

Abstract
HIV exploits cellular proteins during its replicative cycle and latent infection. The positive transcription elongation factor, P-TEFb, is a key cellular transcription factor critical for these viral processes and is a drug target. During viral replication P-TEFb is recruited via interactions of its cyclin T1 subunit with the HIV Tat protein and TAR element. Through RNA silencing and over-expression experiments we discovered that nuclear factor 90 (NF90), a cellular RNA binding protein, regulates P-TEFb expression. NF90 depletion reduced cyclin T1 protein levels by inhibiting translation initiation. Regulation was mediated by the 3′UTR of cyclin T1 mRNA independently of microRNAs. Cyclin T1 induction is involved in the escape of HIV-1 from latency. We show that the activation of viral replication by phorbol ester in latently infected monocytic cells requires the posttranscriptional induction of NF90 and cyclin T1, implicating NF90 in protein kinase C (PKC) signaling pathways. This investigation reveals a novel mechanism of cyclin T1 regulation and establishes NF90 as a regulator of HIV-1 replication during both productive infection and induction from latency.
Keywords: RNA binding protein, P-TEFb, 3′UTR, PMA activation, microRNA, Nuclear Factor 90 (NF90)
Introduction
Human immunodeficiency virus (HIV) replication is dependent on cellular components that interact with viral RNAs and proteins. Understanding these interactions has illuminated many aspects of cell function and holds the prospect of developing antiviral therapies. Earlier work suggested that members of the nuclear factor 90 (NF90) family of RNA binding proteins may influence HIV-1 replication, but the mechanisms and significance of their activities remained to be defined. Here we establish a novel post-transcriptional role for NF90 in controlling the level of the cellular transcription factor P-TEFb (positive transcription elongation factor b) that plays a key role in HIV replication and escape from latency.
The expression of many cellular genes, especially those encoding developmental regulators, is modulated at the transcription elongation step1, and P-TEFb has been implicated in a growing number of physiological and pathological instances (see, for example,2; 3). P-TEFb is a general transcription factor required for the production of mRNA by RNA polymerase II (Pol II). It is composed of the cyclin-dependent kinase CDK9 and a regulatory cyclin, either cyclin T1, T2 or K. Full-length transcription of the integrated HIV-1 proviral genome requires the cooperation of a specific P-TEFb complex, cyclin T1/CDK94; 5. Cyclin T1 binds both the viral transactivator protein (Tat) and transactivation response element RNA (TAR), encoded immediately downstream of the transcription start site in the viral long terminal repeat (LTR). The CDK9 subunit of P-TEFb then phosphorylates Pol II, thereby facilitating transition into a productive elongation mode6. Processive transcriptional elongation dramatically increases the expression of genes driven by the viral promoter and is critical for HIV replication.
P-TEFb is subject to regulation at diverse levels. A fraction of the cell’s P-TEFb is held in enzymatically inactive ribonucleoprotein complexes containing the small nuclear RNA 7SK6; 7, while the overall cellular concentration of P-TEFb appears to be limited by the level of its cyclin T1 subunit3. Cyclin T1 is controlled posttranscriptionally in monocytes, monocyte-derived macrophages and promonocytic cell lines8; 9, whereas in lymphocytes cyclin T1 and CDK9 are induced by mitogens and cytokines through multiple mechanisms9; 10; 11. At least part of the posttranscriptional control observed during the differentiation of monocytes to macrophages is attributed to the microRNA (miRNA) miR-198, which targets sites in the cyclin T1 3′UTR12.
NF90 is encoded by the ILF3 gene, which is widespread in vertebrates13 and essential in mice14; 15. Differential splicing gives rise to two prominent isoforms, NF90 and NF110, that differ at their C termini (Fig. 1A). Both have subsidiary isoforms that carry (NF90/110b) or lack (NF90/110a) a short insert resulting from alternative splicing16; 17. Proteins similar, if not identical, to NF90 and NF110 are known as MPP4/DRBP76/NFAR1/TCP80 and ILF3/NFAR2/TCP110, respectively. They harbor several motifs that confer the ability to interact with nucleic acids, including tandem double-stranded RNA binding motifs (dsRBMs), an RGG domain, and an NF110-specific GQSY-rich region (Fig. 1A). NF90 and NF110 shuttle between the nucleus and cytoplasm18; 19. They are generally isolated in heterodimeric core complexes with nuclear factor 45 (NF45), produced by the ILF2 gene, and they form large complexes in the cell containing additional components including RNA helicases and hnRNPs20. NF90 participates extensively in pathways of mRNA metabolism21; 22, as well as in the metabolism of noncoding and regulatory RNAs such as snaRs23 and miRNAs23; 24.
Figure 1. NF90 modulates HIV-1 gene expression.
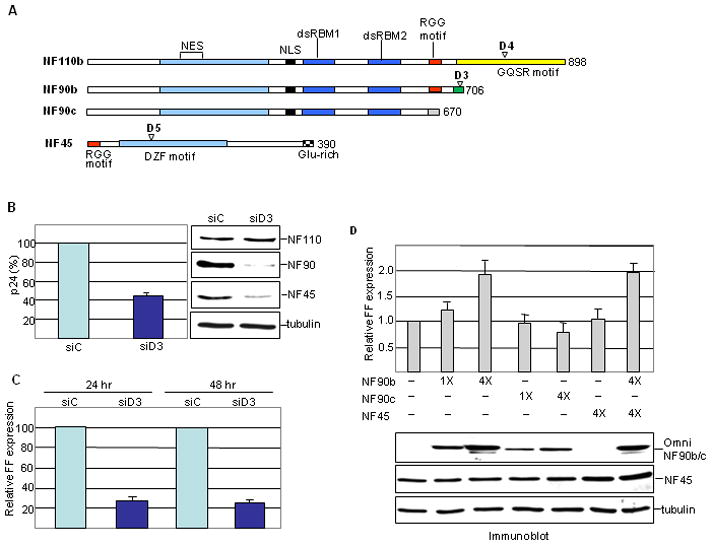
A. Schematic of gene products and siRNAs. Domains and motifs are marked: NES, nuclear export signal; NLS, nuclear localization signal; dsRBM, dsRNA binding motif; RGG, arginine-glycine-glycine; GQSR, glycine, glutamine, serine, arginine; DZF, domain in dsRBM or zinc finger domain containing proteins; Glu-rich, glutamate-rich. D3, D4, D5: siRNA target sites specific for NF90, NF110 and NF45, respectively. The NF90/110 isoforms used in this study contain the tetrapeptide NVKQ between dsRBMs 1 and 2. B. HeLa cells were transfected with control (siC) or NF90 specific (siD3) siRNA, then infected with pseudotyped HIV-1. WCEs were assayed for p24 production (left panel) and by immunoblotting (right panel) at 24 hr post-infection. C. Cells infected as in panel B were assayed for FF luciferase at 24 and 48 hr post-infection. D. HeLa cells were co-transfected with pNL4-3lucE− and pCMV-Renilla together with Omni-NF90b or Omni-NF45 expression vectors (50 or 200 ng) as indicated. FF expression was normalized to Renilla luciferase (bar graph). Protein expression was analyzed by immunoblotting (lower panel).
In the context of viral infection, attention was drawn to NF90 family proteins by the discovery that they bind dsRNA and the non-coding adenovirus transcript VA RNAII25; 26, as well as the RNA-dependent protein kinase PKR involved in the host antiviral response27; 28; 29; 30; 31. NF90 regulates transcription from the interleukin-2 (IL-2) promoter32 and the stability of IL-2 mRNA33. NF90/110 have been implicated in the replication cycles of a number of viruses (see, for example,15; 19; 34) and NF110 over-expression stimulates transcription directed by the HIV-1 promoter17. A variant form, NF90c (also known as NF90ctv), was reported to interact with the HIV-1 regulators Rev and Tat/TAR35; 36 and to regulate viral gene expression by modulating interferon-responsive genes in the antiviral cascade37. However, the physiological relevance of these findings is open to question because the NF90c isoform has not been detected in cells17; 37.
In view of its involvement in both viral replication and cellular antiviral mechanisms, we investigated the function of NF90 in HIV-1 infection and reemergence from latency. We found that depletion of endogenous NF90 reduced viral replication and gene expression, while over-expression of NF90b caused the opposite effect. We determined that P-TEFb, and specifically its cyclin T1 subunit, is critical for this response. Mechanistic analysis revealed that NF90 controls P-TEFb by up-regulating cyclin T1 synthesis at the level of translation initiation. The action of NF90 is exerted via sequences in the cyclin T1 mRNA 3′UTR independently of miRNAs. In promonocytic cells NF90 is highly induced at the posttranscriptional level by treatment with PMA (phorbol 12-myristate 13-acetate), a phorbol ester which activates HIV-1 production in latently infected cells. NF90 knockdown inhibits the posttranscriptional induction of cyclin T1 and the emergence of HIV-1 from latency. These results implicate NF90 in PKC-mediated signaling and the activation of viral replication in latently infected cells, which poses the biggest obstacle to the eradication of HIV-1 infection.
