

Abstract
In bacteria, association of the specialized σN protein with the core RNA polymerase subunits forms a holoenzyme able to bind promoter DNA, but unable to melt DNA and initiate transcription unless acted on by an activator protein. The conserved amino-terminal 50 amino acids of σN (Region I) are required for the response to activators. We have used pre-melted DNA templates, in which the template strand is unpaired and accessible for transcription initiation, to mimic a naturally melted promoter and explore the function of Region I. Our results indicate that one activity of Region I sequences is to inhibit productive interaction of holoenzyme with pre-melted DNA. On pre-melted DNA targets, either activation of σN-holoenzyme or removal of Region I allowed efficient formation of complexes in which melted DNA was sequestered by RNA polymerase. Like natural pre-initiation complexes formed on conventional DNA templates through the action of activator, such complexes were heparin-resistant and transcriptionally active. The inhibitory σN Region I domain functioned in trans to confer heparin sensitivity to complexes between Region I-deleted holoenzyme and pre-melted promoter DNA. Evidence that Region I senses the conformation of the promoter was obtained from protein footprint experiments. We suggest that one function for Region I is to mask a single-strand DNA-binding activity of the holoenzyme. On the basis of extended DNA footprints of Region I-deleted holoenzyme, we also propose that Region I prevents RNA polymerase isomerization, a conformational change necessary for access to and the subsequent stable association of holoenzyme with melted DNA.
Keywords: RNA polymerase, gene activation, σ factors, DNA melting, enhancers
RNA polymerases (RNAPs) are key targets for transcriptional activators that enable processes such as cellular differentiation and adaptation to take place in an ordered manner. Regulated and specific initiation of transcription in bacteria requires a σ protein, which binds to a single common core RNAP (α2ββ′) to form a holoenzyme that directs specific initiation of RNA synthesis (Gross et al. 1992). The major σ class members are similar to the primary Escherichia coli σ70 (Lonetto et al. 1992). The σN class is unrelated (Merrick 1993), and the events leading to initiation at σN-dependent promoters are clearly distinct from those for σ70-dependent initiation.
The σN-holoenzyme is required for enhancer-dependent transcription, characterized as depending on activator γ–β bond hydrolysis of a nucleoside triphosphate (Kustu et al. 1989; Weiss et al. 1991; Austin and Dixon 1992). Activators of the σN-holoenzyme belong to a chaperone-like ATPase protein family, members of which function as oligomers to remodel their substrates in a nucleotide-dependent manner in processes such as transcription, DNA replication, recombination, and restriction (Confalonieri and Duguet 1995; Swaffield et al. 1995; Wyman et al. 1997; Rippe et al. 1998). Occupancy of promoters by the σN-holoenzyme is not limiting for transcription and occurs largely independently of activator (Popham et al. 1991; Wang et al. 1997b) in contrast to other RNA polymerases that are recruited to the promoter (for review, see Ptashne and Gann 1997). The σN-holoenzyme binds promoters in a transcriptionally inactive complex (Sasse-Dwight and Gralla 1988; Kustu et al. 1989; Popham et al. 1989; Lee and Hoover 1995; Wedel and Kustu 1995) without closely contacting the transcription start site (Popham et al. 1991; Cannon et al. 1995a). Because σN interacts with sequences that become melted during activation, σN may participate in DNA melting, acting either positively or negatively (Buck and Cannon 1992; Cannon et al. 1995a; Casaz and Buck 1997; Oguiza and Buck 1997). The σN domain responsible for promoter contact is at the carboxyl terminus, and sequences that promote activation are in the amino-terminal Region I (Merrick 1993; Cannon et al. 1994, 1995b; Wong et al. 1994; Guo and Gralla 1997; Wang et al. 1997a). Region I has a negative function demonstrated by mutants that allow increased activator-independent transcription in vitro (Wang et al. 1997a).
Only the holoenzyme form of RNAP makes specific open promoter complexes on double-stranded DNA (Burgess et al. 1969; Helmann and Chamberlin 1988). The σ70-holoenzyme binds promoters in a transcriptionally active form, and σ70 has a role in DNA melting through contact made with the nontemplate melted DNA strand (for review, see Chan et al. 1996). Current models of activation of the σN-holoenzyme suggest that both kinetic and thermodynamic barriers to open complex formation are overcome by activators (Wedel and Kustu 1995) and that the Region I of σN inhibits the melting function of the holoenzyme (Wang et al. 1997a). Holoenzymes that lack Region I transcribe by use of an unstable intermediate associated with transient DNA melting (Wang et al. 1997a). Protein footprints have shown that the relationship of Region I to core RNAP changes when the holoenzyme is activated, suggesting that changes in Region I may facilitate steps of the activation event (Casaz and Buck 1997).
Melting of DNA is an important regulated activity in processes such as transcription and replication. In this paper, we provide evidence that formation of σN-holoenzyme pre-initiation complexes that are stably bound to melted DNA sequences requires an activator-driven conformational change in the holoenzyme. The σN-holoenzyme isomerization, normally inhibited by Region I, leads to an unmasking of a single-strand DNA-binding activity in the holoenzyme. These results explain how holoenzymes lacking Region I can transcribe without activator under solution conditions favoring transient DNA melting (Wang et al. 1997a) and why efficient and faithful transcription from pre-melted DNA templates still requires activator (Wedel and Kustu 1995). We propose that activators of the σN-holoenzyme act to facilitate a changing interaction between Region I and core to permit polymerase isomerization and allow the subsequent engagement of the holoenzyme with melted DNA so as to form a stable open promoter complex.
Results
Heteroduplexes
Our work focused on the glutamine- and leucine-rich σN Region I implicated in many studies as being key to enhancer-dependent transcription (e.g., see Sasse-Dwight and Gralla 1990; Morris et al. 1994). Previous work demonstrated that σN–holoenzyme binding at several σN-dependent promoters is increased by deletion of Region I (Morris et al. 1994; Cannon et al. 1995b) but that Region I was required for the holoenzyme to stably melt the DNA (Sasse-Dwight and Gralla 1990; Cannon et al. 1995b) and initiate transcription in response to activators (Wang et al. 1997a). We therefore investigated whether pre-opened DNA in the form of heteroduplex promoter DNA molecules (Table 1) could overcome the transcription defect of the Region I-deleted (ΔIσN) holoenzyme. Heteroduplex molecules have been used to demonstrate that the σN-holoenzyme remains transcriptionally inactive at the glnAp2 promoter even though the start site is pre-melted (Wedel and Kustu 1995). Our standard heteroduplex 1 is an 88-bp molecule comprising the −60 to +28 Rhizobium meliloti nifH promoter sequence. It includes the σN-holoenzyme consensus recognition sequence (−28 to −12) and mismatches from −10 to −1 with a mutant top strand and a wild-type bottom template strand to form premelted initiation sequences and preserve the transcription start site (see Table 1). The mismatched region includes the nonconserved sequence from −11 to −5 that interacts with σN within closed complexes (Cannon et al. 1995a) or with holoenzyme in open promoter complexes (Popham et al. 1989; Sasse-Dwight and Gralla 1988; Morett and Buck 1989). To confirm the structure of our heteroduplexes, we challenged them with a variety of footprinting reagents capable of detecting single-strand DNA and observed the expected reactivities across the unpaired sequence (see Fig. 4, below).
Table 1.
R. meliloti nifH promoter homoduplex and heteroduplex DNA promoter fragments
 |
Promoter fragments were sequences from −60 to +28 with the indicated changes from −10 to −1 at −25/−26. Consensus bases within the −28/−12 σN promoter sequence at −25 and −14 are underlined. Sequences downstream of −12 are not conserved among σN-dependent promoters.
Figure 4.



Premelted sequences are sequestered in stable complexes but accessible to KMnO4. (A) Gel mobility-shift assays of heteroduplex 1 DNA complexes. Binding reactions were conducted as in Fig. 1A, treated with S1 nuclease or KMnO4 (see Material and Methods) and loaded onto running native gels. (B) Bound DNA (lanes 3–6); (P) undigested probe (lane 1); (F) digested probe (lane 2); (U) unbound DNA (lanes 3–6). (B,C) The DNA bands were isolated from native gels described in A, and both strands analyzed on sequencing gels [(B) top strand; (C) bottom strand]. The mismatch region between −10 and −1 is indicated. Markers (M) were generated by chemical cleavage of the DNA with piperidine following partial methylation with dimethylsulfate. Probe alone or cleaved with piperidine (without treatment with KMnO4) are lanes marked P− and P+, respectively. Footprints of ΔIσN-holoenzyme plus PspFΔHTH and ddGTP were the same as ΔIσN-holoenzyme alone (not shown).
Removal of Region I allows heparin-resistant holoenzyme complex formation with heteroduplex DNA
The binding of holoenzymes (assembled with σN or ΔIσN) to heteroduplex DNA fragments can be detected by gel mobility-shift assays. The assays included a heparin challenge that disrupts σN-holoenzyme closed complexes at homoduplex promoter sites, whereas open complexes are heparin stable (Popham et al. 1989). We found that removal of Region I allowed the holoenzyme to assemble heparin-resistant complexes on pre-melted DNA independently of activator, whereas the wild-type σN-holoenzyme required activator.
As judged by the amount of bound DNA, heparin-resistant complexes with σN-holoenzyme and heteroduplex promoter DNA formed efficiently only when activator protein PspFΔHTH (Jovanovic et al. 1996) and a hydrolyzable nucleotide (dideoxy nucleotides ddATP and ddGTP were used to prevent formation of the first RNA phosphodiester bond) were present. The number of stable complexes formed in the presence of activator and nucleotide was 5- to 10-fold more than in their absence (Fig. 1A, cf. lanes 1, 2, and 5 with 3, 4, 6, and 7). Both ATP and ddATP supported stable complex formation (lanes 3,4), whereas the non-hydrolyzable analog 5′-adenyl-β,γ-imidodiphosphate did not (data not shown). Therefore, formation of σN-holoenzyme heparin-resistant complexes on heteroduplex DNA shows the same activator and nucleotide requirements as does formation of stable open complexes on homoduplex DNA (Popham et al. 1989; Cannon et al. 1995b) and transcription from heteroduplex glnAp2 templates (Wedel and Kustu 1995).
