

Cellular prion protein neuroprotection against kainate is due to its ability to modulate glutamate receptor 6/7–mediated neurotransmission and JNK3 pathway activation.
Abstract
Cellular prion protein (PrPC) is a glycosyl-phosphatidylinositol–anchored glycoprotein. When mutated or misfolded, the pathogenic form (PrPSC) induces transmissible spongiform encephalopathies. In contrast, PrPC has a number of physiological functions in several neural processes. Several lines of evidence implicate PrPC in synaptic transmission and neuroprotection since its absence results in an increase in neuronal excitability and enhanced excitotoxicity in vitro and in vivo. Furthermore, PrPC has been implicated in the inhibition of N-methyl-d-aspartic acid (NMDA)–mediated neurotransmission, and prion protein gene (Prnp) knockout mice show enhanced neuronal death in response to NMDA and kainate (KA). In this study, we demonstrate that neurotoxicity induced by KA in Prnp knockout mice depends on the c-Jun N-terminal kinase 3 (JNK3) pathway since Prnpo/oJnk3o/o mice were not affected by KA. Pharmacological blockage of JNK3 activity impaired PrPC-dependent neurotoxicity. Furthermore, our results indicate that JNK3 activation depends on the interaction of PrPC with postsynaptic density 95 protein (PSD-95) and glutamate receptor 6/7 (GluR6/7). Indeed, GluR6–PSD-95 interaction after KA injections was favored by the absence of PrPC. Finally, neurotoxicity in Prnp knockout mice was reversed by an AMPA/KA inhibitor (6,7-dinitroquinoxaline-2,3-dione) and the GluR6 antagonist NS-102. We conclude that the protection afforded by PrPC against KA is due to its ability to modulate GluR6/7-mediated neurotransmission and hence JNK3 activation.
INTRODUCTION
The abnormal processing of cellular prion protein (PrPC) gives rise to PrPSC, or pathogenic prion, which is the etiologic agent of several transmissible spongiform encephalopathies (reviewed in Aguzzi et al., 2007; Aguzzi and Calella, 2009; Linden et al., 2008). These devastating encephalopathies are characterized by severe histological changes, including extensive neuronal death, reactive gliosis, and neuroinflammation, concomitant to the accumulation of the aggregated misfolded form of PrPC in affected brains. Although necessary for prion propagation and infectivity (Weissmann et al., 1994), the repertoire of physiological roles of PrPC remains to be fully determined. Indeed, PrPC has been implicated in a wide range of neural functions, such as copper homeostasis (Brown et al., 1997, 1998; Vassallo and Herms, 2003), neurotransmission (Maglio et al., 2004; Prestori et al., 2008; Rangel et al., 2009), stem-cell proliferation and differentiation (Steele et al., 2006), malignant glioma proliferation (Erlich et al., 2007), neurite outgrowth (Chen et al., 2003; Santuccione et al., 2005), and, more recently, putative cross-talk between neurodegenerative diseases (Lauren et al., 2009; Gavin et al., 2010; Gunther and Strittmatter, 2010).
Early in vitro studies with prion protein gene Prnpo/o mice (Zürich I) (Bueler et al., 1992) showed that cultured Prnpo/o neurons were more prone to die than Prnp+/+ neurons after serum withdrawal or other treatments (Kuwahara et al., 1999). Subsequent studies (Brown et al., 2002; Rambold et al., 2008; Aude-Garcia et al., 2011) indicated that PrPC might play a neuroprotective function in the context of excitotoxic lesions. Indeed, it was demonstrated in rats that Prnp is overexpressed in ischemic brains and protects cortical neurons after ischemia (McLennan et al., 2004; Shyu et al., 2005; Spudich et al., 2005; Weise et al., 2006; Mitteregger et al., 2007). In fact, cells overexpressing PrPC show increased toxicity after endoplasmic reticulum stress (Anantharam et al., 2008). Neuroprotective effects of PrPC after central nervous system insults have been associated with both extracellular and intracellular effectors (e.g., the stress-induced-phosphoprotein 1) (Zanata et al., 2002) and are mediated by PI3K-mTOR signaling (Martins et al., 2010; Roffe et al., 2010). However, although it is involved in Alzheimer's disease (Lauren et al., 2009), PrPC does not act as a major modulator in a panel of neurodegenerative disease models (Steele et al., 2009).
In neurons, PrPC has been localized in axons at the synapse (Fournier et al., 2000; Fournier, 2008), which might be consistent with reports of deficits in neurotransmission in Prnp knockout mice (Maglio et al., 2004, 2006; Khosravani et al., 2008; Prestori et al., 2008; Rangel et al., 2009). Indeed, PrPC knockout mice are more prone to cell death after kainate (KA) injections than are Prnp+/+ animals (Walz et al., 1999; Rangel et al., 2007, 2009). In addition, Prnpo/o neurons are also highly susceptible to acute N-methyl-d-aspartic acid (NMDA) or KA treatment in hippocampal slices (Rangel et al., 2007; Khosravani et al., 2008). These findings suggest that PrPC can modulate signaling cascades by activating stress-protective pathways, or by inhibiting cytotoxic pathways (reviewed in Westergard et al., 2007; Nicolas et al., 2009). Although the exact mechanism(s) are unknown, the neuroprotective function may be associated with differential expression of glutamate receptor subunits or the modulation of ligand/receptor affinities (Maglio et al., 2004; Khosravani et al., 2008; Rangel et al., 2009). In particular, it remains to be established whether PrPC alters KA receptor–mediated neurotransmission at the neuron membrane. In fact, PrPC-binding proteins include, among others, membrane receptors (Rutishauser et al., 2009), and two recent studies reported cell-specific effects of glutamate and the role of PrPC in these processes. Indeed, group I metabotropic glutamate receptors are involved in the transduction of cellular signals triggered by PrPC, which modulates neurite outgrowth (Beraldo et al., 2011). In addition, in NMDA-mediated toxicity, the protective role of PrPC is mediated by the silencing of NMDA receptors containing N-methyl-d-aspartate receptor subtype 2 (NR2D) subunits (Khosravani et al., 2008).
In a previous study, we reported the increased presence of phosphorylated c-Jun N-terminal kinase 3 (JNK) in dying pyramidal neurons in the CA1 of the hippocampus of Prnp knockout mice after KA treatment (Rangel et al., 2007). Here we show that KA-induced excitotoxicity in Prnp knockout mice depends on JNK3 activity, by developing a double-knockout mouse lacking PrPC and JNK3 expression and analyzing it by using gene expression and pharmacological approaches. Furthermore, we propose a mechanism by which PrPC regulates the KA receptor function through interaction, at the postsynaptic level, with the glutamate receptor 6/7 (GluR6/7) and the postsynaptic density 95 protein (PSD-95), which in turn modulates JNK3 activity.
RESULTS
Enhanced susceptibility to KA-induced seizures in Prnp knockout mice and its reversion in Prnpo/oJnk3o/o mice
The thresholds for onset of seizure behavior in response to identical intraperitoneal KA injections (6 mg/kg body weight) in the four mouse genotypes Prnpo/oJnk3+/+, Prnp+/+Jnk3o/o, Prnp+/+Jnk3+/+, and Prnpo/oJnk3o/o were analyzed. After the first KA injection, Prnpo/oJnk3+/+ animals developed profound hypoactivity and immobility (grades I–II). After successive injections, hyperactivity (grade III) and scratching (grade IV) were often observed. Some mice progressed to a loss of balance control (grade V) and further chronic whole-body convulsions (grade VI). The bouncing activity commonly referred to as “popcorn” behavior was included in grade VI. After the behavioral study, mice were numbered and kept in separate boxes until histological or biochemical studies; see later discussion. Twelve hours after KA treatment, Prnp+/+Jnk3+/+ and Prnp+/+Jnk3o/o animals showed normal behavior. KA-treated Prnpo/oJnk3o/o mice displayed hypoactivity, immobility, and sensitivity to external stimuli (e.g., box handling) (Table 1).
