

Abstract
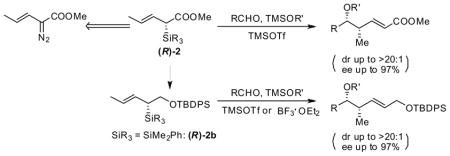
Enantioenriched homoallylic ethers linked to an α,β-unsaturated ester (syn-vinylogous aldol products) were directly accessed by Lewis acid catalyzed crotylation utilizing chiral silane 2. The reagents were prepared by enantioselective Si-H insertion to an α-diazovinylacetates using Davies’ Rh2(DOSP)4 catalyst or chiral Cu(I) Schiff-base complex.
The asymmetric allylation and crotylation of aldehydes utilizing chiral allyl- and crotylmetal reagents as carbon nucleophiles remains an important and useful transformation in orgainc chemistry. 1 In that context, allylsilanes are widely used, owing to their versatility, ease of handling and low toxicity. 2 Well documented studies from our laboratory have established chiral crotylsilane 1 as carbon nucleophiles in highly diastereo- and enantioselective reactions with acetals and aldehydes to construct homoallylic ethers with an isolated E-olefin subunit (Scheme 1). 3
Scheme 1.

Complementary Chrial Silane Reagents
Homoallylic ethers linked to an α,β-unsaturated functional group can be further elaborated to construct amides, acids, and lactones. These “building blocks“ possess exended functionality and therefore are likely to have utility in natural product and complex molecule synthesis.4 Despite recent advances toward the synthesis of polypropionate-like subunits, reagents used to gain access to these structural-types have limited substrate scope.5 In efforts to continue the development of chiral silane reagents capable of delivering useful levels of asymmetric induction, we report the synthesis of syn-homoallylic ethers linked to an α,β-unsaturated ester (vinylogous aldol products). The crotylation takes place with useful dr and ee utilizing chiral silane 2 (Scheme 1).
This study was initiated by establishing a reproducible asymmetric Si-H metal carbenoid insertion to synthesize chiral silane 2a. The known and readily available C2-symmetric copper(I) diimine complexs6 were evaluated for their effectiveness in insertions to α-diazovinylacetates. Although the idea of Cu(I) catalysis has been used to promote Si-H insertions prior to the application of rhodium catalysis, 7, 8 the field remains under developed and few cases of asymmetric variants have been reported.9 In that regard, we reported earlier that useful levels of selectivity with α-diazophenylacetates 10 and anticipated that we could extend the Cu(I) catalysis to α-diazovinylacetates (Table 1, entries 1 and 2), thereby complementing Landais and Davies’ earlier contributions, 11 who were the first to describe examples of Rh(II) promoted asymmetric Si-H insertion to α-diazovinylacetates. In this paper we report an efficient synthesis of chiral allylic silanes with C-centered chirality, and these experiments also allow a comparison of chiral Cu(I) vs Rh(II) catalysis. Consistent with Davies’ studies, the Rh2(DOSP)4 [(R)-6 and (S)-6] catalyst provided both enantiomers of crotylsilane 2a in excellent ee (entry 3).12
Table 1.
Preparation of (R)-2a Bearing Dimethyl Phenyl Silyl Groupc
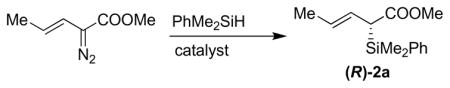 | |||||
|---|---|---|---|---|---|
| entry | catalyst | temperature (°C) | solvent | yield (%)a | ee (%)b |
| 1 | (R, R)-5a (5 mol %) | 0 | benzene | 44–51 | 70–73 |
| 2 | (R, R)-5b (5 mol %) | 0 | benzene | 45–55 | 78 |
| 3 | (S)-6 (1–5 mol %) | −78 | hexane | 65–70 | 88–97 |
|
| |||||
| |||||
Isolated yields were determined after purification over silica gel.