Figure 1.




Gel mobility-shift assays on R. meliloti nifH promoter DNA. (A) Effect of activator and/or nucleotides on heparin-resistant complex formation between σN- and ΔIσN-holoenzymes (EσN and EΔIσN) and heteroduplex 1 DNA (see Table 1). Holoenzymes were at 100 nm, activator PspFΔHTH (lanes 3,4,6,7,10,13,14) at 4 μm, nucleotides at 1 mm and end-labeled DNA at 16 nm. Prior to gel loading, heparin (100 μg/ml) was added for 5 min. Reactions included salmon sperm DNA (455 ng/μl), which helped to reduce background smears. The percentage of DNA shifted is indicated in the histogram. (B) Stability of holoenzyme complexes on heteroduplex DNA. Following holoenzyme binding to DNA and incubation with PspFΔHTH and ddGTP where indicated, samples were taken prior to (time 0) and after addition of heparin for 1, 5, 10, and 20 min, and immediately loaded onto running native gels. Holoenzymes (EσN, EΔIσN) alone (□,█), plus ddGTP (○,●), and plus ddGTP and PspFΔHTH (▵,▴). (C) Formation of heparin-resistant complexes with homoduplex DNA. Holoenzymes were at 250 nm, end-labeled 88-mer homoduplex DNA at 16 nm, PspFΔHTH at 4 μm and GTP at 4 mm (lanes 1,3,6). Prior to gel loading, heparin (lanes 2,3,5,6) was added for 5 min. The level of initial complexes (lanes 1,4) was unaffected by GTP plus PspFΔHTH (data not shown). (D) Region I sequences confer heparin sensitivity to holoenzyme. Holoenzymes (50 nm) containing end-labeled σ proteins (EσN-32P, EΔIσN-32P) were incubated with or without 50 nm 88-mer homoduplex (lanes 3,4,11,12) and heteroduplex 1 (lanes 5–8,13,14), challenged with heparin for 5 min where indicated and loaded onto native gels. PspFΔHTH (4 μm) and GTP (4 mm) were added for activation (lanes 7,8). (H) Holoenzyme; (H+DNA) holoenzyme–DNA complex; (σ) free sigma.
In marked contrast to the σN-holoenzyme, ΔIσN-holoenzyme efficiently formed heparin-stable complexes with heteroduplex DNA independently of activator and nucleotide (Fig. 1A, lane 8). Typically, in the absence of activator, there were 5- to 10-fold more heparin-resistant ΔIσN-holoenzyme complexes than with σN-holoenzyme (cf. lanes 8, 9, and 12 with 1, 2, and 5). We found that stable complexes (formed either by activation or deletion of Region I) accumulated with equal efficiency on heteroduplex 2 (Table 1), which was constructed with a mutant bottom strand −10 to −1 sequence (data not shown), demonstrating this is not a major source of sequence selectivity, as suggested by the lack of conservation in the −10 to −1 region of σN-dependent promoters. Heparin-resistant complexes were inefficiently formed (30-fold reduction) with mutant heteroduplex DNA (heteroduplex 3, see Table 1) in which the consensus −26/−25 GG was changed to AA (data not shown) demonstrating the promoter sequence specificity of complex formation for σN and ΔIσN holoenzymes.
The activator-dependent (σN) and independent (ΔIσN) holoenzyme complexes that formed on heteroduplex DNA in the presence of ddGTP were stable to heparin challenge, a property of open promoter complexes formed on homoduplex DNA (Popham et al. 1989). We directly measured the stabilities of complexes after adding heparin (Fig. 1B). An initial rapid decay was evident, suggesting disruption of unstable complexes, followed by a phase of slower decay allowing an estimate of the half-lives of the stable complexes. For the ΔIσN-holoenzyme (+ddGTP) and the activated σN-holoenzyme (+ddGTP) half-lives were very similar (∼30 min). The presence of ddGTP significantly increased the stability of the ΔIσN-holoenzyme complex as the half-life without nucleotide was only 7 min; activator PspFΔHTH also increased it slightly. For the unactivated σN-holoenzyme (−/+ddGTP) a few complexes (a maximum of 10% of the initial ones) were extremely heparin stable (half-life was >30 min); later experiments indicated that not all of them were transcriptionally active (see Fig. 5, below). Stabilization by ddGTP shows that the ternary complex is accessible to the initiating nucleotide and suggests that the rate of dissociation of the σN-holoenzyme, like the σ70-holoenzyme, is reduced by the first incoming rNTP (see also Gaal et al. 1997). Stabilization by ddGTP implies that phosphodiester bond formation is not required for reducing dissociation of the σN-holoenzyme from the melted DNA template.
Figure 5.
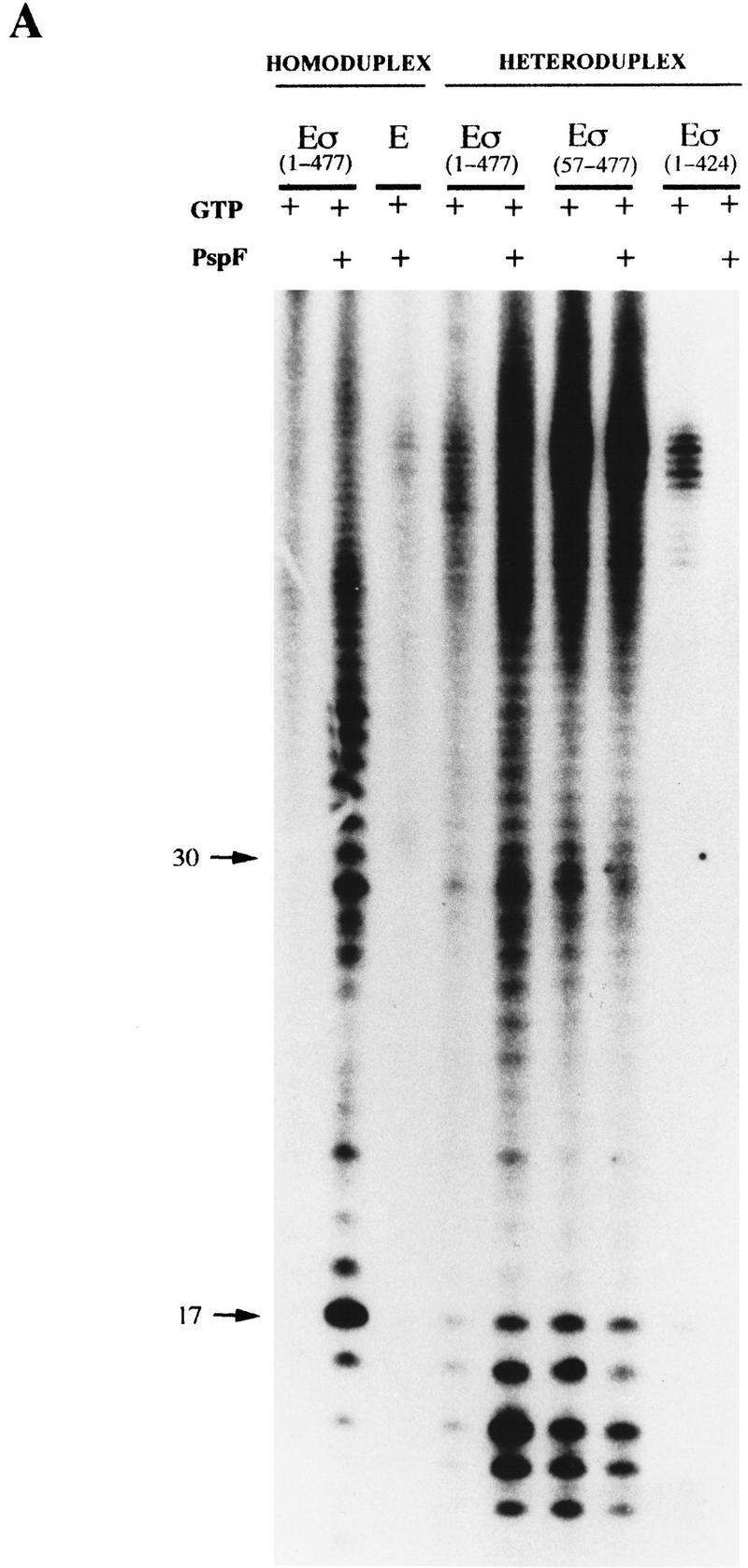
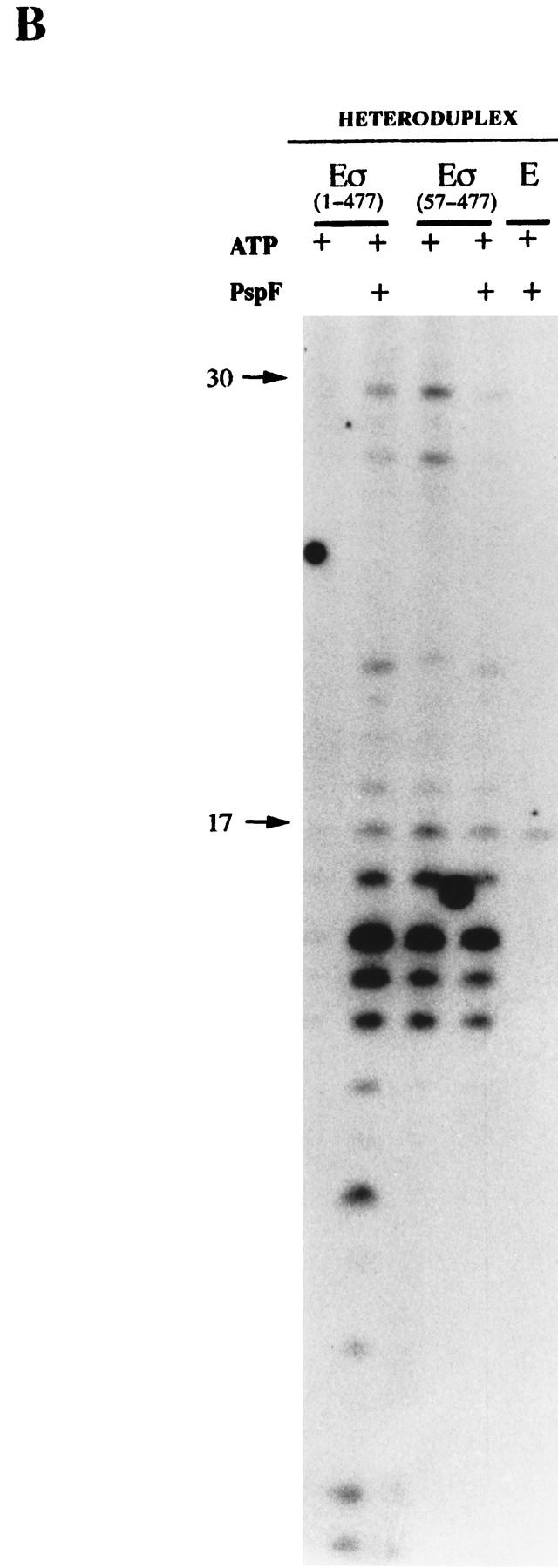
Holoenzyme complexes lacking Region I are transcriptionally active on heteroduplex DNA without activator. Holoenzymes (100 nm) with GTP or ATP (1 mm) in the presence or absence of PspFΔHTH (4 μm) were used to transcribe the 88-mer homoduplex and heteroduplex 1 (16 nm) templates. Core RNAP and holoenzyme containing the DNA nonbinding 1–424 σ derivative (Cannon et al. 1995b) were used as controls. The location of the DNA size markers are shown (arrows). (A) [α-32P]UTP-labeled transcripts with GTP as the hydrolyzable nucleotide used by activator. (B) [γ-32P]GTP-labeled transcripts with ATP as the hydrolyzable nucleotide used by activator.
