P–SIR3::URA3 (lanes 1,5), THC67 ura3-1::GAL10P–SIR3HA::URA3 (lanes 2,3,6,7), and THC70 Δsir3::HIS3 ura3-1::GAL10P–SIR3HA::URA3 (lanes 4,8) were arrested with α-factor. Following induction of SIR3 expression with galactose, cells were treated with formaldehyde. Processed samples were immunoprecipitated with anti-HA antibody, and the precipitated material was probed for the presence of DNA with PCR using primer pairs specific for the a1, GAL1, ACT1, and PHO5 genes. The MATa locus was deleted from all of these strains so that HMRa provided the sole copy of a1 DNA. The positions of DNA mobility standards are marked at left.
Discussion
Silencers are required continuously to maintain silent chromatin
Heritable propagation of the stably repressed state at HM loci requires that (1) a heterochromatin-like structure be re-established following each round of DNA replication, and (2) that the structure be maintained throughout the duration of the cell cycle. A role for silencers in establishment of silencing is already well appreciated. Relocation of silencers to some ectopic loci can result in the silencing of adjacent genes (Lee and Gross 1993; Shei and Broach 1995; Maillet et al. 1996). In this report we have examined the role of silencers in the maintenance of the silent state and found that the elements contribute a critical function at this stage too. By unlinking fragments of preassembled silent chromatin from silencers in vivo, we have shown that silencers are required in cis to preserve silent chromatin; in G1-arrested cells, removal of silencers resulted in reactivation of silenced genes and loss of SIR-dependent alterations in DNA supercoiling (Fig. 2). Similar observations were made in cells arrested at G2/M using nocodazole. These findings demonstrate that silencers act in a continuous manner to maintain the structure and function of silent chromatin.
A protosilencer maintains silencing in the absence of silencers
The conclusion that silencers maintain silent chromatin is supported by our parallel studies of the a and α mating-type genes positioned at HMR. Unlike the situation with the a genes, uncoupling of the α gene pair from flanking silencers resulted in silent DNA rings that were stably repressed in arrested cells (Fig. 3). Persistent silencing in this case relied on a cis-acting silencing element located within the α gene promoter. By point mutation and add-back experiments we showed that the Rap1-binding site constituting UASα was both necessary and sufficient for repression in the absence of silencers. On the basis of this observation, we conclude that silencers, as well as individual silencer binding site sequences, are sufficient to maintain the silent state. Isolated binding sites for silencer binding proteins, termed protosilencers, although lacking intrinsic silencing function, have been shown to interact cooperatively with intact silencers to strengthen silencing (Boscheron et al. 1996). Specifically, repression of a sensitive reporter gene by a sole silencer was aided by tethering either Rap1p, Abf1p, or ORC to a DNA site 4 kb away. By inference, it was posited that the Rap1p-binding site at UASα might contribute similarly to silencing at HML. That UASα performs this function in a near native chromosomal context was demonstrated by comparing wild-type and mutant UASα sequences in a colony color assay for derepression (Fig. 4). The Rap1 protein (repressor/activator protein) derived its name from the discovery that it contributes to either transcriptional repression when bound to silencers or transcriptional activation when bound at the promoters of numerous genes, including MATα (Shore 1994). UASα provides the first example of a specific Rap1p site that possesses dual function, either activating or repressing the associated gene/genes, depending on the chromosomal context of the gene/genes.
Holmes and Broach (1996) have argued that maintenance of repression does not require silencers because derepression of the α genes did not occur when they removed the E and I silencers from HML in G1-arrested cells. This conclusion must now be reconsidered in light of the discovery that UASα functions as a protosilencer. Nonetheless, silencers were shown to be required for inheritance of the silent state; following removal of the elements, reactivation of HML occurred within a single cell cycle, particularly during passage between the G1 and G2 stages of the cell cycle (Holmes and Broach 1996; Bi and Broach 1997). Remarkably, reactivation occurred even in the absence of DNA replication of the silent template. We too have found that nonreplicating DNA rings containing the UASα protosilencer reactivated during this interval (T.-H. Cheng, unpubl.). Although critical constructs of Bi and Broach (1997) lacked UASα, it should be noted that they did contain regions of HML and bacterial DNA fragments not present in our clones. It is possible that a sequence with protosilencer activity resided within this additional DNA. In either case, the results indicate that protosilencers on their own are not capable of propagating the silent state. Whereas the elements can prevent loss of silencing in G1, they do not suffice during subsequent cell cycle progression. In this regard, bona fide silencers appear either to be more efficient or to provide additional functions.