Based on HPLC data of silane alcohol, which was reduced from ester 2a by using LAH.
When (S, S)-5a or (R)-6 Rh2(R-DOSP)4 was used, the opposite enantiomer was obtained in comparable yield and ee.
Once reproducible conditions for the insertion were found, our efforts turned to the use of the enantioenriched silane reagents in Lewis acid promoted reaction with in situ derived oxonium ions. Lewis acid and solvent screening results 13 suggested that TMSOTf and dicloromethane were the optimal choices. The crotylation generally resulted in high yields but moderate diastereoselectivity (Table 2). Ee measurements were carried by HPLC analysis, and select examples showed that the enantioenrichment of the silane reagent was fully transferred into vinylogous-aldol products and were obtained in up to 97% ee when the (R)-2a was used.
Table 2.
Crotylation Using Silane (R)-2a
 | |||||
|---|---|---|---|---|---|
| entry | aldehyde | dra | yield (%)b | ee (%)d | product 3 |
| 1 | benzaldehyde | 3.5:1 | 79 | ND | 3a |
| 2c | 2,5-dimethoxybenzaldehyde | 2.5:1 | 88 | ND | 3b |
| 3 | p-tolualdehyde | 3.0:1 | 73 | ND | 3c |
|
| |||||
| 4 | o-bromobenzaldehyde | 18:1 | 81 | 97 | 3d |
|
| |||||
| 5 | p-bromobenzaldehyde | 4.2:1 | 83 | ND | 3e |
|
| |||||
| 6 | o-nitrobenzaldehyde | 16:1 | 61 | 95 | 3f |
|
| |||||
| 7 | hydrocinnamaldehyde | 4.0:1 | 71 | 91e | 3g |
| 8 | cyclohexanecarboxaldehyde | 5.0:1 | 63 | ND | 3h |
Diastereomeric ratios (dr) were determined by 1H NMR analysis on crude material.
Isolated yields after purification over silica gel.
Using TMSOTf 0.2 equiv.
Selected data basded on chiral HPLC.
Using silane (S)-2a.
As anticipated, the reaction favored the syn product, consistent with the well established anti-SE’ mode of addition,3 where the steric destablizing interaction between the aldehyde substituent and the vinyl methyl group on the allyl silane is minimized in an open transition state model. However, the magnitude of selectivity was dependent on the aldehyde type, activated aromatic aldehydes were less selective than those containing deactivating substituents (Table 2 entries: 3b, 3c vs 3d, 3e, 3f). Additionally, the position of substituents (o, m, p) influenced the magnitude of diastereoselectivity; aldehydes containing an ortho deactivating group afforded excellent syn/anti ratios (3d, 3f). In terms of aliphatic aldehydes, the branched substrate (3h) gave higher selectivity than the straight chain system (3g).
Efforts to improve the syn-selectivity of the crotylation by modification of the silane group were achieved, and eight racemic silane reagents with different nucleophilicities were prepared by carbene insertion.11b As such, p-tolualdehyde and p-bromobenzaldehyde were chosen respectively as representative activated and deactivated aldehydes. As shown in Table 3, reaction of anisyl derivative, which was reported to have greater stability and enhanced nucleophilicity (compared to Ph),14 led to slightly lower selectivity (entries 1 and 2). Increasing the nucleophilicity of silane by incorporating additional TMS groups showed no improvement (entries 3–6). For the cases of silane 2f to 2h, the size of the silicon group did not alter the magnitude of selectivity, as three different alkyl silanes afforded similar levels (entries 7–12), although slightly enhanced with respect to silane 2a. Gratifyingly, when silane reagents with decreased reactivity 15 were used, higher levels of selectivity were obtained. At −78 °C, reactions with triphenyl silane 2i and TrisTMS silane 2j afforded less than 10% conversion after two days. However, increased temperature or concentration drove the reactions to completion with good selectivity (entries 13–16).
Table 3.