Stable complex formation with ΔIσN-holoenzyme requires heteroduplex DNA
To determine the extent to which properties of the ΔIσN-holoenzyme depend on the heteroduplex DNA, we compared the promoter interactions of the σN- and ΔIσN-holoenzymes using an 88-mer homoduplex DNA fragment, equivalent to heteroduplex 1 except with a complementary −10 to −1 sequence (Table 1). Our results demonstrate that σN-holoenzyme can establish a heparin-resistant complex on homoduplex DNA if provided with activator, but that ΔIσN-holoenzyme fails to establish a heparin-resistant complex, irrespective of activator.
Complexes that formed with σN-holoenzyme in the absence of activator and GTP were largely heparin sensitive: Only 2% of the DNA was in a heparin-resistant complex (Fig. 1C, lane 2). Similarly, ΔIσN-holoenzyme shifted 66% of the DNA in the absence of activator and GTP, but only a small fraction of these complexes were heparin resistant (cf. lanes 4 and 5). In contrast, with added PspFΔHTH and GTP, half of the wild-type complexes formed were heparin stable (cf. lanes 1 and 3), whereas the stable fraction with ΔIσN-holoenzyme increased marginally (lane 6).
Comparison of results with the heteroduplex DNA to those with homoduplex DNA (Fig. 1A–C) shows that removal of Region I strongly favors efficient formation of heparin-stable complexes only with heteroduplex promoter DNA. The unpairing of DNA from −10 to +1 may limit stable complex formation by ΔIσN-holoenzyme. Likely DNA melting limits the ability of the ΔIσN-holoenzyme to form stable complexes on homoduplex DNA. Because a similar fraction (about 50%) of unstable σN-holoenzyme complexes is converted to activator-dependent stable complexes with both homoduplex and heteroduplex DNA (see Fig. 1B,C), the same barrier to stable complex formation exists regardless of the template type (see also Wedel and Kustu 1995). Overall, (1) Region I appears to inhibit stable complex formation on DNA templates even when the start site is melted; (2) the inhibitory effect of Region I can be overcome by activator and its associated nucleotide hydrolysis; and (3) removal of Region I bypasses the usual activator requirement to form stable promoter complexes, but only if the DNA is premelted.
Heparin resistance is associated with holoenzyme stability
Previously, we observed that heparin promotes dissociation of the σN-holoenzyme into core RNAP and σ (M.T. Gallegos and M. Buck, in prep.). Using 32P end-labeled σN and ΔIσN, we observed that heparin dissociated the two types of holoenzyme (Fig. 1D, lanes 2,10). However, heteroduplex DNA, but not homoduplex DNA, significantly protected the ΔIσN-holoenzyme from dissociation by heparin (cf. lanes 12 and 10). No obvious protection of the σN-holoenzyme from heparin disruption was afforded by either form of DNA alone (lanes 4 and 6). Activation of the σN-holoenzyme on the heteroduplex DNA did result in the stable association of end-labeled σ with the core polymerase (lane 8). These results are fully congruent with the DNA gel mobility-shift results (Fig. 1A,C) and demonstrate that (1) Region I of σN prevents the holoenzyme from interacting with heteroduplex DNA so as to acquire heparin resistance; (2) activator overcomes this inhibition; and (3) σ is part of the complex that stably associates with the heteroduplex DNA.
Increased binding of ΔIσN-holoenzyme to DNA templates
To examine the basis of the efficient activator-independent stable complex formation observed with the ΔIσN-holoenzyme, we assayed the affinity of σN, ΔIσN and their holoenzymes for homoduplex and heteroduplex DNA molecules without heparin in a gel mobility-shift assay (Fig. 2). The affinity of ΔIσN-holoenzyme for either homoduplex or heteroduplex DNA was higher (about twofold) than that of σN-holoenzyme. Affinities of σ proteins for homoduplex and heteroduplex DNA showed that the ΔIσN bound both with a higher affinity than σN. Thus, removal of Region I increases promoter binding. Because deletion of Region I increases binding of the holoenzyme to both templates, increased binding per se is not sufficient for activator-independent heparin-resistant complex formation on heteroduplex DNA.
Figure 2.

Binding of σ proteins (σ) and their holoenzymes (Eσ) to 88-mer homoduplex and heteroduplex 1 DNA. Binding reactions were essentially as in Fig. 1A, except that 1.6 nm DNA was used and heparin and nonspecific DNA were omitted. (□) σN, (○) ΔIσN, (█) σN-holoenzyme, (●) ΔIσN-holoenzyme.
Region I protease sensitivity in holoenzyme is conditioned by promoter DNA topology
Region I amino acid E36 becomes hyper-reactive to proteolysis in initiated transcription complexes on homoduplex DNA, suggesting a conformational change in Region I associated with initiation (Casaz and Buck 1997). The sensitivity of Region I to endoproteinase Glu-C attack was examined for σN and its holoenzyme bound to homoduplex and heteroduplex DNA. Interaction of core RNA polymerase with σN resulted in a 50% reduction in cleavage at E36 relative to cleavage of the free protein (Fig. 3, lanes 2,5), consistent with previous results (Casaz and Buck 1997). Interaction of σN-holoenzyme with heteroduplex DNA caused a 3.6-fold increase in the rate of cleavage at residue E36 (lane 7). No such change was detected with homoduplex DNA (lane 6) or when σN alone was incubated with either DNA (1.3- to 1.5-fold increase over free σ). Therefore, the hyper-reactivity of E36 seems associated with a reversible interaction of holoenzyme with melted DNA, which may represent a reversible step in the normal initiation pathway before stable DNA melting and heparin resistance have been achieved. It is clear that Region I of σN senses the conformation of the promoter, likely important for a concerted change in holoenzyme conformation necessary for open complex formation (See Discussion).
Figure 3.
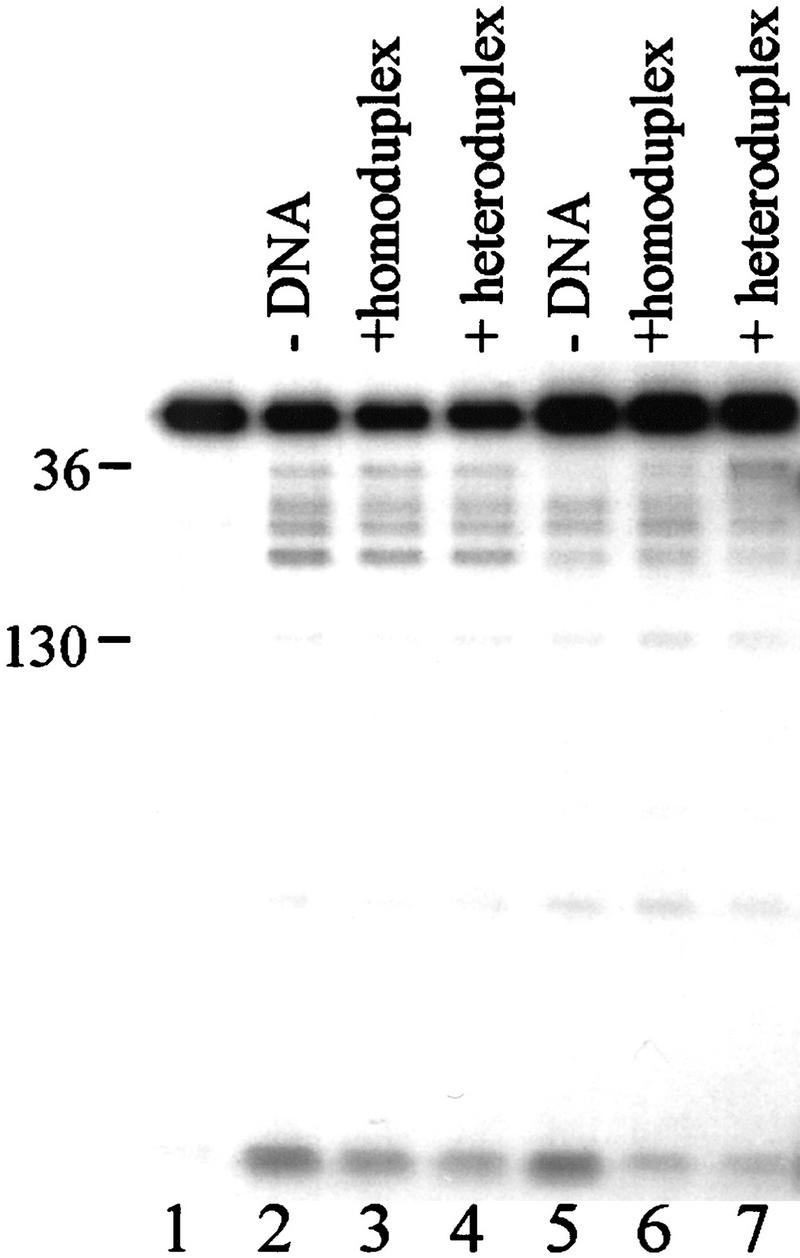
Region I sequences are sensitive to promoter DNA conformation. End-labeled σN (lanes 1–4) or holoenzyme prepared with end-labeled σN (lanes 5–7) were incubated for 10 min without DNA (lanes 2,5), with homoduplex DNA (lanes 3,6) or with heteroduplex DNA (lanes 4,7) and then cleaved with endoproteinase Glu-C (Casaz and Buck 1997). Numbers on the left indicate the amino acid at which cleavage has occurred.
