

Abstract
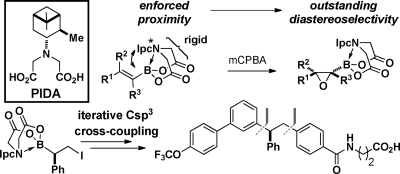
Efficient access to chiral C(sp3) boronates in stereochemically pure form is critical for realizing the substantial potential of such building blocks in complex-molecule synthesis. We herein report that a pinene-derived iminodiacetic acid (PIDA) ligand enables the highly diastereoselective synthesis of a wide range of oxiranyl C(sp3) boronates from the corresponding olefins. These oxiranyl PIDA boronates, in turn, can be readily transformed into unprecedented stable α-boryl aldehydes via a novel 1,2-migration of the boronate group that proceeds with complete maintenance of stereochemical purity. B-Protected haloboronic acids containing dual sp3-hybridized C centers are readily accessible via this platform, and the herein demonstrated capacity for stereocontrolled iterative C(sp3) cross-coupling with this novel type of bifunctional reagent to access a medicinally important chiral small-molecule target in highly enantioenriched form represents a substantial advance for the building-block-based approach to synthesis.
In an idealized form of the building-block approach for small-molecule synthesis, off-the-shelf subunits having all the required functional groups preinstalled in the correct oxidation states and with the desired stereochemical relationships are brought together using a single reaction iteratively.(1) Toward this goal, we have developed N-methyliminodiacetic acid (MIDA) boronates as a highly versatile platform of building blocks.1−3 Recent advances in the stereocontrolled cross-coupling of C(sp3) boronates(4) suggest that the generality of this approach could be substantially increased. C(sp3) boronates also represent highly versatile intermediates for a wide range of other applications.(5) To realize all of this potential, highly stereoselective methods for accessing such intermediates are critical.(6) We herein report the discovery of a pinene-derived iminodiacetic acid (PIDA) ligand that enables the facile synthesis of a wide range of versatile C(sp3) boronate building blocks in a highly stereocontrolled fashion. We further demonstrate that iterative C(sp3) cross-coupling with B-protected haloboronic acids containing dual sp3-hybridized C centers derived from this platform can substantially expand the scope of the building-block-based approach for small-molecule synthesis.
The remarkable stability of the MIDA boronate motif under a wide range of common reaction conditions enables the transformation of simple boron-containing starting materials into many types of complex boronate building blocks.1,2 Importantly, the crystal structures of many MIDA boronates have revealed that the N-methyl substituent is always closely positioned to the organic group appended to the boron atom,1,2 and variable-temperature NMR studies have demonstrated that the iminodiacetic acid framework is conformationally rigid in solution.1,2,7 Collectively, these observations suggested that if the N-alkyl substituent of iminodiacetic acid were chiral, highly effective transfer of stereochemical information might be achieved during functionalizations of the corresponding boronates as a result of the enforced proximity (Figure 1). In view of the exceptional versatility of epoxides in the preparation of many other chiral building blocks, we first questioned whether the epoxidation of alkenylboronates could be rendered asymmetric via such modifications of the MIDA ligand.(8)
Figure 1.

We surveyed a range of iminodiacetic acid ligands derived from different chiral amines and discovered that ligand 1a (PIDA), which can be easily prepared from very cheap and readily available (+)-α-pinene,9,10 is exceptionally effective. Specifically, treatment of the corresponding styrenyl PIDA boronate 2a with m-chloroperoxybenzoic acid (mCPBA) under standard conditions yielded oxiranyl PIDA boronate 3a with outstanding diastereoselectivity (Table 1, entry 1). Alternatively linking the pinene-derived appendage via a conformationally flexible methylene spacer (1b) or employing some other less sterically bulky chiral secondary amine (1c or 1d) resulted in substantially reduced diastereoselectivity (entries 2–4).
Table 1. Diastereoselective Epoxidations of Various Iminodiacetic Acid-Based Alkenylboronates.


Diastereomeric ratios determined via 500 MHz 1H NMR analysis of the unpurified reaction mixtures.