Effect of Silyl Group on Simple Diastereoselection
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | silane | R1 | R2 | R3 | aldehyde | conversion (%)a | dra |
| 1 | rac-2c | Me | Me | anisyl | p-tolualdehyde | 100 | 2.8:1 |
| 2 | rac-2c | Me | Me | anisyl | p-bromobenzaldehyde | 80 | 3.0:1 |
| 3 | rac-2d | Me | Me | TMS | p-tolualdehyde | 100 | 3.2:1 |
| 4 | rac-2d | Me | Me | TMS | p-bromobenzaldehyde | 100 | 4.7:1 |
| 5 | rac-2e | Me | TMS | TMS | p-tolualdehyde | 100 | 3.0:1 |
| 6 | rac-2e | Me | TMS | TMS | p-bromobenzaldehyde | 100 | 3.5:1 |
| 7 | rac-2f | Et | Et | Et | p-tolualdehyde | 45 | 4.2:1 |
| 8 | rac-2f | Et | Et | Et | p-bromobenzaldehyde | 90 | 4.4:1 |
| 9 | rac-2g | Bu | Bu | Bu | p-tolualdehyde | 45 | 3.7:1 |
| 10 | rac-2g | Bu | Bu | Bu | p-bromobenzaldehyde | 91 | 4.1:1 |
| 11 | rac-2h | hexyl | hexyl | hexyl | p-tolualdehyde | 40 | 3.8:1 |
| 12 | rac-2h | hexyl | hexyl | hexyl | p-bromobenzaldehyde | 90 | 4.5:1 |
|
| |||||||
| 13b | rac-2i | Ph | Ph | Ph | p-tolualdehyde | 93 | 5.2:1 |
| 14b | rac-2i | Ph | Ph | Ph | p-bromobenzaldehyde | 95 | 6.8:1 |
| 15b | rac-2j | TMS | TMS | TMS | p-tolualdehyde | 95 | >20:1 |
| 16b | rac-2j | TMS | TMS | TMS | p-bromobenzaldehyde | 98 | >20:1 |
Conversion, syn/anti ratio were based on crude 1H NMR.
Reaction was carried out at −60 °C.
With useful levels of diastereoselectivity obtained in the racemic series, we prepared enantioenriched silane reagents 2f to 2j. Comparable selectivity was achieved with tributylsilane compared with dimethylphenyl silane (Table 4, entry 1). In contrast, Rh2(S-DOSP)4 afforded only moderate ee of 2j16 (entry 2). Owing to the poor solubility of SiPh3H in pentane, the rhodium catalyst was ineffective for preparing enantioenriched 2i (entry 3). Alternatively, our preliminary results showed that using Cu(MeCN)4BF4 and diimine ligand complex (R,R)-5a afforded 2i with good selectivity (>70% ee), which could be improved to 97% ee by recrystallization (2x) from petroleum ether (entry 4).
Table 4.
Enantioselective Si-H Insertion
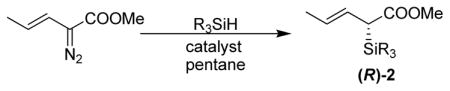 | ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | R3SiH | temperature (°C) | product | yield (%)a | ee (%)b |
| 1 | (S)-6 | nBu3SiH | −78 | (R)-2g | 48 | 86 |
| 2 | (S)-6 | TMS3SiH | −40 | (R)-2j | 30 | 40 |
| 3 | (S)-6 | Ph3SiH | 0 | (R)-2i | NR | NR |
| 4c | (R,R)-5a | Ph3SiH | 0 | (R)-2i | 41/25d | 70/97d |
Isolated yields were determined after purification over silica gel.
Based on HPLC data of silane alcohol, which was reduced from ester by using LAH.
Reaction run in benzene.
Yield and % ee before and after recrystallization from petroleum ether.