Heparin-resistant complexes sequester melted DNA
The interaction of holoenzymes with melted DNA in stable complexes was examined by use of S1 nuclease. We footprinted gel-purified complexes between heteroduplex DNA and the σN-holoenzyme (activating it in the presence of ddGTP) or the ΔIσN-holoenzyme (simply binding it to the DNA). We found that the σN- and ΔIσN-holoenzymes sequester melted DNA in stable complexes, despite the fact that the complexes are formed via different routes. Binding reactions were exposed briefly to S1 nuclease and the heparin-resistant complexes were separated on a native polyacrylamide gel (Fig. 4A). Labeled DNA was eluted from isolated bands and equal amounts (counts per minute) of recovered bound (lanes B) and unbound (lanes U) end-labeled DNA were analyzed on a sequencing gel (Fig. 4B,C). The heparin-resistant complexes formed by either activation or deletion of Region I were indistinguishable by S1 footprinting. The heteroduplex sequence was refractory to S1 cleavage (compare bound and unbound DNA samples), but far more so on the bottom strand (Fig. 4C). The holoenzymes weakly protected the sequence upstream of the mismatched region up to −24. In marked contrast, the footprints of σN or ΔIσN showed that σ alone failed to strongly protect the single-strand region, with only weak protection at the −10 edge that extended upstream to around −30.
To determine the sites of melted DNA and to extend conclusions drawn from the S1 footprints, we probed complexes of holoenzymes stably bound to the heteroduplex DNA with KMnO4 to detect single-stranded sequences (Fig. 4B,C, lanes B). We found that either removal of Region I or activation of the σN-holoenzyme led to the formation of complexes showing almost identical KMnO4 footprints. Top and bottom strands showed a similar KMnO4 reactivity across the mismatched region to that of the free DNA (lane F), even though it was partly or largely inaccessible to S1 attack (see above). Both holoenzymes partially diminished reactivity around −1 on the top strand (Fig. 4C), but slightly increased it around −10. KMnO4 footprints of σN and ΔIσN bound to heteroduplex DNA showed very little difference from free DNA on both strands (Fig. 4B,C).
We conclude that the ΔIσN-holoenzyme and activated σN-holoenzyme interact similarly with melted DNA and that sequestration of the single-strand DNA can occur either through the action of activator or deletion of Region I, suggesting that Region I masks a single-strand DNA-binding activity of the holoenzyme. Together with the DNA binding assays (Fig. 2), we propose that the absence of Region I allows the holoenzyme to establish extra contacts to DNA templates in which sequences are pre-melted, contacts likely responsible for the Region I-dependent heparin stability of holoenzymes on heteroduplex DNA (Fig. 1A,B).
Holoenzyme complexes with heteroduplex DNA are transcriptionally active
Previous work had shown that heparin-resistant holoenzyme complexes formed by activator and NTP hydrolysis on heteroduplex DNA were transcriptionally active (Wedel and Kustu 1995). A transcription assay was performed with the heteroduplex templates to determine whether the heparin-resistant complexes formed on them were active. Using [α-32P]UTP for internal labeling of RNA (Fig. 5A) or end labeling with [γ-32P]GTP (Fig. 5B) to check the starting nucleotide was the same as with conventional DNA (Sundaresan et al. 1983), we found that the patterns of transcripts synthesized on heteroduplex DNA by the ΔIσN-holoenzyme and by the activated σN-holoenzyme were very similar. The σN-holoenzyme without activator transcribed very poorly from homoduplex and heteroduplex DNA (<1% UTP-labeled RNA compared with activator). The short DNA templates employed in this analysis may account for the complex pattern of internally labeled transcripts as the polymerase may not efficiently escape the promoter. The discrepancy in the distribution of products seen in Figure 5A when homoduplex and heteroduplex DNA are utilized by the activated σN-holoenzyme may be related to differences in transcript release and polymerase escape. Controls with core RNAP or with a holoenzyme assembled with a mutant σN lacking the DNA-binding domain (amino acids 1–424, Cannon et al. 1995b) showed that transcripts were specific to holoenzymes able to recognize the σN conserved −12/−24 promoter elements (Fig. 5A). The similarity of transcript patterns occurring with the activated σN-holoenzyme and the ΔIσN form, their size and labeling with [γ-32P]GTP plus the need for the −24 consensus region to allow efficient binding of holoenzymes to the heteroduplex DNA (see above) all argue that the transcription is faithful and occurs from the pre-melted DNA with the expected polarity.
The transcription results complement the DNA binding assays (Figs. 1 and 2) and show that Region I inhibits the formation of transcriptionally competent pre-initiation complexes on homoduplex DNA and that ΔIσN-holoenzyme allows activator-independent transcription from the premelted R. meliloti nifH promoter. Previous work showed that the removal of Region I allowed activator independent transcription from the glnAp2 promoter via a heparin-sensitive pre-initiation complex formed in reactions containing the initiating nucleotide ATP (Wang et al. 1997a). The results of our transcription assays suggest that another pathway distinguished by its insensitivity to heparin can lead to initiation; stable DNA melting is needed for the heparin-resistant pathway to operate.
Region I sequences function in trans
To determine whether Region I could function in isolation and convert the ΔIσN-holoenzyme to a form behaving as though it were a holoenzyme containing full-length σN, we added purified Region I sequences (amino acids 1–51, 1–56, 1–63, and 1–71) at the point of holoenzyme assembly to assays of heparin-resistant heteroduplex complex formation (Fig. 6A). As a control we used a fragment with a disrupted Region I sequence in which amino acids 21–27 are deleted (1–71Δ). We found that Region I peptides were able to inhibit the formation of stable complexes between the ΔσN-holoenzyme and heteroduplex DNA. Specifically, the results showed that the absence of amino acids 1–56 in the ΔIσN-holoenzyme was complemented by the amino terminal fragments 1–51, 1–56, 1–63, and 1–71 (Fig. 6A, cf. lanes 1 and 2 with lanes 4, 5, 6, and 7) but not by the derivative 1–71Δ fragment (cf. lanes 1 and 2 with 3). The number of heparin-resistant complexes forming with heteroduplex DNA and ΔIσN-holoenzyme was diminished by Region I containing peptides to a level seen with σN-holoenzyme. A similar effect of Region I sequences in trans was observed by use of end-labeled ΔIσN-holoenzyme. The dissociation of ΔIσN from holoenzyme was increased (data not shown). The low level of activator-independent heparin-resistant complexes forming with σN-holoenzyme and heteroduplex DNA was unchanged by Region I in trans (data not shown).
Figure 6.


Region I-containing fragments confer heparin sensitivity in trans. (A) Different amino-terminal peptides of σN: 1–51, 1–63, 1–71, 1–71Δ (15 μm) and 1–56 (2.5 μm) were added to ΔIσN (200 nm) prior to core RNAP addition (100 nm) and heteroduplex 1 DNA (16 nm) was added last in a binding reaction. After incubation, heparin was added for 5 min and samples loaded onto native gels. The percentage of heparin-resistant DNA complexes is indicated. (B) Effect of Region I on heparin stability. Gel mobility shifts were conducted with ΔIσN-holoenzyme (100 nm), GTP (1 mm) and heteroduplex 1 DNA (16 nm; █) and Region I peptide (1–56, 2 μm) was added before ΔIσN-holoenyme assembly (▴) or after holoenzyme–heteroduplex complex formation (●). σN-Holoenzyme plus heteroduplex DNA, in which most complexes are heparin sensitive (see Fig. 1B), was used as a control (□). Samples were taken prior to (time 0) and after the addition of heparin for 1, 5, 10, 20 min.
Having established that Region I sequences in trans were able to inhibit stable complex formation by the ΔIσN-holoenzyme when added during holoenzyme formation but prior to DNA binding (Fig. 6A), we conducted order of addition experiments and heparin-stability measurements to examine the mechanism of inhibition. Results (Fig. 6B) showed that Region I did not significantly change the number of initial complexes formed between the ΔIσN-holoenzyme and heteroduplex DNA when added prior to DNA binding, but that it did reduce the number that were heparin stable to a level observed with σN-holoenzyme. It appears that Region I exerts its inhibitory effect on ΔIσN-holoenzyme before it engages with heteroduplex DNA since once stably bound, the ΔIσN-holoenzyme is not destabilized by Region I (Fig. 6B). A modest but consistent stabilization was evident when Region I was added after the ΔIσN-holoenzyme bound the heteroduplex DNA (Fig. 6B). Addition of activator and nucleotide did not reverse the Region I in trans inhibition of stable complex formation by the ΔIσN-holoenzyme nor did Region I inhibit formation of activator-dependent stable complexes between σN-holoenzyme and homoduplex DNA (data not shown). These results suggest that Region I in isolation may not titrate out activator and that activator responsiveness may require that Region I is present in cis. However, Region I functions in trans as an independent domain for its inhibitory effect.
