

Abstract
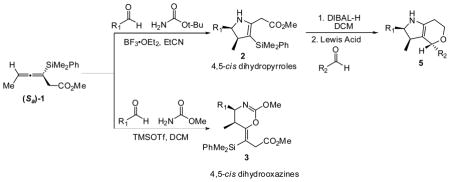
Highly enantioenriched allenylsilanes participate in Lewis acid mediated annulations with in situ generated iminium ions derived from tert-butyl carbamate and methyl carbamate to selectively form functionalized 4,5-dihydropyrroles and 4,5-dihyrooxazines respectively. The dihydropyrrole products were further elaborated in a stereocontrolled vinylsilane terminated cyclization with in situ generated oxonium ions, resulting in pyranopyrroles.
Recently allenes have emerged as an important functional group for a variety of organic transformations.1 In particular, allenylsilanes have proven useful as carbon nucleophiles in addition to carbon-oxygen π-bonds producing homopropargylic alcohols,2 homopropargylic ethers,3 furans,4 and dihydrofurans.5 Many of these reactions occur with excellent diasetereo- and enantioselectivity.
While the additions of allenylsilanes to carbonyl groups have been well developed, there are fewer examples of similar reactivity with activated carbon-nitrogen π-bonds. The addition of crotylsilanes to in situ generated iminium ions for the formation of homoallylic amines and pyrrolidines has been well developed, and generally proceeds with high yield and selectivity.6 Racemic allenes have been used in a variety of additions to imines, resulting in [3 + 2] and [4 + 2] annulation products, many with high diastereoselectivity and enantiomeric excess due to the use of chiral catalysts.7 In addition, a few reports containing examples of allenylsilanes being used in [3 + 2] annulations with iminium ions are available, but they show a limited substrate scope and selectivity. In interest of complete transparency, Danheiser was the first to demonstrate a [3 + 2] annulation of racemic allenylsilanes with β-alkoxylactams, affording pyrrolizinones as the major products.5 More recently, Akiyama reported a Cu(I)-catalyzed cycloaddition to afford dihydroproline derivatives.8 The purpose of this communication is to report our initial studies on the use of chiral allenylsilanes in annulations with N-acyl iminuim ions generated in situ from carbamates and aldehydes.
We have recently reported an efficient synthesis of highly enantioenriched allenylsilane (Sa)−1 and its enantiomer. That sequence, from the corresponding propargylic alcohol, utilized a lipase resolution followed by a Johnson orthoester Claisen rearrangement. These reagents can be accessed on a multi-gram scale (>15g) with >95% ee.3
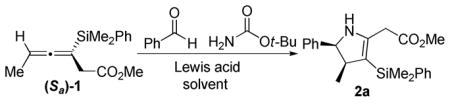
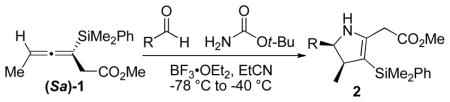
Several conditions were explored for the addition of these allenylsilanes to iminium ions, which were formed in situ by activating an aldehyde in the presence of an amine with a Lewis acid. After screening a number of amines, benzyl amines, hydrazones, and acetamides were found to be unreactive under a variety of conditions, while the use of sulfonamides resulted in a complex mixture of products. Carbamates proved to be the most active amine source for iminium ion formation. When allenylsilane (Sa)−1 was exposed to benzaldehyde in the presence of tert-butyl carbamate and a Lewis acid activator, the major product obtained was dihydropyrrole 2a.
The optimal Lewis acid for iminium ion formation was found to be BF3 · OEt2. Attempts to catalyze the reaction with Sc(OTf)3 resulted in the recovery of the starting materials, while TiCl4, TfOH and TMSOTf provided lower yields (see Table 1). Use of an excess of Lewis acid led to decomposition of the products, while using sub-stoichiometric amounts of Lewis acid resulted in incomplete reactions. No protodesilylation product was isolated under any of the reaction conditions, suggesting that the vinylsilane product is robust. Very polar nitrile solvents (MeCN, EtCN) were found to give the highest yields for the [3 + 2] annulation reactions.9 In all cases a single diastereomer was observed. The best yields were obtained when the iminium ion was formed in situ, by treatment of a solution of aldehyde and carbamate with Lewis acid at −78 °C. The allenylsilane was then added dropwise, and the resulting solution was warmed to −40 °C.
