

Abstract
Development of the DNA gyrase inhibitor, novobiocin, into a selective Hsp90 inhibitor was accomplished through structural modifications to the amide side chain, coumarin ring, and sugar moiety. These species exhibit ~700-fold improved anti-proliferative activity versus the natural product as evaluated by cellular efficacies against breast, colon, prostate, lung, and other cancer cell lines. Utilization of structure–activity relationships established for three novobiocin synthons produced optimized scaffolds, which manifest mid-nanomolar activity against a panel of cancer cell lines and serve as lead compounds that manifest their activities through Hsp90 inhibition.
Introduction
The Hsp90 molecular chaperone has emerged as a promising target for the treatment of cancer.1 While most current therapies are directed at disrupting a single molecular function, Hsp90 is distinctive as it serves to modulate multiple oncogenic pathways. Moreover, Hsp90 is overexpressed in human malignancies and its inhibition provides a unique opportunity to develop small molecules that exhibit high differential selectivity.1–4 There are more than 150 client proteins dependent upon Hsp90 for their folding and conformational maintenance, many of which contribute to cancer cell proliferation and growth.5–12 Small molecule Hsp90 inhibitors transform the normal protein folding machinery into a catalyst for protein degradation via utilization of the ubiquitin-proteasome pathway.13, 14 As such, Hsp90 inhibition is similar to a combinatorial attack, in which multiple signaling pathways that contribute to malignancy are simultaneously disrupted.1, 2, 15–17
Novobiocin (Figure 1), a member of the coumermycin family of antibiotics, manifests potent antimicrobial activity through inhibition of DNA gyrase-mediated ATP-hydrolysis.18–22 Co-crystal structures of DNA gyrase B bound to novobiocin and ADP revealed both molecules to bind in an atypical, bent conformation.23–25 It was this unique binding conformation that led Neckers and coworkers to hypothesize that novobiocin exhibits its anti-tumor activity (~700 µM) against SKBr3 breast cancer cells through Hsp90 inhibition. Through subsequent Western blot analyses, Neckers and coworkers confirmed that novobiocin induces degradation of Hsp90-dependent clients in a concentration-dependent manner, a hallmark of Hsp90 inhibition. In contrast to other Hsp90 inhibitors, it was shown that novobiocin bound to the C-terminus, revealing a previously unrecognized binding site, and perhaps, a new opportunity to modulate the Hsp90 protein folding machinery.26
Figure 1.

Structure of novobiocin.
Although no co-crystal structure of Hsp90 bound to C-terminal inhibitors has been reported, the structure and function of the Hsp90 C-terminal nucleotide binding site are under intense investigation. Compounds exhibiting higher affinity for Hsp90 than novobiocin are likely to provide the tools necessary for elucidation of the Hsp90 C-terminal nucleotide-binding pocket and provide new Hsp90 modulators that manifest chemotherapeutic potential.27 The poor Hsp90 inhibitory activity of novobiocin26 has been greatly improved upon by the preparation of analogues. Yu and co-workers produced a library of compounds that established the first structure–activity relationships for novobiocin as an Hsp90 inhibitor. This focused library explored essential features of the benzamide side chain, coumarin core and noviose sugar, highlighting that attachment of noviose to the 7-position and an amide linker at the 3-position of the coumarin ring are critical for Hsp90 inhibitory activity. Moreover, modifications to the sugar identified the diol as most potent, versus the cyclic carbonate and corresponding carbamates as found in novobiocin.28
Compound 1 (Figure 2) was the most notable compound identified from this library. 1, with a shortened N-acyl side chain, a 4-deshydroxy substituent and no carbamoyl group on noviose, induced degradation of Hsp90-dependent client proteins at ~70-fold lower concentration than novobiocin.28 However, in contrast to N-terminal inhibitors, 1 induced Hsp90 at concentrations 1000–10000-fold lower than that required for client protein degradation, a phenomenon that had not been previously observed. Due to its activity and non-toxic nature, 1 was evaluated as a neuroprotective agent and produced an EC50 of 6 nM in a culture model for Alzheimer’s disease.28
Figure 2.

To confirm SAR trends observed by Yu and co-workers, two natural product analogues, 2 and 3 (Figure 2), were designed and subsequently evaluated by Burlison and co-workers. This study aimed at elucidating functionalities on novobiocin that are responsible for DNA gyrase inhibition and to deconvolute Hsp90-selective inhibitory compounds. Studies with these compounds confirmed that the 4-hydroxyl and the 3’-carbamate are detrimental to Hsp90 inhibitory activity, but critical for DNA gyrase inhibition.29 This study confirmed the trends observed from the 1 library and produced the first selective inhibitors of the Hsp90 C-terminus.
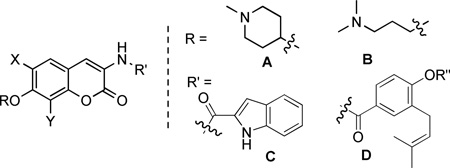
Based on the cytotoxicity of coumermycin A1 (Figure 3), another member of the coumermycin family of antibiotics, it appeared that dimerization could increase Hsp90 inhibitory activity. Burlison and co-workers dimerized 1 in an attempt to transform a non-toxic agent into a more potent analogue. The resulting compound, 5, the most potent identified, exhibited low micromolar anti-proliferative activity, but did not induce the heat shock response. These results suggested that modification of the amide side chain could convert a non-toxic molecule into an anti-proliferative agent.31 Consequently, a series of monomeric species based on 1 was synthesized and evaluated in cell proliferation studies.32 Monomeric analogues were designed to include biaryl and heterocyclic amide derivatives that explored hydrogen-bonding interactions with the putative binding pocket that typically accommodates the prenylated benzamide of novobiocin. This library identified key functionalities and established the first set of SAR for the amide side chain (Figure 4).
Figure 3.

Structures of 5 and coumermycin A1.
Figure 4.

Benzamide SAR.32
The coumarin core of novobiocin was recently explored and SAR established. Derivatives of 1 with variations to the coumarin ring were designed to probe the importance of interactions manifested by the natural substrate purine ring as well the size and nature of the novobiocin binding pocket. The 6- and 8-positions of the coumarin scaffold proved tolerable to substitution, and incorporation of alkoxy groups at these locations led to improved inhibitory activity. Moreover, the lactone moiety of the coumarin ring was found to be dispensable (Figure 5).33
Figure 5.

Coumarin SAR.33
Renoir and co-workers examined the role of noviose in Hsp90 inhibition. Their structure–activity relationship studies demonstrated that Hsp90 inhibition was observed by analogues that lack the noviose moiety, if a tosyl substituent is attached to the C-4 or C-7 position of the coumarin. These analogues manifested mid-micromolar IC50 values.34 In a subsequent paper, the same group suggested that Hsp90 inhibition can be enhanced by removal of C7/C8 substituents in denoviose analogues bearing a tosyl group at the 4-position. These studies produced inhibitors with simplified coumarins that also exhibited mid-micromolar IC50 values.35 The compounds produced in these two publications represented the first novobiocin analogues that lacked the noviose sugar while manifesting Hsp90 inhibition.
The noviose sugar found in novobiocin is complex and is prepared via laborious efforts. Even the most efficient syntheses of noviose require more than 10 steps and the overall yields are less than optimal.36 In an effort to gain further insight into this region of novobiocin, simplified noviose derivatives were attached to the novobiocin core. This study, which resulted in compounds that exhibit a 700-fold improvement in activity over novobiocin, indicated that while the sugar moiety plays a critical role in maintaining binding interactions with Hsp90, not all functionalities are necessary.
Sugar mimics were originally attached to the 7-position of coumarin containing an 8-methyl substituent and a biaryl benzamide. The most potent coumarin scaffolds obtained from the coumarin studies were coupled with the optimal biaryl side chain to yield a scaffold upon which sugar surrogates were appended.32, 33 Based on the inhibitory activity manifested by the biaryl analogues, the most promising sugar surrogates were coupled with coumarins containing the 2-indole side chain.32 The simplified sugars and related azasugars were found to conserve structural units present in noviose, but lacked the complexity of multi-step synthesis. These sugar surrogates were designed to probe potential hydrogen bonds as well as to determine the dimensions of the pocket. In addition to simplified sugars, several surrogates containing heteroatoms and various appendages were also explored. These diverse sugar and non-sugar analogues simplify the preparation of novobiocin analogues, while providing a handle to improve both solubility and efficacy. The design, synthesis and biological evaluation of these optimized compounds are reported herein.
Results and Discussion
Design, synthesis and evaluation of modified sugar analogues of novobiocin
Numerous biologically active natural products contain carbohydrates appended to their scaffolds that serve to increase solubility and provide interactions with their cognate receptor. In many cases, removal of the carbohydrate moiety renders the aglycon inactive, whereas alteration of the sugar ring size can alter affinity of the compounds toward specific targets.37–39 Modifications to the noviose pyranose ring were proposed to elucidate functionalities required for inhibitory activity, as well as to determine whether different sized sugars can be utilized as replacements for noviose. Thus, 5-, 6- and 7-membered sugars were synthesized and coupled to the aforementioned scaffolds to determine optimal interactions. When available, the α- and β-anomers were also evaluated. A set of protected mono-, di- and trihydroxylated furanoses, pyranoses and oxepanose sugars previously synthesized by Yu and coworkers40 (E–L, Figure 6) was chosen based on these considerations.
Figure 6.