Deletion of Region I alters the interaction of holoenzyme with start site and early transcribed sequences
We exploited the single-strand and weak double-strand nuclease activity of S1 nuclease to determine whether the ΔIσN-holoenzyme interacted with the −10 to −1 sequence of homoduplex DNA with which it fails to establish a stable complex (see Fig. 1C). We found that the ΔIσN-holoenzyme does footprint this sequence, and, surprisingly, that the footprint extends beyond the transcription start site.
S1 nuclease partially cleaved the target R. meliloti nifH promoter DNA template at nonrandom positions (Fig. 7, lane 0). Predominant positions of cleavage generally coincided with sequences displaying a background level reactivity to KMnO4, which detects unstacked T residues, and with sites sensitive to attack by the single-strand nuclease P1 and mung bean nuclease (data not shown). We conclude that the S1 nuclease is attacking sequences with a local conformation related to some DNA distortion. Changes in S1 nuclease cutting when σ and holoenzymes were incubated with DNA allowed us to footprint their binding, extending the set of DNA-reactive reagents used to date (Morris et al. 1994; Sasse-Dwight and Gralla 1988, 1990), and to probe for conformational changes in the promoter DNA associated with the pathway leading to stable DNA melting within the open complex.
Figure 7.
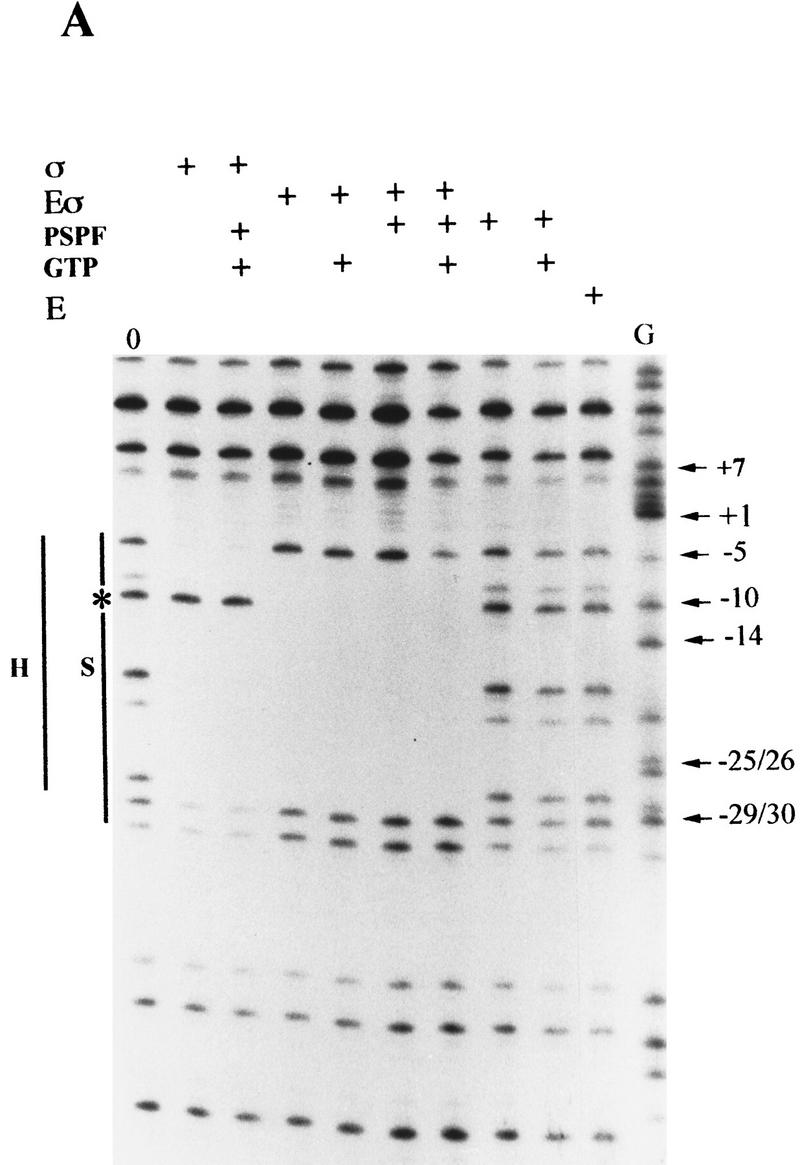
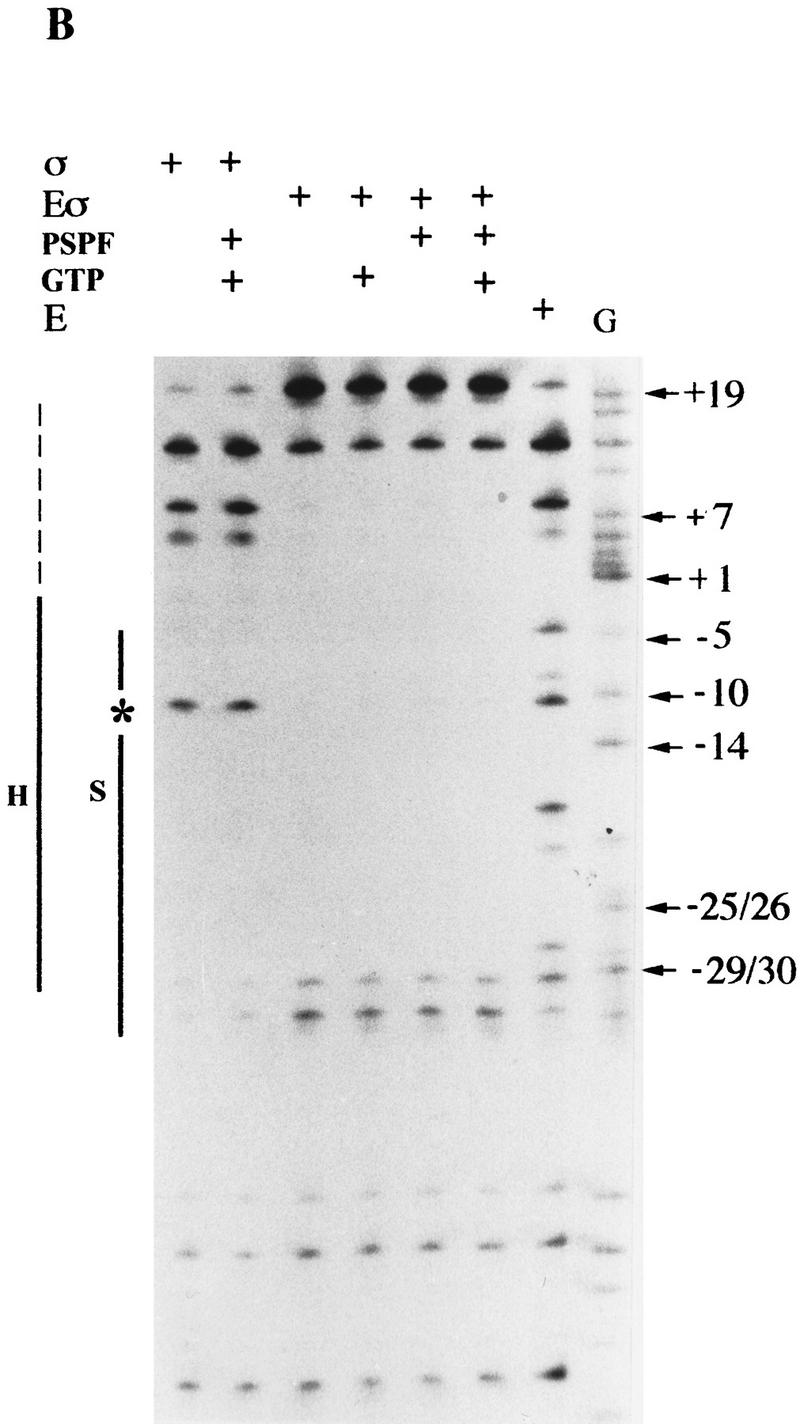
S1 nuclease footprints on linear DNA. Linear end-labeled templates prepared by primer extension were used. (A) σN and holoenzyme (amino acids 1–477); (B) ΔIσN and holoenzyme (amino acids 57–477). (Solid line) The extent of protection seen with holoenzyme (H) or σ (S); (broken line) weaker protection; (star) −10 cutting lost on holoenzyme binding. Markers were chemical cleavage G reactions. σ Proteins were at 1 μm, holoenzymes at 100 nm, activator PspFΔHTH at 4 μm, and GTP at 1 mm. Compared to σN-holoenzyme, the ΔIσN-holoenzyme displays an extended footprint in the direction of transcription (A vs. B). Activation of the σN-holoenzyme by PspFΔHTH (A) results in an extended footprint with increased protection from −5 to +7. The extended footprint to +20 observed with ΔIσN-holoenzyme is independent of activator and nucleotide. Reproducible footprints and cutting of promoter template DNA was observed with several different batches of S1 nuclease. A control with core RNAP did not produce a S1 footprint.
On homoduplex DNA, σN and ΔIσN had similar footprints, characterized by protection across sequences from about −5 to −33 with sequences around −10 being susceptible to cleavage by S1 nuclease (Fig. 7, cf. A and B). In contrast, σN- and ΔIσN-holoenzymes protected the residue at −10 from S1 cleavage and their footprints differed markedly at start-site-proximal bases (but not at the upstream −33 footprint edge; Fig. 7, cf. A and B). Specifically, the σN-holoenzyme footprinted to −5 and the ΔIσN-holoenzyme to around +20, with the residue at +20 showing high reactivity to cleavage. The extended S1 footprint of the ΔIσN-holoenzyme was observed with three other σN-dependent promoters (Klebsiella pneumoniae nifH, nifH049, and nifU promoters, data not shown). S1 nuclease footprints of holoenzymes containing σN or ΔIσN were insensitive to the addition of the initiating nucleotide or all four nucleotides, indicating that the complexes were largely inactive for transcription initiation on linear templates (data not shown). The extended footprint of the ΔIσN-holoenzyme compared to the σN-holoenzyme suggests that the holoenzyme has undergone a conformational change when Region I is absent allowing the holoenzyme to cover early transcribed sequences. However, the DNA covered is not stably melted as judged by KMnO4 reactivity measurements (Cannon et al. 1995b and data not shown). Clearly the absence of Region I changes the properties of the holoenzyme independently of an interaction with stably melted DNA. This suggests that binding to the heteroduplex DNA may not exclusively drive the ΔIσN-holoenzyme to the conformation able to establish a stable association with melted DNA.