Results
Stimulation of HIV-1 gene expression by NF90
To examine the effect of NF90 depletion on HIV-1 infection, HeLa cells were infected with a pseudotyped virus (NL4-3LucE−) carrying the firefly luciferase gene (FF). Selective knockdown of NF90 by small interfering (si) RNA D3 reduced production of viral p24 protein by ~60% compared to cells treated with control siRNA, siC (Fig. 1B). Correspondingly, virus-directed FF activity was reduced by ~70% (Fig. 1C). These results implied that NF90 exerts a positive effect on HIV-1 gene expression. Accordingly, over-expression of NF90b stimulated FF expression by ~2 fold in HeLa cells transfected with the pNL4-3LucE− molecular clone (Fig. 1D). Similar results were obtained in transfected 293 cells, and in the d3 cell line20 which is NF90-depleted (data not shown).
NF90 is coordinately regulated at the post-translational level with its heterodimeric partner NF4520. Accordingly, NF90 knockdown resulted in depletion of NF45 (Fig. 1B), reflecting destabilization of the NF90/NF45 complex. Nevertheless, over-expression of NF45 on its own had no effect on FF expression (Fig. 1D). Furthermore, NF45 over-expression did not elicit a further stimulation when co-expressed with NF90b (Fig. 1D). The NF90c variant, which is not commonly found in cells and has an anomalous, highly acidic C terminus, was slightly inhibitory to FF expression (Fig. 1D). This indicates that the NF90b C terminus is critical for its ability to stimulate the expression of HIV-1.
NF90 regulates HIV-1 transcription
Two viral genes, tat and rev, are critical for HIV-1 gene expression. While Tat functions in transcription, Rev plays a post-transcriptional role by modulating the splicing and nuclear export of viral RNAs. To determine whether either of these viral proteins is involved in the action of NF90, we first monitored the effect of NF90 knockdown on FF expression from a minimal promoter-reporter construct that lacks viral protein-coding genes. This HIV-FF construct was tested in the presence or absence of the HIV-1 transactivator protein Tat. As with the pNL4-3-LucE− molecular clone, which expresses Tat in cis, Tat-stimulated expression from HIV-FF was reduced ~70% by NF90 depletion mediated by D3. Basal expression from HIV-FF (in the absence of Tat) was unaffected, however, suggesting that NF90 acts at the level of Tat-dependent transcription (Fig. 2A). Tat levels were unaffected by NF90 depletion (Fig. 2B). We therefore proceeded to assay reporter gene expression at the RNA level. NF90 knockdown reduced the abundance of FF mRNA from HIV-FF in the presence of Tat by 70% (Fig. 2C), consistent with an action at the level of transcription. No change in FF mRNA abundance was detected after knockdown of NF110 or of NF45, indicating that the response is specific to NF90. Moreover, depletion of these proteins together with NF90 did not influence the reduction in FF mRNA expression brought about by NF90 depletion (Supplementary Fig. S1A).
Figure 2. NF90 knockdown inhibits activated transcription from the HIV promoter.
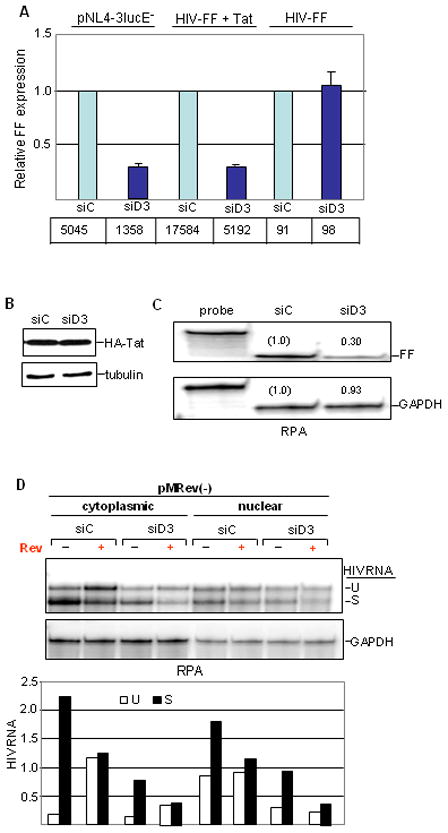
A. HeLa cells were transfected with siRNAs followed by pNL4-3lucE− or HIV-FF reporter plasmid with Tat expression vector where indicated. WCEs were analyzed for FF expression, tabulated in arbitrary units and plotted relative to control values. B. HeLa cells were transfected with siRNA followed by HA tagged-Tat expression vector. Tat expression was analyzed by immunoblotting. C. HeLa cells were transfected with siRNA followed by co-transfection with HIV-FF and Tat expression vector as in panel A. RNA was analyzed by RNase protection assay (RPA). Radioactivity was detected and quantified using a Typhoon phosphorimager and the imagequant program: relative levels of fragments derived from FF and GAPDH RNA are shown. D. siRNA transfected HeLa cells were co-transfected with pMRev(−) without or with Rev expression vector as indicated. Cytoplasmic and nuclear RNA was analyzed by RPA (top panel) and quantified as in panel C. Radioactivity of fragments derived from spliced (S) and unspliced (U) HIV-1 RNA was normalized to GAPDH.
For a direct test of Rev’s involvement in the NF90 depletion phenotype, we transfected cells with pMRev(−). This molecular clone contains the entire HIV-1 genome, but rev expression is defective causing impaired splicing and nucleo-cytoplasmic transport of viral mRNA38. Rev was supplied to some cultures in trans, and HIV-1 RNA was analyzed (Fig. 2D). As expected, in control cultures there was little unspliced RNA in the cytoplasm in the absence of Rev although substantial levels were present in the nucleus, and Rev expression increased the level of unspliced RNA in the cytoplasm. NF90 depletion reduced the overall level of viral RNAs in both subcellular compartments regardless of the presence or absence of Rev, without discernibly altering the ratio of spliced to unspliced transcripts. Furthermore, the FF reporter in NL4-3-LucE− replaces the nef gene, whose mRNA is spliced. Taken together, these results indicate that gene expression from two HIV-1 molecular clones is dependent on NF90, and that the action of NF90 is related to Tat-dependent transcription.
NF90 modulates transcription via P-TEFb
Tat’s principal role in HIV transcription is to recruit the elongation factor P-TEFb, composed of CDK9 and cyclin T1, which normally entails interactions of its cyclin T1 subunit with Tat and TAR. To determine whether Tat is directly involved in the NF90 effect, we used a well-characterized tethering system39 to recruit P-TEFb to the promoter independently of Tat. The HIV-1 promoter is modified by insertion of Gal4 binding sites that interact with Gal4-cyclin T1 or Gal4-CDK9 fusion proteins. FF expression in this system is dependent on assembly of the fusion protein with its endogenous partner (CDK9 or cyclin T1) to form Gal4-tagged P-TEFb. NF90 knockdown reduced FF expression by 2–6 fold in the tethering system (Fig. 3A) and this inhibition was exerted at the RNA level (Fig. 3B). Evidently Tat is dispensable for the NF90 effect when P-TEFb is recruited to the modified HIV-1 promoter via Gal4 fusion proteins (Fig. 3C), suggesting that NF90 affects the level or activity of P-TEFb.
Figure 3. P-TEFb dependence of NF90 action.
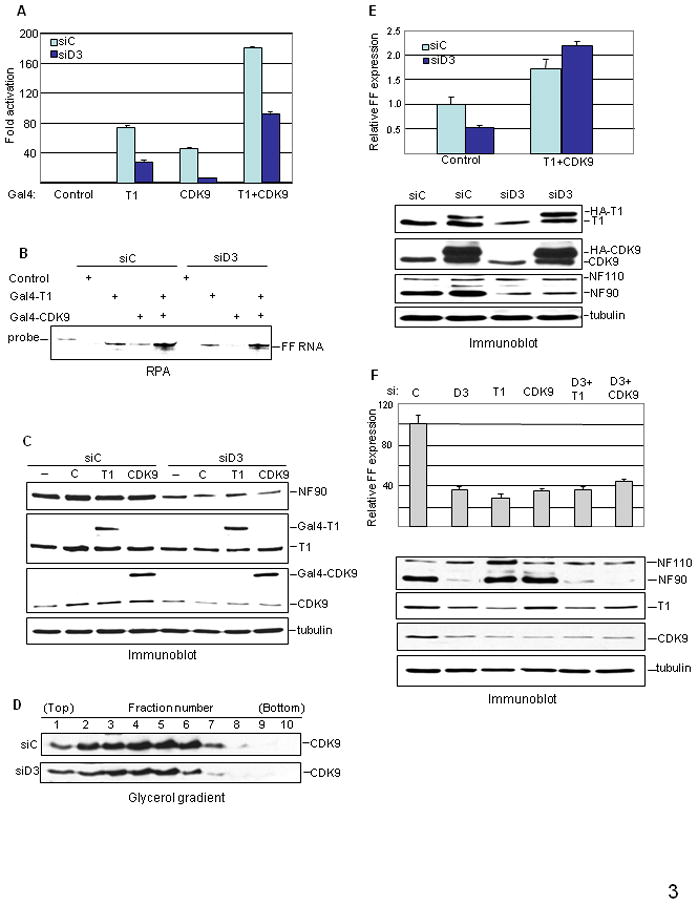
A–C. Results from tethering system. HeLa cells were transfected with siRNA followed by G5-HIV-FF and Gal4-cyclin T1, Gal4-CDK9, or both, as indicated. Cell extracts were assayed (A) for luciferase activity as in Fig. 2A, (B) for RNA by RPA, and (C) for protein expression by immunoblotting. D. WCEs from siRNA-transfected cells (equal amounts of protein) were resolved in glycerol gradients and fractions were analyzed by immunoblotting. E. HeLa cells were transfected with siRNA, then with pNL4-3lucE− together with HA-cyclin T1 and HA-CDK9 expression vectors or control vector. WCEs were analyzed for FF expression (top panel) and protein expression by immunoblotting (bottom panel). F. HeLa cells were transfected with control siRNA (C), siRNA specific for NF90 (D3), cyclin T1 (T1) or CDK9, or the combinations indicated, and were transfected with pNL4-3lucE− 48 hr later. Cell extracts were analyzed for FF production (top panel) and protein expression by immunoblotting (bottom panel).