Table 1.
Optimization of the Annulation of (Sa)-1 with Benzaldehyde and tert-Butyl Carbamate
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acid | time (h) | solvent | yield of 2aa | drc |
| 1 | TMSOTf | 48 | DCM | 9% | >20:1 |
| 2 | TMSOTf | 48 | EtCN | 41% | >20:1 |
| 3 | TiCl4 | 48 | DCM | <5% | >20:1 |
| 4 | TiCl4 | 48 | EtCN | 33% | >20:1 |
| 5 | TfOH | 48 | DCM | 44% | >20:1 |
| 6 | TfOH | 48 | EtCN | 47% | >20:1 |
| 7 | BF3 · OEt2 | 72 | DCM | 14% | >20:1 |
| 8 | BF3 · OEt2 | 72 | MeCN | 56% | >20:1 |
| 9 | BF3 · OEt2 | 72 | EtCN | 67% | >20:1 |
Isolated yields after purification over silica gel. All reactions run with 1.2 equiv Lewis acid at −78 to −40 °C except for entry 8 run at −40 °C.
Diastereomeric ratios were determined by 1H NMR analysis on crude material.
The stereochemical course of the [3 + 2] annulation can be described by either an antiperiplanar or synclinal transition state, where the axial chirality of the allenylsilane (Sa)−1 is transferred to the si face of the iminium ion (see Figure 1). The antiperiplanar transition state limits the gauche interactions, while the synclinal transition state places the R group of the iminium ion furthest from the incoming allenylsilane. The absolute sterechemistry of the products is based of the addition to the allene being anti to the carbon-silicon bond.10
Figure 1.

Open Transition State Modela
aFor detailed analysis on the proposed transition states see Supporting Information.
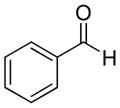
The [3 + 2] annulations were successful for a variety of aldehydes (see Table 2). Aromatic aldehydes typically provided the highest yields, while straight chain aliphatic substrates provided moderate yields and branched aliphatic aldehydes provided slightly lower yields. For the cases examined, the annulation products were formed as a single diastereomer. Stereochemical assignment of the annulation products was confirmed with the aid of NOE measurements and clearly indicated a cis relationship between the C-4 and C-5 stereocenters.11
Table 2.
[3 + 2] Annulations with tert-Butyl Carbamate
 | ||||
|---|---|---|---|---|
| entry | aldehyde | yielda | drb | product 2 |
| 1 |
|
67% | >20:1 | 2a |
| 2 |
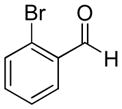
|
82% | >20:1 | 2b |
| 3 |
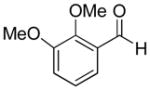
|
60% | >20:1 | 2c |
| 4 |
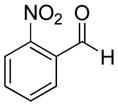
|
50% | >20:1 | 2d |
| 5 |
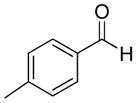
|
67% | >20:1 | 2e |
| 6 |
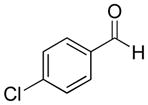
|
69% | >20:1 | 2f |
| 7 |
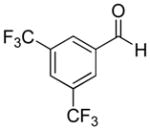
|
58% | >20:1 | 2g |
| 8 |
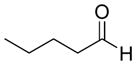
|
57% | >20:1 | 2h |
| 9 |
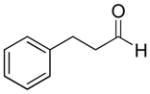
|
48% | >20:1 | 2i |
| 10 |
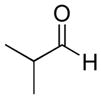
|
33% | >20:1 | 2j |
| 11 |
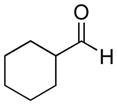
|
37% | >20:1 | 2k |
Isolated yields after purification over silica gel.
Diastereomeric ratios determined by 1H NMR analysis on crude material.
In reactions with tert-butyl carbamate, the only observed product was the dihydropyrrole bearing a free secondary amine; no BOC protected product was isolated. Presumably, the combination of Lewis acidic conditions and/or the equivalent of water produced in the formation of the iminium ion promoted decomposition of the acid labile BOC protecting group.