Silent chromatin is dynamic
How do silencers function in the preservation of silent chromatin? The answer may lie in the relationships between Sir protein availability, recruitment, and silent chromatin stability. Sir proteins have been shown previously to be limiting for repression at HM loci due to competition for the factors by telomeres where Sir-mediated repression also occurs (Aparicio et al. 1991; Buck and Shore 1995; Marcand et al. 1996). In a number of studies, alteration of the level of free Sir2p, Sir3p, or Sir4p influenced either the efficiency of the silencing of reporter constructs or the span of the silenced domain (Renauld et al. 1993; Sussel et al. 1993; Maillet et al. 1996; Enomoto and Berman 1998). Here, persistence of silencing in DNA rings was shown to be extended by increasing the levels of Sir3p and Sir4p (Figs. 5 and 6). The striking feature was that stabilization by elevated Sir3p did not require passage through S phase. Moreover, stabilization appeared to be the result of direct incorporation of the protein into the repressive structure (Fig. 7). These findings demonstrate that recruitment of Sir factors can occur during maintenance of the silent state, as well as during its establishment. Together with the facile loss of silent chromatin upon removal of silencers, the observations indicate that silent chromatin is dynamic with critical components, such as Sir3p, equilibrating on and off the structure. Therefore, a reasonable role for silencers during maintenance may be to continually recruit new Sir proteins. For example, if silencing components turn over naturally, due either to dissociation or degradation, cis-acting elements would promote replenishment of the depleted components. Recruitment could be facilitated by direct protein-protein interactions with silencer-bound proteins, as described above, as well as by silencers targeting silent chromatin to regions of the nucleus that are enriched in Sir proteins (Andrulis et al. 1998).
Silencers are not likely to act alone in recruitment of the Sir proteins. A large network of protein–protein interactions could favor binding of free Sir proteins by those already bound. Sir3p and Sir4p form homomeric and heteromeric complexes (Chien et al. 1991; Moretti et al. 1994), and Sir2p and Sir4p also form a complex (Moazed and Johnson 1996; Strahl-Bolsinger et al. 1997). In addition, Sir3p and Sir4p bind preferentially to hypoacetylated amino-terminal tails of histones H3 and H4 (Hecht et al. 1995), which are enriched at the silent loci (Braunstein et al. 1993). Finally, tethering Sir3p and Sir4p to DNA directly leads to repression of adjacent genes (Lustig et al. 1996; Marcand et al. 1996). This last example demonstrates that silencers can be bypassed if a high local concentration of Sir protein is maintained. Conceivably, pre-existing silent chromatin could be propagated by self-recruitment if the intracellular concentration of Sir proteins was elevated. In flies and humans, such a mechanism has been proposed to explain the epigenetic behavior of kinetochores, the specialized chromatin-based structures that segregate chromosomes (Murphy and Karpen 1998; Wiens and Sorger 1998, and references therein). Functional kinetochores sometimes assemble on noncentromeric locations, where they are propagated in a heritable fashion despite the lack of discernible centromeric DNA sequences. Perpetuation of this class of kinetochores may rely entirely on self-templating by the pre-existing chromatin structure. In yeast, Sir2p and Sir3p must be maintained at low levels because they are toxic, potentially due to the promiscuous silencing of critical genes (Holmes et al. 1997). Therefore, silencers may have evolved to recruit Sir proteins efficiently and specifically to the HM loci in an environment where telomeres and other sites, such as rDNA (Smith et al. 1998), compete for limited pools of Sir proteins.
Enomoto and Berman (1998) showed that maintenance of silent chromatin was influenced by mutations in a replication-coupled chromatin assembly factor encoded by the CAC genes. In G1-arrested cac mutants, derepression of the mating-type loci in individual cells was recorded with a sensitive time-lapse microscopy assay. These investigators argued that a defect during silent chromatin assembly was manifest in a metastable repressive structure that could not be maintained appropriately. Given our results, an alternative testable hypothesis is that the chromatin assembly factor acts directly on preassembled silent chromatin in G1-arrested cells to maintain the silent state. In support of this notion, the mouse Cac1p homolog CAF-1 p150 was shown recently to associate with heterochromatin in non-S-phase cells (Murzina et al. 1999).
Maintenance of repressed chromosomal domains in other organisms
Variegated repression of genes adjacent to heterochromatin is thought to occur by the stochastic but stable spread of heterochromatic structure into adjacent DNA. It was shown recently that reporter genes subject to this form of repression in Drosophila reactivated upon excision from the genome, even in mitotically quiescent cells (Ahmad and Golic 1996). This observation indicates that maintenance of heterochromatic repression in flies, like yeast, requires preservation of proper genomic context. It is not clear whether loss of continuity with chromosomal heterochromatin or nuclear compartmentalization accounts for reactivation in the Drosophila studies. However, recombination-based studies with engineered excision cassettes hold promise of identifying cis-acting sequences sufficient for maintenance of heterochromatic repression.