We next explored the substrate scope with silanes 2i and 2g (Table 5). Crotylation with 2i and aromatic aldehydes gave useful levels of selectivity. However, with less reactive aliphatic aldehydes, only trace amounts of produts were observed spectroscopically. To solve this problem, we evaluated tri-n-butyl silane 2g, which exhibited slightly higher selectivity than silane 2a in the crotylation. Branched aliphatic aldehydes 3h-3l gave the homoallylic ethers with higher selectivity than aliphatic aldehydes 3g.
Table 5.
Crotylation Using Chiral Silane (R)-2g and (R)-2i
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | silane | aldehyde | TMSOR′ | dra | yield (%)b | ee (%)d | product 3 |
| 1 | (R)-2g | hydrocinnamaldehyde | TMSOMe | 5.3:1 | 57 | ND | 3g |
| 2 | (R)-2g | cyclohexanecarboxaldehyde | TMSOMe | 7.6:1 | 61 | ND | 3h |
| 3 | (R)-2g | isobutyraldehyde | TMSOMe | 6.6:1 | 41c | ND | 3i |
| 4 | (R)-2g | isobutyraldehyde | TMSOBn | 7.0:1 | 63 | ND | 3j |
| 5 | (R)-2g | trimethylacetaldehyde | TMSOMe | 11:1 | 39c | ND | 3k |
| 6 | (R)-2g | trimethylacetaldehyde | TMSOBn | 15:1 | 79 | 85 | 31 |
| 7 | (R)-2i | benzaldehyde | TMSOMe | 6.3:1 | 79 | 97 | 3a |
| 8 | (R)-2i | benzaldehyde | TMSOBn | 6.4:1 | 68 | ND | 3m |
| 9 | (R)-2i | p-tolualdehyde | TMSOMe | 5.2:1 | 75 | ND | 3c |
| 10 | (R)-2i | p-bromobenzaldehyde | TMSOMe | 6.8:1 | 67 | ND | 3e |
| 11 | (R)-2i | o-bromobenzaldehyde |
|
15:1 | 75 | ND | 3n |
Diastereomeric ratios were determined by 1H NMR analysis on crude material.
Isolated yields after purification over silica gel.
Low yields due to volatility of products.
Selected data basded on chiral HPLC, ND = not determoned.
The parent silane 2a was converted to a primary TBDPS ether 2b17 by reduction of the ester group and silylation of the resulting alcohol (two steps 91% yield) in an attempt to increase selectivity. Subsequent reaction of 2b with a variety of aliphatic and aromatic aldehydes exhibited good to excellent syn-selectivity and typically good yields (Table 6). Notably, even the straight chain aliphatic systems (4k, 4l), which normally gave poor diastereoselectivity, afforded satisfactory syn/anti ratios. Aliphatic aldehydes often produced tetrahydrofuran byproducts17a that were not observed with silane 2a. This pathway was minimized by adding excess TMSOMe (3 equiv).
Table 6.
Crotylation Using Chiral Silane (R)-2b
 | ||||||
|---|---|---|---|---|---|---|
| entry | aldehyde | TMSOR′ | dra | yield (%)b | ee (%)e | product 4 |
| 1c | 2,3-dimethoxybenzaldehyde | TMSOMe | >20:1 | 92 | ND | 4a |
| 2c | 2,5-dimethoxybenzaldehyde | TMSOMe | 8:1 | 91 | ND | 4b |
| 3 | p-tolualdehyde | TMSOMe | 11:1 | 79 | ND | 4c |
| 4 | benzaldehyde | TMSOMe | 15:1 | 77 | ND | 4d |
| 5 | p-bromobenzaldehyde | TMSOMe | 16:1 | 55 | ND | 4e |
| 6 | o-bromobenzaldehyde | TMSOMe | >20:1 | 58 | ND | 4f |
| 7 | 2-naphthaldehyde | TMSOMe | 10:1 | 73 | ND | 4g |
| 8c | 2,5-dimethoxybenzaldehyde | TMSOBn | >20:1 | 87 | ND | 4h |
| 9 | benzaldehyde | TMSOBn | 11:1 | 51 | 97 | 4i |
| 10d | cyclohexanecarboxaldehyde | TMSOMe | >20:1 | 76 | ND | 4j |
| 11d | valeraldehyde | TMSOMe | >20:1 | 61 | ND | 4k |
| 12d | hydrocinnamaldehyde | TMSOMe | 16:1 | 47 | ND | 4l |
| 13 | benzaldehyde |
|
>20:1 | 72 | ND | 4m |
| 14 | benzaldehyde |
|
ND | trace | ND | 4n |
Diastereomeric ratios were determined by 1H NMR analysis on crude material.