5-, 6-, and 7-membered noviose alternatives.
The analogues were assembled in modular fashion, allowing sequential coupling of sugars and the biaryl acid side chain with the desired scaffold. As shown in the retrosynthetic analysis (Scheme 1), the biaryl acid side chain was assembled through a Suzuki coupling reaction, as described previously.33 Next, the biaryl acid was converted to its corresponding acid chloride and then coupled with the protected coumarin, 9. Finally, Mitsunobu etherification between coumarin phenols 6a–c and sugars E–L yielded the desired analogues in good yields.
Scheme 1.

Retrosynthesis of novobiocin biaryl analogues with sugars E–L.
Synthesis of these analogues, as described in Scheme 2, began via protection of phenol 732, 33 as the corresponding ester, 8. Next, hydrogenolysis was employed to liberate vinylogous amide 9, which was subsequently coupled with biaryl acyl chloride, 11. The biaryl acid chloride was generated from the corresponding biaryl acid 10, as described previously.33 Finally, solvolysis of ester 12 afforded phenol 6a–c in good yield.
Scheme 2.

Synthesis of phenol 6.
Coumarin phenols 6a–c and protected sugar F were coupled via a Mitsunobu etherification to yield an inseparable mixture of diastereomers, 13, in a 3:2 ratio, respectively (Scheme 3). The cyclic carbonates of the resulting mixture were solvolyzed to afford a mixture of diols 14 and 15, which were separated by silica chromatography. The stereochemistry at the anomeric carbon was assigned by 2D-NOESY spectroscopy.
Scheme 3.

Synthesis of pyranose-containing biaryl analogues.
An anomeric mixture of compound 13 was synthesized via Mitsunobu etherification between sugar F and coumarin 6. Likewise, the Mitsunobu conditions were used to append sugar G to coumarin 6 and furnish the anomeric mixture of compound 16, and to attach sugar H to coumarin 6 to yield anomeric mixture 19. Solvolysis of the cyclic carbonate present in 13 furnished a mixture of separable diols, 13 and 14. Moreover, benzoyl (Bz) deprotection of 16 yielded analogues 17 and 18 in a 3:2 ratio,5 while TIPS removal of 19 furnished analogues 20 and 21 in a 4:3 ratio. Not surprisingly, a single diastereomer of 22 was exclusively formed when coumarin 6 and protected sugar E were subjected to Mitsunobu conditions. Subsequent deprotection of the acetonide and benzyl protecting groups afforded triol 23 in good yield (Scheme 3).
As shown in Scheme 4, protected furanose sugars J–L were coupled with coumarin 6 enlisting Mitsunobu etherification to afford the corresponding analogues in good yields. Mitsunobu coupling of protected sugar J with coumarin 6 yielded exclusively diastereomer 24, which was subsequently hydrolyzed to afford diol 25. Mitsunobu etherification was used to append sugar K to yield analogue 26, which, after silyl removal, furnished separable compounds 27 and 28. Likewise, the same conditions were applied to append sugar L to 29, which, after silyl cleavage, yielded analogues 30 and 31. Protected oxepanose I was coupled to coumarin 6 using Mitsunobu conditions to afford 32 as the major product, which was hydrolyzed to give 33.
Scheme 4.

Synthesis of furanose- and oxepanose-containing biaryl analogues.
In addition to the variable-sized sugar-containing analogues, cyclohexyl analogues were designed to examine if a sugar was necessary. Additionally, to probe the tolerance of steric bulk, analogues with and without alkyl substituents were pursued. Many of the simplified cyclohexyl sugar mimics were accessible using common procedures (Scheme 5), such as reduction with lithium aluminum hydride to produce compounds 37 and 38, or Luche reduction to yield 39. Compounds containing a double bond were designed to be coupled with the scaffold and subsequently oxidized to the corresponding diols (e.g., 43–45).
Scheme 5.