Maintenance of the inactive X chromosome (Xi) in female mammals is notably different from examples in yeast and flies. Like yeast, establishment of the repressed state requires a cis-acting regulatory element, the X inactivation center (XIC). However, inactivation of Xi is heritably propagated following removal of XIC, indicating that the element is not required for maintenance of the repressed state (Brown and Willard 1994). Xi is structurally distinct in numerous ways, including differential DNA methylation, histone acetylation, and the presence of an RNA chromatin component (Lee and Jaenisch 1997, and references therein). Any one of these features could participate in a self-templating mechanism to propagate heterochromatic repression. It also seems possible that the X chromosome contains stabilization elements, like protosilencers of yeast, that promote maintenance of repression but that lack the ability to establish repression on their own. Based on Xi-autosome translocation data, Gartler and Riggs (1983) postulated early on that the X chromosome contains stabilization elements that serve as “booster” sites to help spread heterochromatic structure throughout Xi. It was posited recently that long interspersed nuclear elements (LINEs), which are enriched in the X chromosome (Boyle et al. 1990), function in this way (Lyon 1998). Precedent for booster sites is found in yeast where native protosilencers within subtelomeric repeat sequences propagate silencing away from chromosomal termini (Fourel et al. 1999; Pryde and Louis 1999). Although the mechanisms of booster site action may differ widely between humans and yeast, the underlying requirement for amplification of a silencing signal along the chromosome appears to be conserved.
Materials and methods
Strain and plasmid constructions
pΔhmra::rHMRα was constructed by replacing the entire a gene fragment within the excision cassette of phmr::rA1A2 (Cheng et al. 1998) with a PCR fragment containing the α genes from HML (chromosome III coordinates 11695–14018). pΔhmra::rHMRα(rap1pm) was derived from pΔhmra::rHMRα by using overlap PCR to introduce a C-to-A transversion in the Rap1p-binding site of UASα (see Results). pΔhmr::rHMRa/α was obtained by inserting a PCR fragment containing the α gene divergent promoters (coordinates 12909–13332) into phmr::rA1A2Δp at a BamHI site that replaced the a gene promoters (Cheng et al. 1998). pRS406GAL10P–SIR3 was obtained by ligating a EcoRV–BamHI fragment that contained a GAL10P–SIR3 chimeric gene from pAR42 (S. Holmes, Wesleyan University, Middletown, CT) into the multiple cloning site of pRS406. pRS406GAL10P–SIR3HA was generated by replacing the carboxyl terminus of SIR3 in pRS406GAL10P–SIR3 (XbaI–XhoI) with an HA-tagged version from pRS416–SIR3HA (Ansari and Gartenberg 1999). phmlα::URA3P–ADE2 was derived from plasmid puc19–HML (D. Shore, University of Geneva, Switzerland) by replacing the EagI–ClaI fragment of HML with a 2.0-kb, PCR-amplified fragment that contained the URA3P–ADE2 chimeric gene from plasmid pDR859 (D. Rivier, University of Illinois, Urbana-Champagne). phmlα(rap1pm)::URA3P–ADE2 was constructed by replacing the EagI–BlpI fragment of phmlα::URA3P–ADE2 with the mutagenized version from pΔhmr::rHMRα(rap1pm). pΔmata::TRP1 was created by replacing the ApaI–SmaI fragment of pΔmat::URA3 with a 1.6-kb fragment (ApaI–SnaBI) containing the TRP1 gene from pRS414 (Cheng et al. 1998). Plasmids pJR910, pJR69, pJR273, and pJR368 were constructed in the laboratory of J. Rine (UC, Berkeley) and provided by J. Berman (University of Minnesota, St. Paul).
Unless specified otherwise, all strains were constructed by the one-step gene disruption method and confirmed by Southern hybridization. THC42 was derived from THC23 in three steps: (1) LYS2 was disrupted with plasmid pUC18–Δlys2 (Cheng et al. 1998); (2) BAR1 was disrupted with plasmid pTM47 (Menees and Sandmeyer 1994); and (3) MATa was disrupted with pΔmata::TRP1. THC78 was derived from THC42 by replacing the α genes at HMLα with a PCR-amplified kanMX gene from plasmid pUG6 (Wach et al. 1994). THC51 was derived from THC42 by replacing the a genes at HMRa with URA3, as described previously (Cheng et al. 1998). THC59, THC68, and THC75 were derived from THC51 by replacing the Δhmr::rURA3 locus with modified hmr loci from plasmids pΔhmra::rHMRα, pΔhmra::rHMRα(rap1pm), and pΔhmra::rHMRa/α, respectively. THC43, THC62, THC69, and THC77 were derived from THC42, THC59, THC68, and THC75, respectively, by regeneration of chromosomal SIR3 using pAR3. THC57 and THC67 were derived from THC43 by targeted integration of one copy of pRS406GAL10P–SIR3 and pRS406GAL10P–SIR3HA, respectively, into ura3-1. THC70 was derived from THC78 by targeted integration of one copy of pRS406GAL10P–SIR3HA into ura3-1. THC37 was derived from YCL1 (Cheng et al. 1998) by disrupting MATa with pΔmat::URA3. THC79 was derived from YCL2 (Cheng et al. 1998) by disrupting MATa with pΔmat::URA3 and replacing the α genes at HMLα with kanMX. Strains THC74 and THC76 were derived from PJ1 (W303-1A URA3) by replacing HMLα with modified loci in plasmids phmlα::URA3P–ADE2 and phmlα(rap1pm)::URA3P–ADE2, respectively. Selection of PJ1 derivatives was aided by prior transformation with an “anti-sir” plasmid, pCTC23 (Chien et al. 1998), which was subsequently evicted. Note that chromosomal excision cassettes hmr::rHMRa and hmr::rHMRa+EI previously were named hmr::rA1A2 and hmr::rHMR, respectively (Cheng et al. 1998).