Isolated yields after purification over silica gel.
Using TMSOTf 0.2 equiv.
Using excess TMSOMe, see Supporting Information.
Selected data basded on chiral HPLC, ND = not determined.
In summary, we have extended the use of Jacobsen’s C2-symmetric copper(I) diimine complexes to carbene insertions with α-diazovinylacetates, resulting in the formation of crotylsilanes bearing C-centered chirality with high enantioenrichment. The silanes described in this work complement our earlier work to afford vinylogous-aldol products (syn-polypoprionate building blocks) with high levels of diastereo- and enantioselectivity. Presently, the Davies’ catalyst Rh2(DOSP)4 provides slightly higher levels of selectivity, as such, development of more selective Cu(I) catalysts as an effective approach to chiral silane reagents and their use in complex molecule synthesis are currently underway and will be reported at a latter time.
Supplementary Material
Acknowledgments
Financial support was obtained from NIH CA 53604. The authors are grateful to Professor Scott Schaus (Boston University) and Professor Hisashi Yamamoto (University of Chicago), Dr. Yun Zhang, Dr. Paul Ralifo and Dr. Norman Lee at Boston University for helpful discussions and assistance with NMR and HRMS measurements.
Footnotes
Supporting Information Available Experimental details and new selected spectral for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Chapter 10 Wiley-VCH; Weinheim: 2000. [Google Scholar]; (b) Chemler SR, Roush WR. In: In Modern Carbonyl Chemistry. Otera J, editor. Chapter 11 Wiley-VCH; Weinheim: 2000. [Google Scholar]; (c) Yamamoto Y, Asao N. Chem Rev. 1993;93:2207. [Google Scholar]; (d) Denmark SE, Fu J. J Am Chem Soc. 2001;123:9488. doi: 10.1021/ja016552e. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chabaud L, James P, Landais Y. Eur J Org Chem. 2004:3173–3199. [Google Scholar]; (b) Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL. J Am Chem Soc. 2002;124:7920–7921. doi: 10.1021/ja0264908. [DOI] [PubMed] [Google Scholar]; (c) Grote RE, Jarvo ER. Org Lett. 2009;11:485–488. doi: 10.1021/ol8026297. [DOI] [PubMed] [Google Scholar]; (d) Kubota K, Leighton JL. Angew Chem Int Ed. 2003;42:946–948. doi: 10.1002/anie.200390252. [DOI] [PubMed] [Google Scholar]; (e) Hackman BM, Lombardi PJ, Leighton JL. Org Lett. 2004;6:4375–4377. doi: 10.1021/ol0480731. [DOI] [PubMed] [Google Scholar]
- 3.(a) Masse CE, Panek JS. Chem Rev. 1995;95:1293–1316. [Google Scholar]; (b) Panek JS, Yang M. J Am Chem Soc. 1991;113:9868–9870. [Google Scholar]; (c) Panek JS, Yang M, Feng X. J Org Chem. 1992;57:5790–5792. [Google Scholar]