Additional carbamate sources were explored in an effort to expand the scope of the annulation and allow for the formation of N-acylated dihydropyrroles. However, when the amine source was changed to methyl carbamate, the major product was determined to be the dihydrooxazine. The optimal conditions for the specific formation of 4,5-cis-dihydrooxazines were found when TMSOTf was employed as the Lewis acid and DCM as the solvent (3a to 3g, table 3). These products are similar to a byproduct observed by Danheiser.5 2D NMR studies have confirmed that the major product, is the oxazine bearing an E-vinyl silane, and not the anticipated methyl carbamate protected pyrrole.11
Table 3.
Annulations with Methyl Carbamate: Dihydrooxazines
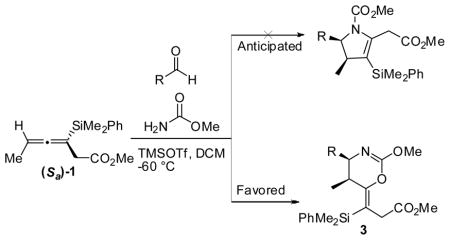 | ||||
|---|---|---|---|---|
| entry | aldehyde | yielda | drb | product 3 |
| 1 | Benzaldehyde | 63% | >20:1 | 3a |
| 2 | 2-Bromobenzaldehyde | 81% | >20:1 | 3b |
| 3 | 2, 3-Dimethoxybenzaldehyde | 54% | >20:1 | 3c |
| 4 | 2-Nitrobenzaldehyde | 58% | >20:1 | 3d |
| 5 | 4-Chlorobenzaldehyde | 59% | >20:1 | 3e |
| 6 | Valeraldehyde | 61% | >20:1 | 3f |
| 7 | Hydrocinnamaldehyde | 38% | >20:1 | 3g |
| 8 | Isobutyraldehyde | 47% | >20:1 | 3h |
| 9 | Cyclohexanecarboxaldehyde | 48% | >20:1 | 3i |
Isolated yields after purification over silica gel.
Diastereomeric ratios determined by 1H NMR analysis on crude material.
HPLC traces of select examples show that the axial chirality of the allenylsilane is fully transferred into central chirality in the dihydropyrroles, which are obtained in >99% ee when the (Sa) allene is used.12
The combined activating effects of the enamine and vinylsilane functional groups of the dihydropyrrole products were exploited as illustrated in Scheme 1. Reduction of the methyl ester in 2 to alcohol 4 proceeds well with DIBAL-H in DCM. Primary alcohols 4a–4c can then form hemiacetals using a series of aldehydes, mediated by TMSOTf or BF3 · OEt2. The resulting oxonium ion undergoes vinylsilane terminated cyclization,13 resulting in the formation of pyranopyrrole products 5a–5h, in high yields as a single diastereomer. The relative stereochemistry of the pyranopyrrole products has been assigned by NOE studies.10
Scheme 1.

Formation of Pyranopyrrolesa
Reaction conditions A (1.2 equiv TMSOTf, DCM) used for 5a–5e and B (1.2 equiv BF3 · OEt2, MeCN) used for 5g–5h.
Isolated yields after purification over silica gel. All products were obtained in >20:1 dr, as determined by 1H NMR analysis on crude material.
Exposure of dihydropyrrole 2a to m-CPBA in DCM results in the formation of epoxide 6 as a single diastereomer. The stereochemistry of this product has not been confirmed, but it is tentatively assigned as the α-epoxide, as epoxidation likely takes place from opposite the sterically hindered face containing the aromatic ring and methyl group.
In closing, we have documented the use of chiral allenylsilanes in enantioselective annulations that assemble vinylsilane substituted 4,5-cis-dihydropyrrole and 4,5-cis-dihydrooxazine building blocks. We have illustrated that these these dihydropyrroles can be used in vinylsilane terminated cyclizations, generating pyranopyrroles with good functional group variation. Ongoing experiments seek to apply this annulation strategy in complex molecule and natural product synthesis.
Supplementary Material
Scheme 2.