Cell growth and analysis of nucleic acids
Strains were transformed with a recombinase expression vector (pHM153) and grown at 30°C in synthetic dropout media containing 2% raffinose. At mid-log phase, cells were treated for 3 hr with 2 μg/ml α-factor (Sigma) for Δbar1 strains and 10 μg/ml pheromone for BAR1 strains. Persistent α-factor-mediated arrest in G1 was confirmed by flow cytometry and/or visual inspection of cell morphology. Nocodazole-mediated arrest at G2/M was achieved by treatment with 10 μg/ml nocodazole (Sigma) for 3 hr. Approximately 90% of the cells arrested with large buds (dumbbell shaped), indicative of G2/M block, and remained in this configuration for the duration of the experiment.
DNA ring formation was induced with galactose (Cf = 2%), and parallel aliquots of culture were used for both Northern blots and DNA supercoiling analyses. Isolation, electrophoresis, and detection of nucleic acids were described previously (Cheng et al. 1998). Topoisomer distributions were evaluated by the Gaussian method following electrophoresis in gels containing 2 μg/ml chloroquine, such that more negatively supercoiled rings migrated more rapidly (Depew and Wang 1975).
Colony color assays
Uniformly red colonies of strains THC74 and THC76 were grown overnight in nonselective liquid media (YPDA) and plated on SC plates that contained limiting adenine (6 μg/ml). Following 2 days of incubation at 30°C, plates were stored for 3 days at 4°C for enhanced color development.
Characterization of integrated GAL10P–SIR3 fusions
Overexpression of SIR2 and SIR3 from high copy vectors is cytotoxic (Holmes et al. 1997). To verify that expression of a single integrated GAL10P–SIR3 fusion gene was not overtly deleterious, SIR3 strains containing or lacking the chimera were grown side by side on plates containing galactose. In both cases, equivalent numbers of colonies appeared and grew at equivalent rates. Galactose-induced expression of either the GAL10P–SIR3 or GAL10P–SIR3HA was sufficient to suppress the mating defect in a sir3 strain. When the strains were grown in raffinose, however, no silencing could be detected with a quantitative mating assay and Sir3HAp could not be visualized by Western blot analysis. Together, these data show that the integrated GAL10P–SIR3 chimeras are functional, tightly regulated, yet not harmful.
ChImp assays and PCR analysis
ChImp assays were performed essentially as described in Aparicio et al. (1997), with the following exceptions. Following cell cycle arrest with α-factor, GAL10P–SIR3HA expression was induced by galactose addition. After a 45-min incubation, cells were treated with formaldehyde and chromatin-containing extracts were prepared. Epitope-tagged Sir3p was immunoprecipitated with anti-HA monoclonal antibody HA.11 (BAbCO, Richmond, CA) bound to protein A–Sepharose CL-4B beads (Pharmacia, Piscataway, NJ). PCR reactions were performed with either 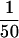 of the precipitated DNA or
of the precipitated DNA or 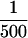 of the input material. Two pairs of gene-specific primers were used simultaneously in each reaction (A1-1/A1-2 for the a1 gene and GAL1-1/GAL1-2 for the GAL1 gene, or ACT-1/ACT1-2 for the ACT1 gene and PHO5-1/PHO5-2 for the PHO5 gene). Primer sequences are as follows: A1-1 (5′-ATGGATGATATTTGTAGTATGGCG-3′); A1-2 (5′-GGTGGTATATTTCTAACCTATTGTTAG-3′); GAL1-1 (5′- CTGCAAGTCTTCTGTGAGG-3′); GAL1-2 (5′-GATACAACAAGGGTGTTCGC-3′); ACT1-1 (5′-AGACCAAGACACCAAGGTATC-3′); ACT1-2 (5′-GAGTACAAGGACAAAACGGCT-3′); PHO5-1 (5′-ACTTGACCTCAACTGACGC-3′); and PHO5-2 (5′-AGGATATCGGTATCGTGGG-3′). Twenty-five cycles of PCR were performed with an annealing temperature of 51°C. PCR products were separated by agarose gel electrophoresis, stained with EtBr, and photographed using Polaroid 665 film.