- 4.(a) O’Neil GW, Phillips AJ. J Am Chem Soc. 2006;128:5340–5341. doi: 10.1021/ja0609708. [DOI] [PubMed] [Google Scholar]; (b) Shin Y, Fournier JH, Fukui Y, Bruckner AM, Curran DP. Angew Chem Int Ed. 2004;43:4634–4637. doi: 10.1002/anie.200460593. [DOI] [PubMed] [Google Scholar]; (c) Fukui Y, Bruckner AM, Shin Y, Balachandran R, Day BW, Curran DP. Org Lett. 2006;8:301–304. doi: 10.1021/ol0526827. [DOI] [PubMed] [Google Scholar]; (d) Shin Y, Choy N, Balachandran R, Madiraju C, Day BW, Curran DP. Org Lett. 2002;4:4443–4446. doi: 10.1021/ol026942l. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hasfeld J, Christmann M, Kalesse M. Org Lett. 2001;3:3561–3564. doi: 10.1021/ol016677o. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Beutner GL. J Am Chem Soc. 2003;125:7800–7801. doi: 10.1021/ja035448p. [DOI] [PubMed] [Google Scholar]; (c) Kalesse M, Simsek S. Tetrahedron Lett. 2009;50:3485–3488. [Google Scholar]
- 6.Larrow JF, Jacobsen EN. J Org Chem. 1994;59:1939–1942. [Google Scholar]
- 7.Rijkens F, Janssen MJ, Drenth W, van derKerk GJM. J Organomet Chem. 1964;2:347. [Google Scholar]
- 8.Selected examples for chiral Cu-mediated X-H insertions. see: Maier TC, Fu GC. J Am Chem Soc. 2006;128:4594–4595. doi: 10.1021/ja0607739.Lee EC, Fu GC. J Am Chem Soc. 2007;129:12066–12067. doi: 10.1021/ja074483j.Liu B, Zhu SF, Zhang W, Chen C, Zhou QL. J Am Chem Soc. 2007;129:5834. doi: 10.1021/ja0711765.Chen C, Zhu SF, Liu B, Wang LX, Zhou QL. J Am Chem Soc. 2007;129:12616. doi: 10.1021/ja074729k.
- 9.Zhang YZ, Zhu SF, Wang LX, Zhou QL. Angew Chem Int Ed. 2008;47:8496–8498. doi: 10.1002/anie.200803192. [DOI] [PubMed] [Google Scholar]
- 10.(a) Dakin LA, Schaus SE, Jacobsen EN, Panek JS. Tetrahedron Lett. 1998;39:8947–8950. [Google Scholar]; (b) Dakin LA, Ong PC, Panek JS, Staples RJ, Stavropoulos P. Organometallics. 2000;19:2896–2908. [Google Scholar]
- 11.(a) Davies HM, Hansen T, Rutberg J, Bruzinski PR. Tetrahedron Lett. 1997;38:1741–1744. [Google Scholar]; (b) Bulugahapitiya P, Landais Y, Rapado LP, Planchenault D, Weber V. J Org Chem. 1997;62:1630–1641. [Google Scholar]
- 12.For reaction modification and optimization, see Supporting Information.
- 13.TMSOTf, BF3·OEt2, Sc(OTf)3, In(OTf)3, TiCl4 were screened as Lewis acid in the reaction with benzaldehyde and silane (R)-2a. Solvents screening included DCM, toluene, pentane and THF.
- 14.Franz AK, Dreyfuss PD, Schreiber SL. J Am Chem Soc. 2007;129:1020–1021. doi: 10.1021/ja067552n. [DOI] [PubMed] [Google Scholar]
- 15.Hagen G, Mayr H. J Am Chem Soc. 1991;113:4954–4961. [Google Scholar]
- 16.Freeze pump thaw degassed pentane was required to avoid oxidation of TTMSSH.
- 17.Related silane reagents, see: Heitzman CL, Lambert WT, Mertz E, Shotwell JB, Tinsley JM, Va P, Roush WR. Org Lett. 2005;7:2405–2408. doi: 10.1021/ol0506821.Komatsu K, Tanino K, Miyashita M. Angew Chem Int Ed. 2004;43:4341 –4345. doi: 10.1002/anie.200460434.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.