Synthesis of cyclohexyl-containing biaryl analogues.
Upon construction of the modified sugar analogues of novobiocin, the compounds were evaluated for anti-proliferative activity against SKBr3 (estrogen receptor negative, HER2 over-expressing breast cancer cells), MCF-7 (estrogen receptor positive breast cancer cells), LNCaP-LN3 (androgen-dependent human prostate cancer cells) and PC3-MM2 (androgen-independent prostate cancer cells) cell lines. As shown in Table 1, analogues containing six-membered pyranose moieties (scaffold A) were found to be the most active compounds against the two breast cancer cell lines. Analogues containing dihydroxyl groups consistently exhibited good to modest anti-proliferative activities against both breast cancer cell lines. In contrast, analogues that contained a single hydroxyl at the 2’-position proved inactive, while most analogues with a 3’-hydroxyl exhibited modest activity. These data indicate that the 3’-position hydroxy group is essential for anti-proliferative activity. Interestingly, the natural substrate for the C-terminal pocket of Hsp90, ATP/ADP, also contains a 3’-hydroxyl. Although the β epimer exhibited better activity than its α counterpart (14a–c vs. 15a–c) for most analogues, this was not a general trend observed throughout the series (120a–c vs. 21a–c). In addition, the majority of the compounds from this series demonstrated modest activity against the prostate cancer cell lines. Notably, however, some compounds (18a) that were completely inactive against both breast cancer cells lines were efficacious against prostate cancer.
Table 1.
Anti-proliferative activity of various sugar analogues.
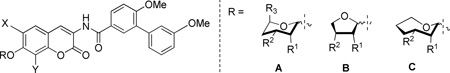 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound (IC50, µM) |
R | R1 | R2 | R3 | Anomer | X | Y | SKBr3 | MCF-7 | LNCAP-LN3 | PC3-MM2 |
| 14a | A | OH | OH | H | β | H | CH3 | 6.23 ± 0.52a,c | 2.56 ± 0.08a | 3.59 ± 3.40b | NTb |
| 15a | A | OH | OH | H | α | H | CH3 | 6.71± 0.80 | 42.56 ± 2.48 | 1.20 ± 0.83 | 12.88 ± 9.30 |
| 14b | A | OH | OH | H | β | H | OCH3 | 1.46 ± 0.46 | 17.46 ± 1.22 | 2.29 ± 1.25 | 10.72 ± 10.85 |
| 15b | A | OH | OH | H | α | H | OCH3 | 11.10 ± 0.43 | 10.85 ± 0.18 | NT | NT |
| 14c | A | OH | OH | H | β | OCH3 | CH3 | 7.18 ± 0.20 | 9.50 ± 0.16 | 12.76 ± 5.12 | 24.03 ± 17.14 |
| 15c | A | OH | OH | H | α | OCH3 | CH3 | 12.52 ± 0.54 | 64.51 ± 3.52 | 14.93 ± 9.24 | 25.37 ± 7.26 |
| 17a | A | OH | H | H | β | H | CH3 | >100 | >100 | 3.75 ± 0.01d | 10.05 ± 0.01d |
| 18a41 | A | OH | H | H | α | H | CH3 | >100 | >100 | 7.34 ± 11.82 | NT |
| 20a | A | H | OH | H | β | H | CH3 | >100 | >100 | 3.11 ± 2.58 | >100 |
| 21a41 | A | H | OH | H | α | H | CH3 | 9.28 ± 0.04 | 11.20 ± 0.49 | 8.56 ± 9.00 | NT |
| 20b | A | H | OH | H | β | H | OCH3 | 8.75 ± 0.49 | 95.77 ± 3.21 | 11.50 ± 5.66 | NT |
| 20c | A | H | OH | H | β | OCH3 | CH3 | >100 | >100 | 1.49 ± 0.51 | 1.24d |
| 21c | A | H | OH | H | α | OCH3 | CH3 | 1.37 ± 0.14 | >100 | 3.27 ± 0.09d | 2.35 ± 1.11 |
| 23a | A | OH | OH | CH2OH | β | H | CH3 | 9.59 ± 0.42 | 11.07 ± 0.47 | 3.23 ± 3.76 | 4.58 ± 2.78 |
| 23b | A | OH | OH | CH2OH | β | H | OCH3 | 11.76 ± 0.06 | 14.34 ± 0.16 | 6.78 ± 2.25 | 17.94 ± 10.33 |
| 23c | A | OH | OH | CH2OH | β | OCH3 | CH3 | 9.45 ± 0.19 | 3.37 ± 0.06 | 4.66 ± 1.48 | 5.90 ± 2.58 |
| 25a | B | OH | OH | H | β | H | CH3 | 12.46 ± 0.52 | 37.17 ± 1.68 | 3.65 ± 2.83 | NT |
| 25b | B | OH | OH | H | β | H | OCH3 | 7.57 ± 1.05 | 11.73 ± 0.78 | 2.60 ± 1.51 | 10.64 ± 20.83 |
| 25c | B | OH | OH | H | β | OCH3 | CH3 | 9.45 ± 0.19 | 3.37 ± 0.07 | 4.72 ± 1.48 | 9.56 ± 2.58 |
| 27a | B | OH | H | -- | β | H | CH3 | >100 | >100 | NT | NT |
| 28a | B | OH | H | -- | α | H | CH3 | >100 | >100 | 5.34 ± 4.76 | NT |
| 30a | B | H | OH | -- | β | H | CH3 | 35.24 ± 1.76 | >100 | 3.72d | 10.05 ± 6.75d |
| 31a | B | H | OH | -- | α | H | CH3 | >100 | >100 | 4.96 ± 10.58 | 26.79 ± 58.24 |
| 33a | C | OH | OH | -- | β | H | CH3 | 2.38 ± 0.06 | >100 | >100 | NT |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Values represent mean ± standard deviation from dose response curves for at least two separate experiments performed in duplicate.
All error values listed represent 95% confidence intervals throughout the manuscript except where indicated
Error values listed as standard deviations
Analogues containing a cyclohexyl moiety in lieu of the sugar demonstrated a range of activities. While analogues containing an 8-methyl group (43a, 44a, 45a) were inactive against breast cancer, those with an 8-methoxy group (43b, 44b, 45b) exhibited an IC50 value of ~10 µM, which is comparable to its noviosylated counterpart.33 Interestingly, the gem-dimethyl group is well tolerated in the case of the 8-methoxy coumarin (43b), but detrimental in the case of the 6-methoxy coumarin (43c). This finding suggests a mutually exclusive orientation between the gem-dimethyl group and substituents on the coumarin, which may preclude proper binding orientation.
Overall, this library containing sugar surrogates and cyclohexyl groups confirmed the 3’-hydroxy as the most important functional group. Moreover, the 4’-methoxy, 5’-gem-dimethyl and anomeric oxygen found in noviose were identified as dispensable moieties. Several analogues (15b, 24c and 37a) exhibited low micromolar anti-proliferative activity against SKBr3 cells, which makes them ~500-fold more active than novobiocin.
Design, synthesis and biological evaluation of azasugar analogues of novobiocin
After evaluation of the initial library, it was proposed that other six-membered heteroatom-containing ring systems may serve as replacements for noviose. N-heterocycles are found in a wide variety of biologically active compounds, imparting solubility to otherwise insoluble aglycons. Several heterocycles were designed to probe hydrogen-bonding interactions with the binding pocket as well as to improve solubility.
In modular fashion, the analogues were assembled similarly to those described previously, in modular fashion, allowing sequential coupling of various sugar surrogates and the biaryl side chain. As shown in the retrosynthetic analysis depicted in Scheme 6, the biaryl acid chloride, prepared from the corresponding biaryl acid, was coupled with coumarin 12. Following deprotection, coumarin phenol 6 was etherified using Mitsunobu conditions, to yield the desired analogues.
Scheme 6.

Retrosynthesis of azasugar-containing biaryl analogues.
In order to avoid synthetic complications, only a single hydroxyl group was installed on the piperidine ring prior to Mitsunobu coupling and the amine was masked as the carbamate, 46. With compound 6 and 46 in hand, compound 47 was synthesized via Mitsunobu etherification, followed by deprotection to yield amine 48 in quantitative yield. Acetylation of 48 gave amide 50, while treatment with methyl iodide furnished the tertiary amine, 51, which underwent dihydroxylation with OsO4/NMO to give 55. Dihydroxylation of 47 afforded compound 49, which, upon removal of Boc, afforded 52. Compounds 53 and 54 were synthesized from compound 50 via reduction or dihydroxylation conditions, respectively (Scheme 7).
Scheme 7.

Synthesis of 1,3-azasugar-containing biaryl analogues.
In addition, compounds lacking the diol functionality and containing a transposed nitrogen within the heterocycle were selected for investigation. Boc-protected 1,3- or 1,4-azasugars were coupled with phenol 6 to afford compounds 56 and 58, which were subsequently deprotected to yield amines 57 and 59. Similarly, N-methylated analogues 60 and 61 were synthesized via Mitsunobu etherification with commercially available piperidines in good yields (Scheme 8).
Scheme 8.