of the input material. Two pairs of gene-specific primers were used simultaneously in each reaction (A1-1/A1-2 for the a1 gene and GAL1-1/GAL1-2 for the GAL1 gene, or ACT-1/ACT1-2 for the ACT1 gene and PHO5-1/PHO5-2 for the PHO5 gene). Primer sequences are as follows: A1-1 (5′-ATGGATGATATTTGTAGTATGGCG-3′); A1-2 (5′-GGTGGTATATTTCTAACCTATTGTTAG-3′); GAL1-1 (5′- CTGCAAGTCTTCTGTGAGG-3′); GAL1-2 (5′-GATACAACAAGGGTGTTCGC-3′); ACT1-1 (5′-AGACCAAGACACCAAGGTATC-3′); ACT1-2 (5′-GAGTACAAGGACAAAACGGCT-3′); PHO5-1 (5′-ACTTGACCTCAACTGACGC-3′); and PHO5-2 (5′-AGGATATCGGTATCGTGGG-3′). Twenty-five cycles of PCR were performed with an annealing temperature of 51°C. PCR products were separated by agarose gel electrophoresis, stained with EtBr, and photographed using Polaroid 665 film.
Table 1.
Yeast strains
| W303-1A | MATa HMLα HMRa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 |
| THC23 | W303-1A hmr::rHMRa Δsir3::HIS3 |
| THC37 | W303-1A Δmat::URA3 hmr::rHMRa + EI |
| THC42 | W303-1A Δmat::TRP1 hmr::rHMRa Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC43 | W303-1A Δmat::TRP1 hmr::rHMRa Δbar1::hisG Δlys2 |
| THC51 | W303-1A Δmat::TRP1 Δhmr::rURA3 Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC57 | W303-1A Δmat::TRP1 hmr::rHMRa ura3-1::GAL10P–SIR3::URA3 Δbar1::hisG Δlys2 |
| THC59 | W303-1A Δmat::TRP1 Δhmr::rHMRα Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC62 | W303-1A Δmat::TRP1 Δhmr::rHMRα Δbar1::hisG Δlys2 |
| THC67 | W303-1A Δmat::TRP1 hmr::rHMRa ura3-1::GAL10P–SIR3HA::URA3 Δbar1::hisG Δlys2 |
| THC68 | W303-1A Δmat::TRP1 Δhmr::rHMRα(rap1pm) Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC69 | W303-1A Δmat::TRP1 Δhmr::rHMRα(rap1pm) Δbar1::hisG Δlys2 |
| THC70 | W303-1A Δmat::TRP1 hmr::rHMRa Δhml::kanMX ura3-1::GAL10P–SIR3HA::URA3 Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC74 | W303-1A URA3 HMLα::URA3P–ADE2 |
| THC75 | W303-1A Δmat::TRP1 Δhmr::rHMRa/α Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC76 | W303-1A URA3 hml(rap1pm)::URA3P–ADE2 |
| THC77 | W303-1A Δmat::TRP1 Δhmr::rHMRa/α Δbar1::hisG Δlys2 |
| THC78 | W303-1A Δmat::TRP1 hmr::rHMRa Δhml::kanMX Δbar1::hisG Δlys2 Δsir3::HIS3 |
| THC79 | W303-1A Δmat::URA3 hmr::rHMRa + EI Δhml::kanMX Δsir3::HIS3 |
Acknowledgments
We thank David Shore, Scott Holmes, Judith Berman, and David Rivier for kindly providing plasmids, Danny Reinberg for critical comments on the manuscript, and Ken Irvine for use of digital photography equipment. We also thank Bruce Howard for pointing out valuable references, Prahba Joy for technical assistance, and the rest of the Gartenberg laboratory for stimulating discussions. This work was funded by a grant from the NIH (GM51402).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gartenbe@UMDNJ.edu; FAX (732) 235-4073.