P-TEFb can be sequestered in inactive complexes containing cellular HEXIM proteins and 7SK RNA. Although this RNA also forms complexes with NF9040; 41, we did not observe any effect of NF90 knockdown on the level of 7SK (Supplementary Fig. S2). On the other hand, glycerol gradient sedimentation analysis showed that siRNA D3 reduced the overall level of P-TEFb complexes in cell extracts by about half (Fig. 3D). The distribution of P-TEFb in the gradient between active (slower-sedimenting, 7SK-free) and inactive (faster-sedimenting, 7SK-containing) complexes was unaltered, however, implying that NF90 regulates the cellular level of P-TEFb.
This conclusion was supported by the results of experiments in which the cellular levels of cyclin T1 and CDK9 were manipulated. Ectopic expression of HA-tagged cyclin T1 and CDK9 increased gene expression from the molecular clone and rescued the inhibitory effect of D3 on FF expression from pNL4-3-LucE− (Fig. 3E). Conversely, depletion of cyclin T1 or CDK9 by specific siRNAs did not exacerbate the inhibition of FF expression from HIV-FF brought about by D3 (Fig. 3F, upper panel). Similar results were obtained in cells infected with pseudotyped virus (data not shown). These data argue that the action of NF90 on HIV-1 transcription is mediated exclusively by its effect on P-TEFb.
Posttranscriptional control of cyclin T1 and CDK9 by NF90
NF90 knockdown resulted in reduced levels of both cyclin T1 and CDK9 (Fig. 3C), and these proteins were depleted to similar extents by siRNAs directed against cyclin T1 or CDK9 (Fig. 3F, lower panel). Semi-quantitative immunoblotting analysis confirmed that both cyclin T1 and CDK9 levels decreased by >50% in NF90-depleted cells (Fig. 4A). As with FF expression from pNL4-3LucE−, no such reduction was seen when NF110 or NF45 were depleted (Supplementary Fig. S1B). The NF90-dependence of cyclin T1 and CDK9 could, in principle, be due to effects on the proteins themselves or on their mRNAs. To elucidate the function of NF90 in controlling P-TEFb levels, we first examined the mRNAs encoding cyclin T1 and CDK9. The levels of these mRNAs were unchanged (Fig. 4B), as was their nucleo-cytoplasmic distribution (Fig. 4C), implying that NF90 affects the synthesis and/or stability of these proteins.
Figure 4. NF90 controls cyclin T1 synthesis.
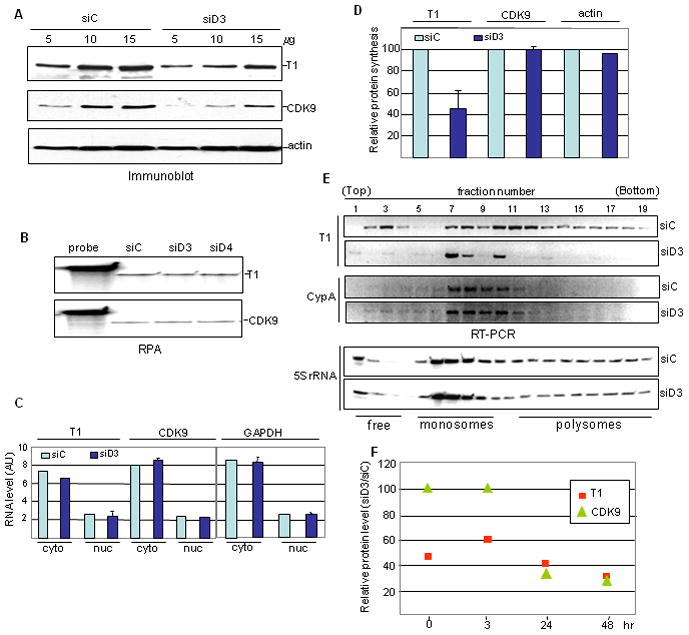
A. Increasing amounts of WCEs of siRNA-transfected HeLa cells were analyzed by immunoblotting. B. Total RNA from siRNA-transfected HeLa cells was analyzed by RPA. C. Cytoplasmic (cyto) and nuclear (nuc) RNA isolated from siRNA-transfected cells was analyzed by RPA and quantified (AU, arbitrary units). D. HeLa cells transfected with siRNA as indicated were incubated with [35S]-Met/Cys for 2 hr. Proteins immunoprecipitated with anti-CDK9 or anti-actin antibody were separated in denaturing gels and cyclin T1, CDK9 and actin were quantified. E. Polysome profile analysis of control and D3 siRNA treated cells. RNA from each fraction was assayed for cyclin T1 (T1) and cyclophilin A (CypA) mRNA by RT-PCR and for 5S rRNA by northern blot. F. Pulse-chase analysis of siRNA transfected cells. HeLa cells were pulse-labeled for 2 hr then chased with unlabeled amino acids at the times shown. WCEs were assayed as in panel D. Cyclin T1, CDK9 and actin signals were quantified using a Typhoon phosphorimager and the Image Quant program. Radioactivity in cyclin T1 and CDK9 was normalized to that in actin. Results are presented as siD3/siC ratio at each time point.
We next measured the synthesis of cyclin T1 and CDK9 proteins. Cells were labeled with 35S-amino acids and P-TEFb was immunoprecipitated. Quantitation showed that D3 treatment reduced the labeling of cyclin T1 by ~3 fold, while the labeling of CDK9 and of a control protein (actin) were unaffected (Fig. 4D). Reduced synthesis of cyclin T1 was observed with labeling times as short as 30 min (Supplementary Fig. S3), ruling out the possibility of rapid protein turnover. Changes in translation rates are generally accompanied by changes in the association of mRNA with ribosomes, which can be visualized by sucrose gradient sedimentation. As seen in Figure 4E (top panels), NF90 depletion caused a marked shift of cyclin T1 mRNA from large polysomes to monosomes and smaller polysomes. The distribution of a control mRNA (cyclophilin A; middle panels) was unaffected, as was the overall polysome profile (represented by 5S rRNA; bottom panels). These data imply that NF90 regulates cyclin T1 synthesis at the level of translation initiation.
Despite its reduced accumulation (Fig. 4A), the synthesis of CDK9 was unaffected in NF90-depleted cells (Fig. 4D). CDK9 turnover is accelerated when cyclin T1 is deficient in HeLa cells42. Accordingly, we hypothesized that CDK9 is destabilized in NF90-depleted cells as a result of the reduced level of cyclin T1. Pulse-chase experiments were conducted to test this hypothesis: CDK9 and cyclin T1 labeling was measured in siD3-treated cells relative to siC-treated cells (Fig. 4F). Consistent with previous results, the amount of pulse-labeled CDK9 was unaffected by NF90 knockdown, whereas cyclin T1 labeling was reduced by ~50% (Fig. 4F; 0 hr). CDK9 was stable for the first 3 hr of chase, but subsequently fell to <40% of the level in control cells. After an extended chase period, the decrease in labeled CDK9 matched that in labeled cyclin T1 in NF90-depleted cells (Fig. 4F; 24–48 hr). We conclude that NF90 regulates P-TEFb posttranscriptionally, by controlling the synthesis of cyclin T1 and consequently the stability of CDK9.
Regulation of cyclin T1 translation via its 3′UTR
Unlike endogenous CDK9 and cyclin T1, the accumulation of Gal4 fusion proteins appeared to be unaffected by NF90 depletion (Fig. 3C). This observation was confirmed in cells transfected with a vector expressing HA-tagged cyclin T1. While the level of endogenous cyclin T1 was reduced by almost 50% in NF90-depleted cells, HA-cyclin T1 levels were undiminished (Fig. 5A). Both the Gal4 and HA vectors lack the 5′ and 3′ flanking regions of the endogenous mRNA, which contain potential regulatory sequences, suggesting that sensitivity is conferred by UTR sequences of the endogenous cyclin T1 mRNA that are absent from the ectopically expressed cyclin T1 vectors. To examine this inference, we inserted the cyclin T1 5′UTR (~323 nt) and/or 3′UTR (~4.7 kb) into a test vector containing the FF gene under the control of the PCNA promoter which displays little dependence on P-TEFb43; 44. Sensitivity to NF90 knockdown was conferred exclusively by the cyclin T1 3′UTR (Fig. 5B). Consistent with an action at the translational level, the UTRs had little effect on FF mRNA accumulation in D3 treated cells (± <20%). Four subfragments of the 3′UTR were also tested (Fig. 5C). All of the subfragments conferred a degree of inhibition, which was greatest with the distal subfragment (adjacent to polyA; ~55% inhibition) and least with the proximal subfragment (adjacent to the coding region; ~20% inhibition). Evidently more than one site in the 3′UTR of cyclin T1 mRNA can contribute to NF90 sensitivity.