Synthesis of 1,4-azasugar biaryl analogues.
Upon construction of the azasugar-containing analogues, they were evaluated for anti-proliferative activity against two breast and two prostate cancer cell lines. As shown in Table 3, the activity against breast cancer cells exhibited by the majority of these secondary and tertiary amines varied between 1–3 µM, making them ~700-fold more active than novobiocin. Although dihydroxylation of the piperidine ring is well-tolerated when appended to the 8-methyl scaffold (52a), this is not the case for the other two scaffolds (52b and 52c). These data suggest that dihydroxylation of the piperidine ring is not essential for anti-proliferative activity. Inclusion of unsaturation within the piperidine ring is also well tolerated (scaffold A vs. C and D, Table 3) while methylation of the amine within this unsaturated ring decreases activity (48 vs. 51). It was observed that acetylation of the amine functionality (50a, 53a, 54a) severely decreased solubility, and produced inactive compounds. Although insolubility can be overcome through dihydroxylation of the piperidine ring (54a), the resulting compound is ~5-fold less active than secondary or tertiary amines. There is no significant effect when the nitrogen is transposed within the piperidine ring, as all of the analogues exhibited activity between ~1–3 µM. In addition, secondary amines exhibited comparable activity to methylated tertiary amines (57 vs. 60, 59 vs. 61). Overall, these azasugar analogues consistently exhibited low micromolar anti-proliferative activity, representing an improved sugar mimic.
Table 3.
Anti-proliferative activity of azasugar analogues.
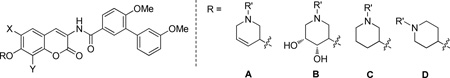 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound (IC50, µM) |
R | X | Y | R’ | SKBr3 | MCF-7 | LNCAP-LN3 | PC3-MM2 |
| 48a | A | H | CH3 | H | 1.61 ± 0.05a | 1.73a | 4.27 ± 0.05b,d | 4.40 ± 0.06b,d |
| 48b | A | H | OCH3 | H | 3.07 ± 0.78 | 1.43 ± 0.37 | 6.45 ± 2.70d | 4.88 ± 2.20 |
| 48c | A | OCH3 | CH3 | H | 1.21 ± 0.07 | 1.68 ± 0.04 | 5.40 ± 9.99 | 3.57 ± 0.71d |
| 50a | A | H | CH3 | Ac | >50 | >50 | 0.90 ± 0.59 | 3.73 ± 0.67 |
| 51a | A | H | CH3 | CH3 | 2.92 ± 1.33 | 5.29 ± 0.23 | 1.22d | 1.73 ± 1.80 |
| 51b | A | H | OCH3 | CH3 | 3.42 ± 0.45 | 1.65 ± 0.28 | 0.36 ± 0.14 | 0.98 ± 0.40 |
| 51c | A | OCH3 | CH3 | CH3 | 1.96 ± 0.48 | 5.27 ± 0.22 | 4.49 ± 0.17d | 3.87 ± 0.03d |
| 52a | B | H | CH3 | H | 2.91 ± 0.90 | 2.07 ± 0.86 | 4.90 ± 9.16 | 6.33 ± 4.12 |
| 52b | B | H | OCH3 | H | 8.98 ± 0.44 | 10.17 ± 0.02 | 12.19 ± 6.30 | 14.43 ± 7.39 |
| 52c | B | OCH3 | CH3 | H | 3.65 ± 0.22 | 3.34 | 9.45 ± 5.90 | 6.84 ± 2.80 |
| 53a | C | H | CH3 | Ac | >50 | >50 | 1.23 ± 0.67 | 3.42 ± 1.24 |
| 54a | B | H | CH3 | Ac | 10.79 ± 0.08 | 9.18 ± 0.60 | 0.71 ± 0.54 | 2.22 ± 1.04 |
| 55a | B | H | CH3 | CH3 | 3.92 ± 0.32 | 1.85 ± 0.02 | 0.59 ± 0.54 | 2.99 ± 1.27 |
| 55b | B | H | OCH3 | CH3 | 6.64 ± 0.54 | 11.02 ± 1.12 | 1.50 ± 0.62 | 3.52 ± 1.43 |
| 55c | B | OCH3 | CH3 | CH3 | 2.77 ± 0.90 | 2.01 ± 0.46 | 4.69 ± 0.57d | 4.17 ± 0.16d |
| 57a | C | H | CH3 | H | 1.16 ± 0.16 | 1.63 ± 0.28 | 3.02 ± 0.97d | 2.57 ± 1.13 |
| 57b | C | H | OCH3 | H | 2.61 ± 0.37 | 3.29 ± 0.42 | 12.72 ± 3.25 | 10.43 ± 2.15 |
| 57c | C | OCH3 | CH3 | H | 2.27 ± 0.61 | 2.90 ± 0.67 | 3.66d | 2.59 ± 13.91 |
| 59a | D | H | CH3 | H | 1.19 ± 0.06 | 1.47 ± 0.02 | 3.38 ± 1.25 | 4.12 ± 1.29 |
| 59b | D | H | OCH3 | H | 1.79 | 3.23 ± 0.20 | 10.85 ± 5.92 | 8.47 ± 4.28 |
| 59c | D | OCH3 | CH3 | H | 1.20 ± 0.08 | 2.82 ± 1.10 | 5.27 ± 22.18 | 4.02 ± 2.13 |
| 60a | C | H | CH3 | CH3 | 2.06 ± 0.57 | 5.04 ± 0.02 | 1.22 ± 0.17d | 4.23 ± 1.68 |
| 60b | C | H | OCH3 | CH3 | 1.40 ± 0.14 | 1.38 ± 0.14 | 1.78 ± 0.80 | 2.16 ± 1.28 |
| 60c | C | OCH3 | CH3 | CH3 | 1.49 ± 0.06 | 1.41 ± 0.13 | 10.48 ± 5.46d | 6.86 ± 2.92d |
| 61a41 | D | H | CH3 | CH3 | 1.34 ± 0.18 | 1.51 ± 0.24 | 4.12 ± 0.16d | 3.13 ± 0.67 |
| 61b | D | H | OCH3 | CH3 | 0.97 ± 0.07 | 1.39 ± 0.28 | 4.75 ± 2.03 | 3.71 ± 1.27 |
| 61c | D | OCH3 | CH3 | CH3 | 1.19 ± 0.17 | 1.79 ± 0.09 | 4.17 ± 0.15d | 2.84 ± 1.88 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Values represent mean ± standard deviation from dose response curves for at least two separate experiments performed in duplicate.
Design, synthesis and biological evaluation of novobiocin analogues with acyclic sugar replacements
The promising results obtained when azasugars were appended to these scaffolds encouraged the synthesis of acyclic nitrogen-containing replacements. To probe the importance of a constrained ring system, a library of ring-opened, amine-containing sugar surrogates was designed. A series of heteroatom-containing aliphatic chains were synthesized to impart flexibility and explore the potential of additional interactions with the binding pocket. Aliphatic amines, either commercially available or prepared over a modest number of steps, were appended to coumarin core 6 through standard Mitsunobu coupling to yield compounds 64–66. The dihydroxylated aliphatic chain was attached via treatment with allyl bromide followed by dihydroxylation to afford compound 68 (Scheme 9).
Scheme 9.

Synthesis of alkyl sugar biaryl analogues.
In addition to the aliphatic amine analogues synthesized, several simplified non-sugar molecules were appended in lieu of the noviose sugar. These non-sugars represent simplified functionalities manifested by compound 4,30 a recently described inhibitor of the Hsp90 C-terminus (Figure 2).30 Substitutions were selected to probe hydrogen-bonding interactions, dimensions, and improve solubility. Carbamates (69–71) with variable substitution prepared through standard conditions. Phosphate ester 71 was introduced through an esterification reaction to increase the hydrophilicity of the inhibitor and finally, methyl and toluene sulfonic esters (73, 74) were incorporated using the corresponding sulfonic chlorides to explore both the hydrogen bonding network and dimensions of the pocket similarly to that reported by Renoir and co-workers34 (Scheme 10).
Scheme 10.