References
- Ahmad K, Golic KG. Somatic reversion of chromosomal position effects in Drosophila melanogaster. Genetics. 1996;144:657–670. doi: 10.1093/genetics/144.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrulis E, Neiman AM, Zappulla DC, Sternglanz R. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature. 1998;394:592–595. doi: 10.1038/29100. [DOI] [PubMed] [Google Scholar]
- Ansari A, Gartenberg MR. Persistence of an alternate chromatin structure at silenced loci in vitro. Proc Natl Acad Sci. 1999;96:343–348. doi: 10.1073/pnas.96.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio OM, Gottschling DE. Overcoming telomeric silencing: A trans-activator competes to establish gene expression in a cell-cycle dependent way. Genes & Dev. 1994;8:1133–1146. doi: 10.1101/gad.8.10.1133. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in Saccharomyces cerevisiae. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in Saccharomyces cerevisiae: Redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- Astell CR, Ahlstrom-Jonasson L, Smith M, Tatchell K, Nasmyth KA, Hall BD. The sequence of the DNAs coding for the mating-type loci of Saccharomyces cerevisiae. Cell. 1981;27:15–23. doi: 10.1016/0092-8674(81)90356-1. [DOI] [PubMed] [Google Scholar]
- Bell SP, Kobayashi R, Stillman B. Yeast origin recognition complex functions in transcription silencing and DNA replication. Science. 1993;262:1844–1849. doi: 10.1126/science.8266072. [DOI] [PubMed] [Google Scholar]
- Bi X, Broach JR. DNA in transcriptionally silent chromatin assumes a distinct topology that is sensitive to cell cycle progression. Mol Cell Biol. 1997;17:7077–7087. doi: 10.1128/mcb.17.12.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscheron C, Maillet L, Marcand S, Tsai-Pflugfelder M, Gasser SM, Gilson E. Cooperation at a distance between silencers and proto-silencers at the yeast HML locus. EMBO J. 1996;15:2184–2195. [PMC free article] [PubMed] [Google Scholar]
- Boyle AL, Ballard SG, Ward DC. Differential distribution of long and short interspersed element sequences in the mouse genome: Chromosome karyotyping by fluorescence in situ hybridization. Proc Natl Acad Sci. 1990;87:7757–7761. doi: 10.1073/pnas.87.19.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Breeden L, Abraham J, Sternglanz R, Nasmyth K. Characterization of a “silencer” in yeast: A DNA sequence with properties opposite those of a transcriptional enhancer. Cell. 1985;41:41–48. doi: 10.1016/0092-8674(85)90059-5. [DOI] [PubMed] [Google Scholar]
- Braunstein M, Rose AB, Holmes SG, Allis CD, Broach JR. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes & Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Willard HF. The human X-inactivation center is not required for maintenance of X-chromosome inactivation. Nature. 1994;368:154–156. doi: 10.1038/368154a0. [DOI] [PubMed] [Google Scholar]
- Buck SW, Shore D. Action of a RAP1 carboxy-terminal silencing domain reveals an underlying competition between HMR and telomeres in yeast. Genes & Dev. 1995;9:370–384. doi: 10.1101/gad.9.3.370. [DOI] [PubMed] [Google Scholar]
- Cheng T-H, Li Y-C, Gartenberg MR. Persistence of an alternate chromatin structure at silenced loci in the absence of silencers. Proc Natl Acad Sci. 1998;95:5521–5526. doi: 10.1073/pnas.95.10.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien C-T, Bartel PL, Sternglanz R, Fields S. The two-hybrid system: A method to identify and clone genes for proteins that interact with a protein of interest. Proc Natl Acad Sci. 1991;88:9578–9582. doi: 10.1073/pnas.88.21.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depew DE, Wang JC. Conformational fluctuations of the DNA double helix. Proc Natl Acad Sci. 1975;72:4275–4279. doi: 10.1073/pnas.72.11.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin SCR. Heterochromatin and gene regulation in Drosophila. Curr Opin Genet Dev. 1996;6:193–202. doi: 10.1016/s0959-437x(96)80050-5. [DOI] [PubMed] [Google Scholar]
- Enomoto S, Berman J. Chromatin assembly factor I contributes to the mainenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes & Dev. 1998;12:219–232. doi: 10.1101/gad.12.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourel G, Revardel E, Koering CE, Gilson E. Cohabitiation of insulators and silencing elements in yeast subtelomeric regions. EMBO J. 1999;18:2522–2537. doi: 10.1093/emboj/18.9.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CA, Loo S, Dillin A, Rine J. The origin recognition complex has essential functions in transcriptional silencing and chromosomal replication. Genes & Dev. 1995;9:911–924. doi: 10.1101/gad.9.8.911. [DOI] [PubMed] [Google Scholar]
- Fox CA, Ehrenhofer-Murray AE, Loo S, Rine J. The origin of recognition complex, SIR1, and the S phase requirement for silencing. Science. 1997;276:1547–1551. doi: 10.1126/science.276.5318.1547. [DOI] [PubMed] [Google Scholar]