Epoxidation of Dihydropyrrole 2a
Acknowledgments
Financial support was obtained from NIH CA 53604. J. S. P. is grateful to Amgen, Johnson & Johnson, Merck Co., Novartis, Pfizer, and GSK for financial support. The authors are grateful to Dr. Jason Lowe, Dr. John Snyder, Dr. Jonathan Lee, and Dr. Paul Ralifo at Boston University for helpful discussions and assistance with 2D NMR experiments.
Footnotes
Supporting Information Available Experimental data and selected spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For recent reviews on allene chemistry see: Hassam HHAM. Curr Org Syn. 2007;4:413–439.Ma S. Chem Rev. 2005;105:2829–2872. doi: 10.1021/cr020024j.Zimmer R, Dinesh CU, Nandanan E, Khan FA. Chem Rev. 2000;100:3067–3125. doi: 10.1021/cr9902796.
- 2.(a) Danheiser RL, Carini DJ, Kwasigroch CA. J Org Chem. 1986;51:3870–3878. [Google Scholar]; (b) Marshall JA, Maxson K. J Org Chem. 2000;65:630–633. doi: 10.1021/jo991543y. [DOI] [PubMed] [Google Scholar]
- 3.Brawn RA, Panek JS. Org Lett. 2007;9:2689–2692. doi: 10.1021/ol070936d. [DOI] [PubMed] [Google Scholar]
- 4.Danheiser RL, Stoner EJ, Koyama H, Yamashita DS, Klade CA. J Am Chem Soc. 1989;111:4407–4413. [Google Scholar]
- 5.Danheiser RL, Kwasigroch CA, Tsai YM. J Am Chem Soc. 1985;107:7233–7235. [Google Scholar]
- 6.(a) Panek JS, Jain NF. J Org Chem. 1994;59:2674–2675. [Google Scholar]; (b) Schaus JV, Jain NF, Panek JS. Tetrahedron. 2000;56:10263–10274. [Google Scholar]; (c) Lipomi DJ, Panek JS. Org Lett. 2005;7:4701–4704. doi: 10.1021/ol051885s. [DOI] [PubMed] [Google Scholar]; (d) Restorp P, Fischer A, Somfai P. J Am Chem Soc. 2006;128:12646–12647. doi: 10.1021/ja0647102. [DOI] [PubMed] [Google Scholar]
- 7.(a) Depature M, Grimaldi J, Hatem J. Eur J Org Chem. 2001:941–946. [Google Scholar]; (b) Ma S, Gao W. Org Lett. 2002;4:2989–2992. doi: 10.1021/ol026406t. [DOI] [PubMed] [Google Scholar]; (c) Kaden S, Brockmann M, Reissig HU. Helv Chim Acta. 2005;88:1826–1838. [Google Scholar]; (d) Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234–12235. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]; (e) Castellano S, Fiji HDG, Kinderman SS, Watanabe M, de Leon P, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843–5845. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fuchibe K, Hatemata R, Akiyama T. Tetrahedron Lett. 2005;46:8563–8566. [Google Scholar]; (g) Fang YQ, Jacobsen EN. J Am Chem Soc. 2008;130:5660–5661. doi: 10.1021/ja801344w. [DOI] [PubMed] [Google Scholar]
- 8.Daidouji K, Fuchibe K, Akiyama T. Org Lett. 2005;7:1051–1053. doi: 10.1021/ol047343c. [DOI] [PubMed] [Google Scholar]
- 9.All reactions were run until the allene was consumed, as determined by TLC analysis. Reactions run in a variety of other solvent systems, including THF, diethyl ether, toluene and hexanes produced little or no desired product.
- 10.Marshall JA, Maxson K. J Org Chem. 2000;65:630–633. doi: 10.1021/jo991543y. [DOI] [PubMed] [Google Scholar]
- 11.See Supporting information for details of NOE and 2D NMR experiments. The authors are grateful to a referee for suggesting the possibility of dihydrooxazine formation.
- 12.See Supporting information for HPLC traces and ee analysis.
- 13.(a) Blumenkopf TA, Overman LE. Chem Rev. 1986;86:857–873. [Google Scholar]; (b) Zhang X, Li X, Lanter JC, Sui Z. Org Lett. 2005;7:2043–2046. doi: 10.1021/ol050623n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.