Synthesis of non-sugar biaryl analogues.
Upon construction of these alkyl and non-sugar compounds, they were evaluated for anti-proliferative activity against various cancer cell lines. The anti-proliferative data are summarized in Tables 4 and 5. All of the tertiary amine analogues demonstrated notable anti-proliferative activity of ~1 µM, while the secondary amine analogues maintained activity of ~3–5 µM against both breast cancer cell lines. The tertiary amine analogues homologated by two methylene groups (65a–c) exhibited activity of 1–3 µM, which is comparable to the piperidine analogues. Moreover, homologation by three methylene groups (66a–c) resulted in compounds that manifested anti-proliferative activity in the mid-nanomolar range, a finding that is consistent with the piperidine ring analogues. In contrast, the diol-containing analogues, when appended to the 8-methyl (68a) or 8-methoxy (68b) coumarin scaffolds were inactive, but resulted in modest activity when appended to the 6-methoxy coumarin (68c). Furthermore, the aliphatic amine and corresponding dihydroxylated analogues manifested modest activities against the prostate cancer cell lines as well.
Table 4.
Anti-proliferative activity of novobiocin analogues with aliphatic sugar replacement.
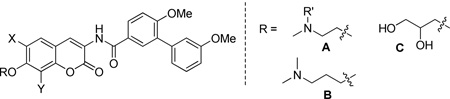 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound (IC50, µM) |
R | X | Y | R’ | SKBr3 | MCF-7 | LNCAP-LN3 | PC3-MM2 |
| 64a | A | H | CH3 | H | 5.36 ± 0.08a | 9.80 ± 0.11a | >100b | 14.01 ± 0.28b,d |
| 64b | A | H | OCH3 | H | 3.60 ± 0.28 | 4.32 ± 0.32 | 15.24 ± 7.96 | 9.12 ± 5.92 |
| 64c | A | OCH3 | CH3 | H | 3.15 ± 0.54 | 6.23 ± 0.14 | >100 | 11.77 ± 5.94 |
| 65a | A | H | CH3 | CH3 | 1.02 ± 0.13 | 1.46 ± 0.08 | 6.65 ± 12.43 | 4.17 ± 19.64 |
| 65b | A | H | OCH3 | CH3 | 1.77 ± 0.12 | 3.08 ± 0.08 | 5.22 ± 3.25 | 6.20 ± 2.22 |
| 65c | A | OCH3 | CH3 | CH3 | 1.42 ± 0.21 | 1.29 ± 0.17 | 6.28 ± 2.22d | 4.87 ± 2.91 |
| 66a | B | H | CH3 | -- | 0.60 ± 0.01 | 0.50 ± 0.03 | 37.51 ± 54.24 | 12.85 ± 11.42 |
| 66b | B | H | OCH3 | -- | 0.91 ± 0.14 | 1.53 ± 0.14 | 8.03 ± 7.93 | 4.08 ± 1.83 |
| 66c | B | OCH3 | CH3 | -- | 0.49 ± 0.20 | 0.70 ± 0.15 | 4.71 ± 0.21d | 5.40 ± 3.15 |
| 68a | C | H | CH3 | -- | >50 | >50 | NT | 0.19 ± 0.33 |
| 68b | C | H | OCH3 | -- | >50 | >50 | 0.96 ± 1.20 | 1.66 ± 6.05 |
| 68c | C | OCH3 | CH3 | -- | >50 | >50 | NT | NT |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Values represent mean ± standard deviation from dose response curves for at least two separate experiments performed in duplicate.
Table 5.
Anti-proliferative activity of non-sugar analogues.
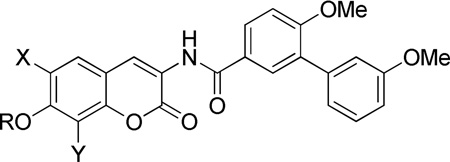 | |||||||
|---|---|---|---|---|---|---|---|
| Compound (IC50, µM) |
X | Y | R | SKBr3 | MCF-7 | LNCAP-LN3 | PC3-MM2 |
| 6a41 | H | CH3 | H | 8.88 ± 0.48a | 6.93 ± 0.48a | 3.21 ± 1.78b | 5.36 ± 2.71b |
| 12a41 | H | CH3 | COCH3 | 0.98 ± 0.02 | 1.40 | 1.50 ± 1.00 | 2.85 ± 1.66 |
| 8532 | H | CH3 | Noviose | 7.50 ± 0.80 | 18.70 ± 1.44 | NT | NT |
| 73a41 | H | CH3 | Ms | 7.33 ± 0.96 | 8.85 ± 0.88 | 19.96 ± 42.67 | >100 |
| 74a41 | H | CH3 | Ts | > 100 | > 100 | 0.58 ± 1.34 | >100 |
| 69a | H | CH3 | CONH2 | 3.02 ± 0.56 | 1.16 ± 0.08 | 2.61 ± 1.09 | 6.37 ± 4.31 |
| 70a41 | H | CH3 | CONHMe | 2.40 ± 0.16 | 1.72 ± 0.16 | 3.75 ± 0.76 | 5.22 ± 2.16 |
| 71a | H | CH3 | CONMe2 | 39.85 ± 0.48 | 74.35 ± 3.92 | 5.40 ± 6.58 | >100 |
| 72a | H | CH3 | PO(OMe)2 | > 100 | > 100 | 3.65 ± 5.73 | >100 |
| 6b | OCH3 | CH3 | H | 3.23 ± 1.20 | 11.70 ± 1.92 | 15.79 ± 32.60 | 11.23 ± 30.70 |
| 12b | OCH3 | CH3 | COCH3 | 5.72 ± 0.02 | 1.50 ± 0.24 | 1.05 ± 0.59 | 1.69 ± 2.05 |
| 8633 | OCH3 | CH3 | Noviose | 58.80 ± 1.04 | > 100 | 26.57 ± 67.44 | >100 |
| 73b | OCH3 | CH3 | Ms | > 100 | > 100 | >100 | NT |
| 74b | OCH3 | CH3 | Ts | > 100 | > 100 | NT | >100 |
| 69b | OCH3 | CH3 | CONH2 | 6.83 ± 0.24 | 6.29 ± 0.88 | 3.55 ± 3.44 | 2.62 ± 2.04 |
| 70b | OCH3 | CH3 | CONHMe | 1.53 ± 0.16 | 7.78 ± 1.68 | 4.77 ± 8.37 | 3.13 ± 2.34 |
| 71b | OCH3 | CH3 | CONMe2 | 6.17 ± 1.68 | 34.4 ± 7.20 | 12.75 ± 0.32d | 4.04 ± 16.68 |
| 72b | OCH3 | CH3 | PO(OMe)2 | 26.48 ± 0.80 | 24.22 ± 2.40 | 1.36 ± 2.80 | 31.32 ± 54.87 |
| 6c | H | OCH3 | H | > 100 | 5.32 ± 0.08 | 6.18 ± 7.43 | >100 |
| 12c | H | OCH3 | COCH3 | 20.50 ± 1.28 | 8.62 ± 2.00 | 14.62 ± 23.63 | 19.33 ± 50.90 |
| 8733 | H | OCH3 | Noviose | 13.90 ± 0.96 | 9.00 ± 4.32 | 0.11 ± 0.02 | 1.44 ± 0.32 |
| 73c | H | OCH3 | Ms | >100 | >100 | 0.87 ± 1.32 | NT |
| 74c | H | OCH3 | Ts | >100 | >100 | 0.11 ± 4.68 | NT |
| 69c | H | OCH3 | CONH2 | 76.83 ± 1.84 | 5.56 ± 0.24 | 4.83 ± 3.87 | >100 |
| 70c | H | OCH3 | CONHMe | >100 | 6.54 ± 1.84 | 2.37 ± 1.44 | 1.68 ± 1.89 |
| 71c | H | OCH3 | CONMe2 | >100 | >100 | 15.73 ± 14.09 | 11.57 ± 10.04 |
| 72c | H | OCH3 | PO(OMe)2 | 18.07 ± 2.88 | >100 | 6.58 ± 4.14 | 11.46 ± 6.54 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Values represent mean ± standard deviation from dose response curves for at least two separate experiments performed in duplicate.
The anti-proliferative data for the non-sugar analogues revealed several interesting trends. The free phenol of each scaffold was nearly equal in activity to its noviosylated counterpart. Moreover, the acetylated phenols, in the case of the 8-methyl and 6-methoxy coumarins, displayed activities of ~1 µM against breast cancer cells. Similarly, the carbamate and methylated carbamate were well tolerated when appended to 8-methyl and 6-methoxy coumarins, resulting in low micromolar analogues. Addition of another methyl group to the carbamate severely compromised activity. The methyl sulfonic ester was only tolerated when appended to the 8-methyl scaffold, while the toluene sulfonic ester was not tolerated by any analogue. Likewise, addition of the phosphate ester decreased activity of the analogues. Overall, hydrogen bonding and limited hydrophobic bulk are essential features for optimal binding.
Synthesis of optimized scaffolds using most promising noviose replacements
Based on the biological evaluation discussed thus far, we identified the N-methyl-4-hydroxy-piperidine and 3-(dimethylamino)propan-1-ol as optimal noviose replacements. Therefore, these surrogates were appended to three previously identified and optimized coumarin scaffolds, containing either the 2-indole or prenylated benzamide side chain in lieu of the biaryl system.32, 42 As detailed earlier, the analogues were assembled in a modular fashion, allowing sequential coupling of sugar mimics and the indole acid chloride or benzoic acid chloride with the desired scaffold (Scheme 11). Unlike previously discussed scaffolds, the Mitsunobu ether coupling was performed prior to amide coupling due to solubility issues arising from the indole-containing free phenol.
Scheme 11.