Figure 5. Role of the cyclin T1 mRNA 3′UTR.
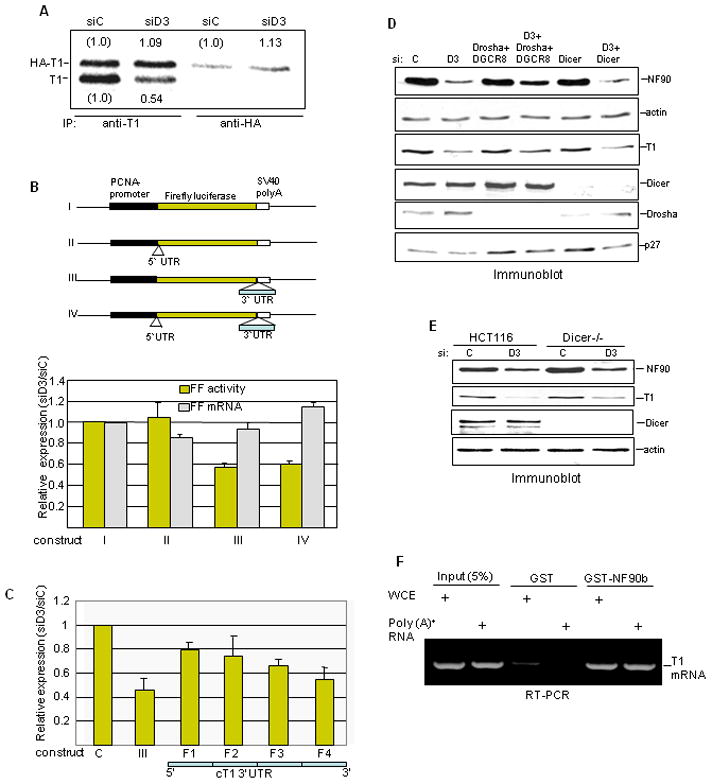
A. HeLa cells transfected with siRNA and then with HA-cyclin T1 expression vector were pulse-labeled with [S35]-Met/Cys 24 hr later. Proteins were immunoprecipitated with anti-cyclin T1 or anti-HA antibody, resolved in denaturing gels, and detected and quantified by phosphorimager. Radioactivity in HA-cyclin T1 (HA-T1) and in endogenous cyclin T1 (T1) in NF90-depleted cells is reported relative to that in siC-treated cells. B. siRNA-treated HeLa cells were transfected with PCNA promoter-driven FF reporter construct (I) or constructs carrying the cyclin T1 mRNA 5′UTR (II), 3′UTR (III), or both UTRs (IV) as diagrammed (top panel). Cell extracts were analyzed for FF activity and FF RNA by RPA. Data were normalized to total protein and GAPDH RNA and are presented as the siD3/siC ratio (bottom panel). C. Four 3′UTR sub-fragments (F1–F4) were inserted downstream of FF and assayed for luciferase activity in comparison with construct III as in panel B. D. HeLa cells were transfected twice with the siRNAs indicated and extracts were analyzed by immunoblotting. E. HCT116 wild-type and HCT116 Dicer−/− cells were transfected with D3 siRNA and protein was analyzed by immunoblotting at 72 hr post-transfection. F. RT-PCR analysis of RNA bound to GST proteins was performed using primers specific for cyclin T1 3′UTR.
This finding was suggestive of a mechanism involving miRNA. Recent reports indicated that miR-198 inhibits cyclin T1 translation in primary monocytes via several sequences in its 3′UTR12 and that NF90 inhibits miRNA maturation in HeLa cells24. Furthermore PMA has been shown to regulate miRNA expression45 and cellular miRNAs regulate HIV replication and latency46; 47; 48. To evaluate the involvement of miRNA in cyclin T1 regulation by NF90, we inhibited miRNA processing by silencing Drosha (alone or with DGCR8) or Dicer. Neither treatment prevented the reduction of cyclin T1 by NF90 depletion in HeLa cells (Fig. 5D). Correspondingly, siRNA D3 suppressed cyclin T1 in colon carcinoma cells containing a homozygous Dicer disruption (Dicer−/−), as in the parental HCT116 cells (Fig. 5E). None of these experiments offered support for a miRNA role in NF90’s action on cyclin T1. In addition, transfection of HeLa cells with a miR-198 antagomir failed to rescue the inhibition of cyclin T1 by siD3 (data not shown). On the other hand, as an RNA binding protein, NF90 could act by directly binding to cyclin T1 mRNA. In support of this mechanism, GST-NF90 pulled down cyclin T1 mRNA from HeLa cell extract and from total HeLa poly(A)+ RNA (Fig. 5F). These results favor the view that NF90 acts by binding to one or more sites in the cyclin T1 mRNA 3′UTR and facilitating the recruitment of factors required for translation initiation.
Role of NF90 during HIV-1 induction in latently infected cells
Production of HIV-1 is induced when latently-infected cells are activated by agents such as PMA49. We and others found that PMA stimulates cyclin T1 levels in U937 promonocytic cells50 and P-TEFb activity in their latently-infected U1 derivatives51. U1 cells harbor an HIV-1 variant encoding a defective Tat protein52, and it is thought that P-TEFb induction compensates for the low activity of the mutant Tat50. These observations led us to consider the possibility that NF90 is involved in the induction of cyclin T1 and virus production after PMA stimulation.
PMA activation of U1 cells resulted in a large stimulation in p24 expression beginning about 9 hr after treatment (Fig. 6A), and cyclin T1 levels were induced concomitantly (Fig. 6B). As reported previously53, the induction of cyclin T1 in U937 cells occurred with no change in the cyclin T1 mRNA level (Fig. 6C). Similar post-transcriptional regulation of cyclin T1 was observed in U1 cells (Fig. 6C). Strikingly, NF90 was greatly induced by PMA in both U1 and U937 cells (Fig. 6B) and this induction also occurred without an increase in its mRNA level (Fig. 6C). Treatment with D3 siRNA repressed the induction by PMA of cyclin T1 as well as of NF90 in both U1 and U937 cells (Fig. 6B; right panels). Consistent with the reduction in cyclin T1 levels, the production of viral p24 by U1 cells was inhibited by NF90 knockdown (Fig. 6D). These results indicate that NF90 regulates cyclin T1 in U1 and U937 cells, as in HeLa and HCT116 cells, and imply that NF90 induction plays a key role in the activation of HIV-1 by PMA in latently infected cells.
Figure 6. Induction of NF90, cyclin T1 and HIV-1 p24 in promonocytic cells.
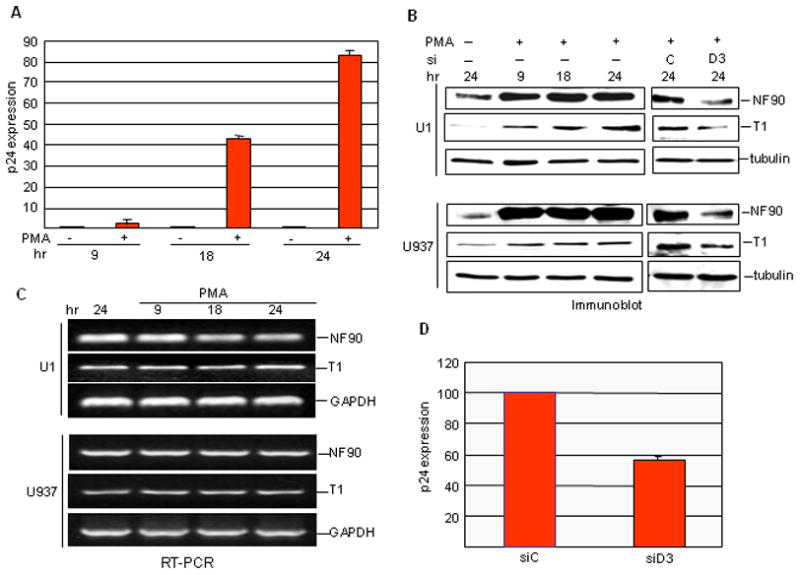
A. Production of p24 in U1 cells assayed as a function of PMA treatment time. B. U1 (top) or U937 (bottom) cells were treated with PMA for the times shown, or left untreated for 24 h, and WCEs were analyzed by immunoblotting (left panels). PMA treated cells were transfected with siRNA (C or D3) and analyzed in the same way (right panels). C. Total RNA from PMA-treated and untreated U1 or U937 cells was analyzed by semi quantitative RT-PCR. D. Effect of siRNA transfection on p24 production in U1 cells.
Discussion
NF90 is an RNA-binding protein that serves as a molecular adapter between nucleic acids and proteins. We show here that it plays a major role in the replication of HIV-1 in productively infected cells and in the activation of quiescent virus in latently infected cells by phorbol ester. NF90 acts via the essential transcription factor P-TEFb, regulating the synthesis of its cyclin T1 subunit and, consequently, the stability of its CDK9 subunit. Specifically, NF90 enhances translation initiation via the cyclin T1 3′UTR (Fig. 7A). Furthermore, NF90 itself is induced posttranscriptionally by PMA in monocytes implying a role in PKC signaling (Fig. 7B).