Holoenzyme closed promoter complexes with full-length σN were incubated with activator PspFΔHTH in the presence of GTP, which served as the initiating and hydrolyzable nucleotide (Fig. 7A). Controls showed that neither GTP nor PspFΔHTH alone modified the S1 footprint of the template DNA. However activator and GTP in the presence of σN-holoenzyme generated an altered footprint across early transcribed sequences to +7, where cleavage sites between −5 and +7 showed partial protection from S1 nuclease attack (30%). The footprint resembled that of the ΔIσN-holoenzyme without added activator (Fig. 7B) in that residues to +7 were protected. The extent of −5 to +7 protection with activated σN-holoenzyme likely reflects the incomplete conversion of closed to open complexes on linear templates (Fig. 1C). Footprints of ΔIσN-holoenzyme did not respond to activator plus GTP (Fig. 7B), consistent with prior data indicating such complexes do not efficiently form KMnO4-reactive melted DNA on linear templates (Cannon et al. 1995b; Wang et al. 1997a). Since activation of the σN-holoenzyme produces KMnO4-reactive sequences (Cannon et al. 1995b) diagnostic of DNA melting, the protection pattern of activated complexes seen with S1 nuclease (Fig. 7A) suggests that the top DNA strand of the melted sequences is sequestered, as seen with the heteroduplex DNA (Fig. 4). By dividing an activation assay on homoduplex DNA and probing half with S1 nuclease and half with KMnO4, we directly confirmed that KMnO4-reactive sequences were refractory to S1 attack (data not shown).
Discussion
The σN-holoenzyme is distinct among bacterial RNAPs in its requirement for γ–β nucleoside triphosphate hydrolysis to convert closed complexes to open promoter complexes in which the template DNA strand is revealed by DNA melting (Kustu et al. 1989). The barrier to open complex formation appears to be both kinetic and thermodynamic and to involve Region I of σN (Morris et al. 1994; Wedel and Kustu 1995; Wang et al. 1995, 1997a). The DNA melting function of the σN-holoenzyme was suggested to be inhibited by Region I (Wang et al. 1995). We now provide evidence that Region I controls the interaction of holoenzyme with melted DNA. Key sequences in Region I needed for inhibition appear to be within a small leucine patch (Wang et al. 1995, 1997a). Our data indicate that Region I acts as an intramolecular repressor preventing a conformational change in holoenzyme that is needed to unmask a holoenzyme single-strand DNA-binding activity (see Fig. 8). We note that in σ70 systems, repressor action at a step following promoter binding of RNAP requires a specific separate repressor protein (for review, see Choy et al. 1997).
Figure 8.

Gel mobility-shift, footprinting, and transcription assays (see Figs. 1, 4, and 7) show activator and nucleotide hydrolysis are required to efficiently form stable heparin-resistant σN-holoenzyme complexes on homoduplex (open complex, in which DNA is melted) and heteroduplex DNA. In contrast, ΔIσN-holoenzyme bypasses the activator requirement for stable complex formation on heteroduplex DNA but fails on homoduplex, regardless of the presence of activator. We propose that the inhibition of DNA melting associated with Region I (Wang et al. 1995, 1997a) is due to prevention of polymerase isomerization, necessary to reveal a single-strand DNA-binding activity in the holoenzyme required for its stable association with melted DNA (Fig. 4). The thermodynamic barrier to open complex formation (Wedel and Kustu 1995) at σN-dependent promoters is suggested to be associated with energetically unfavorable DNA strand separation.
Because σN-holoenzyme open complexes decay to closed complexes and do not efficiently accumulate without activator (Wedel and Kustu 1995), it seems that Region I in the absence of activator normally drives the holoenzyme into a conformation in which contact with melted DNA is strongly disfavored, consistent with the data showing that DNA strand denaturation per se is not the rate-limiting step in open complex formation (Wedel and Kustu 1995). Nucleotide hydrolysis by activators of the σN-holoenzyme may promote the holoenzyme conformational change, inhibited by Region I, which can take place prior to DNA melting for ΔIσN-holoenzyme (Fig. 7). However, if the conformational change is normally physically coupled through changing DNA–holoenzyme contacts to the energetically unfavorable opening of the DNA, one component of the thermodynamic barrier to open complex formation at σN-dependent promoters (Wedel and Kustu 1995) is likely to be the activator and NTP hydrolysis-driven overriding of Region I inhibition.
We show that the acquisition of heparin stability by holoenzyme in promoter complexes requires an interaction of holoenzyme with stably melted DNA, consistent with transcription from transiently melting templates being heparin sensitive (Wang et al. 1995, 1997a). In the two-step model for initiation at σN-dependent promoters (Wang and Gralla 1996), a heparin-sensitive intermediate is proposed. Our results suggest that the conversion of this unstable intermediate to the heparin-resistant complex involves establishing contact between the isomerized holoenzyme and stably melted DNA. Below, we discuss the role of Region I in a pathway leading to formation of the stable open promoter complex from the closed complex.
Closed complexes
The S1 footprints of σN- and ΔIσN-holoenzymes on conventional DNA templates suggest that different types of closed complex may from. For σN-holoenzyme, a short complex extending from −33 to −5 was detected by DNase I, hydroxyl radical, and S1 footprinting (Buck and Cannon 1992; Cannon et al. 1995a; see also Popham et al. 1989 and this paper). For the ΔIσN-holoenzyme, the S1 footprint extended to +20 without stable melting (Cannon et al. 1995b; this paper). Isomerization of RNA polymerase is predicted to occur before DNA melting for σ70-holoenzymes (Gross et al. 1992). The extended footprint detected when Region I is deleted suggests that the ΔIσN-holoenzyme has undergone a significant change with respect to DNA and has isomerized. Whether the conformational change in the σN-holoenzyme can occur prior to or concomitant with DNA melting, remains unknown. We suggest that S1 nuclease detects an extended footprint for the ΔIσN-holoenzyme not readily discernible with DNase I or hydroxyl radical because the on rate of the S1 is slower than that of the other footprinting reagents, hence holoenzyme that transiently interacts with DNA sequences ahead of −5 will protect them from S1 attack.
Role of Region I of σN
We were able to efficiently form transcriptionally active heparin-resistant complexes using heteroduplex DNA in the absence of activator but only when Region I was absent. When Region I was present, the usual activator and nucleotide requirements for stable complex formation and transcriptional activity had to be met. Together with protein footprinting experiments that detect changes in the relationship of Region I and core upon activation (Casaz and Buck 1997), it seems that Region I is part of the interface between σ and core where changing interactions driven by the activator are needed to allow polymerase isomerization. Evidence for a physical coupling between conformational changes in Region I and DNA melting comes from the finding that, when heteroduplex DNA binds the σN-holoenzyme, the protease reactivity of Region I increases (this paper). Certain single substitutions in the DNA-binding domain of σN give rise to holoenzymes with the same activator-independent stable complex forming activity as the ΔIσN-holoenzyme (M.K. Chaney and M. Buck, unpubl.), suggesting that Region I could interact with the DNA-binding domain and sense DNA conformation indirectly. Therefore, communication between the core RNA polymerase and the promoter may be mediated by Region I of σN and could influence several sequential events that lead to formation of a stable open promoter complex. Some steps will involve alteration of the interaction of Region I with core to relieve inhibition and allow isomerization, some may depend on σN sensing of promoter DNA structure, and some will involve the creation or stabilization of DNA conformations that favor DNA melting. Region I is known to be required for one early step, the nucleation of DNA melting (Morris et al. 1994). Multiple roles for Region I including a potential contact site for activator likely explain why corruption of Region I to allow polymerase isomerization is insufficient to allow the isomerized holoenzyme to cause stable melting (Wang et al. 1997a; Cannon et al. 1995b; Morris et al. 1994). Although it is clear that Region I controls the interaction of holoenzyme with melted DNA, precisely how melting occurs and at how many steps the activator contributes remains to be determined. The ability of Region I to act as an independent domain and function in trans to inhibit binding of the ΔIσN-holoenzyme to heteroduplex DNA argues for the reversibility of the conformational change in the σN-holoenzyme normally inhibited by Region I and is consistent with the finding that stable open promoter complexes decay to closed complexes (Wedel and Kustu 1995).
Organization of open complexes
Within stable promoter complexes the greater protection from S1 nuclease attack of the template strand versus the nontemplate strand argues that the template strand is located within the catalytic site for RNA synthesis and that the nontemplate strand is somewhat displaced. Establishment of the above relationship of the melted DNA to the polymerase depends on either removal of σN Region I or activation of the holoenzyme. The extents of S1 and KMnO4 reactivities seen in the stable complexes were equivalent regardless of the presence of Region I, suggesting that Region I does not establish any major direct contacts with the melted DNA. Unless the binding of the core to σ significantly increases contact that σ makes with the melted promoter sequences, the patterns of S1 protection seen with holoenzymes are largely due to contacts that the core subunits make with the melted DNA regions. Although σN has a role in establishing the nucleation of DNA strand separation (Morris et al. 1994) it may have a lesser role than σ70 in directly contacting the melted DNA (Chan et al. 1996) to form open complexes. Because σN touches DNA from −10 to −5 in the closed complex (Cannon et al. 1995) some modification of contacts with DNA may be necessary to allow formation of open complexes in which the −10 to −5 DNA is melted out. Interestingly, the roles of the amino-terminal domains of the two σ classes (which are unrelated by primary amino acid sequence) appear opposite. Amino-terminal sequences favor polymerase isomerization for the σ70-holoenzyme (Wilson and Dombroski 1997) but inhibit isomerization for the σN-holoenzyme (this paper).