Single-crystal X-ray analysis of 3a (Table 1, entry 1) revealed that despite N-alkylation with a very sterically bulky substituent, the [3.3.0]-bicyclic structure of the iminodiacetic acid motif was preserved, with the chiral alkyl group positioned <2.4 Å from the newly formed epoxide. Moreover, variable-temperature NMR analysis confirmed that the iminodiacetic acid framework of the PIDA ligand was conformationally rigid in both the starting material 2a and the product 3a.(10) Collectively, these findings are consistent with the conclusion that highly effective transfer of stereochemical information in this system is attributable to the enforced proximity between the chiral appendage and the site of reactivity in the transition state of the epoxidation reaction.
Fortunately, both enantiomers of α-pinene are cheap and readily available on a very large scale, so PIDA has the potential to serve as a very practical chiral auxiliary.(11) We therefore explored the capacity of this ligand to enable the diastereoselective epoxidation of a variety of alkenylboronates. As shown in Table 2, the series of trans-1,2-disubstituted olefins 2a and 2e–i all were efficiently epoxidized in good yields and with outstanding stereocontrol (entries 1–6). PIDA boronate 2a could also be epoxidized on a 15 mmol scale and isolated via simple crystallization (entry 1). Cis-1,2-disubstituted and trisubstituted olefins were also very effective substrates (entries 7 and 8). Somewhat diminished but still synthetically useful diastereoselectivities were observed with 1,1-disubstituted olefins(12) (entries 9 and 10). Strikingly, even the smallest olefin, vinyl PIDA boronate 2n, was epoxidized with outstanding diastereoselectivity (entry 11). Importantly, all of the oxiranyl PIDA boronates 3 were produced as crystalline free-flowing solids that were completely stable toward silica gel chromatography and benchtop storage under air, making them highly desirable chiral building blocks for many applications in complex-molecule synthesis.
Table 2. Highly Diastereoselective Epoxidations of a Wide Range of Alkenyl PIDA Boronates.


Isolated yields after silica gel chromatography.
The stereochemistries of epoxides 3a, 3g, and 3n were all determined unambiguously via single-crystal X-ray analysis. The remaining product configurations were assigned by analogy.
Diastereomeric ratios determined via 500 MHz 1H NMR analysis of the unpurified reaction mixtures.
Conducted on a 15 mmol scale and isolated by crystallization.
In this vein, preliminary studies also revealed that oxiranyl PIDA boronates can be transformed into previously inaccessible C(sp3) boronate building blocks. For example, Mg(ClO4)2 promotes a very interesting Meinwald rearrangement(13) of 3a to generate air-stable α-boryl aldehyde 4 (Scheme 1). To the best of our knowledge, stable α-boryl aldehydes have not been previously reported in the literature,(14) and the stability of 4 is likely attributable to unique properties of the iminodiacetic acid boronate motif. Importantly, this rearrangement also occurs with complete maintenance of stereochemical purity. Moreover, ketalization of 4 and X-ray analysis of the resulting C(sp3) boronate 5 (Scheme 1) enabled the stereochemical assignment of 4, which was found to be consistent with exclusive migration of the boronate group during the rearrangement of 3a.(14) To the best of our knowledge, this type of 1,2-boryl migration has not been previously reported in the literature either.13,14
Scheme 1.

Stable α-boryl aldehydes such as 4 represent a new type of nucleophilic/electrophilic bifunctional reagent akin to those under development by Yudin and co-workers,14b,15 and we have also begun to explore the substantial potential of this motif to generate a range of new types of C(sp3) boronate building blocks (Scheme 1). In addition to the transformation of 4 to complex ketal 5 described above, reduction of 4 with sodium triacetoxyborohydride cleanly provided primary alcohol 6. Subsequent iodide exchange also provided convenient access to a novel type of B-protected haloboronic acid, 7, in which both C termini are sp3-hybridized.
To explore the potential of this novel type of bifunctional building block to provide access to important chiral targets via a previously undescribed iterative C(sp3) cross-coupling approach, we pursued the synthesis of 13, a glucagon receptor antagonist under evaluation for treatment of type-II diabetes (Scheme 2).(16) The iodide terminus of PIDA boronate 7 was first transformed into its organozinc counterpart, which smoothly underwent cross-coupling with aryl iodide 8 to provide 9 in 73% yield as a single diastereomer. Subsequent transesterification of the PIDA boronate motif in 9 to the corresponding pinacol boronic ester 10 proceeded in 84% yield with complete maintenance of stereochemical purity.(17) Importantly, the PIDA ligand was easily recovered from this reaction in 96% yield. Finally, cross-coupling of this chiral secondary boronic ester 10 with aryl iodide 11 employing the conditions developed by Crudden4a,4b provided 12 with nearly perfect maintenance of stereochemical purity (94:6 e.r.).17,18 Deprotection of 12 completed a very efficient, modular, and highly stereocontrolled synthesis of 13.