TABLE 1:
Effect of KA-induced status epilepticus and death in Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, Prnpo/oJnk3o/o, and Prnp+/+Jnk3o/o mice.
| Genotype. | Animal | Number of seizures | Number of blinkings | Behavioral stages | Prioritary stage |
|---|---|---|---|---|---|
| Prnp+/+Jnk3+/+ | 1 | 1 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 2 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 3 | 1 | 1 | I–II | II |
| Prnp+/+Jnk3+/+ | 4 | 1 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 5 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 6 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 7 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 8 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 9 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 10 | 1 | 0 | I–III | II |
| Prnp+/+Jnk3+/+ | 11 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 12 | 0 | 0 | I–II | II |
| Prnpo/oJnk3+/+ | 13 | 8 | 4 | I–VI | † |
| Prnpo/oJnk3+/+ | 14 | 5 | 3 | I–VI | IV–VI |
| Prnpo/oJnk3+/+ | 15 | 6 | 4 | I–VI | V–VI |
| Prnpo/oJnk3+/+ | 16 | 6 | 3 | I–VI | † |
| Prnpo/oJnk3+/+ | 17 | 5 | 2 | I–VI | V–VI |
| Prnpo/oJnk3+/+ | 18 | 6 | 3 | I–VI | † |
| Prnpo/oJnk3+/+ | 19 | 6 | 4 | I–VI | V–VI |
| Prnpo/oJnk3+/+ | 20 | 5 | 2 | I–VI | IV–VI |
| Prnpo/oJnk3+/+ | 21 | 4 | 2 | I–VI | V–VI |
| Prnpo/oJnk3+/+ | 22 | 8 | 5 | I–VI | V–VI |
| Prnpo/oJnk3+/+ | 23 | 6 | 3 | I–VI | IV–VI |
| Prnpo/oJnk3+/+ | 24 | 7 | 5 | I–VI | V–VI |
| Prnpo/oJnk3o/o | 25 | 1 | 1 | I–II | II |
| Prnpo/oJnk3o/o | 26 | 2 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 27 | 1 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 28 | 0 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 29 | 0 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 30 | 0 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 31 | 0 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 32 | 2 | 2 | I–III | II |
| Prnpo/oJnk3o/o | 33 | 0 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 34 | 1 | 1 | I–II | II |
| Prnpo/oJnk3o/o | 35 | 0 | 0 | I–II | II |
| Prnpo/oJnk3o/o | 36 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 37 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 38 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 39 | 0 | 0 | I–II | II |
| Prnp+/+Jnk3+/+ | 40 | 0 | 0 | I–II | II |
See Results for stage classification.
Prnpo/oJnk3+/+ mice were highly susceptible to KA, showing a greater number (from five to eight) of severe seizures (grade VI). In addition, they maintained grade IV–VI seizures for 2–3 h after the first episode, whereas and Prnp+/+Jnk3+/+ and Prnpo/oJnk3o/o mice displayed only grade III seizure. Furthermore, three Prnpo/oJnk3+/+ animals died during the experiments. These data corroborate previous results (Walz et al., 1999; Rangel et al., 2007, 2009) indicating increased sensitivity to KA in Prnp knockout mice. We established that the minimal concentration of KA required to induce seizures in the Prnp+/+Jnk3+/+ animals was 35–40 mg/kg body weight, which is similar to that required for Prnp+/+Jnk3+/o. At this concentration, all Prnpo/oJnk3+/+ animals died shortly after a second injection. These results suggested an active role of JNK3 in Prnpo/o susceptibility to KA. Indeed, Prnp+/+Jnk3o/o and Prnp+/+Jnk3+/o mice were not affected by KA treatment as described (Yang et al., 1997).
Decreased seizures in Prnpo/oJnk3o/o mice correlated with lower number of dying cells in the hippocampus
Protein expression was analyzed by Western blot (Figure 1A), which showed that PrPC expression was similar in Prnp+/+Jnk3+/+ and Prnp+/+Jnk3o/o mice. In addition, JNK3 expression was similar in Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ mice, and neither of these proteins was detected in the double-knockout mice, as expected (Figure 1A). Next we analyzed in more detail the time course of the seizure score after KA injection (Figure 1B). Prnpo/oJnk3+/+ mice showed maximum scores (V–VI) between 90 and 180 min after the first KA injection. To determine whether the severity of seizure observed in Prnpo/oJnk3+/+ correlates with neuronal loss and reactive glial changes in the hippocampus after KA injection, we carried out several histochemical and immunohistochemical analyses (Figure 1, C and D).
FIGURE 1:

KA-dependent sensitivity, seizure behavior, neurotoxicity and apoptosis in the different genotypes studied. (A) Western blot of JNK3, PrPC, and tubulin in protein extract from the hippocampi of the different mouse strains used in this study (Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, Prnpo/oJnk3o/o, and Prnp+/+Jnk3o/o); tubulin was used as a loading control. (B) Comparison of seizure responses in littermates of Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, Prnpo/oJnk3o/o, and Prnp+/+Jnk3o/o mice to intraperitoneal injection of KA (6 mg/kg body weight) or 0.1 M PBS. KA-injection timing is indicated below the graph. Seizures were scored as indicated in Materials and Methods. Eight mice in each group were observed and scored to determinate the time-dependent seizure score. (C) Photomicrographs showing examples of the pattern of Fluoro-Jade B and DAPI staining of hippocampal region of Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice after 24 h of KA treatment. Dying cells positive for Fluoro-Jade B are located in the pyramidal cell layer of Prnpo/oJnk3+/+ (arrows). (D) Examples of TUNEL-positive cells in CA1–CA3 hippocampal regions of Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice after 24 h of KA treatment. (E) Quantification of Fluoro-Jade B and TUNEL-positive cells in the CA1 and CA3 regions of the hippocampus in Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice after 24 h of KA treatment. Results are obtained from nine mice per genotype. DG, dentate gyrus; CA1–3, hippocampal regions 1 and 3; gl, granule cell layer; h, hilus; ml, molecular layer; sl, stratum lucidum; slm, stratum lacunosum-moleculare; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bar, C, top, 200 μm; C, bottom, 100 μm; D, 100 μm. Asterisks in E indicate statistical significance (***p < 0.001, ANOVA test).
First, we analyzed the pattern of neurodegeneration in the Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice 24 h after KA treatment by using Fluoro-Jade B staining (Schmued and Hopkins, 2000; Figure 1C). Numerous Fluoro-Jade B–positive pyramidal neurons were observed in the CA1 and CA3 regions of Prnpo/oJnk3+/+ hippocampus. In the other genotypes Fluoro-Jade B labeling was rare and generally pale (Figure 1C). To further corroborate the death of these neurons, we applied in parallel sections the in situ nick-translation technique with biotinylated-UTP to determine the DNA fragmentation level (TUNEL technique, by using ApopTag kit; Heatwole, 1999; Figure 1D). Distribution of TUNEL-positive nuclei largely correlated with the pattern of Fluoro-Jade B labeling observed in the CA1 and CA3 of Prnpo/oJnk3+/+ mice (Figure 1, D and E). Cell counts showed statistical significance (see Materials and Methods for details) when comparing data of Prnpo/oJnk3+/+ mice with any of the other two genotypes.
Acute down-regulation of PrPC expression in Prnp+/+Jnk3+/+ primary neuronal cultures increased susceptibility to KA
We examined whether the neurotoxic effects of KA are modified when PrPC expression levels are acutely altered in Prnp+/+Jnk3+/+ neurons. Thus, Prnp+/+Jnk3+/+ primary cortical neurons were infected with a lentivirus carrying a small interfering RNA (siRNA) interference sequence for Prnp. Cells infected with lentivirus showed an 81% decrease in PrPC expression (Figure 2A). After exposure to KA, cells infected with lentivirus showed a statistically significant 29% decrease in survival (Figure 2B) compared with parallel enhanced green fluorescent protein (eGFP)–infected cultures (Figure 2B). Thus, although similar data were previously described in neuroblastoma cells (N2A) using interference RNA against Prnp (Rangel et al., 2007), this is the first observation of KA-mediated cell death after acute PrPC down-regulation in Prnp+/+Jnk3+/+ neurons. This finding reinforces the notion that decreased expression of PrPC compromises neuronal viability after acute KA treatment.
FIGURE 2:
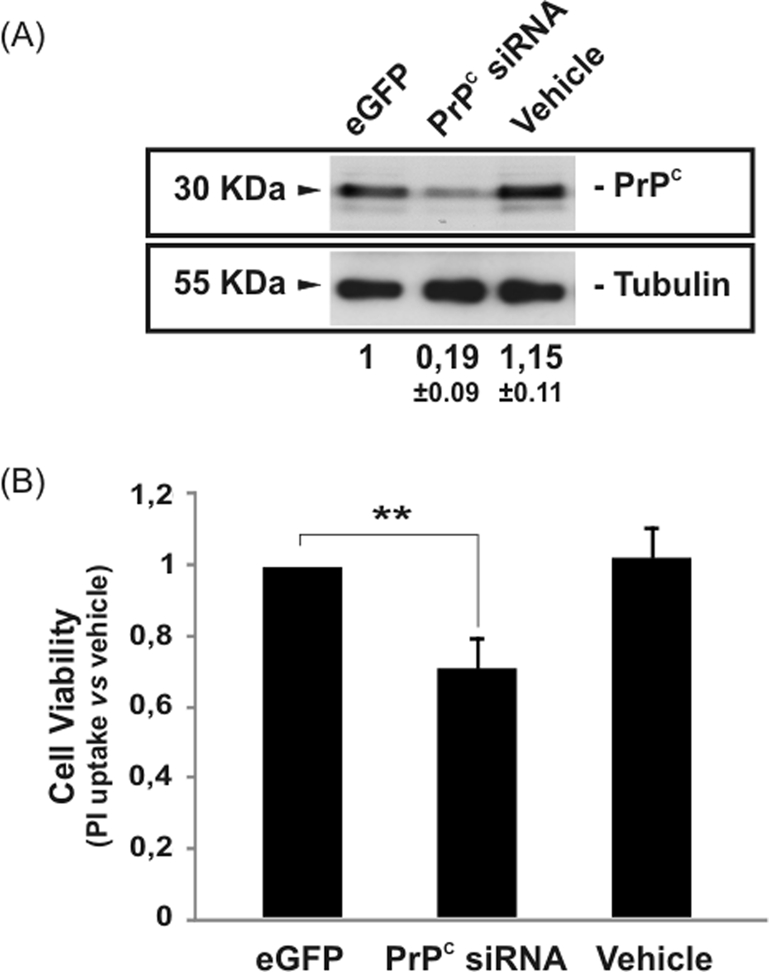
Decrease in PrPC expression–enhanced KA neurotoxicity in primary hippocampal neurons. Primary hippocampal neurons were infected with lentivirus containing eGFP or siRNA for Prnp for 24 h. Next, neurons were treated with 150 μM KA for 8 h, and cell viability was analyzed after further 24 h with the WST-1 method as indicated in Materials and Methods. (A) Western blot for PrPC and tubulin expression after lentiviral infection with eGFP and siRNA Prnp. Note the decrease of the PrPC expression after lentiviral infection vs. vehicle and eGFP. (B) Histogram illustrating cell viability infection and KA exposure. No differences were observed between vehicle and eGFP. Data are expressed as fold increase ± SEM of three independent experiments. Asterisks in B indicate statistical significance (**p < 0.01, ANOVA test).