Figure 7. Role of NF90 in HIV-1 replication.
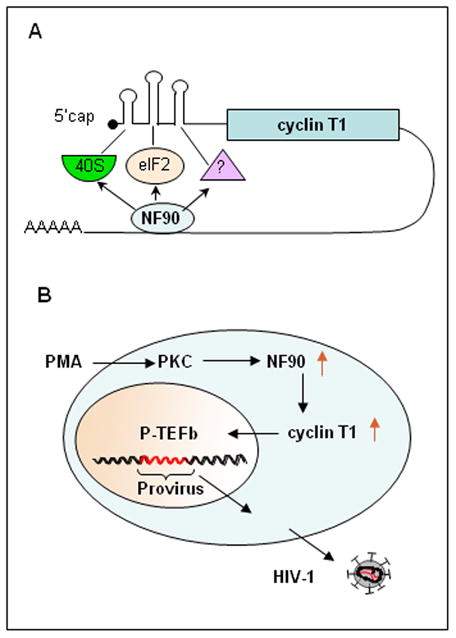
A. Models for NF90’s action on cyclin T1 translation. B. Pathway of posttranscriptional PMA signaling and HIV-1 reactivation in latently infected cells. See text for details.
Our study is the first to observe that NF90 exerts a positive effect on cyclin T1 protein levels, in cells of diverse lineages, and to demonstrate a positive action of NF90 on translation initiation. NF90’s ability to bind structured RNA and a growing list of proteins, especially RNA binding proteins, makes it a pleiotropic modulator of mRNA metabolism. It has been reported to act posttranscriptionally to regulate the nuclear export15, stability33;14; 54; 55 and translation of certain mRNAs54; 56. In contrast to its action on cyclin T1 reported here, most of NF90’s known translational effects are inhibitory. For example, NF90 negatively regulates the translation of a set of mRNAs through a 3′UTR motif, as well as at least one mRNA that does not contain the motif57. We found that positive regulation of cyclin T1 mRNA translation by NF90 is also conferred by its 3′UTR. This response, unlike that seen when monocytes differentiate into macrophages12, is not mediated via miRNAs in the cell types examined (HeLa and HCT116), but the miRNA- and NF90-based mechanisms could act in concert in other cells. At least four segments of the cyclin T1 3′UTR are able to confer sensitivity to NF90 upon a reporter gene, implying that multiple NF90 response elements exist in the mRNA. While the sequences and structures of these elements await identification, the well-documented RNA binding activity of NF90 is consistent with the ability of GST-NF90 to pull down cyclin T1 mRNA and is suggestive of a direct interaction between NF90 and the mRNA.
How does this interaction lead to an up-regulation of initiation? The closed-loop model for active mRNA translation, originally visualized as a linkage of the mRNA 5′-cap to its 3′-polyA tail via specific binding proteins58, has expanded with the recognition of 5′-3′ end interactions mediated by other proteins that bind mRNA UTRs59. NF90 has been shown to circularize viral RNAs34 and we speculate that it serves to facilitate 5′-3′ end interactions on cyclin T1 mRNA (Fig. 7A). NF90 participates in complexes containing a variety of additional proteins including RNA binding proteins60; 61, ribosomes15; 27 and ribosomal protein S19 (RPS19)62. The RPS19 interactome includes RNA helicases, some of which were identified in our search for NF90 binding partners20. NF90 is found in the ribosomal salt wash27 which is enriched in translation initiation factors, and it binds to the α subunit of eIF229; 63. Lending further support to the involvement of this initiation factor, NF90 shares a sequence element with eIF2α and both are substrates for the eIF2 kinase PKR27; 28; 31. While the cyclin T1 5′UTR is not essential for the action of NF90, it is notable that it contains an internal ribosome entry site (IRES)64. This IRES is activated by the polypyrimidine tract binding protein (PTB) suggesting that loop formation may involve direct or indirect PTB-NF90 interactions.
Enhanced cyclin T1 translation appears to require the C-terminal domain of NF90 (Fig. 1A). This domain contains an RGG motif, originally described as an RNA binding sequence in hnRNP proteins65; 66. The NF90c variant, which lacks the RGG motif, is modestly inhibitory, suggesting that it exerts a dominant negative effect with respect to endogenous NF90. Although not found in human cell lines17; 37, it remains possible that NF90c is a physiological repressor of NF90 function in specific tissues or circumstances. Indirect evidence indicates that NF110 also exerts a negative effect on HIV transcription (data not shown). This isoform has the RGG motif within an extended C-terminal domain, including a GQSR motif which may counteract the stimulatory action of NF90. Additional NF90 domains involved in its translational control function remain to be defined. Even though NF45 is complexed with NF90/110 and other proteins in large multi-protein complexes20, neither over-expression or knockdown of NF45 elicited a discernible effect on cyclin T1 in HeLa cells, suggesting that NF45 is not part of the NF90 complex involved in translational control of cyclin T1. Dissociation of NF90/110 from NF45 in the cytoplasm has been reported19. Alternatively, it may be present in the complex but not critical for NF90’s translational function. On the other hand, it is notable that NF45 enhanced translation of the mRNA encoding the inhibitor of apoptosis protein cIAP1 via interactions with its IRES67.
P-TEFb is a critical HIV-1 co-factor and an important limiting factor in populations of latently infected primary T cells. As reported previously42; 68 and shown here, P-TEFb depletion impairs HIV gene expression and replication. P-TEFb is present at very low levels in PBLs, monocytes and monocytic cell lines, restricting viral replication to activated cells9; 69; 70. P-TEFb is greatly induced by stimulation of the T-cell receptor, contributing to viral replication and the emergence from latency71. The monocytic cell line U937 and its latently infected derivative, the U1 cell line, recapitulate many of the characteristics of uninfected and infected primary monocytic cells, including a dramatic increase in cyclin T1 levels after exposure to PMA, differentiation into macrophages, and viral production72. Strikingly, we found that NF90 protein levels, which are very low in both U937 and U1 cells, are greatly induced after exposure to PMA. As with cyclin T1, the induction of NF90 cells is also posttranscriptional, and possibly translational. Depletion of NF90 reduced the level of cyclin T1 in U937 and U1 cells, arguing that NF90 is positioned upstream of cyclin T1 in the signal transduction pathway triggered by PMA (Fig. 7B).
PMA signals by activating PKC73. We therefore propose that PMA up-regulates the translation of cyclin T1 mRNA, and probably of NF90 mRNA, in U937 and U1 cells via PKC (Fig. 7B). Several lines of evidence support this view. First, PKC is required for Tat-mediated transactivation of the HIV-1 promoter74, and the increase in cyclin T1 levels seen in PMA-activated PBLs is prevented by a PKC inhibitor11. Second, translational effects have been reported for PKC75; 76. Third, NF90 is a phosphoprotein18; 77, and recent work demonstrated that PMA induces the PKC-dependent phosphorylation of NF90 and enhances its export into the cytoplasm78. Finally, the closely related protein TCP80 is also a PKC substrate and its translation regulatory properties are drastically affected by its phosphorylation status79. Our data implicate NF90 induction in viral transcription and proviral activation, but whether NF90 phosphorylation is also a prerequisite remains to be determined.
The findings presented here establish the role of NF90 in regulating cyclin T1 synthesis, HIV-1 replication and emergence from latency. Currently almost all HIV-1 antivirals target viral proteins, which are subject to rapid mutation leading to the evolution of drug resistant viruses. Agents are currently being developed against cellular proteins that are critical for the virus, including P-TEFb which appears to be a particularly sensitive target80; 81; 82. Our findings highlight NF90 as a potential target for therapeutics against HIV-1 and expand the scope of translational control as a growing area of interest for therapeutic intervention.
Materials and methods
Cell culture
HeLaS3 and 293T cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). U937 cells and U1 cells (U1/HIV; NIH AIDS Research and Reference Reagent Program, Germantown, MD) were grown in Roswell Park Memorial Institute (RPMI) medium with 8% FBS. HCT116 and HCT116 Dicer−/− cells (83,84), provided by Dr. F. Kashanchi, were grown in McCoy’s 5a medium with 10% FBS.
HIV-1
Pseudotyped NL4-3-LucE− virus85 was prepared as described previously86, concentrated using Microcon (Millipore, Billerica, MA), and estimated by p24 assay (ZeptoMetrix Corporation, Buffalo, NY). Cells were infected 72 hr after siRNA treatment at a multiplicity of 10.
Plasmids
pMRev (−) and pCMV-Rev were obtained from the NIH AIDS Research and Reference Reagent Program. The full-length cyclin T1 3′UTR and its F1–F4 fragments were sub-cloned from pCMV-luc cT1 3′UTR and derivatives (Sung and Rice, 2009) into the pGL3-PCNA vector downstream of the firefly luciferase gene. pGL3-PCNA was generated by inserting the minimal PCNA promoter between the Kpn I and Hind III sites of pGL3. The cyclin T1 5′UTR sequence was PCR amplified and inserted into the pGL3-PCNA plasmid between Hind III and Nco I sites. To generate a pGL3-PCNA expression vector carrying both 5′ and 3′UTRs, the CMV promoter of pCMV-luc cT1 3′UTR was replaced with a fragment containing the PCNA promoter linked to the cyclin T1 5′UTR. Other plasmids have been described previously: pcDNA3.1+HisB-NF90b, -NF110b, and -NF90c87; pcDNA3.1+HisB-NF4588; pGL2TAR (HIV-FF) and pNL4-3-LucE−86; Gal4-cyclin T1 and -CDK9, and G5-HIV firefly luciferase89; and pcDNA3-HA- cyclin T1, -Tat and -CDK9, and pCMV-Renilla90.