Retrosynthesis of azasugar indole novobiocin analogues.
As seen in Scheme 12, phenol 732, 33 was coupled with the azasugar using Mitsunobu conditions to yield protected scaffold 75. The vinylogous amide was liberated via hydrogenolysis and coupled with indole acyl chloride 76, which was generated from commercially available indole acid, to yield 78.32 The analogues containing a prenylated benzamide side chain (77) were assembled via the same sequence (Scheme 12), to yield alkyl amine analogues 79 and 80. The prenylated side chain, 77, was assembled as described previously by Burlison and co-workers.29
Scheme 12.

Synthesis of compound optimized compounds.
These compounds were evaluated for their anti-proliferative activity. The piperidine-containing analogues with an indole side chain (78a–c) exhibited increased anti-proliferative activity when attached to the 8-methyl and 6-methoxy coumarin (78a and 78c). 78c was notably 10-fold more active against SKBr3 cells when compared to its biaryl counterpart 61c. In contrast, attachment of this sugar surrogate to the 8-methoxy coumarin (78b) compromised its activity versus the biaryl-containing analogue 61b. In the case of the prenylated benzamide side chain (79a–c and 80a–c), attachment of the azasugar increased activity against both cell lines, with the 6-methoxy coumarin manifesting superior activity (79c and 80c). In addition, the intermediate acetylated phenols 79a–c demonstrated comparable activity to the hydrolyzed phenol products 80a–c. Based on the results obtained with the biaryl containing analogues, it was proposed that appendage of the aliphatic amine to the coumarins with an indole side chain would result in increased activity (see Table 4). However, the anti-proliferative activity of these indole analogues (82a–c) was maintained or slightly decreased in nearly every case. When these alkyl sugars were attached to the coumarins containing a prenylated benzamide side chain, the compounds manifested anti-proliferative activity in the mid-nanomolar range against SKBr3 cells. Moreover, compounds built upon the 6-methoxy coumarin scaffold (83c and 84c) exhibited the best activity against both cell lines.
Finally, several analogues from each of the libraries were selected for testing against a panel of melanoma and head and neck squamous cell carcinoma cell lines. In preliminary studies, C-terminal Hsp90 inhibitors have demonstrated modest activity against melanoma cells and have shown promise in treating head and neck cancers as well. Unpublished in vivo data produced by the Cohen lab has demonstrated tumor regression upon sustained treatment with a C-terminal Hsp90 inhibitor previously synthesized.33 Analogues were selected that exhibited diverse structural features to probe the efficacy of these analogues against these cancers. B16F10 (murine melanoma), SKMEL28 (human melanoma), MDA1986 (human head and neck squamous cell carcinoma (HNSCC)), and JMAR (human oral squamous cell carcinoma) were selected for this panel of testing. The majority of the analogues tested manifested mid to low micromolar activity against these cell lines. While some of the compounds exhibited better activity against melanoma than head and neck cancers (61b), others were potent in a specific melanoma (57b) or a specific head and neck (17a) cell line. The low micromolar activity of 78c and 82c against all of the cell lines warrant further investigation.
Validation of Hsp90 inhibition via Western blot analysis
To confirm that the anti-proliferative activities exhibited by the optimized sugar analogues result from Hsp90 inhibition, analogues 80c and 83c were evaluated by Western blot analyses (Figure 7). Notably, compounds 61a, 12b, 70a, and 87 have already been shown to exert their anti-proliferative activities through an Hsp90-dependent mechanism, as confirmed through previously executed Western blot analyses.30, 33, 41 It was essential that these optimized derivatives demonstrate selective Hsp90-dependent client protein degradation, versus a loading control, to confirm the compounds reported herein also manifest Hsp90 inhibition. Figure 7 shows that in MCF-7 cells, the Hsp90-dependent client proteins Akt, Raf-1, and Her2 were degraded in a concentration-dependent manner upon treatment with the compounds. Hsp90 client protein degradation occurred at concentrations that paralleled the observed anti-proliferative IC50 values of 0.54 ± 0.03 µM, for 80c and 0.70 ± 0.04 µM for 83c confirming that Hsp90-dependent client protein degradation is causative for inhibition of cell growth. Furthermore, Hsp90 protein levels remained constant at all concentrations tested, which is consistent with C-terminal Hsp90 inhibition. Since the non-Hsp90-dependent protein, actin, was not affected by these analogues, it was concluded that selective degradation of Hsp90-dependent proteins occurred.
Figure 7.