Differential activation of JNK3 and extracellular regulated kinases 1/2 in Prnpo/oJnk3+/+ mouse hippocampus treated with KA
Because genetic deletion of Jnk3 on a Prnpo/o background overcomes the intrinsic susceptibility to KA of PrPC-deficient mice, we next developed several experiments to identify changes in JNK3 activation in the hippocampus after KA treatment (Figure 3). Mice were injected with KA as described and killed 6 h later (see Materials and Methods for details). Activation of extracellular regulated kinase (ERK) 1/2, JNK3, c-Jun, and p53 was analyzed in protein samples from the hippocampus of treated mice by Western blot and compared with vehicle-injected mice. In the absence of KA, ERK1/2 was activated in Prnpo/oJnk3+/+ and Prnpo/oJnk3o/o hippocampus compared with Prnp+/+Jnk3+/+ mice (Figure 3A). These data corroborate the reported data of ERK1/2 activation in Prnp knockout mice (Brown et al., 2002). In parallel to the phosphorylation of ERK1/2 in all genotypes after KA treatment, an increase in p-JNK3 and p-c-Jun Ser63 was observed in Prnpo/oJnk3+/+ hippocampus (Figure 3A). This phosphorylation was undetectable in vehicle-treated mice and in Prnp+/+Jnk3+/+, as well as in Prnpo/oJnk3o/o mice (Figure 3A) and Prnp+/+Jnk3o/o (unpublished data), as expected. Total c-Jun protein was slightly increased in Prnp+/+Jnk3+/+ but strongly increased in both Prnpo/oJnk3+/+ and Prnpo/oJnk3o/o after KA treatment (Figure 3A). Finally, p53, which was recently reported to be activated in a JNK-dependent manner in dying hippocampal neurons after glutamate and/or KA (Choi et al., 2011), was phosphorylated at Ser-15 and activated in Prnpo/oJnk3+/+ hippocampus after KA treatment.
FIGURE 3:

KA-induced neurotoxicity in Prnp knockout hippocampal mouse correlates with JNK3 activation. (A) Western blot for p-JNK, JNK3, p-ERK1/2, ERK1/2, p-c-Jun, c-Jun, p-p53, and tubulin from hippocampus extracts of Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice after 6 h of KA treatment (6 mg/kg body weight). KA-induced neurotoxicity in the Prnpo/oJnk3+/+ hippocampus is cell-specific and dependent on JNK3 activity. (B) Photomicrograph illustrating the pattern of p-ERK1/2 in the different phenotypes after 24 h of KA treatment. Notice the relevant increase of p-ERK1/2 in Prnpo/oJnk3+/+ and Prnpo/oJnk3o/o hippocampus. (C) High magnification of the CA1 region of hippocampus from Prnpo/oJnk3+/+, after 24 h of KA treatment, immunostained with p-ERK1/2 and GFAP antibodies. Note that numerous cells are double labeled (arrows). (D) Photomicrograph illustrating examples of the CA1 region of the three genotypes analyzed after 24 h of KA treatment and immunostained for GFAP. Levels of reactive glia are not completely abolished by the genetic deletion of Jnk3. Abbreviations as in Figure 1. Scale bars, B, 150 μm; C, 100 μm; and D, 100 μm.
These results suggest that signaling pathways other than the JNK3 pathway are activated after KA treatment. Indeed, we determined the cellular expression of ERK1/2 in detail in treated mice (Figure 3, B and C). p-ERK1/2 staining was strong in hippocampal mossy fibers at the stratum lucidum in Prnpo/oJnk3+/+ and Prnpo/oJnk3o/o, with low levels in Prnp+/+Jnk3+/+, after 24 h of KA treatment. Double-labeling techniques with glial fibrillary acidic protein (GFAP) and p-ERK1/2 revealed a second group on p-ERK1/2–stained cells at CA1 and CA3 regions (Figure 3C). However, detailed observation of astroglial cells in these mutants revealed an increased reactivity of astrocytes in Prnpo/oJnk3+/+ hippocampus (relevant GFAP content and thick glial expansions), in contrast to moderate reactivity in Prnpo/oJnk3o/o and absence in Prnp+/+Jnk3+/+ mice (Figure 3D). Thus these data indicate that most p-ERK1/2 staining observed in Prnpo/oJnk3+/+ and Prnpo/oJnk3o/o after KA treatment is mainly associated with mossy fiber staining with the participation of GFAP-reactive cells.
The JNK3 pathway is implicated in c-fos and c-jun activation after glutamate treatment (Brami-Cherrier et al., 2007). To discriminate the particular contribution of JNK3 to c-jun and c-fos expression in our experiments, we performed quantitative real-time PCR (RT-qPCR; Figure 4). Data obtained demonstrate that both c-jun and c-fos were overexpressed in the hippocampus of Prnpo/oJnk3+/+ mice 6 h after KA administration (4.10- and 49.2-fold increase, respectively; Figure 4A). Deletion of Jnk3 in Prnpo/o mice offset the c-jun and c-fos increase by 34 and 25% respectively, compared with Prnpo/oJnk3+/+ levels. These results support the idea that other KA-activated pathways are the main contributors to the overexpression of both early genes. To examine this hypothesis, we analyzed the differential expression of cyclooxygenase-2 (cox-2), a molecular target of JNK activation in neurodegeneration and inflammation activated by activator protein-1 (AP-1; Hunot et al., 2004). RT-qPCR of cox-2 revealed statistically significant overexpression in the Prnpo/oJnk3+/+ hippocampus after KA treatment. Furthermore, stronger decrease of cox-2 was observed in Prnpo/oJnk3o/o (60% compared with those reported by c-jun and c-fos) (Figure 4B). The results indicate that the absence of Jnk3 is not the only factor mediating the early changes in gene expression mediated by KA.
FIGURE 4:

RT-qPCR of (A) c-jun and c-fos and (B) cox-2 from hippocampal RNA extracts of Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice treated with KA (6 mg/kg body weight) or buffer for 6 h. Data (mean ± SEM) represent mean fold induction obtained in three independent experiments. GAPDH was used as the housekeeping gene. Data are referred with Prnp+/+Jnk3+/+ in each histogram in the absence or presence of KA. Data are expressed as fold change ± SEM. Asterisks in A and B indicate statistical significance (** p < 0.01, *** p < 0.001, ANOVA test).
Pharmacological inhibition of JNK in Prnpo/oJnk3+/+ hippocampal slices overcame the neurotoxic effect of KA
To ascertain whether neurotoxic differences are cell specific or associated with an increased glutamatergic input from the entorhinal cortex, we performed a parallel study in isolated hippocampal slices (Figure 5). Prnpo/oJnk3+/+ and Prnp+/+Jnk3+/+ hippocampal slices were treated with KA or phosphate-buffered saline (PBS), following the addition (when indicated) of two different JNK inhibitors: the pharmacological inhibitor SP600125 and the peptide TAT-JIP, a specific inhibitor derived from the minimal JNK-binding region of the scaffold protein JNK-interacting protein 1 (JIP-1) coupled to the short cell-permeable TAT sequence (Bonny et al., 2001; Figure 5A). After propidium iodide (PI) incubation, we observed few randomly distributed, labeled cells in the CA1–CA3 of Prnp+/+Jnk3+/+–derived hippocampal slices. However, Prnpo/oJnk3+/+ slices showed a statistically significant 71% increase of PI fluorescence compared to Prnp+/+Jnk3+/+ cultures (Figure 5B). The inhibition of JNK activity with SP600125 and TAT-JIP in Prnpo/oJnk3+/+–derived slices decreased the fluorescence levels by 54 and 42%, respectively, compared with parallel KA-treated slices without the inhibitors (Figure 5, B and C). These data indicate that increased cell death of hippocampal Prnpo/oJnk3+/+ neurons is associated with intrinsic synaptic properties.