The binding of σN to promoter DNA is insensitive to heparin (Oguiza and Buck 1997), and the acquisition of heparin resistance by σN-holoenzyme–DNA complexes seems to be associated with a change in holoenzyme conformation, occurring either upon activation or upon deletion of Region I. A simple explanation is that Region I normally inactivates a single-strand DNA-binding site in holoenzyme, a site closely associated with the polymerase catalytic center and accessible to heparin prior to polymerase isomerization but inaccessible afterward when the subsequent binding to melted DNA has occurred.
Summary
Overall, it seems that the σN-holoenzyme is able to maintain extremely low levels of transcription (Wedel and Kustu 1995; Wang and Gralla 1996; this paper) without use of additional silencing factors such as repressors because Region I blocks polymerase isomerization (see also Wang et al. 1997a). Region I can be viewed as a protein domain that functions to control an equilibrium between alternate states of the holoenzyme that either favor maintainenance of either the closed or the open complex through the masking and unmasking of an activity allowing the holoenzyme to bind to DNA sequences that are melted out. Activator and associated nucleotide hydrolysis drive the holoenzyme into the conformation in which the single-strand binding activity of the holoenzyme is unmasked.
Materials and methods
Proteins
Proteins were prepared as described previously (Cannon et al. 1995b, 1997; Casaz and Buck 1997). The K. pneumoniae σN protein (amino acids 1–477), ΔIσN (amino acids 57–477), and the carboxy-terminally truncated σN (amino acids 1–424) were used to form holoenzymes. Purified amino-terminal sequences of σN were obtained by overproduction of amino-terminal (1–56) and carboxy-terminal (1–51, 1–63, 1–71, 1–71Δ) histidine-tagged fragments as soluble sequences in E. coli (M.T. Gallegos and M. Buck, in prep.). A purified carboxy-terminally deleted form of activator, PspFΔHTH (Jovanovic et al. 1996) was used in activation assays. Purified core RNAP was from E. coli (a generous gift from A. Kolb, Pasteur Institute, Paris). Working protein solutions were stored at −20°C, stocks at −70°C, in TGED [10 mm Tris-HCl (pH 8.0), 50% vol/vol glycerol, 0.1 mm EDTA, 1 mm DTT] containing 50–250 mm NaCl.
End labeling of σ proteins
σ Proteins (σN or ΔIσN, Cannon et al. 1995b) modified by precise addition of a carboxy-terminal heart muscle kinase tag were prepared and end labeled with 32P as described previously (Casaz and Buck 1997).
DNA heteroduplex formation
Mismatched R. meliloti nifH promoter DNA heteroduplexes were prepared by annealing of 5′ 32P-end-labeled oligonucleotides (Table 1). The extent of nonduplex DNA from −10 to −1 was chosen by reference to the known start of DNA melting at the nifH promoter (Morett and Buck 1989; Cannon et al. 1995b) and previous work with the σN-dependent glnAp2 promoter (Popham et al. 1989; Sasse-Dwight and Gralla 1988; Wedel and Kustu 1995). Sequences were chosen to prevent purine–pyrimidine mismatch base-pairing and preserve the +1 G initiation site. Oligonucleotides (supplied by MWG-Biotech) were size purified on 8% denaturing polyacrylamide gels (detection was by UV shadowing) prior to kinasing. Pairs of DNA strands with either the unlabeled strand present at a twofold molar excess (10 pmoles in 20 μl), or for preparing unlabeled heteroduplex with both strands at equal concentration, were heated at 95°C for 3 min in 10 mm Tris-HCl (pH 8.0), 10 mm MgCl2 and then chilled rapidly in iced water for 5 min to allow heteroduplex formation.
DNA homoduplex formation
For DNA footprinting, homoduplexes were prepared by extension of R. meliloti nifH M13mp19 single-stranded DNA with 5′-32P-labeled universal primer as described previously (Cannon et al. 1995a). Fully complementary end-labeled oligonucleotides (Table 1) for preparing short 88-mer −60 to +28 homoduplexes were annealed together as described above.
Native gel complex formation assays
A gel mobility-shift assay (Casaz and Buck 1997; Wang et al. 1997b) was used to detect σN and its holoenzyme bound to a radioactively labeled R. meliloti nifH homoduplex or heteroduplex promoter DNA fragment (substituted by unlabeled DNA as appropriate, see figure legends 1 and 6). Typical holoenzyme interaction assays included 100 nm core RNAP plus 200 nm σ (substituted by end-labeled σ as appropriate, see figure legends 1 and 6), and 1.6 or 16 nm homoduplex or heteroduplex DNA assembled from purified oligonucleotides (50 nm when using end-labeled proteins), plus where necessary nucleotide as indicated, in STA buffer [25 mm Tris-acetate (pH 8.0), 8 mm Mg-acetate, 10 mm KCl, 1 mm DTT, 3.5% w/vol PEG 6000]. For activation, 4 μm PspFΔHTH activator protein (Casaz and Buck 1997) and 1 mm nucleotide (see figure legends 1 and 6) was also added. To optimize complex formation with the 88-mer homoduplex fragment 250 nm holoenzyme (core:σ 1 : 2), and, where indicated, 4 μm PspFΔHTH and 4 mm GTP was used. Core RNAP, σ proteins, and DNA were pre-incubated at 30°C for 10 min and, then, if necessary, nucleotide and activator were added for 10–15 min followed by a glycerol bromophenol blue loading dye (final concentration 10% glycerol) and, if required, heparin (final concentration 100 μg/ml). Then, samples were loaded on 4.5% native polyacrylamide gels and run at 60 V for 80 min at room temperature in 25 mm Tris, 200 mm glycine buffer (pH 8.6). Free DNA and proteins from protein–DNA complexes were detected by autoradiography and/or silver staining. Quantitative data were from PhosphorImager analysis.
Protein footprints
Protein footprint assays were conducted with end-labeled σN as described previously (Casaz and Buck 1997). Briefly, labeled σN as free protein (25 nm) or as holoenzyme (50 nm) was incubated in TGED buffer with or without promoter DNA (50 nm) and challenged with 75 ng of endoproteinase Glu-C for 3 min at 30°C. Reactions were terminated with 2× SDS loading buffer [4% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.125 m Tris-HCl (pH 6.8), 0.004% bromophenol blue] containing 0.5 mm 3,4-dichloroisocoumarin. Digestion products were separated on 12.5% denaturing polyacrylamide gels and detected by autoradiography. The amino termini of the products have been mapped previously (Casaz and Buck 1997). The product of cleavage at residue E36 was quantitated by PhosphorImager analysis with the product of cleavage at residue 130 as an internal control. Values for stimulation or reduction given in the text are relative to cleavage of free σ without added DNA.
DNA footprinting assays
S1 nuclease footprinting was used to examine binding of σN, ΔIσN and their holoenzymes to homoduplex and heteroduplex DNA. For S1 footprints on homoduplex (extended M13mp19 with R. meliloti nifH promoter), reactions (50 μl) were conducted in STA buffer at 30°C and contained 1.6 nm DNA together with 1 μm σ or 100 nm holoenzyme (core:σ at 1 : 2) and activator PspFΔHTH (4 μm) and GTP (1 mm) where needed. Holoenzymes and DNA were incubated for 10 min and, for activation assays, PspFΔHTH and GTP were added for a further 10 min. S1 nuclease (1400 units, Pharmacia) was added for 5 min before reactions were terminated by the addition of 10 mM EDTA followed by rapid phenol extraction. DNA was recovered by ethanol precipitation and run on 6% denaturing polyacrylamide gels. Markers were generated by chemical cleavage of the DNA with piperidine following partial methylation with dimethylsulfate. For S1 footprints on heteroduplex DNA (heteroduplex 1), binding reactions (15 μl) were conducted as above but with 100 nm holoenzyme or 2 μm σ and ddGTP (1 mm) instead of GTP. S1 nuclease (300 units) was added for 1 min before the reaction was terminated with 10 mm EDTA and 100 μg/ml heparin. After 5 min, reactions were run on 4.5% native gels, and all labeled DNA bands were excised and the DNA eluted into 10 mm Tris (pH 8.0), 0.1 mm EDTA for 2 hr at 50°C and dried under vacuum. For KMnO4 footprints on heteroduplex DNA, binding reactions were treated with 4 mm fresh KMnO4 for 30 sec, stopped with 50 mm β-mercaptoethanol and complexes resolved on native gels. DNA from excised bands was eluted into water and cleaved with piperidine (10% vol/vol) at 90°C for 30 min. Recoveries of isolated DNA were determined by dry Cerenkov counting and equal numbers of counts loaded (hence lanes are directly comparable) on 10% denaturing polyacrylamide gels. Quantitative data were obtained by PhosphorImager analysis.
In vitro transcription assay
Nonradioactively labeled heteroduplex or homoduplex promoter DNA fragments (16 nm) and holoenzyme (100 nm) were incubated in a 40-μl reaction for 10 min at 30°C in STA buffer containing 40 units of RNase inhibitor (RNasin, Promega) prior to the addition of 1 mm GTP or 1 mm ATP (for [α-32P]UTP and [γ-32P]GTP transcript assays respectively) and 4 μm PspFΔHTH activator protein to allow open complexes to form. After a further 10 min, heparin (100 μg/ml) and elongation mix {ATP, CTP, UTP at 0.1 mm and 12.5 μCi of [α-32P]UTP (Amersham, 800 Ci/mmole) or GTP, CTP, UTP at 0.1 mm and 10 μCi of [γ-32P]GTP (Amersham, 5000 Ci/mmole)} were added to allow synthesis of transcripts, which were allowed to accumulate for 10 min before phenol extraction and ethanol precipitation. Samples were run on 15% denaturing polyacrylamide gels and transcripts were visualized by autoradiography.