Biological reagents
siRNA against NF45 (D5), NF90 (D3) and NF110 (D4) were described previously20. Antibodies against cyclin T1, CDK9, HA, actin and Omni were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); against tubulin, from Sigma (St. Louis, MO; and against NF90/110 (DRBP76), from BD Bioscience (Franklin Lakes, NJ). Anti-NF45 antibody was described previously18. Radiochemicals were from MP Biochemicals (Santa Ana, CA).
Transfection
HeLa cells were transfected, 1 day after seeding, with siRNA at a final concentration of 50 nM using HiPerFect (Qiagen, Valencia, CA) or INTERFERin™ (Polyplus transfection, New York, NY). At 24 hr posttransfection cells were reseeded for plasmid transfection using JetPEI (polyplus transfection™). Except where noted, cells were harvested 24 hr later for analysis of RNA, protein and luciferase activity (Dual luciferase kit; Promega, Madison, WI). In some experiments, the cells were re-transfected with siRNA 24 hr after reseeding and were transfected with plasmid 6 hr later. U937 and U1 cells were transfected with siRNA using N-TER nanoparticle siRNA transfection reagent (Sigma, St. Louis, MO) and maintained at a density of 5×104 cells/ml for 72 hr, then activated with PMA (1 ng/ml) for 24 hr before harvesting.
RNA analysis
Nuclear and cytoplasmic RNA was isolated as described previously86 and assayed by RNase protection using the RPAIII kit (Ambion, Austin, TX) and radiolabeled antisense probes for firefly and Renilla luciferase and GAPDH90 and for spliced and unspliced HIV RNA86. Cyclin T1 and CDK9 antisense probes were generated by subcloning the N-terminal 300 and 250 nt into the pcDNA3.1 vector.
Metabolic labeling
HeLa cells transfected with siRNA were labeled for 2 hr with [35S]-Met/Cys (MP Biochemicals) and chased without label as described previously20. Extracts were analyzed by immunoprecipitation. To analyze recombinant HA-cyclin T1 synthesis, cells were transfected with cyclin T1 or CDK9 expression vectors 48 hr after siRNA transfection.
Sedimentation analysis
HeLa WCE was fractionated for P-TEFb in glycerol gradients as described previously91. HeLa cell polysomes were fractionated in sucrose gradient as described previously92.
mRNA pulldown
GST proteins (5 μg of GST-NF90 or an equimolar amount of GST) produced in bacteria were coupled to GSH beads and mixed with HeLa whole cell extract (WCE; 500 μg protein) or equivalent purified poly(A)+ RNA. After incubation for 2 hr at 4°C with mild rocking and extensive washing in lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 20 U/ml RNasin, 1x protease inhibitor cocktail, and 0.5 mM DTT), RNA was isolated from the beads by Trizol extraction (Invitrogen, Carlsbad, CA) and analyzed by semi-quantitative RT-PCR One-step RT kit (Qiagen).
Supplementary Material
Acknowledgments
We thank Dr. Andrew P. Rice for plasmids carrying the cyclin T1 3′UTR, Dr. Fatah Kashanchi for HCT116 wild-type and Dicer−/− cells, and Ms. Anita Antes for help with cell culture.
Abbreviations used
- HIV
human immunodeficiency virus
- P-TEFb
positive transcription elongation factor b
- NF90
nuclear factor 90
- NF110
nuclear factor 110
- NF45
nuclear factor 45
- Pol II
RNA polymerase II
- CDK
cyclin-dependent kinase
- Tat
transactivator of transcription
- TAR
transactivation response
- miRNA
microRNA
- LTR
long terminal repeat
- 3′UTR
3′ untranslated region
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- siRNA
small interfering RNA
- FF
firefly luciferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mainul Hoque, Email: hoquema@umdnj.edu.
Raghavendra A. Shamanna, Email: shamanra@umdnj.edu.
Deyu Guan, Email: guandeyu@yahoo.com.
Tsafi Pe’ery, Email: peeryts@umdnj.edu.
References
- 1.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lis JT, Mason P, Peng J, Price DH, Werner J. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000;14:792–803. [PMC free article] [PubMed] [Google Scholar]
- 3.Espinoza-Derout J, Wagner M, Salciccioli L, Lazar JM, Bhaduri S, Mascareno E, Chaqour B, Siddiqui MA. Positive transcription elongation factor b activity in compensatory myocardial hypertrophy is regulated by cardiac lineage protein-1. Circ Res. 2009;104:1347–54. doi: 10.1161/CIRCRESAHA.108.191726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–62. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 6.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev. 2006;70:646–59. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liou LY, Herrmann CH, Rice AP. Human immunodeficiency virus type 1 infection induces cyclin T1 expression in macrophages. J Virol. 2004;78:8114–9. doi: 10.1128/JVI.78.15.8114-8119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice AP, Herrmann CH. Regulation of TAK/P-TEFb in CD4+ T lymphocytes and macrophages. Curr HIV Res. 2003;1:395–404. doi: 10.2174/1570162033485159. [DOI] [PubMed] [Google Scholar]
- 10.Garriga J, Peng J, Parreno M, Price DH, Henderson EE, Grana X. Upregulation of cyclin T1/CDK9 complexes during T cell activation. Oncogene. 1998;17:3093–102. doi: 10.1038/sj.onc.1202548. [DOI] [PubMed] [Google Scholar]
- 11.Marshall RM, Salerno D, Garriga J, Grana X. Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J Immunol. 2005;175:6402–11. doi: 10.4049/jimmunol.175.10.6402. [DOI] [PubMed] [Google Scholar]
- 12.Sung TL, Rice AP. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009;5:e1000263. doi: 10.1371/journal.ppat.1000263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. The double-stranded-RNA-binding motif: interference and much more. Nat Rev Mol Cell Biol. 2004;5:1013–23. doi: 10.1038/nrm1528. [DOI] [PubMed] [Google Scholar]
- 14.Shi L, Zhao G, Qiu D, Godfrey WR, Vogel H, Rando TA, Hu H, Kao PN. NF90 regulates cell cycle exit and terminal myogenic differentiation by direct binding to the 3′-untranslated region of MyoD and p21WAF1/CIP1 mRNAs. J Biol Chem. 2005;280:18981–9. doi: 10.1074/jbc.M411034200. [DOI] [PubMed] [Google Scholar]
- 15.Pfeifer I, Elsby R, Fernandez M, Faria PA, Nussenzveig DR, Lossos IS, Fontoura BM, Martin WD, Barber GN. NFAR-1 and -2 modulate translation and are required for efficient host defense. Proc Natl Acad Sci U S A. 2008;105:4173–8. doi: 10.1073/pnas.0711222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duchange N, Pidoux J, Camus E, Sauvaget D. Alternative splicing in the human interleukin enhancer binding factor 3 (ILF3) gene. Gene. 2000;261:345–53. doi: 10.1016/s0378-1119(00)00495-9. [DOI] [PubMed] [Google Scholar]
- 17.Reichman TW, Parrott AM, Fierro-Monti I, Caron DJ, Kao PN, Lee CG, Li H, Mathews MB. Selective regulation of gene expression by nuclear factor 110, a member of the NF90 family of double-stranded RNA-binding proteins. J Mol Biol. 2003;332:85–98. doi: 10.1016/s0022-2836(03)00885-4. [DOI] [PubMed] [Google Scholar]
- 18.Parrott AM, Walsh MR, Reichman TW, Mathews MB. RNA binding and phosphorylation determine the intracellular distribution of nuclear factors 90 and 110. J Mol Biol. 2005;348:281–93. doi: 10.1016/j.jmb.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 19.Harashima A, Guettouche T, Barber GN. Phosphorylation of the NFAR proteins by the dsRNA-dependent protein kinase PKR constitutes a novel mechanism of translational regulation and cellular defense. Genes Dev. 2010;24:2640–53. doi: 10.1101/gad.1965010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan D, Altan-Bonnet N, Parrott AM, Arrigo CJ, Li Q, Khaleduzzaman M, Li H, Lee CG, Pe’ery T, Mathews MB. Nuclear factor 45 (NF45) is a regulatory subunit of complexes with NF90/110 involved in mitotic control. Mol Cell Biol. 2008;28:4629–41. doi: 10.1128/MCB.00120-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barber GN. The NFAR’s (nuclear factors associated with dsRNA): evolutionarily conserved members of the dsRNA binding protein family. RNA Biol. 2009;6:35–9. doi: 10.4161/rna.6.1.7565. [DOI] [PubMed] [Google Scholar]
- 22.Reichman TW, Mathews MB. The NF90 Family of Double-stranded RNA Binding Proteins: Regulators of Viral and Cellular Function. In: Bradshaw R, Dennis E, editors. Cell Signaling Handbook. Academic Press; 2003. pp. 335–342. [Google Scholar]