- Gartler SM, Riggs AD. Mammalian X-chromosome inactivation. Ann Rev Genet. 1983;17:155–190. doi: 10.1146/annurev.ge.17.120183.001103. [DOI] [PubMed] [Google Scholar]
- Giesman D, Best L, Tatchell K. The role of RAP1 in the regulation of the MATα locus. Mol Cell Biol. 1991;11:1069–1079. doi: 10.1128/mcb.11.2.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, Grunstein M. Histone H3 and H4 N-termini interact with Sir3 and Sir4 proteins: A molecular model for the formation of heterochromatin in yeast. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, Grunstein M. Spreading of transcriptional repressor Sir3 from telomeric heterochromatin. Nature. 1996;383:92–95. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- Herskowitz I, Rine J, Strathern JN. Mating-type determination and mating-type interconversion in Saccharomyces cerevisiae. In: Broach JR, Jones EW, Pringle JR, editors. The molecular and cellular biology of yeast Saccharomyces: Gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1991. pp. 583–656. [Google Scholar]
- Holmes SG, Broach JR. Silencers are required for inheritence of the repressed state in yeast. Genes & Dev. 1996;10:1021–1032. doi: 10.1101/gad.10.8.1021. [DOI] [PubMed] [Google Scholar]
- Holmes SG, Rose AB, Steuerle K, Saez E, Sayegh S, Lee YM, Broach JR. Hyperactivation of the silencing proteins, Sir2p and Sir3p, causes chromosome loss. Genetics. 1997;145:605–614. doi: 10.1093/genetics/145.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Fanti L, Pak DT, Botchan MR, Pimpinelli S, Kellum R. Distinct cytoplasmic and nuclear fractions of Drosophila heterochromatin protein 1: Their phosphorylation levels and associations with origin recognition complex proteins. J Cell Biol. 1998;142:307–318. doi: 10.1083/jcb.142.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs CW, Adams AEM, Szaniszlo PJ, Pringle JR. Functions of microtubules in the Saccharomyces cerevisiae cell cycle. J Cell Biol. 1988;107:1409–1426. doi: 10.1083/jcb.107.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Shore D. RAP1 protein activates and silences transcription of mating-type genes in yeast. Genes & Dev. 1991;5:616–628. doi: 10.1101/gad.5.4.616. [DOI] [PubMed] [Google Scholar]
- Lee JT, Jaenisch R. The (epi)genetic control of mammalian X-chromosome inactivation. Curr Opin Genet Dev. 1997;7:274–280. doi: 10.1016/s0959-437x(97)80138-4. [DOI] [PubMed] [Google Scholar]
- Lee S, Gross DS. Conditional silencing: The HMRE mating-type silencer exerts a rapidly reversible position effect on the yeast HSP82 heat shock gene. Mol Cell Biol. 1993;13:727–738. doi: 10.1128/mcb.13.2.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo S, Rine J. Silencers and domains of generalized repression. Science. 1994;264:1768–1771. doi: 10.1126/science.8209257. [DOI] [PubMed] [Google Scholar]
- ————— Silencing and heritable domains of gene expression. Annu Rev Cell Dev Biol. 1995;11:519–548. doi: 10.1146/annurev.cb.11.110195.002511. [DOI] [PubMed] [Google Scholar]
- Lustig AJ, Liu C, Zhang C, Hanish JP. Tethered Sir3p nucleates silencing at telomeres and internal loci in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2483–2495. doi: 10.1128/mcb.16.5.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. X-chromosome inactivation: A repeat hypothesis. Cytogenet Cell Genet. 1998;80:133–137. doi: 10.1159/000014969. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Marquardt R, Shei G-J, Rose AB, Broach JR. Mutations in HML E silencer of Saccharomyces cerevisiae yield metastable inheritence of transcriptional repression. Genes & Dev. 1991;5:605–615. doi: 10.1101/gad.5.4.605. [DOI] [PubMed] [Google Scholar]
- Maillet L, Boscheron C, Gotta M, Marcand S, Gilson E, Gasser SM. Evidence of silencing compartments within the yeast nucleus: A role for telomere proximity and Sir protein concentration in silencer-mediated repression. Genes & Dev. 1996;10:1796–1811. doi: 10.1101/gad.10.14.1796. [DOI] [PubMed] [Google Scholar]
- Marcand S, Buck SW, Moretti P, Gilson E, Shore D. Silencing of genes at nontelomeric sites in yeast is controlled by sequestration of silencing factors at telomeres by Rap1 protein. Genes & Dev. 1996;10:1297–1309. doi: 10.1101/gad.10.11.1297. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Nakajima R, Nishiyama J, Araki H, Oshima Y. Chromosome engineering in Saccharomyces cerevisiae by using a site-specific recombination system of a yeast plasmid. J Bacteriol. 1990;172:610–618. doi: 10.1128/jb.172.2.610-618.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menees TM, Sandmeyer SB. Transposition of the yeast retrovirus-like element Ty3 is dependent on the cell cycle. Mol Cell Biol. 1994;14:8229–8240. doi: 10.1128/mcb.14.12.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AM, Nasmyth KA. Role of DNA replication in the repression of silent mating type loci in yeast. Nature. 1984;312:247–251. doi: 10.1038/312247a0. [DOI] [PubMed] [Google Scholar]