Scheme 2.

These findings have collectively established the substantial potential of the PIDA platform to access a wide array of novel C(sp3) boronate building blocks. Moreover, the demonstrated capacity to transform stereochemically pure halo PIDA boronates containing dual sp3-hybridized C termini into a medicinally important chiral small-molecule target via highly efficient and flexible iterative C(sp3) cross-coupling represents a major step toward generalizing this building-block approach to synthesis.
Acknowledgments
Dr. Danielle Gray is gratefully acknowledged for X-ray analysis. We also thank the NSF (CAREER 0747778) and Bristol-Myers Squibb for funding. M.D.B. is an HHMI Early Career Scientist, Beckman Young Investigator, and Sloan Foundation Fellow. J.L. is a UIUC Department of Chemistry Graduate Fellow.
Supporting Information Available
Procedures, spectral data, and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- a Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2007, 129, 6716–6717. [DOI] [PubMed] [Google Scholar]; b Gillis E. P.; Burke M. D. Aldrichimica Acta 2009, 42, 17–27. [PMC free article] [PubMed] [Google Scholar]
- a Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2008, 130, 14084–14085. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee S. J.; Gray K. C.; Paek J. S.; Burke M. D. J. Am. Chem. Soc. 2008, 130, 466–468. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Knapp D. M.; Gillis E. P.; Burke M. D. J. Am. Chem. Soc. 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Uno B. E.; Gillis E. P.; Burke M. D. Tetrahedron 2009, 65, 3130–3138. [Google Scholar]; e Woerly E. M.; Cherney A. H.; Davis E. K.; Burke M. D. J. Am. Chem. Soc. 2010, 132, 6941–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lee S. J.; Anderson T. M.; Burke M. D. Angew. Chem., Int. Ed. 2010, 49, 8860–8863. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Struble J. R.; Lee S. J.; Burke M. D. Tetrahedron 2010, 66, 4710–4718. [Google Scholar]; h Dick G. D.; Knapp D. M.; Gillis E. P.; Burke M. D. Org. Lett. 2010, 12, 2314–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Woerly E. M.; Struble J. R.; Palyam N.; O’Hara S. P.; Burke M. D. Tetrahedron 2011, 67, 4333–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Fujii S.; Chang S. Y.; Burke M. D.. Angew. Chem., Int. Ed. [Online early access]. DOI: 10.1002/anie.201102688. Published Online: June 16, 2011. [Google Scholar]; k More than 75 MIDA boronates are now commercially available.
- For some other recent applications of MIDA boronates, see: ; a Brak K.; Ellman J. A. J. Org. Chem. 2010, 75, 3147–3150. [DOI] [PubMed] [Google Scholar]; b Brak K.; Ellman J. A. Org. Lett. 2010, 12, 2004–2007. [DOI] [PubMed] [Google Scholar]; c Brenzovich W. E.; Brazeau J.-F.; Toste F. D. Org. Lett. 2010, 12, 4728–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Burns A. R.; McAllister G. D.; Shanahan S. E.; Taylor R. J. K. Angew. Chem., Int. Ed. 2010, 49, 5574–5577. [DOI] [PubMed] [Google Scholar]; e Han J.; Xu B.; Hammond G. B. J. Am. Chem. Soc. 2010, 132, 916–917. [DOI] [PubMed] [Google Scholar]; f Gustafson J. L.; Lim D.; Barrett K. T.; Miller S. J. Angew. Chem., Int. Ed. 2011, 50, 5125–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Chan M. W. J.; Amarante G. W.; Toste F. D. Tetrahedron 2011, 67, 4306–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Grob J. E.; Nunez J.; Dechantsreiter M. A.; Hamann L. G. J. Org. Chem. 2011, 76, 4930–4940. [DOI] [PubMed] [Google Scholar]; i Colgin N.; Finn T.; Cobb S. L. Org. Biomol. Chem. 2011, 9, 1864–1870. [DOI] [PubMed] [Google Scholar]; j Mohamed Y. M. A.; Hansen T. V. Tetrahedron Lett. 2011, 52, 1057–1059. [Google Scholar]