FIGURE 5:

KA-enhanced neurotoxicity in Prnp knockout organotypic slices correlates with JNK3 activation. (A) Schematic diagram illustrating the experimental procedure. The hippocampi of Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ animals were dissected and cultured in transwells until drug treatment and afterward analyzed by PI uptake. (B) Examples of PI uptake in organotypic hippocampal slice cultures from Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ mice treated with 0.1 M PBS or 150 μM KA for 2 h in the presence or absence of JNK inhibitors SP600125 (20 μM) and TAT-JIP (10 μM) as indicated. Twenty-four hours after treatment slices were incubated with PI. Note the incorporation of PI in dying cells in KA-treated slices from Prnpo/oJnk3+/+ mice. (C) Histogram illustrating quantitative results of A. Histograms represent the mean ± SEM of three independent experiments. Asterisks in C indicate statistical significance (***p < 0.001, ANOVA test). Abbreviations as in Figure 1. Scale bars, B, 100 μm.
Coimmunoprecipitation of PrPC with PSD-95 and GluR6/7 in the PSD fractions
To investigate the participation of PrPC in the neurotoxic effects of KA, we examined whether PrPC exerts its neuroprotective function by modulating the intracellular signaling triggered by KA receptor after ligand binding. Because neuronal depolarization mediated by KA requires the interaction of the GluR6/7 subunit of KA receptor with PSD-95 and MLK3 to form a trimeric complex (Pei et al., 2006; Li et al., 2010), we analyzed biochemically the distribution of GluR6/7 and PrPC in the Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o hippocampus and whether changes in JNK3 alter the normal distribution of PrPC. Enriched PSD fractions were isolated from the hippocampus of Prnp+/+Jnk3+/+, Prnpo/oJnk3+/+, and Prnpo/oJnk3o/o mice 6 h after administration of KA and processed for GluR6/7, PSD-95, and PrPC by Western blot (Figure 6A). Correct purification of the PSD fractions was corroborated since the percentage of PSD-95 in this fraction was 91.6% of total PSD-95. Results revealed no changes in subcellular distribution of these proteins among genotypes. As expected, PrPC was detected in all cell compartments but enriched in PSD fractions, where GluR6/7 and PSD-95 were also found.
FIGURE 6:

PrPC interacts with PSD-95 and glutamate receptor 6/7. (A) Hippocampus from KA-injected Prnp+/+Jnk3+/+, Prnpo/o Jnk3+/+, and Prnpo/oJnk3o/o mice was fractionated by differential centrifugation in order to purify PSD fraction. Fractions were separated by SDS–PAGE, followed by immunoblotting with the indicated antibodies. (B) Extracts from total hippocampus or hippocampal PSD fraction from Prnp+/+Jnk3+/+ mouse were immunoprecipitated using two PrPC antibodies (6H4 and SAF61). Immunoprecipitated samples were separated by SDS–PAGE, followed by immunoblotting with the indicated antibodies. (C) Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ mice were injected with KA (6 mg/kg body weight) or PBS and analyzed for 4 h. Hippocampi were dissected, and cellular extracts were immunoprecipitated with control and anti–PSD-95 antibodies. Immunoprecipitated samples were developed by Western blot with the anti–GluR6/7 antibody. Densitometry values are standardized with Prnp+/+Jnk3+/+ in untreated cultures, and quantification was represented as fold change ± SEM. Notice the increase of GluR6/7 labeling after PSD immunoprecipitation in Prnpo/oJnk3+/+ after KA treatment.
These results suggested a putative interaction between GluR6/7 and PrPC. To explore this possibility, we performed coimmunoprecipitation experiments from whole Prnp+/+Jnk3+/+ hippocampus protein extracts and PSD fractions using two different PrPC antibodies (6H4 and SAF61; see Materials and Methods; Figure 6B). Results indicated that PrPC interacts with PSD-95 and GluR6/7 both in whole-hippocampus extracts and in purified PSD. As expected, both interactions were stronger in the PSD fraction (Figure 6B). Taken together, these results implicate PrPC in the binding of PSD-95 with the KA receptor containing GluR6/7. This would allow the formation of the ternary complex (GluR6/7–PSD-95–MLK3), which in turn activates the JNK3 pathway. Thus, to determine this, we immunoprecipitated PSD-95 in hippocampal protein extracts from Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ mice to measure the amount of GluR6/7 coimmunoprecipitated in the presence or absence of KA (Figure 6C). Immunoblots showed that the interaction between GluR6/7 and PSD-95 was higher in the hippocampus of Prnpo/oJnk3+/+ compared with Prnp+/+Jnk3+/+ mice in the presence of KA (1.01 vs. 2.13, respectively; Figure 6C). Taken together, these data indicate that, in the absence of PrPC, KA treatment increased the binding of GluR6/7-containing KA receptors to PSD-95, which correlates with increased activation of JNK3.
Increased KA-mediated neurotoxicity in Prnpo/o mice was reversed by 6,7-dinitroquinoxaline-2,3-dione and NS-102
To examine the role of GluR6/7 in KA-induced neurotoxicity in Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ mice, we injected the AMPA/KA inhibitor 6,7-dinitroquinoxaline-2,3-dione (DNQX) and the GluR6 antagonist NS-102 (Verdoorn et al., 1994) prior to the intraperitoneal injection with KA (Figure 7). DNQX inhibits AMPA/KA receptors in vivo (Alford and Grillner, 1990) and inhibits JNK3 activation after excitotoxic insults (Tian et al., 2005), and NS-102 is a good functional antagonist of GluR6 in vivo in response to excitotoxic insults (Tian et al., 2005; Hu et al., 2008; Chen et al., 2009). First, we counted the seizures and the blinking episodes in treated mice (Figure 7A). Pharmacological treatments indicate that preinjection with DNQX and, more effectively, with NS-102 reduced the number of all of the episodes counted in Prnpo/oJnk3+/+ mice. Prnp+/+Jnk3+/+ mice were almost insensitive to KA, as described earlier (Figure 7A). Again, both treatments reduced the number of Fluoro-Jade B–positive cells in the hippocampus (Figure 7B) and the presence of p-c-Jun in Prnpo/oJnk3+/+ hippocampus in Western blots (Figure 7C). In addition, we examined the effect of DNQX and NS-102 on c-Fos expression, which was overexpressed in the hippocampus of Prnpo/oJnk3+/+ mice after KA injections (see earlier discussion). Blockage of both AMPA/KA receptors and GluR6 prevented c-Fos overexpression in CA1 (Figure 7D) and increased immunostaining of p-ERK1/2 in mossy fibers of Prnpo/oJnk3+/+ (Figure 7E). Measure of the fluorescence in the mossy fibers with ImageJ software (square regions of interest of 75 × 75 μm, six measures per section) indicates a statistically significant reduction of 80 and 75% of p-ERK1/2 staining after DNQX and NS-102 treatments respective (Figure 7F).
FIGURE 7:

AMPA/kainate receptor inhibition decreased the KA-enhanced neurotoxicity in Prnpo/oJnk3+/+ mice. (A) Seizures and blinking events for KA-injected mice. Injections were carried out with KA (6 mg/kg body weight), PBS, and the inhibitors DNQX and NS-102 as indicated on the timeline. (B) Examples of Fluoro-Jade B and DAPI staining in hippocampal CA1 region of Prnpo/oJnk3+/+ mice 24 h after injection of KA in the presence or absence of DNQX and NS-102. (C) Western blots of p-c-Jun, c-Jun, and tubulin from the hippocampus of the animals 4 h after injection of KA in presence or absence of DNQX and NS-102. (D) Photomicrographs of c-Fos immunoreactivity in the hippocampal CA1 region of Prnpo/oJnk3+/+ mice 24 h after injection of KA in the presence or absence of DNQX and NS-102. (E) Photomicrographs of p-ERK1/2 immunoreactivity in the hippocampus of Prnpo/oJnk3+/+ mice 24 h after injection of KA in the presence or absence of DNQX and NS-102. Representative images from three different experiments are shown. (F) Histogram illustrating quantitative results of the fluorescence levels analyzed in E. Data are represented as mean ± SEM. Asterisks indicate statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001; ANOVA test). Abbreviations as in Figure 1. Scale bars, B, 100 μm; E, 200 μm.
DISCUSSION
Differential participation of ERK1/2, JNK3 kinases, and early gene expression in KA-mediated neuronal death in Prnpo/o mice
In this study we demonstrate that abolition of JNK3 activity (either genetically or pharmacologically) blocks the neurotoxic activity induced by KA in Prnpo/o mice. These data indicate that the signal for neuronal cell death in Prnp knockout mice is mainly associated with JNK3 activity. In addition, to avoid the effects of the increased oxidative phenotype in absence of Prnp (Wong et al., 2001; Brown et al., 2002), we show that an acute decrease in PrPC expression in cultured neurons from Prnp+/+Jnk3+/+ mice also increased their susceptibility to KA. In fact, absence of Jnk3 completely blocked the epileptogenic effects of KA in Prnp+/+ mice, and the original Jnk3o/o mice were characterized as being resistant to KA, with similar scores to those reported here (Yang et al., 1997; de Lemos et al., 2010).