Western blot analyses of Hsp90 client protein degradation assays against MCF-7 cells. Concentrations (in µM) of 80c (top) and 83c (bottom) are indicated above each lane. G (geldanamycin, 500 nM) and D (DMSO) were respectively employed as positive and negative controls.
Conclusion
Several libraries of novobiocin analogues with various structural features were designed, synthesized and evaluated. From the library of sugar mimics, we identified the pyranose as optimal and that a 2’-hydroxy group is indispensable. Several low micromolar analogues containing modified sugars were also identified. From the series of azasugar-containing analogues, we concluded that replacement of noviose with a piperidine ring resulted in consistent low micromolar anti-proliferative activities. The piperidine sugars confirmed that interactions made between noviose and the binding pocket could also be maintained with azasugars. In addition, the acyclic library produced several aliphatic sugar surrogates that manifested mid-nanomolar activity. This library confirmed that flexibility is tolerated and that noviose can be replaced with a simple acyclic moiety to maximize the potency of novobiocin analogues. Finally, the synthesis of optimized scaffolds with various cytotoxic benzamide side chains identified several novobiocin analogues that exhibited low nanomolar anti-proliferative activity. These analogues confirmed that results obtained from earlier studies could be applied to these appendages and produce compounds that warrant further study.
Supplementary Material
Table 2.
Anti-proliferative activity of cyclohexyl analogues.
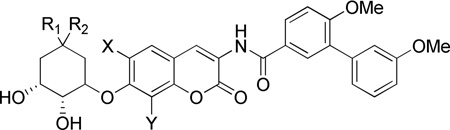 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound (IC50, µM) |
R1 | R2 | X | Y | SKBr3 | MCF-7 | LNCAP-LN3 | PC3-MM2 |
| 43a | CH3 | CH3 | H | CH3 | >100a | >100a | 1.24 ± 0.17b,d | 2.49 ± 1.70b |
| 43b | CH3 | CH3 | H | OCH3 | 3.45 ± 1.73 | 1.56 ± 0.03 | NT | NT |
| 43c | CH3 | CH3 | OCH3 | CH3 | >100 | >100 | NT | NT |
| 44a | H | CH3 | H | CH3 | >100 | >100 | 1.14 ± 0.67 | 4.09 ± 1.63 |
| 44b | H | CH3 | H | OCH3 | 6.38 ± 0.71 | 8.52 ± 0.36 | NT | NT |
| 44c | H | CH3 | OCH3 | CH3 | >100 | >100 | NT | NT |
| 4541 | H | H | H | CH3 | >100a | >100 | 1.58 ± 0.75 | 4.04 ± 0.38d |
| 45b | H | H | H | OCH3 | 7.44 ± 0.36 | 5.46 ± 0.36 | NT | NT |
| 45c | H | H | OCH3 | CH3 | 8.18 ± 0.79 | 10.13± 1.04 | NT | NT |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Values represent mean ± standard deviation from dose response curves for at least two separate experiments performed in duplicate.
Table 6.
Anti-proliferative activity of optimized analogues.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound (IC50, µM) |
R | X | Y | R’ | R” | SKBr3 | MCF-7 | LNCAP-LN3 | PC3-MM2 |
| 78a | A | H | CH3 | C | -- | 0.48 ± 0.09a | 0.57 ± 0.03a | 11.83 ± 0.54b | 11.40 ± 5.25b |
| 78b | A | H | OCH3 | C | -- | 2.58 ± 0.28 | 1.86 ± 0.08 | 12.64 ± 0.32d | 7.93 ± 4.18 |
| 78c | A | OCH3 | CH3 | C | -- | 0.11 ± 0.01 | 0.52 ± 0.04 | 1.47 ± 0.53 | 0.87 ± 0.46 |
| 79a | A | H | CH3 | D | Ac | 0.58 ± 0.04 | 1.18 ± 0.16 | NT | 2.12 ± 3.32 |
| 79b | A | H | OCH3 | D | Ac | 1.07 ± 0.14 | 1.64 ± 0.24 | NT | 3.98 ± 0.06d |
| 79c | A | OCH3 | CH3 | D | Ac | 0.42 ± 0.01 | 0.58 ± 0.02 | NT | 1.41 ± 0.04d |
| 80a | A | H | CH3 | D | H | 0.76 ± 0.14 | 1.09 ± 0.08 | NT | 1.37 ± 1.42 |
| 80b | A | H | OCH3 | D | H | 0.92 ± 0.01 | 1.54 ± 0.21 | NT | 3.53 ± 0.01d |
| 80c | A | OCH3 | CH3 | D | H | 0.42 ± 0.01 | 0.54 ± 0.02 | NT | 2.26 ± 1.43 |
| 82a | B | H | CH3 | C | -- | 1.13 ± 0.01 | 5.23 ± 0.22 | NT | 13.69 ± 0.18d |
| 82b | B | H | OCH3 | C | -- | 1.50 ± 0.13 | 1.41 ± 0.09 | 4.71 ± 1.23d | 8.95 ± 6.11 |
| 82c | B | OCH3 | CH3 | C | -- | 0.57 ± 0.09 | 0.56 | NT | 2.58 ± 4.47 |
| 83a | B | H | CH3 | D | Ac | 0.46 ± 0.15 | 1.18 ± 0.02 | NT | 1.42 ± 0.05d |
| 83b | B | H | OCH3 | D | Ac | 0.78 ± 0.17 | 2.14 ± 0.22 | NT | 4.59 ± 4.23 |
| 83c | B | OCH3 | CH3 | D | Ac | 0.36 ± 0.03 | 0.70 ± 0.03 | NT | 1.46 ± 0.03d |
| 84a | B | H | CH3 | D | H | 0.44 ± 0.02 | 1.35 ± 0.30 | NT | 1.81 ± 1.22 |
| 84b | B | H | OCH3 | D | H | 0.77 ± 0.08 | 3.26 ± 0.26 | NT | 9.24 ± 17.79 |
| 84c | B | OCH3 | CH3 | D | H | 0.39 ± 0.06 | 0.80 ± 0.07 | NT | 1.38 ± 0.02d |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Values represent mean ± standard deviation from dose response curves for at least two separate experiments performed in duplicate.
Table 7.
Anti-proliferative activity of selected analogues against melanoma and HNSCC.
| Compound (IC50, µM) |
B16F10 | SKMEL28 | MDA1986 | JMAR |
|---|---|---|---|---|
| 6b | 5.77 ± 0.96a | 12.40 ± 0.64a | 6.70 ± 1.20a | 24.60 ± 1.92a |
| 12a | 2.30 ± 0.32 | 2.14 ± 0.08 | 1.67 ± 0.16 | 12.60 ± 0.40 |
| 14a | 37.50 ± 6.56 | 14.90 ± 2.88 | 13.40 ± 4.24 | 2.79 ± 0.64 |
| 15a | 8.56 ± 2.16 | 15.80 ± 0.48 | 3.70 ± 0.64 | 6.14 ± 1.28 |
| 17a | 1.01 ± 0.48 | 1.47 ± 0.24 | 0.81 ± 0.40 | 16.10 ± 2.16 |
| 18a | 10.40 ± 1.20 | 19.60 ± 2.48 | 18.80 ± 3.68 | 3.60 ± 0.40 |
| 23a | 9.60 ± 1.12 | 10.65 ± 0.40 | 5.70 ± 0.56 | 5.80 ± 1.04 |
| 25a | 4.55 ± 1.20 | 5.74 ± 2.08 | 1.94 ± 0.40 | 12.64 ± 1.28 |
| 25b | 1.30 ± 0.16 | 6.41 ± 0.56 | 5.73 ± 0.96 | 2.71 ± 0.32 |
| 27a | 42.70 ± 3.36 | 18.40 ± 2.96 | 21.40 ± 3.12 | 14.90 ± 3.68 |
| 30a | 12.70 ± 1.44 | 21.40 ± 3.28 | 6.90 ± 2.08 | 24.10 ± 3.28 |
| 31a | 9.37 ± 1.04 | 12.40 ± 2.48 | 18.80 ± 3.76 | 6.19 ± 2.48 |
| 33a | >50 | 18.50 ± 5.76 | 22.90 ± 4.96 | 7.40 ± 1.68 |
| 40a | >50 | 23.80 ± 2.88 | 24.40 ± 2.00 | 6.70 ± 2.32 |
| 41a | >50 | 20.60 ± 2.16 | 21.80 ± 4.32 | 24.90 ± 2.80 |
| 42a | >50 | 18.60 ± 2.00 | 22.40 ± 3.36 | 7.50 ± 1.84 |
| 43a | 15.90 ± 2.16 | 3.47 ± 1.20 | 18.80 ± 0.64 | 8.50 ± 1.68 |
| 44a | 2.50 ± 0.48 | 11.41 ± 1.28 | 6.74 ± 2.08 | 2.73 ± 0.32 |
| 45a | 14.10 ± 2.08 | 2.94 ± 1.20 | 17.90 ± 1.44 | 13.90 ± 2.00 |
| 48a | 2.94 ± 0.08 | 5.41 ± 0.08 | 6.54 ± 0.56 | 3.40 ± 0.16 |
| 48b | 3.41 ± 0.32 | 5.49 ± 1.12 | 5.97 ± 0.40 | 6.71 ± 0.24 |
| 48c | 2.86 ± 0.24 | 5.41 ± 0.16 | 5.90 ± 0.40 | 11.60 ± 0.32 |
| 50c | 21.40 ± 1.28 | 27.60 ± 1.20 | 25.10 ± 2.08 | 35.10 ± 1.20 |
| 51a | 6.40 ± 1.92 | 10.79 ± 0.64 | 7.70 ± 0.56 | 11.40 ± 1.20 |
| 51b | 6.47 ± 2.80 | 1.48 ± 0.48 | 2.36 ± 0.08 | 1.94 ± 0.16 |
| 51c | 6.14 ± 0.64 | 31.50 ± 1.84 | 16.40 ± 1.68 | 18.40 ± 2.56 |
| 52a | 2.80 ± 0.40 | 6.14 ± 1.28 | 5.60 ± 1.04 | 5.89 ± 0.48 |
| 52b | 11.75 ± 0.40 | 11.40 ± 2.56 | 21.70 ± 1.12 | 11.60 ± 1.92 |
| 52c | 5.04 ± 0.32 | 13.80 ± 2.56 | 13.80 ± 3.44 | 11.50 ± 0.72 |
| 53a | 12.40 ± 2.00 | 20.9 ± 2.56 | 22.40 ± 1.36 | 15.14 ± 1.84 |
| 53c | 23.90 ± 2.88 | 29.40 ± 3.36 | 36.40 ± 1.20 | 32.80 ± 3.36 |
| 55b | 6.58 ± 0.48 | 5.90 ± 1.68 | 5.90 ± 0.48 | 6.70 ± 0.24 |
| 55c | 2.81 ± 0.32 | 5.73 ± 0.32 | 12.80 ± 1.28 | 24.50 ± 0.72 |
| 57a | 1.90 ± 0.24 | 2.79 ± 0.96 | 23.60 ± 2.88 | >50 |
| 57b | 0.64 ± 0.08 | 14.67 ± 0.40 | 12.80 ± 1.92 | 12.70 ± 1.44 |
| 57c | 1.84 ± 0.08 | 7.19 ± 0.24 | 6.40 ± 0.96 | 5.70 ± 0.32 |