- 23.Parrott AM, Walsh MR, Mathews MB. Analysis of RNA:protein interactions in vivo: identification of RNA-binding partners of nuclear factor 90. Methods Enzymol. 2007;429:243–60. doi: 10.1016/S0076-6879(07)29012-3. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto S, Aoki K, Higuchi T, Todaka H, Morisawa K, Tamaki N, Hatano E, Fukushima A, Taniguchi T, Agata Y. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol Cell Biol. 2009;29:3754–69. doi: 10.1128/MCB.01836-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bass BL, Hurst SR, Singer JD. Binding properties of newly identified Xenopus proteins containing dsRNA-binding motifs. Curr Biol. 1994;4:301–316. doi: 10.1016/s0960-9822(00)00069-5. [DOI] [PubMed] [Google Scholar]
- 26.Liao HJ, Kobayashi R, Mathews MB. Activities of adenovirus virus-associated RNAs: purification and characterization of RNA binding proteins. Proc Natl Acad Sci USA. 1998;95:8514–9. doi: 10.1073/pnas.95.15.8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langland JO, Kao PN, Jacobs BL. Nuclear factor-90 of activated T-cells: A double-stranded RNA-binding protein and substrate for the double-stranded RNA-dependent protein kinase, PKR. Biochemistry. 1999;38:6361–8. doi: 10.1021/bi982410u. [DOI] [PubMed] [Google Scholar]
- 28.Parker LM, Fierro-Monti I, Mathews MB. Nuclear factor 90 is a substrate and regulator of the eukaryotic initiation factor 2 kinase double-stranded RNA-activated protein kinase. J Biol Chem. 2001;276:32522–30. doi: 10.1074/jbc.M104408200. [DOI] [PubMed] [Google Scholar]
- 29.Parker LM, Fierro-Monti I, Reichman TW, Gunnery S, Mathews MB. Double-stranded RNA-binding proteins and the control of protein synthesis and cell growth. Cold Spring Harb Symp Quant Biol. 2001;66:485–97. doi: 10.1101/sqb.2001.66.485. [DOI] [PubMed] [Google Scholar]
- 30.Patel RC, Vestal DJ, Xu Z, Bandyopadhyay S, Guo W, Erme SM, Williams BR, Sen GC. DRBP76, a double-stranded RNA-binding nuclear protein, is phosphorylated by the interferon-induced protein kinase, PKR. J Biol Chem. 1999;274:20432–7. doi: 10.1074/jbc.274.29.20432. [DOI] [PubMed] [Google Scholar]
- 31.Saunders LR, Perkins DJ, Balachandran S, Michaels R, Ford R, Mayeda A, Barber GN. Characterization of two evolutionarily conserved, alternatively spliced nuclear phosphoproteins, NFAR-1 and -2, that function in mRNA processing and interact with the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem. 2001;276:32300–12. doi: 10.1074/jbc.M104207200. [DOI] [PubMed] [Google Scholar]
- 32.Corthésy B, Kao PN. Purification by DNA affinity chromatography of two polypeptides that contact the NF-AT DNA binding site in the interleukin 2 promoter. J Biol Chem. 1994;269:20682–20690. [PubMed] [Google Scholar]
- 33.Shim J, Lim H, J RY, Karin M. Nuclear export of NF90 is required for interleukin-2 mRNA stabilization. Mol Cell. 2002;10:1331–44. doi: 10.1016/s1097-2765(02)00730-x. [DOI] [PubMed] [Google Scholar]
- 34.Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. Nuclear factors are involved in hepatitis C virus RNA replication. RNA. 2007;13:1675–92. doi: 10.1261/rna.594207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agbottah ET, Traviss C, McArdle J, Karki S, St Laurent GC, 3rd, Kumar A. Nuclear Factor 90(NF90) targeted to TAR RNA inhibits transcriptional activation of HIV-1. Retrovirology. 2007;4:41. doi: 10.1186/1742-4690-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urcuqui-Inchima S, Castano ME, Hernandez-Verdun D, St-Laurent G, 3rd, Kumar A. Nuclear Factor 90, a cellular dsRNA binding protein inhibits the HIV Rev-export function. Retrovirology. 2006;3:83. doi: 10.1186/1742-4690-3-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krasnoselskaya-Riz I, Spruill A, Chen YW, Schuster D, Teslovich T, Baker C, Kumar A, Stephan DA. Nuclear factor 90 mediates activation of the cellular antiviral expression cascade. AIDS Res Hum Retroviruses. 2002;18:591–604. doi: 10.1089/088922202753747941. [DOI] [PubMed] [Google Scholar]
- 38.Sadaie MR, Benter T, Wong-Staal F. Site-directed mutagenesis of two trans-regulatory genes (tat-III, trs) of HIV-1. Science. 1988;239:910–3. doi: 10.1126/science.3277284. [DOI] [PubMed] [Google Scholar]
- 39.Majello B, Napolitano G, Giordano A, Lania L. Transcriptional regulation by targeted recruitment of cyclin-dependent CDK9 kinase in vivo. Oncogene. 1999;18:4598–605. doi: 10.1038/sj.onc.1202822. [DOI] [PubMed] [Google Scholar]
- 40.Parrott AM, Walsh MR, Mathews MB. Analysis of RNA:protein interactions in vivo: Identification of RNA binding partners of nuclear factor 90. In: Lorsch JR, editor. Methods in Enzymology. Vol. 429. Elsevier Inc; San Diego, CA: 2007. pp. 243–260. [DOI] [PubMed] [Google Scholar]
- 41.Van Herreweghe E, Egloff S, Goiffon I, Jady BE, Froment C, Monsarrat B, Kiss T. Dynamic remodelling of human 7SK snRNP controls the nuclear level of active P-TEFb. Embo J. 2007;26:3570–80. doi: 10.1038/sj.emboj.7601783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiu YL, Cao H, Jacque JM, Stevenson M, Rana TM. Inhibition of human immunodeficiency virus type 1 replication by RNA interference directed against human transcription elongation factor P-TEFb (CDK9/CyclinT1) J Virol. 2004;78:2517–29. doi: 10.1128/JVI.78.5.2517-2529.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoque M, Tian B, Mathews MB, Pe’ery T. Granulin and granulin repeats interact with the Tat.P-TEFb complex and inhibit Tat transactivation. J Biol Chem. 2005;280:13648–57. doi: 10.1074/jbc.M409575200. [DOI] [PubMed] [Google Scholar]
- 44.Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M, Wahl LM, Harris ED, Mikovits JA, Monks AP, Hollingshead MG, Sausville EA, Staudt LM. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:0041.1–11. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeung ML, Yasunaga J, Bennasser Y, Dusetti N, Harris D, Ahmad N, Matsuoka M, Jeang KT. Roles for microRNAs, miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressor in cell growth dysregulation by human T-cell lymphotrophic virus 1. Cancer Res. 2008;68:8976–85. doi: 10.1158/0008-5472.CAN-08-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–7. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 47.Triboulet R, Mari B, Lin YL, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, Reynes J, Corbeau P, Jeang KT, Benkirane M. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315:1579–82. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 48.Chable-Bessia C, Meziane O, Latreille D, Triboulet R, Zamborlini A, Wagschal A, Jacquet JM, Reynes J, Levy Y, Saib A, Bennasser Y, Benkirane M. Suppression of HIV-1 replication by microRNA effectors. Retrovirology. 2009;6:26. doi: 10.1186/1742-4690-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Folks TM, Justement J, Kinter A, Schnittman S, Orenstein J, Poli G, Fauci AS. Characterization of a promonocyte clone chronically infected with HIV and inducible by 13-phorbol-12-myristate acetate. J Immunol. 1988;140:1117–22. [PubMed] [Google Scholar]
- 50.Reza SM, Rosetti M, Mathews MB, Pe’ery T. Differential activation of Tat variants in mitogen-stimulated cells: implications for HIV-1 postintegration latency. Virology. 2003;310:141–56. doi: 10.1016/s0042-6822(03)00106-5. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Gold MO, Tang DN, Lewis DE, Aguilar-Cordova E, Rice AP, Herrmann CH. TAK, an HIV Tat-associated kinase, is a member of the cyclin-dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proc Natl Acad Sci USA. 1997;94:12331–6. doi: 10.1073/pnas.94.23.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emiliani S, Fischle W, Ott M, Van Lint C, Amella CA, Verdin E. Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the U1 cell line. J Virol. 1998;72:1666–70. doi: 10.1128/jvi.72.2.1666-1670.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J Virol. 1998;72:9881–8. doi: 10.1128/jvi.72.12.9881-9888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuwano Y, Kim HH, Abdelmohsen K, Pullmann R, Jr, Martindale JL, Yang X, Gorospe M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol Cell Biol. 2008;28:4562–75. doi: 10.1128/MCB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vumbaca F, Phoenix KN, Rodriguez-Pinto D, Han DK, Claffey KP. Double-stranded RNA-binding protein regulates vascular endothelial growth factor mRNA stability, translation, and breast cancer angiogenesis. Mol Cell Biol. 2008;28:772–83. doi: 10.1128/MCB.02078-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu YH, Busald C, Grabowski GA. Reconstitution of TCP80/NF90 translation inhibition activity in insect cells. Mol Genet Metab. 2000;70:106–15. doi: 10.1006/mgme.2000.3010. [DOI] [PubMed] [Google Scholar]