Although basal p-ERK1/2 is higher in Prnpo/o mice (Brown et al., 2002; Chiarini et al., 2002; Nicolas et al., 2007; Rangel et al., 2007; and present results), treatment with KA induces overphosphorylation of ERK1/2, which is not affected by the absence of Jnk3. In contrast, ERK1/2 activation is abolished in Jnk3o/o mice 3 h after KA treatment (de Lemos et al., 2010). Thus it is reasonable to consider that changes in p-ERK1/2 in Prnpo/oJnk3o/o mice after KA treatment are mainly associated with the Prnpo/o background. p-ERK1/2 is expressed by mossy fibers of granule cells, as well as by reactive, GFAP-positive astrocytes, after KA treatment; in contrast, JNK activation mainly takes place in dying neurons (CA1–CA3 pyramidal neurons; Rangel et al., 2007). Our histological and biochemical analyses demonstrate that an increased number of GFAP-positive cells can be seen in the double-knockout hippocampus without relevant cell death. These data also contrast with those obtained with Jnk3o/o mice (Yang et al., 1997; de Lemos et al., 2010). Although we cannot rule out an effect of activated p-ERK1/2/GFAP–positive astrocytes on JNK-activated dying neurons, it seems that intrinsic ERK1/2 phosphorylation (which is not blocked by deletion of Jnk3) in granule cells could participate in the sustained c-jun overexpression in response to KA in a Prnpo/o background. On the other hand, c-jun was upregulated in Prnpo/oJnk3o/o mice after KA treatment, but no neurodegeneration was observed when compared with both Prnp+/+Jnk3+/+ and Prnpo/oJnk3+/+ mice in brain sections when examined by different methods (Fluoro-Jade B or TUNEL). Thus c-Jun overexpression cannot be associated with cell death and needs to be phosphorylated by JNK3 to induce cell death. Indeed, JNK3 phosphorylates c-Jun at Ser-63 and Ser-73, and abrogation of c-Jun phosphorylation using alanine mutants for c-Jun at Ser-63 and Ser-73 confers resistance to epileptic seizures and neuronal apoptosis induced by KA (Behrens et al., 1999). On the other hand, c-fos was upregulated only in dying Prnpo/oJnk3+/+ cells, and Jnk3 deletion did not totally decrease c-fos levels, suggesting its implication in different KA-signaling mechanisms. In addition, our data also implicate p53 in these results since phosphorylation of p53 follows the activity pattern of JNK3 (only activated in Prnpo/oJnk3+/+ after KA treatment). This is compatible with the recent proposal that p53 activation after glutamatergic stimulus depends on JNK activity (Choi et al., 2011).
JNK3-mediated signaling activates the AP-1 complex, which triggers the expression of early response genes such as c-fos or c-jun, as well as the up-regulation of cox-2, which is involved in the inflammatory response after KA treatment (Tu and Bazan, 2003). In fact, pharmacological inhibition or genetic deletion of cox-2, but not Cox-1, increases susceptibility to KA-induced hippocampal excitotoxicity (Toscano et al., 2008). In our study, cox-2 was up-regulated after KA treatment in Prnpo/oJnk3+/+, whereas in Prnpo/oJnk3o/o, cox-2 expression was largely reduced (up to 60%), indicating a reduction in inflammatory responses in the double-knockout mice. Indeed, a direct relation between JNK activity, c-Jun phosphorylation, and cox-2 up-regulation has been demonstrated in several models (Waetzig et al., 2005), and the up-regulation of Cox-2 mediated by JNK is necessary and required for neurodegeneration in a mouse model of Parkinson disease (Hunot et al., 2004).
A putative role of PrPC at the neuronal membrane as modulator of GluR6/7–mediated neurotransmission and JNK3 activity
GluR6 knockout and Jnk3o/o mice are resistant to KA treatment. In fact, it has been reported that GluR6 binds to PSD-95, and this binding is necessary to trigger GluR6-dependent neurotransmission (Savinainen et al., 2001) due to the formation of a stable trimeric complex GluR6–PSD-95–MLK3 (Li et al., 2010). In this study we found large colocalization of PrPC with GluR6/7 in the PSD. Moreover, Prnp deletion favors the interaction between PSD-95 and GluR6/7 in the presence of KA. Taken together, our results suggest that absence of PrPC enhances the interaction of the ternary complex PSD-95 and GluR6 and most probably MLK3 at the synapse, thus enabling GluR6-mediated intracellular signaling. This in turn leads to JNK3 activation among that of other kinases.
As mentioned, PrPC has been detected in axon and synaptic contacts, although whether its location is presynaptic or postsynaptic is not fully determined (Fournier et al., 2000; Fournier, 2008). Here we report that a fraction of PrPC localized to the PSD, where neurotransmitter receptors are located. Recent studies indicate that PrPC is a constitutive basal protector against NMDA-mediated toxicity. Deletion of the Prnp gene induces enhanced prolonged NMDA-evoked currents in hippocampal neurons, which increases neuronal excitability and glutamate excitotoxicity (Khosravani et al., 2008). The same authors described changes in some parameters associated with AMPA/KA–mediated miniature inhibitory postsynaptic currents and γ-aminobutyric acid A–mediated miniature inhibitory postsynaptic currents (Khosravani et al., 2008). These findings are in concordance with our data (Rangel et al., 2007, 2009), which demonstrated that enhanced neurotoxicity to KA in hippocampal slices of Prnpo/o mice has both a KA receptor and a NMDA receptor component. However, the molecular mechanism by which PrPC inhibits NMDA and KA receptors is unclear. PrPC may either influence agonist binding, favor the closed state of the channel, or interfere with the signal transmission, thus forming a negative modulator complex (Steele, 2008). In any case PrPC expression does not appear to be critical for neuronal glutamate transport (Thais et al., 2006). Irrespective of the molecular mechanisms responsible for these effects, our data indicate that PrPC (both for KA and NMDA receptors) inhibits glutamatergic neurotransmission and that the absence of PrPC enhances neurotoxic signaling. Furthermore, it is tempting to speculate on a similar protective effect in other brain injuries that share a similar activation mechanism through the receptor complex GluR6/7–PSD-95–MLK3, where signaling is transmitted downstream into the JNK3 pathway. Further studies would be needed to confirm this hypothesis. In this regard, increased levels of PrPC in plasma have been found after acute ischemia in rats and in human stroke patients (Mitsios et al., 2007). Indeed, Weise et al. (2004) suggested that the extent of the up-regulation of PrPC in ischemic brains depends on the severity of ischemia and may therefore reflect the extent of ischemia-induced neuronal damage. We believe that discerning the functional roles of PrPC at the synapse, as well as its specific interacting partners in normal and pathological conditions at the synapse, would help us to understand neurotoxicity in synapse-associated neurodegenerative diseases and ischemic processes. Figure 8 shows a general scheme depicting the role of the GluR6/7–JNK3 signaling pathway in mediating the neurotoxic effects of KA in mouse hippocampus in the absence of PrPC.
FIGURE 8:

Proposed scheme illustrating the neurotoxic effects of KA in mouse hippocampus in the absence of PrPC. In the absence of Prnp (knockout, siRNA), KA activates neurotoxic signaling in the hippocampal cells, favoring the interaction of PSD-95 with GluR6/7, inducing c-Jun and c-Fos overexpression and JNK3 activation, which, in turn, provokes the phosphorylation of c-Jun, leading to neurotoxicity and cell death.
MATERIALS AND METHODS
Animals
Prnp knockout mice Zürich I (Prnpo/o) were purchased from the European Mouse Mutant Archive (EMMA, Monterotondo, Italy). Prnpo/o mice were backcrossed with C57BL6J mice from 10 generations up to obtain 92–95% of C57BL6J microsatellite markers (Charles River background analysis) from the previous 46–48% of the Zürich I mice. Jnk3 knockout mice were generated as described elsewhere (Yang et al., 1997). Double-knockout animals (Prnpo/oJnk3o/o) were generated by crossing backcrossed Prnpo/o mice with Jnk3o/o mice (with 70–75% of C57BL6J microsatellite markers). To avoid putative background-specific differences between mice, all of the experiments were conducted using littermates derived from heterozygous (Prnp+/oJnk3+/o) parents. Specific primers for Prnp genotyping were designed in our laboratory based on the original P10 and P3 primers described elsewhere (Bueler et al., 1992). The Jnk3 knockout mice were genotyped as described elsewhere (Kuan et al., 1999). A total of 45 litters were used in the present study. All experimental procedures were carried out in accordance with the guidelines of the Spanish Ministry of Science and Technology following European standards.