- Moazed D, Johnson AD. A deubiquinating enzyme interacts with SIR4 and regulates silencing in Saccharomyces cerevisiae. Cell. 1996;86:667–677. doi: 10.1016/s0092-8674(00)80139-7. [DOI] [PubMed] [Google Scholar]
- Moretti P, Freeman K, Coodly L, Shore D. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes & Dev. 1994;8:2257–2269. doi: 10.1101/gad.8.19.2257. [DOI] [PubMed] [Google Scholar]
- Murphy TD, Karpen GH. Centromeres take flight: Alpha satellite and the quest for the human centromere. Cell. 1998;93:317–320. doi: 10.1016/s0092-8674(00)81158-7. [DOI] [PubMed] [Google Scholar]
- Murzina N, Verreault A, Laue E, Stillman B. Heterochromatin dynamics in mouse cells: interaction between Chromatin Assembly Factor 1 and HP1 proteins. Mol Cell. 1999;4:529–540. doi: 10.1016/s1097-2765(00)80204-x. [DOI] [PubMed] [Google Scholar]
- Pillus L, Rine J. Epigenetic inheritence of transcriptional states in Saccharomyces cerevisiae. Cell. 1989;59:637–647. doi: 10.1016/0092-8674(89)90009-3. [DOI] [PubMed] [Google Scholar]
- Pryde FE, Louis EJ. Limitations of silencing at native telomeres. EMBO J. 1999;18:2538–2550. doi: 10.1093/emboj/18.9.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renauld H, Aparicio OM, Zierath PD, Billington BL, Chhablani SK, Gottschling DE. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes & Dev. 1993;7:1133–1145. doi: 10.1101/gad.7.7a.1133. [DOI] [PubMed] [Google Scholar]
- Rivier DH, Ekena JL, Rine J. HMR-I is an origin of replication and a silencer in Saccharomyces cerevisiae. Genetics. 1999;151:521–529. doi: 10.1093/genetics/151.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman H. Studies of gene mutation in Saccharomyces. Cold Spring Harbor Symp Quant Biol. 1957;21:175–184. doi: 10.1101/sqb.1956.021.01.015. [DOI] [PubMed] [Google Scholar]
- Schnell R, Rine J. A position effect on the expression of a tRNA gene mediated by the SIR genes in Saccharomyces cerevisiae. Mol Cell Biol. 1986;6:494–501. doi: 10.1128/mcb.6.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shei G-J, Broach JR. Yeast silencers can act as orientation-dependent gene inactivation centers that respond to environmental signals. Mol Cell Biol. 1995;15:3496–3506. doi: 10.1128/mcb.15.7.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D. RAP1: A protean regulator in yeast. Trends Genet. 1994;10:408–412. doi: 10.1016/0168-9525(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Singh J, Klar AJS. Active genes in budding yeast display enhanced in vivo accessibility to foreign DNA methylases: A novel in vivo probe for chromatin structure of yeast. Genes & Dev. 1992;6:186–196. doi: 10.1101/gad.6.2.186. [DOI] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Pillus L, Boeke JD. Distribution of a limited Sir2 protein pool regulates the strength of yeast rDNA silencing and is modulated by Sir4p. Genetics. 1998;149:1205–1219. doi: 10.1093/genetics/149.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Pillus L. Activation of an MAP kinase cascade leads to Sir3p hyperphosphorylation and strengthens transcriptional silencing. J Cell Biol. 1996;135:571–583. doi: 10.1083/jcb.135.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. Sir2 and Sir4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes & Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- Sussel L, Vannier D, Shore D. Epigenetic switching of transcriptional states: cis- and trans-acting factors affecting establishment of silencing at the HMR locus in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3919–3928. doi: 10.1128/mcb.13.7.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terleth C, van Sluis CA, van de Putte P. Differential repair of UV damage in Saccharomyces cerevisiae. Nucleic Acids Res. 1989;17:4433–4439. doi: 10.1093/nar/17.12.4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JS, Johnson LM, Grunstein M. Specific repression of the yeast silent mating locus HMR by an adjacent telomere. Mol Cell Biol. 1994;14:446–455. doi: 10.1128/mcb.14.1.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triolo T, Sternglanz R. Role of interactions between the origin recognition complex and SIR1 in transcriptional silencing. Nature. 1996;381:251–253. doi: 10.1038/381251a0. [DOI] [PubMed] [Google Scholar]
- Vignais M-L, Sentenac A. Asymmetric DNA bending induced by the yeast multifunctional factor TUF. J Biol Chem. 1989;264:8463–8466. [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wallrath LL, Elgin SCR. Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes & Dev. 1995;9:1263–1277. doi: 10.1101/gad.9.10.1263. [DOI] [PubMed] [Google Scholar]
- Weiss K, Simpson RT. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating type locus HMLα. Mol Cell Biol. 1998;18:5392–5403. doi: 10.1128/mcb.18.9.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens GR, Sorger PK. Centromeric chromatin and epigenetic effects in kinetochore assembly. Cell. 1998;93:313–316. doi: 10.1016/s0092-8674(00)81157-5. [DOI] [PubMed] [Google Scholar]