- a Imao D.; Glasspole B. W.; Laberge V. S.; Crudden C. M. J. Am. Chem. Soc. 2009, 131, 5024–5025. [DOI] [PubMed] [Google Scholar]; b Crudden C. M. Abstr. Pap.—Am. Chem. Soc. 2011, 241, ORGN 323. [Google Scholar]; c Owston N. A.; Fu G. C. J. Am. Chem. Soc. 2010, 132, 11908–11909. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lundin P. M.; Fu G. C. J. Am. Chem. Soc. 2010, 132, 11027–11029. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Saito B.; Fu G. C. J. Am. Chem. Soc. 2008, 130, 6694–6695. [DOI] [PubMed] [Google Scholar]; f Ohmura T.; Awano T.; Suginome M. J. Am. Chem. Soc. 2010, 132, 13191–13193. [DOI] [PubMed] [Google Scholar]; g Sandrock D. L.; Jean-Gerard L.; Chen C.-Y.; Dreher S. D.; Molander G. A. J. Am. Chem. Soc. 2010, 132, 17108–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257–9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Crudden C. M.; Glasspoole B. W.; Lata C. J. Chem. Commun. 2009, 6704–6716. [DOI] [PubMed] [Google Scholar]; b Hall D. G.Boronic Acids; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- For some recent advances in the stereoselective synthesis of C(sp3) boronates, see: ; a Matteson D. S. Tetrahedron 1998, 54, 10555–10607. [Google Scholar]; b Crudden C. M.; Hleba Y. B.; Chen A. C. J. Am. Chem. Soc. 2004, 126, 9200–9201. [DOI] [PubMed] [Google Scholar]; c Pelz N. F.; Woodward A. R.; Burks H. E.; Sieber J. D.; Morken J. P. J. Am. Chem. Soc. 2004, 126, 16328–16329. [DOI] [PubMed] [Google Scholar]; d Kliman L. T.; Mlynarski S. N.; Morken J. P. J. Am. Chem. Soc. 2009, 131, 13210–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]; e O’Brien J. M.; Lee K.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 10630–10633. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chen I.-H.; Yin L.; Itano W.; Kanai M.; Shibasaki M. J. Am. Chem. Soc. 2009, 131, 11664–11665. [DOI] [PubMed] [Google Scholar]; g Lee J. C. H.; Hall D. G. J. Am. Chem. Soc. 2010, 132, 5544–5545. [DOI] [PubMed] [Google Scholar]; h Bagutski V.; French R. M.; Aggarwal V. K. Angew. Chem., Int. Ed. 2010, 49, 5142–5145. [DOI] [PubMed] [Google Scholar]; i Thomas S. P.; French R. M.; Jheengut V.; Aggarwal V. K. Chem. Rec. 2009, 9, 24–39. [DOI] [PubMed] [Google Scholar]; j Noh D.; Chea H.; Ju J.; Yun J. Angew. Chem., Int. Ed. 2009, 48, 6062–6064. [DOI] [PubMed] [Google Scholar]; k Gonzalez A. Z.; Román J. G.; Gonzalez E.; Martinez J.; Medina J. R.; Matos K.; Soderquist J. A. J. Am. Chem. Soc. 2008, 130, 9218–9219. [DOI] [PubMed] [Google Scholar]
- a Mancilla T.; Contreras R.; Wrackmeyer B. J. Organomet. Chem. 1986, 307, 1–6. [Google Scholar]; b Mancilla T.; Zamudio-Rivera L. S.; Beltrán H. I.; Santillan R.; Farfán N. ARKIVOC 2005, (vi), 366–376. [Google Scholar]
- In the absence of chiral reagents, low diastereoselectivities (1.2:1 to 1.7:1) were recently reported for the epoxidation of various alkenylboronic esters derived from the divalent (2R,3R)-1,4-dimethoxy-1,1,4,4-tetraphenyl-2,3-butanediol ligand. See: ; a Fernandez E.; Frey W.; Pietruszka J. Synlett 2009, 9, 1386–1388. [Google Scholar]; Variable diastereoselectivities have been observed for cyclopropanations and Johnson rearrangements of alkenylboronic esters derived from this same ligand. See: ; b Luithle J. E. A.; Pietruszka J. J. Org. Chem. 1999, 64, 8287–8297. [DOI] [PubMed] [Google Scholar]; c Pietruszka J.; Schöne N. Angew. Chem., Int. Ed. 2003, 42, 5638–5641. [DOI] [PubMed] [Google Scholar]; For a recent example of diastereoselective epoxidation of vinyl boronic esters bearing an allylic alcohol, see: ; d Hussain M. H.; Toribio J. H.; Carroll P. J.; Walsh P. J. Angew. Chem., Int. Ed. 2011, 50, 6337–6340. [DOI] [PubMed] [Google Scholar]; For the non-stereoselective epoxidation of alkenyltrifluoroborates, see: ; e Molander G. A.; Ribagorda M. J. Am. Chem. Soc. 2003, 125, 11148–11149. [DOI] [PubMed] [Google Scholar]; f For the non-stereoselective epoxidation of MIDA boronates, see ref (2d).