Reagents and antibodies
KA, PI, SP600125, TAT-JIP peptide, DNQX, NS-102, and antitubulin antibody were from Sigma (Poole, United Kingdom). Anti–p-ERK1/2, total anti–ERK1/2, p-c-Jun (Ser-63), anti–p-JNK, anti–p-p53 (Ser-15). and total anti-JNK3 antibodies were from Cell Signaling (Beverly, MA). Anti–c-Jun antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–c-Fos was from Calbiochem (Darmstadt, Germany). Postsynaptic density 95 (anti–PSD-95) and glutamate receptor 6/7 (anti–GluR6/7) antibodies and Fluoro-Jade B were from Millipore (Billerica, MA). SAF61 antibody was from Spi-Bio (Cayman Chemical, Massy, France), and 6H4 antibody was from Prionics (Schlieren, Switzerland). SYBR green was from Roche (Basel, Switzerland).
KA injections
To induce convulsive nonlethal seizures in mice, we developed a progressive KA treatment by administering several consecutive intraperitoneal (i.p.) injections of the glutamate agonist KA dissolved in 0.1 M PBS, pH 7.2 (Rangel et al., 2007). PBS 0.1 M, pH 7.2 (vehicle), was injected as control. Adult animals (Prnpo/oJnk3+/+; 2–3 mo old) were weighed and i.p. injected with two injections of KA (6 mg/kg body weight) at time 0 and 30 min. Seizure intensity after KA injections was evaluated as described previously during the 4 h after the first KA injection (Peng et al., 1997; Lee et al., 2000). Tissue samples were analyzed at the indicated times.
Immunohistochemistry
After KA treatment, mice were perfused with phosphate buffered 4% paraformaldehyde, pH 7.3, postfixed overnight in the same fixative, and cryoprotected in 30% sucrose. Coronal sections (30 μm thick) were obtained in a freezing microtome (Leica, Wetzlar, Germany). Free-floating sections were rinsed in 0.1 M PBS, and endogenous peroxidases were blocked with 3% H2O2 and 10% methanol dissolved in 0.1 M PBS. After extensive rinsing, sections were incubated in 0.1 M PBS containing 0.2% gelatin, 10% normal serum, 0.2% glycine, and 0.2% Triton X-100 for 1 h at room temperature. Sections were then incubated overnight at 4°C with the distinct primary p-ERK1/2 and GFAP antibodies. After washing in 0.1 M PBS containing 0.2% Triton X-100, sections were incubated with Alexa Fluor 488– and/or Alexa Fluor 568–tagged secondary antibodies (1:200 diluted; Molecular Probes, Eugene, OR), washed in 0.1 M, PBS counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μM in 0.1 M PBS; 10 min), and mounted in Fluoromount (Vector Labs, Burlingame, CA). Dying cells in histological sections were measured using the S7100 ApopTag peroxidase in situ apoptosis (TUNEL) detection kit, following the manufacturer's instructions (Chemicon, Temecula, CA).
Fluoro-Jade B staining of dying neurons in brain sections
Coronal brain sections were obtained as described and rinsed for 2 h in 0.1 M Tris, pH 7.4, mounted, and air dried at room temperature overnight. The next day, sections were pretreated for 3 min in absolute ethanol, followed by 1 min in 70% ethanol and 1 min in distilled water. They were then oxidized in a solution of 0.06% KMnO4 for 15 min. After three rinses of 1 min each in distilled water, the sections were incubated for 30 min in a solution of 0.001% Fluoro-Jade B (Chemicon) containing 0.01% of DAPI (Sigma) in 0.1% acetic acid. The slides were rinsed in deionized water for 3 min each, dried overnight, cleared in xylene, and cover-slipped with Eukitt (Merck, Darmstadt, Germany) and examined using an Olympus (Hamburg, Germany) BX61 epifluorescence microscope.
Neuronal primary cultures and treatments
Primary hippocampal neurons were prepared from the hippocampus of perinatal (embryological day 18 to day of birth) animals. Briefly, hippocampal neurons were grown on dishes coated with poly-d-lysine (30 μg/ml) in Neurobasal medium supplemented with B27, 0.5 mM glutamine, 12.5 mM glutamate, and antibiotics (all purchased from Invitrogen, Carlsbad, CA). At 9 d in vitro (DIV), hippocampal neurons were infected with lentivirus carrying the eGFP construct (Morales et al., 2008) or the siRNA for Prnp. Lentiviral particles were produced by transient transfection of 293FT cells with Lipofectamine 2000 (Invitrogen), using Mission short hairpin RNA prion protein NM_011170 (Sigma), the second-generation packaging construct psPAX2 (Tronolab, Lausanne, Switzerland), and the envelope plasmid pMD2G (Tronolab). 293FT cells were cultured in DMEM supplemented with 10% fetal calf serum and without antibiotics before transfection. Medium was changed and supplemented with antibiotics after 6 h. Vector supernatants containing viral particles were harvested ∼24 and 48 h later and concentrated by ultracentrifugation (2 h at 26,000 × g at 4°C). After 5 d of infection, cultured neurons were treated with KA (150 μM dissolved in 0.1 M PBS; for 8 h). After treatment, cultures were rinsed twice in KA-free culture medium. The next day, cultures were incubated for 2 h with WST-1 reagent (Roche), and the absorbance (450 nm) was measured in a multiwell plate reader (Merck ELISA System MIOS).
Organotypic slice cultures of hippocampus
Hippocampal slice cultures were prepared from day of birth as described elsewhere (Stoppini et al., 1991, 1993; del Rio and Soriano, 2010). Animals were anaesthetized, and brains were removed aseptically from the skull. Horizontal slices (325–350 μm thick) of the hippocampus were obtained using a McIlwain tissue chopper (Mickle Laboratory, Guildford, United Kingdom). Sections were maintained and selected in cold MEM supplemented with glutamine (2 mM; MEM dissecting salt solution) for 45 min. Selected slices were then cultured using the membrane interface method (Stoppini et al., 1991). Slices were placed on 30-mm-diameter sterile membranes (Millicell-CM, Millipore) and transferred to six-well tissue culture plates (Nunc, Roskilde, Denmark). Cultures were incubated with 1.2 ml of culture medium (50% MEM, 25% horse serum, 25% Hank's buffered salt solution, containing 2 mM glutamine and 0.04% NaHCO3, adjusted to pH 7.3). The membrane cultures were maintained in a humidified incubator at 37ºC in a 5% CO2 atmosphere. The medium was changed every 2 d. All culture reagents were purchased from Invitrogen-Life Technologies (Merelbeke, Belgium).
Neuronal death after treatments in organotypic slice cultures was monitored by PI uptake (Rangel et al., 2007). After 9 d of culture, hippocampal slice cultures were serum deprived for 3 h, and 150 μM of KA was added. Cultured were exposed to protein JNK inhibitors (SP600125 20 μM or TAT-JIP 10 μM) for 45 min before KA treatment. After treatment, slices were rinsed twice in KA-free culture medium. The next day, slices were incubated for 2 h with 1 μg/ml of PI dissolved in culture medium. PI-treated slices were fixed for 1 h with 4% buffered paraformaldehyde, pH 7.2. After rinsing, cultures were mounted in Fluoromount and photodocumented in an Olympus confocal microscope. To determine the maximum PI fluorescence for quantification, total fluorescence in the pyramidal layer of the hippocampus was measured in a 500-μm segment of the layer in the CA1–CA3 regions after incubation with 10 mM glutamate for 10 min, using Quantity One Imaging and Analysis software (Bio-Rad Laboratories, Hemel Hempstead, United Kingdom).
PSD purification from mice hippocampus
The PSD fraction was purified from adult mice (10–12 mo old) as indicated (Coba et al., 2009). Briefly, tissue was weighed, and 1 ml of homogenization buffer (0.32 M sucrose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 2 mM EDTA, 2.5 mM N-ethylmaleimide, containing protease and phosphatase inhibitors) was added for every 125 mg of tissue. Samples were homogenized in a glass–Teflon Dounce homogenizer (40 strokes) and centrifuged at 800 × g for 10 min at 4°C. The supernatant was centrifuged at 10,000 × g for 15 min at 4°C, and pellet was resuspended in 0.5 ml of Triton buffer: 50 mM HEPES, pH 7.4, 2 mM EDTA, 5 mM EGTA, 1% Triton X-100, 2.5 mM N-ethylmaleimide, containing protease and phosphatase inhibitors) for every 125 mg of tissue. Samples were centrifuged at 50,000 × g for 30 min at 4°C, and the pellet was resuspended in 125 μl of resuspension buffer—50 mM Tris, pH 7.4, 1% SDS (when used for Western blotting) or 50 mM Tris, pH 8.5, 1% deoxycholate (when used for immunoprecipitation experiments)—for every 125 μg of tissue. Samples were incubated for 10 min at room temperature and centrifuged at 50,000 × g for 15 min at 4°C.