- 57.Kuwano Y, Pullmann R, Jr, Marasa BS, Abdelmohsen K, Lee EK, Yang X, Martindale JL, Zhan M, Gorospe M. NF90 selectively represses the translation of target mRNAs bearing an AU-rich signature motif. Nucleic Acids Res. 2010;38:225–38. doi: 10.1093/nar/gkp861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–40. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 59.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Merz C, Urlaub H, Will CL, Luhrmann R. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA. 2007;13:116–28. doi: 10.1261/rna.336807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tran H, Schilling M, Wirbelauer C, Hess D, Nagamine Y. Facilitation of mRNA deadenylation and decay by the exosome-bound, DExH protein RHAU. Mol Cell. 2004;13:101–11. doi: 10.1016/s1097-2765(03)00481-7. [DOI] [PubMed] [Google Scholar]
- 62.Orru S, Aspesi A, Armiraglio M, Caterino M, Loreni F, Ruoppolo M, Santoro C, Dianzani I. Analysis of the ribosomal protein S19 interactome. Mol Cell Proteomics. 2007;6:382–93. doi: 10.1074/mcp.M600156-MCP200. [DOI] [PubMed] [Google Scholar]
- 63.Ting NS, Kao PN, Chan DW, Lintott LG, Lees-Miller SP. DNA-dependent protein kinase interacts with antigen receptor response element binding proteins NF90 and NF45. J Biol Chem. 1998;273:2136–2145. doi: 10.1074/jbc.273.4.2136. [DOI] [PubMed] [Google Scholar]
- 64.Bushell M, Stoneley M, Kong YW, Hamilton TL, Spriggs KA, Dobbyn HC, Qin X, Sarnow P, Willis AE. Polypyrimidine tract binding protein regulates IRES-mediated gene expression during apoptosis. Mol Cell. 2006;23:401–12. doi: 10.1016/j.molcel.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 65.Kiledjian M, Dreyfuss G. Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J. 1992;11:2655–64. doi: 10.1002/j.1460-2075.1992.tb05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cobianchi F, Karpel RL, Williams KR, Notario V, Wilson SH. Mammalian heterogeneous nuclear ribonucleoprotein complex protein A1. Large-scale overproduction in Escherichia coli and cooperative binding to single-stranded nucleic acids. J Biol Chem. 1988;263:1063–71. [PubMed] [Google Scholar]
- 67.Graber TE, Baird SD, Kao PN, Mathews MB, Holcik M. NF45 functions as an IRES trans-acting factor that is required for translation of cIAP1 during the unfolded protein response. Cell Death Differ. 2010;17:719–29. doi: 10.1038/cdd.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, Xiong Y, Peng Y, Pan J, Chen Y, Wu X, Hussain S, Tien P, Guo D. Specific inhibition of HIV-1 replication by short hairpin RNAs targeting human cyclin T1 without inducing apoptosis. FEBS Lett. 2005;579:3100–6. doi: 10.1016/j.febslet.2005.04.074. [DOI] [PubMed] [Google Scholar]
- 69.Ghose R, Liou LY, Herrmann CH, Rice AP. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J Virol. 2001;75:11336–43. doi: 10.1128/JVI.75.23.11336-11343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liou LY, Haaland RE, Herrmann CH, Rice AP. Cyclin T1 but not cyclin T2a is induced by a post-transcriptional mechanism in PAMP-activated monocyte-derived macrophages. J Leukoc Biol. 2006;79:388–96. doi: 10.1189/jlb.0805429. [DOI] [PubMed] [Google Scholar]
- 71.Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–37. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cassol E, Alfano M, Biswas P, Poli G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J Leukoc Biol. 2006;80:1018–30. doi: 10.1189/jlb.0306150. [DOI] [PubMed] [Google Scholar]
- 73.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jakobovits A, Rosenthal A, Capon D. Trans-activation of HIV-1 LTR-directed gene expression requires protein kinase C. EMBO J. 1990;9:1165–1170. doi: 10.1002/j.1460-2075.1990.tb08223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ceci M, Gaviraghi C, Gorrini C, Sala LA, Offenhauser N, Marchisio PC, Biffo S. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;426:579–84. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 76.Grosso S, Volta V, Sala LA, Vietri M, Marchisio PC, Ron D, Biffo S. PKCbetaII modulates translation independently from mTOR and through RACK1. Biochem J. 2008;415:77–85. doi: 10.1042/BJ20080463. [DOI] [PubMed] [Google Scholar]
- 77.Pei Y, Zhu P, Dang Y, Wu J, Yang X, Wan B, Liu JO, Yi Q, Yu L. Nuclear export of NF90 to stabilize IL-2 mRNA is mediated by AKT-dependent phosphorylation at Ser647 in response to CD28 costimulation. J Immunol. 2008;180:222–9. doi: 10.4049/jimmunol.180.1.222. [DOI] [PubMed] [Google Scholar]
- 78.Zhu P, Jiang W, Cao L, Yu W, Pei Y, Yang X, Wan B, Liu JO, Yi Q, Yu L. IL-2 mRNA stabilization upon PMA stimulation is dependent on NF90-Ser647 phosphorylation by protein kinase CbetaI. J Immunol. 2010;185:5140–9. doi: 10.4049/jimmunol.1000849. [DOI] [PubMed] [Google Scholar]
- 79.Xu YH, Grabowski GA. Translation modulation of acid beta-glucosidase in HepG2 cells: participation of the PKC pathway. Mol Genet Metab. 2005;84:252–64. doi: 10.1016/j.ymgme.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 80.Zhou M, Deng L, Lacoste V, Park HU, Pumfery A, Kashanchi F, Brady JN, Kumar A. Coordination of transcription factor phosphorylation and histone methylation by the P-TEFb kinase during human immunodeficiency virus type 1 transcription. J Virol. 2004;78:13522–33. doi: 10.1128/JVI.78.24.13522-13533.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ali A, Ghosh A, Nathans RS, Sharova N, O’Brien S, Cao H, Stevenson M, Rana TM. Identification of flavopiridol analogues that selectively inhibit positive transcription elongation factor (P-TEFb) and block HIV-1 replication. Chembiochem. 2009;10:2072–80. doi: 10.1002/cbic.200900303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coley W, Kehn-Hall K, Van Duyne R, Kashanchi F. Novel HIV-1 therapeutics through targeting altered host cell pathways. Expert Opin Biol Ther. 2009;9:1369–82. doi: 10.1517/14712590903257781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA, Jr, Sjoblom T, Barad O, Bentwich Z, Szafranska AE, Labourier E, Raymond CK, Roberts BS, Juhl H, Kinzler KW, Vogelstein B, Velculescu VE. The colorectal microRNAome. Proc Natl Acad Sci U S A. 2006;103:3687–92. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carpio L, Klase Z, Coley W, Guendel I, Choi S, Van Duyne R, Narayanan A, Kehn-Hall K, Meijer L, Kashanchi F. microRNA machinery is an integral component of drug-induced transcription inhibition in HIV-1 infection. J RNAi Gene Silencing. 2010;6:386–400. [PMC free article] [PubMed] [Google Scholar]
- 85.Chen BK, Saksela K, Andino R, Baltimore D. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol. 1994;68:654–60. doi: 10.1128/jvi.68.2.654-660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hoque M, Hanauske-Abel HM, Palumbo P, Saxena D, D’Alliessi Gandolfi D, Park MH, Pe’ery T, Mathews MB. Inhibition of HIV-1 gene expression by Ciclopirox and Deferiprone, drugs that prevent hypusination of eukaryotic initiation factor 5A. Retrovirology. 2009;6:90. doi: 10.1186/1742-4690-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reichman TW, Mathews MB. RNA binding and intramolecular interactions modulate the regulation of gene expression by nuclear factor 110. RNA. 2003;9:543–54. doi: 10.1261/rna.2181103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reichman TW, Muniz LC, Mathews MB. The RNA binding protein nuclear factor 90 functions as both a positive and negative regulator of gene expression in mammalian cells. Mol Cell Biol. 2002;22:343–56. doi: 10.1128/MCB.22.1.343-356.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoque M, Young TM, Lee CG, Serrero G, Mathews MB, Pe’ery T. The Growth Factor Granulin Interacts with Cyclin T1 and Modulates P-TEFb-Defendent Transcription. Mol Cell Biol. 2003;23:1688–702. doi: 10.1128/MCB.23.5.1688-1702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Young TM, Wang Q, Pe’ery T, Mathews MB. The Human I-mfa Domain-Containing Protein, HIC, Interacts with Cyclin T1 and Modulates P-TEFb-Dependent Transcription. Mol Cell Biol. 2003;23:6373–84. doi: 10.1128/MCB.23.18.6373-6384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoque M, Mathews MB, Pe’ery T. Progranulin (granulin/epithelin precursor) and its constituent granulin repeats repress transcription from cellular promoters. J Cell Physiol. 2010;223:224–33. doi: 10.1002/jcp.22031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parrott AM, Tsai M, Batchu P, Ryan K, Ozer HL, Tian B, Mathews MB. The evolution and expression of the snaR family of small non-coding RNAs. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.