- Brown H. C.; Jadhav P. K. J. Am. Chem. Soc. 1983, 105, 2092–2093. [Google Scholar]; Rathke M. W.; Millard A. A.. Organic Syntheses; Wiley: New York, 1988; Collect. Vol. VI, p 943. [Google Scholar]
- See the Supporting Information.
- Both enantiomers of 1a will soon be commercially available from Sigma-Aldrich: (R,R,R,S)-PIDA 752428, (S,S,S,R)-PIDA 752436.
- 1,1-Disubstituted alkenes are classically challenging substrates for asymmetric hydroboration and epoxidation. For a discussion of recent advances, see: ; Thomas S. P.; Aggarwal V. K. Angew. Chem., Int. Ed. 2009, 48, 1896–1898. [DOI] [PubMed] [Google Scholar]
- a Meinwald J.; Labana S. S.; Chadha M. S. J. Am. Chem. Soc. 1963, 85, 582–585. [Google Scholar]; Similar migration of a trialkylsilane has been reported. See:; b Eisch J. J.; Trainor J. T. J. Org. Chem. 1963, 28, 2870–2876. [Google Scholar]
- Contemporaneous unpublished studies in the Yudin group have revealed a very similar rearrangement of oxiranyl MIDA boronates to stable and highly versatile α-boryl aldehydes promoted by BF3·OEt2. These authors first demonstrated with deteurium-labeling studies that this rearrangement also proceeds with exclusive migration of the boronate group. See: ; a Yudin A. K. Private communication.; b He Z.; Yudin A. K. J. Am. Chem. Soc., in press; DOI: 10.1021/ja205910d. [Google Scholar]
- a Hili R.; Rai V.; Yudin A. K. J. Am. Chem. Soc. 2010, 132, 2889–2891. [DOI] [PubMed] [Google Scholar]; b Hili R.; Yudin A. K. J. Am. Chem. Soc. 2009, 131, 16404–16406. [DOI] [PubMed] [Google Scholar]; c Li X.; Yudin A. K. J. Am. Chem. Soc. 2007, 129, 14152–14153. [DOI] [PubMed] [Google Scholar]; d Hili R.; Yudin A. K. J. Am. Chem. Soc. 2006, 128, 14772–14773. [DOI] [PubMed] [Google Scholar]; e Baktharaman S.; Hili R.; Yudin A. K. Aldrichimica Acta 2008, 41, 109–118. [Google Scholar]; f Afagh N. A.; Yudin A. K. Angew. Chem., Int. Ed. 2010, 49, 262–310. [DOI] [PubMed] [Google Scholar]
- Stelmach J. E.; Keith R.; Parmee E. R.; Tata J. R.. PCT/US2006/009694, Sep 28, 2006.
- Enantiomeric ratios (e.r.) were determined via chiral HPLC using independently synthesized racemic standards; see the Supporting Information for details.
- The major stereoisomer of 12 was assigned as that resulting from retention of configuration at the benzylic center by analogy with recent reports by Crudden and co-workers.4a,4b
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.