Western blotting
Tissue samples from the hippocampus of KA-treated or vehicle-treated mice were homogenized (10% wt/vol) in ice-cold lysis buffer—50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% (wt/vol) Triton X-100, 0.5% (wt/vol) Nonidet P-40 (IGEPAL; Sigma), glycerol 10%, 1 mM EDTA, 1 mM EGTA, and protease and phosphatase inhibitors—using a motor-driven, glass–Teflon homogenizer in ice and centrifuged at 15,000 × g for 20 min. After protein quantification, tissue extracts were boiled in Laemmli sample buffer at 100ºC for 5 min, followed by 6–10% SDS–PAGE electrophoresis, electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore), and processed for immunoblotting and the ECL Plus kit (Amersham-Pharmacia Biotech, GE Healthcare Bio-Sciences, Piscataway, NJ).
Immunoprecipitation
Cell extracts from total hippocampus or hippocampal PSD fractions were used for immunoprecipitation experiments. For total hippocampus, cell extracts were processed as indicated for Western blot. After protein quantification, 1 mg of hippocampal protein extract was mixed with 10 μl of protein A/G–Sepharose (Sigma) previously equilibrated with lysis buffer—50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1.5 mM MgCl2, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, and protease and phosphatase inhibitors—for 2 h (preclearing). Samples were centrifuged, and the supernatant was mixed with the indicated immunoprecipitation antibodies plus 10 μl of protein A/G–Sepharose overnight at 4°C. The immunocomplexes were washed in lysis buffer and once in lysis buffer plus 0.25 M NaCl. For immunoprecipitation from PSD hippocampal fractions, PSD fractions were diluted with RIPA buffer: 150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0, up to 500 μl. Samples were processed as described earlier but using RIPA buffer instead of lysis buffer for immunoprecipitation washes. Proteins attached to Sepharose beads were eluted with SDS–PAGE sample buffer, subjected to SDS–PAGE, transferred to PVDF membranes, and probed with the GluR6/7 and PSD-95 antibodies.
RT-qPCR
RT-qPCR was performed on total RNA extracted with the mirVana isolation kit (Ambion, Austin, TX) from the hippocampus of analyzed mice. Purified RNAs were used to generate the corresponding cDNAs, which served as PCR templates for mRNA quantification. The primers used in this study for RT-qPCR validation are given in Supplemental Figure S1. PCR amplification and detection were performed with the Roche LightCycler 480 detector, using 2x SYBR Green Master Mix (Roche) as reagent, following the manufacturer's instructions. The reaction profile was as follows: denaturation–activation cycle (95°C for 10 min) followed by 40 cycles of denaturation–annealing–extension (95°C, 10 min; 72°C, 1 min; 98°C, continuous). mRNA levels were calculated using the LightCycler 480 software. The data were analyzed using the ΔΔCt method, which provides the target gene expression values as fold changes in the problem sample compared with a calibrator sample. Both problem and calibrator samples were normalized by the relative expression of a housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]).
Statistical analysis
All data are presented as mean ± SEM. Pairwise one-way analysis of variance (ANOVA) with Newman–Keuls multiple comparisons test was used for comparison. The operations were performed using Systat Software (Chicago, IL) or Statgraphics (StatPoint Technologies, Warrenton, VA). Statistical significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Acknowledgments
We thank R. Rycroft for English language advice and D. Reginensi, I. Jimenez, and G. Tormen for technical assistance. This study was supported by the FP7-PRIORITY, the Spanish Ministry of Science and Innovation (BFU2009-10848), and the Generalitat de Catalunya (SGR2009-366) (J.A.D.R.) and grants from the Instituto Salud Carlos III (Madrid, Spain) (J.A.D.R. and I.F.) and SAF2010-21682 (C.C.). F.L. was supported by the FP7-PRIORITY. P.C. is supported by an FPI fellowship from the Spanish Ministry of Science and Innovation. A.B. is a Sara Borrell Postdoctoral Researcher of the Instituto Salud Carlos III.
Abbreviations used:
- AP-1
activator protein-1
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- ERK
extracellular regulated kinase
- GFAP
glial fibrillary acidic protein
- GluR6/7
glutamate receptor 6/7
- JNK3
c-Jun N-terminal kinase 3
- KA
kainate
- NMDA
N-methyl-d-aspartic acid
- NR2D
N-methyl-d-aspartate receptor subtype 2
- PI
propidium iodide
- Prnp
prion protein gene
- PrPC
cellular prion protein
- PSD-95
postsynaptic density 95 protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-04-0321) on July 14, 2011.
REFERENCES
- Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009;89:1105–1152. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Alford S, Grillner S. CNQX and DNQX block non-NMDA synaptic transmission but not NMDA-evoked locomotion in lamprey spinal cord. Brain Res. 1990;506:297–302. doi: 10.1016/0006-8993(90)91266-j. [DOI] [PubMed] [Google Scholar]
- Anantharam V, Kanthasamy A, Choi CJ, Martin DP, Latchoumycandane C, Richt JA, Kanthasamy AG. Opposing roles of prion protein in oxidative stress- and ER stress-induced apoptotic signaling. Free Radic Biol Med. 2008;45:1530–1541. doi: 10.1016/j.freeradbiomed.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aude-Garcia C, Villiers C, Candeias SM, Garrel C, Bertrand C, Collin V, Marche PN, Jouvin-Marche E. Enhanced susceptibility of T lymphocytes to oxidative stress in the absence of the cellular prion protein. Cell Mol Life Sci. 2011;68:687–696. doi: 10.1007/s00018-010-0477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- Beraldo FH, et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011;25:265–279. doi: 10.1096/fj.10-161653. [DOI] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- Brami-Cherrier K, Lavaur J, Pages C, Arthur JS, Caboche J. Glutamate induces histone H3 phosphorylation but not acetylation in striatal neurons: role of mitogen- and stress-activated kinase-1. J Neurochem. 2007;101:697–708. doi: 10.1111/j.1471-4159.2006.04352.x. [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res. 2002;67:211–224. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- Brown DR, et al. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, Kretzschmar HA. Effects of copper on survival of prion protein knockout neurons and glia. J Neurochem. 1998;70:1686–1693. doi: 10.1046/j.1471-4159.1998.70041686.x. [DOI] [PubMed] [Google Scholar]
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Coba MP, Pocklington AJ, Collins MO, Kopanitsa MV, Uren RT, Swamy S, Croning MD, Choudhary JS, Grant SG. Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci Signal. 2009;2:ra19. doi: 10.1126/scisignal.2000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. GluR6-containing KA receptor mediates the activation of p38 MAP kinase in rat hippocampal CA1 region during brain ischemia injury. Hippocampus. 2009;19:79–89. doi: 10.1002/hipo.20479. [DOI] [PubMed] [Google Scholar]
- Chen S, Mange A, Dong L, Lehmann S, Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci. 2003;22:227–233. doi: 10.1016/s1044-7431(02)00014-3. [DOI] [PubMed] [Google Scholar]
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Kang KS, Fukui M, Zhu BT. Critical role of the JNK-p53-GADD45alpha apoptotic cascade in mediating oxidative cytotoxicity in hippocampal neurons. Br J Pharmacol. 2011;162:175–192. doi: 10.1111/j.1476-5381.2010.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lemos L, Junyent F, Verdaguer E, Folch J, Romero R, Pallas M, Ferrer I, Auladell C, Camins A. Differences in activation of ERK1/2 and p38 kinase in Jnk3 null mice following KA treatment. J Neurochem. 2010;114:1315–1322. doi: 10.1111/j.1471-4159.2010.06853.x. [DOI] [PubMed] [Google Scholar]
- del Rio JA, Soriano E. Regenerating cortical connections in a dish: the entorhino-hippocampal organotypic slice co-culture as tool for pharmacological screening of molecules promoting axon regeneration. Nat Protoc. 2010;5:217–226. doi: 10.1038/nprot.2009.202. [DOI] [PubMed] [Google Scholar]
- Erlich RB, Kahn SA, Lima FR, Muras AG, Martins RA, Linden R, Chiarini LB, Martins VR, Moura Neto V. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia. 2007;55:1690–1698. doi: 10.1002/glia.20579. [DOI] [PubMed] [Google Scholar]
- Fournier JG. Cellular prion protein electron microscopy: attempts/limits and clues to a synaptic trait. Implications in neurodegeneration process. Cell Tissue Res. 2008;332:1–11. doi: 10.1007/s00441-007-0565-5. [DOI] [PubMed] [Google Scholar]
- Fournier JG, Escaig-Haye F, Grigoriev V. Ultrastructural localization of prion proteins: physiological and pathological implications. Microsc Res Tech. 2000;50:76–88. doi: 10.1002/1097-0029(20000701)50:1<76::AID-JEMT11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Gavin R, Ferrer I, del Rio JA. Involvement of Dab1 in APP processing and beta-amyloid deposition in sporadic Creutzfeldt-Jakob patients. Neurobiol Dis. 2010;37:324–329. doi: 10.1016/j.nbd.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Gunther EC, Strittmatter SM. Beta-amyloid oligomers and cellular prion protein in Alzheimer's disease. J Mol Med. 2010;88:331–338. doi: 10.1007/s00109-009-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatwole VM. TUNEL assay for apoptotic cells. Methods Mol Biol. 1999;115:141–148. doi: 10.1385/1-59259-213-9:141. [DOI] [PubMed] [Google Scholar]