Acknowledgments
The work was supported by funding from the Wellcome Trust and the Biotechnology and Biological Sciences Research Council. M.T.G. was supported by a Spanish Ministry of Education and Culture postdoctoral fellowship and an European Community Biotechnology Training and Mobility of Researchers fellowship. We thank D. Studholme for comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL m.buck@ic.ac.uk; FAX 44-(0) 171-59-45419.
References
- Austin S, Dixon RA. The prokaryotic enhancer binding protein NtrC has ATPase activity which is phosphorylation and DNA dependent. EMBO J. 1992;11:2219–2228. doi: 10.1002/j.1460-2075.1992.tb05281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M, Cannon W. Specific binding of the transcription factor σ-54 to promoter DNA. Nature. 1992;358:422–424. doi: 10.1038/358422a0. [DOI] [PubMed] [Google Scholar]
- Burgess R, Travers A, Dunn J, Bautz E. Factor stimulating transcription by RNA polymerase. Nature. 1969;221:43–46. doi: 10.1038/221043a0. [DOI] [PubMed] [Google Scholar]
- Cannon W, Claverie-Martin F, Austin S, Buck M. Identification of a DNA-contacting surface in the transcription factor σ54 (σN) Mol Microbiol. 1994;11:227–236. doi: 10.1111/j.1365-2958.1994.tb00303.x. [DOI] [PubMed] [Google Scholar]
- Cannon W, Austin S, Moore M, Buck M. Identification of close contacts between σ54 (σN) protein and promoter DNA in closed promoter complexes. Nucleic Acids Res. 1995a;23:351–356. doi: 10.1093/nar/23.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon W, Missailidis S, Smith C, Cottier A, Austin S, Moore M, Buck M. Core RNA polymerase and promoter DNA interactions of purified domains of σN: Bipartite functions. J Mol Biol. 1995b;248:781–803. doi: 10.1006/jmbi.1995.0260. [DOI] [PubMed] [Google Scholar]
- Cannon WV, Chaney MK, Wang X-Y, Buck M. Two domains within σN (σ54) cooperate for DNA-binding. Proc Natl Acad Sci. 1997;94:5006–5011. doi: 10.1073/pnas.94.10.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaz P, Buck M. Probing the assembly of transcription initiation complexes through changes in σN protease sensitivity. Proc Natl Acad Sci. 1997;94:12145–12150. doi: 10.1073/pnas.94.22.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CL, Lonetto MA, Gross CA. Sigma domain structure: One down, one to go. Structure. 1996;4:1235–1238. doi: 10.1016/s0969-2126(96)00131-1. [DOI] [PubMed] [Google Scholar]
- Choy HE, Hanger RR, Aki T, Mahoney M, Murakami K, Ishihama A, Adhya S. Repression and activation of promoter-bound RNA polymerase activity by gal repressor. J Mol Biol. 1997;272:293–300. doi: 10.1006/jmbi.1997.1221. [DOI] [PubMed] [Google Scholar]
- Confalonieri F, Duguet M. A 200-amino acid ATPase module in search of a basic function. BioEssays. 1995;17:639–650. doi: 10.1002/bies.950170710. [DOI] [PubMed] [Google Scholar]
- Gaal T, Bartlett MS, Ross W, Turnbough CL, Jr, Gourse RL. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science. 1997;278:2092–2097. doi: 10.1126/science.278.5346.2092. [DOI] [PubMed] [Google Scholar]
- Gross CA, Lonetto M, Losick R. Sigma factors. In: Yamamoto K, McKnight S, editors. Transcriptional regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 129–176. [Google Scholar]
- Guo Y, Gralla JD. DNA-binding determinants of sigma-54 as deduced from libraries of mutants. J Bacteriol. 1997;179:1239–1245. doi: 10.1128/jb.179.4.1239-1245.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Jovanovic G, Weiner L, Model P. Identification, nucleotide sequence, and characterization of PspF, the transcriptional activator of the Escherichia coli stress-induced psp operon. J Bacteriol. 1996;178:1936–1945. doi: 10.1128/jb.178.7.1936-1945.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustu S, Santero E, Keener J, Popham D, Weiss D. Expression of σN (ntrA)-dependent genes is probably united by a common mechanism. Microbiol Rev. 1989;53:367–376. doi: 10.1128/mr.53.3.367-376.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Hoover TR. Protein Cross-linking studies suggest that Rhizobium meliloti C-4-dicarboxylic acid transport protein-D, a sigma(54)-dependent transcriptional activator, interacts with sigma(54)-subunit and the beta-subunit of RNA-polymerase. Proc Natl Acad Sci. 1995;92:9702–9706. doi: 10.1073/pnas.92.21.9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross CA. The σ70 family: Sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick MJ. In a class of its own—the RNA polymerase sigma factor σ54 (σN) Mol Microbiol. 1993;10:903–909. doi: 10.1111/j.1365-2958.1993.tb00961.x. [DOI] [PubMed] [Google Scholar]
- Morett E, Buck M. In vivo studies on the interaction of RNA polymerase-σ54 with the Klebsiella pneumoniae and Rhizobium meliloti nifH promoters. J Mol Biol. 1989;210:65–77. doi: 10.1016/0022-2836(89)90291-x. [DOI] [PubMed] [Google Scholar]
- Morris L, Cannon W, Claverie-Martin F, Austin S, Buck M. DNA distortion and nucleation of local DNA unwinding within sigma-54 (σN) holoenzyme closed promoter complexes. J Biol Chem. 1994;269:11563–11571. [PubMed] [Google Scholar]
- Oguiza JA, Buck M. DNA-binding domain mutants of sigma-N (σN, σ54) defective between closed and stable open promoter complex formation. Mol Microbiol. 1997;26:655–664. doi: 10.1046/j.1365-2958.1997.5861954.x. [DOI] [PubMed] [Google Scholar]
- Popham D, Szesto D, Keener J, Kustu S. Function of a bacterial activator protein that binds to transcriptional enhancers. Science. 1989;243:629–635. doi: 10.1126/science.2563595. [DOI] [PubMed] [Google Scholar]
- Popham D, Keener J, Kustu S. Purification of the alternative sigma factor, sigma 54, from Salmonella typhimurium and characterization of sigma 54-holoenzyme. J Biol Chem. 1991;266:19510–19518. [PubMed] [Google Scholar]
- Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- Rippe K, Mucke N, Schulz A. Association states of the transcription activator protein NtrC from E. coli determined by analytical ultracentrifugation. J Mol Biol. 1998;278:915–933. doi: 10.1006/jmbi.1998.1746. [DOI] [PubMed] [Google Scholar]
- Sasse-Dwight S, Gralla JD. Probing the Escherichia coli glnALG upstream activation mechanism in vivo. Proc Natl Acad Sci. 1988;85:8934–8938. doi: 10.1073/pnas.85.23.8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse-Dwight S, Gralla JD. Role of eukaryotic-type functional domains found in the prokaryotic enhancer receptor factor σ54. Cell. 1990;62:945–954. doi: 10.1016/0092-8674(90)90269-k. [DOI] [PubMed] [Google Scholar]
- Sundaresan V, Jones JD, Ow DW, Ausubel FM. Klebsiella pneumoniae nifA product activates the Rhizobium meliloti nitrogenase promoter. Nature. 1983;301:728–732. doi: 10.1038/301728a0. [DOI] [PubMed] [Google Scholar]
- Swaffield JC, Melcher K, Johnston SA. A highly conserved ATPase protein as a mediator between acidic activation domains and the TATA-binding protein. Nature. 1995;374:88–91. doi: 10.1038/374088a0. [DOI] [PubMed] [Google Scholar]
- Wang JT, Syed A, Hsieh M, Gralla JD. Converting Escherichia coli RNA polymerase into an enhancer-responsive enzyme: Role of an NH2-terminal leucine patch in σ54. Science. 1995;270:992–994. doi: 10.1126/science.270.5238.992. [DOI] [PubMed] [Google Scholar]
- Wang JT, Gralla JD. The transcription initiation pathway of sigma 54 mutants that bypass the enhancer protein requirement. Implications for the mechanism of activation. J Biol Chem. 1996;271:32707–32713. doi: 10.1074/jbc.271.51.32707. [DOI] [PubMed] [Google Scholar]
- Wang JT, Syed A, Gralla JD. Multiple pathways to bypass the enhancer requirements of sigma 54 RNA polymerase: Roles for DNA and protein determinants. Proc Natl Acad Sci. 1997a;94:9538–9543. doi: 10.1073/pnas.94.18.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-Y, Kolb A, Cannon W, Buck M. Nucleoprotein complex formation by the enhancer binding protein NifA. Nucleic Acids Res. 1997b;25:3478–3485. doi: 10.1093/nar/25.17.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel A, Kustu S. The bacterial enhancer-binding protein NtrC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes & Dev. 1995;9:2042–2052. doi: 10.1101/gad.9.16.2042. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Batut J, Klose KE, Keener J, Kustu S. The phosphorylated form of the enhancer-binding protein NtrC has an ATPase activity that is essential for activation of transcription. Cell. 1991;67:155–167. doi: 10.1016/0092-8674(91)90579-n. [DOI] [PubMed] [Google Scholar]
- Wilson C, Dombroski AJ. Region I of σ70 is required for efficient isomerization and initiation of transcription by Escherichia coli RNA polymerase. J Mol Biol. 1997;267:60–74. doi: 10.1006/jmbi.1997.0875. [DOI] [PubMed] [Google Scholar]
- Wong C, Tintut Y, Gralla JD. The domain structure of sigma-54 as determined by analysis of a set of deletion mutants. J Mol Biol. 1994;236:81–90. doi: 10.1006/jmbi.1994.1120. [DOI] [PubMed] [Google Scholar]
- Wyman C, Rombel I, North AK, Bustamante C, Kustu S. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science. 1997;275:1658–1661. doi: 10.1126/science.275.5306.1658. [DOI] [PubMed] [Google Scholar]