- Hu WW, Du Y, Li C, Song YJ, Zhang GY. Neuroprotection of hypothermia against neuronal death in rat hippocampus through inhibiting the increased assembly of GluR6-PSD95-MLK3 signaling module induced by cerebral ischemia/reperfusion. Hippocampus. 2008;18:386–397. doi: 10.1002/hipo.20402. [DOI] [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravani H, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Park J, Kim YH, Kim DH, Kim CG, Koh JY. Induction by synaptic zinc of heat shock protein-70 in hippocampus after kainate seizures. Exp Neurol. 2000;161:433–441. doi: 10.1006/exnr.1999.7297. [DOI] [PubMed] [Google Scholar]
- Li C, Xu B, Wang WW, Yu XJ, Zhu J, Yu HM, Han D, Pei DS, Zhang GY. Coactivation of GABA receptors inhibits the JNK3 apoptotic pathway via disassembly of GluR6-PSD-95-MLK3 signaling module in KA-induced seizure. Epilepsia. 2010;51:391–403. doi: 10.1111/j.1528-1167.2009.02270.x. [DOI] [PubMed] [Google Scholar]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- Maglio LE, Martins VR, Izquierdo I, Ramirez OA. Role of cellular prion protein on LTP expression in aged mice. Brain Res. 2006;1097:11–18. doi: 10.1016/j.brainres.2006.04.056. [DOI] [PubMed] [Google Scholar]
- Maglio LE, Perez MF, Martins VR, Brentani RR, Ramirez OA. Hippocampal synaptic plasticity in mice devoid of cellular prion protein. Brain Res Mol Brain Res. 2004;131:58–64. doi: 10.1016/j.molbrainres.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MM, Linden R. Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol. 2010;12:63–86. [PubMed] [Google Scholar]
- McLennan NF, et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsios N, et al. Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J Neurosci Res. 2007;85:602–611. doi: 10.1002/jnr.21142. [DOI] [PubMed] [Google Scholar]
- Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nolting S, Hamann GF, Kretzschmar HA. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007;17:174–183. doi: 10.1111/j.1750-3639.2007.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Riss M, Wang L, Gavin R, Del Rio JA, Alcubilla R, Claverol-Tinture E. Integrating multi-unit electrophysiology and plastic culture dishes for network neuroscience. Lab Chip. 2008;8:1896–1905. doi: 10.1039/b802165a. [DOI] [PubMed] [Google Scholar]
- Nicolas O, Gavin R, Braun N, Urena JM, Fontana X, Soriano E, Aguzzi A, del Rio JA. Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. FASEB J. 2007;21:3107–3117. doi: 10.1096/fj.06-7827com. [DOI] [PubMed] [Google Scholar]
- Nicolas O, Gavin R, del Rio JA. New insights into cellular prion protein (PrPc) functions: the “ying and yang” of a relevant protein. Brain Res Rev. 2009;61:170–184. doi: 10.1016/j.brainresrev.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Pei DS, Wang XT, Liu Y, Sun YF, Guan QH, Wang W, Yan JZ, Zong YY, Xu TL, Zhang GY. Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain. 2006;129:465–479. doi: 10.1093/brain/awh700. [DOI] [PubMed] [Google Scholar]
- Peng YG, Clayton EC, Means LW, Ramsdell JS. Repeated independent exposures to domoic acid do not enhance symptomatic toxicity in outbred or seizure-sensitive inbred mice. Fundam Appl Toxicol. 1997;40:63–67. doi: 10.1006/faat.1997.2360. [DOI] [PubMed] [Google Scholar]
- Prestori F, Rossi P, Bearzatto B, Laine J, Necchi D, Diwakar S, Schiffmann SN, Axelrad H, D'Angelo E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J Neurosci. 2008;28:7091–7103. doi: 10.1523/JNEUROSCI.0409-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Muller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27:1974–1984. doi: 10.1038/emboj.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel A, Burgaya F, Gavin R, Soriano E, Aguzzi A, Del Rio JA. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: role of AMPA/kainate receptors. J Neurosci Res. 2007;85:2741–2755. doi: 10.1002/jnr.21215. [DOI] [PubMed] [Google Scholar]
- Rangel A, Madronal N, Gruart A, Gavin R, Llorens F, Sumoy L, Torres JM, Delgado-Garcia JM, Del Rio JA. Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS One. 2009;4:e7592. doi: 10.1371/journal.pone.0007592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffe M, et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc Natl Acad Sci USA. 2010;107:13147–13152. doi: 10.1073/pnas.1000784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser D, Mertz KD, Moos R, Brunner E, Rulicke T, Calella AM, Aguzzi A. The comprehensive native interactome of a fully functional tagged prion protein. PLoS One. 2009;4:e4446. doi: 10.1371/journal.pone.0004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinainen A, Garcia EP, Dorow D, Marshall J, Liu YF. Kainate receptor activation induces mixed lineage kinase-mediated cellular signaling cascades via post-synaptic density protein 95. J Biol Chem. 2001;276:11382–11386. doi: 10.1074/jbc.M100190200. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade: novel fluorochromes for detecting toxicant-induced neuronal degeneration. Toxicol Pathol. 2000;28:91–99. doi: 10.1177/019262330002800111. [DOI] [PubMed] [Google Scholar]
- Shyu WC, et al. Hypoglycemia enhances the expression of prion protein and heat-shock protein 70 in a mouse neuroblastoma cell line. J Neurosci Res. 2005;80:887–894. doi: 10.1002/jnr.20509. [DOI] [PubMed] [Google Scholar]
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005;20:442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Steele AD. All quiet on the neuronal front: NMDA receptor inhibition by prion protein. J Cell Biol. 2008;181:407–409. doi: 10.1083/jcb.200803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci USA. 2006;103:3416–3421. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Zhou Z, Jackson WS, Zhu C, Auluck P, Moskowitz MA, Chesselet MF, Lindquist S. Context dependent neuroprotective properties of prion protein (PrP) Prion. 2009;3:240–249. doi: 10.4161/pri.3.4.10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. Lesion-induced neurite sprouting and synapse formation in hippocampal organotypic cultures. Neuroscience. 1993;57:985–994. doi: 10.1016/0306-4522(93)90043-f. [DOI] [PubMed] [Google Scholar]
- Thais ME, et al. Synaptosomal glutamate release and uptake in mice lacking the cellular prion protein. Brain Res. 2006;1075:13–19. doi: 10.1016/j.brainres.2005.12.045. [DOI] [PubMed] [Google Scholar]
- Tian H, Zhang QG, Zhu GX, Pei DS, Guan QH, Zhang GY. Activation of c-Jun NH2-terminal kinase 3 is mediated by the GluR6.PSD-95.MLK3 signaling module following cerebral ischemia in rat hippocampus. Brain Res. 2005;1061:57–66. doi: 10.1016/j.brainres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Toscano CD, Kingsley PJ, Marnett LJ, Bosetti F. NMDA-induced seizure intensity is enhanced in COX-2 deficient mice. Neurotoxicology. 2008;29:1114–1120. doi: 10.1016/j.neuro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B, Bazan NG. Hippocampal kindling epileptogenesis upregulates neuronal cyclooxygenase-2 expression in neocortex. Exp Neurol. 2003;179:167–175. doi: 10.1016/s0014-4886(02)00019-5. [DOI] [PubMed] [Google Scholar]
- Vassallo N, Herms J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J Neurochem. 2003;86:538–544. doi: 10.1046/j.1471-4159.2003.01882.x. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Johansen TH, Drejer J, Nielsen EO. Selective block of recombinant glur6 receptors by NS-102, a novel non-NMDA receptor antagonist. Eur J Pharmacol. 1994;269:43–49. doi: 10.1016/0922-4106(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, Hanisch UK. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia. 2005;50:235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia. 1999;40:1679–1682. doi: 10.1111/j.1528-1157.1999.tb01583.x. [DOI] [PubMed] [Google Scholar]
- Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bahr M, Zerr I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci Lett. 2004;372:146–150. doi: 10.1016/j.neulet.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bahr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37:1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4. [DOI] [PubMed] [Google Scholar]
- Weissmann C, Bueler H, Fischer M, Sailer A, Aguzzi A, Aguet M. PrP-deficient mice are resistant to scrapie. Ann N Y Acad Sci. 1994;724:235–240. doi: 10.1111/j.1749-6632.1994.tb38913.x. [DOI] [PubMed] [Google Scholar]
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta. 2007;1772:629–644. doi: 10.1016/j.bbadis.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BS, et al. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J Neurochem. 2001;76:565–572. doi: 10.1046/j.1471-4159.2001.00028.x. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Zanata SM, et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.