| 59a | 2.76 ± 0.32 | 5.19 ± 0.16 | 2.74 ± 0.08 | 5.91 ± 0.40 |
| 59b | 2.14 ± 0.08 | 16.45 ± 1.36 | 10.90 ± 0.64 | 12.60 ± 0.32 |
| 59c | 1.84 ± 0.16 | 5.75 ± 0.24 | 14.60 ± 1.20 | 10.90 ± 0.48 |
| 60a | 2.60 ± 0.32 | 6.14 ± 0.56 | 4.50 ± 0.08 | 12.60 ± 0.32 |
| 60b | 1.51 ± 0.24 | 7.14 ± 0.40 | 11.40 ± 2.56 | 7.50 ± 0.64 |
| 60c | 2.67 ± 0.32 | 5.86 ± 1.20 | 13.20 ± 0.72 | 19.10 ± 2.56 |
| 61b | 0.41 ± 0.08 | 6.14 ± 0.32 | 13.10 ± 1.68 | 13.10 ± 0.64 |
| 61c | 36.73 ± 0.64 | 3.74 ± 0.24 | 1.54 ± 0.08 | 1.84 ± 0.08 |
| 64a | 37.40 ± 2.16 | 25.60 ± 2.96 | 21.90 ± 2.56 | 35.10 ± 2.16 |
| 65a | 15.70 ± 4.56 | 14.70 ± 1.92 | 17.50 ± 3.28 | 18.40 ± 2.00 |
| 78c | 1.19 ± 0.32 | 1.47 ± 0.08 | 1.42 ± 0.16 | 1.24 ± 0.24 |
| 82c | 2.03 ± 0.08 | 1.35 ± 0.08 | 1.81 ± 0.32 | 2.65 ± 0.24 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Acknowledgment
Authors gratefully acknowledge support of this project by the NIH/NCI (CA120458) and the ACS Division of Organic Chemistry Fellowship (A.D.).
Abbreviations
- Hsp90
90 kilodalton heat shock protein
- ATP
Adenosine triphosphate
- DNA
Deoxyribonucleic acid
- SAR
Structure–activity relationships
- Akt
Protein kinase B
- Her2
Human epidermal growth factor receptor 2
Footnotes
Supporting Information Available. Experimental procedures and characterization for all compounds.
References
- 1.Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocrine-Related Cancer. 2006;Vol. 13:S125–S135. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 2.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin. Biol. Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 3.Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Letters. 2004;562:11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 4.Brandt GEL, Blagg BSJ. Alternative strategies of Hsp90 modulation for the treatment of cancer and other diseases. Curr. Med. Chem. 2009;9:1447–1461. doi: 10.2174/156802609789895683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 6.Terasawa K, Minami M, Minami Y. Constantly updated knowledge of Hsp90. J. Biochem. 2005;137:443–447. doi: 10.1093/jb/mvi056. [DOI] [PubMed] [Google Scholar]
- 7.Buchner J. Hsp90 & co.-a holding for folding. Trends Biochem. Sci. 1999;24:136–141. doi: 10.1016/s0968-0004(99)01373-0. [DOI] [PubMed] [Google Scholar]
- 8.Picard D. Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. Heat-induced chaperone activity of Hsp90. J. Biol. Chem. 1996;271:2641–2645. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 10.Xiao L, Lu X, Ruden DM. Effectiveness of Hsp90 inhibitors as anti-cancer drugs. Mini Rev. Med. Chem. 2006;6:1137–1143. doi: 10.2174/138955706778560166. [DOI] [PubMed] [Google Scholar]
- 11.Zhao R, Houry WA. Hsp90: a chaperone for protein folding and gene regulation. Biochem. Cell Biol. 2005;83:703. doi: 10.1139/o05-158. [DOI] [PubMed] [Google Scholar]
- 12.Issacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 13.Blagg BSJ, Kerr TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med. Res. Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 14.Peterson LB, Blagg BSJ. To fold or not to fold: modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009;1:267–283. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Letters. 2004;206:149–157. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 16.Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 2008;15:2702–2717. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishop SC, Burlison JA, Blagg BSJ. Hsp90: a novel target for the disruption of multiple signaling cascades. Curr. Cancer Drug Targets. 2007;7:369–388. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]
- 18.Lewis RJ, Tsai FT, Wigley DB. Molecular mechanisms of drug inhibition of DNA gyrase. BioEssays. 1996;18:661–671. doi: 10.1002/bies.950180810. [DOI] [PubMed] [Google Scholar]
- 19.Reece RJ, Maxwell A. DNA gyrase: structure and function. Crit. Rev. Biochem. Mol. Biol. 1991;26:335–375. doi: 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- 20.Laurin P, Ferroud D, Schio L, Klich M, Dupuis-Hamelin C, Mauvais P, Lassaigne P, Bonnefoy A, Musicki B. Structure-activity relationship in two series of aminoalkyl substituted coumarin inhibitors of gyrase B. Bioorg. Med. Chem. Lett. 1999;9:2875–2880. doi: 10.1016/s0960-894x(99)00492-8. [DOI] [PubMed] [Google Scholar]
- 21.Ali JA, Jackson AP, Howells AJ, Maxwell A. The 43-kilodalton N-terminal fragment of the DNA gyrase B protein hydrolyzes ATP and binds coumarin drugs. Biochemistry. 1993;32 doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 22.Amolins MW, Blagg BSJ. Natural product inhibitors of Hsp90: potential leads for drug discovery. Mini Rev. Med. Chem. 2009;9:140–152. doi: 10.2174/138955709787316056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holdgate GA, Tunnicliffe A, Ward WHJ, Weston SA, Rosenbrock G, Barth PT, Taylor IWF, Paupit RA, Timms D. The entropic penalty of ordered water accounts for weaker binding of the antibiotic novobiocin to a resistant mutant of DNA gyrase: a thermodynamic and crystallographic study. Biochemistry. 1997;36:9663–9673. doi: 10.1021/bi970294+. [DOI] [PubMed] [Google Scholar]
- 24.Lewis RJ, Singh OMP, Smith CV, Skarzyknski T, Maxwell A, Wonacott AJ, Wigley DB. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai FTF, Singh OMP, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O'Brien R, Ladbury JE, Wigley DB. The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin. Proteins. 1997;28:41–52. [PubMed] [Google Scholar]
- 26.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 27.Allan RK, Mok D, Ward BK, Ratajczak T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90. J. Biol. Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- 28.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. Hsp90 inhibitors identified from a library of novobiocin analogues. J. Am. Chem. Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 29.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of Hsp90. J. Am. Chem. Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 30.Shelton SN, Shawgo ME, Comer SB, Lu Y, Donnelly AC, Szabla K, Tanol M, Vielhauer GA, Rajewski RA, Matts RL, Blagg BS, Robertson JD. KU135, a novel novobiocin-derived C-terminal inhibitor of Hsp90, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 2009;76:1314–1322. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burlison JA, Blagg BSJ. Synthesis and evaluation of coumermycin A1 analogues that inhibit the Hsp90 protein folding machinery. Org. Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 32.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. J. Org. Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 33.Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BSJ. The design, synthesis, and evaluation of coumarin ring derivatives of the novobiocin scaffold that exhibit antiproliferative activity. J. Org. Chem. 2008;73:8901–8920. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Bras G, Radanyi C, Peyrat J-F, Brio J-D, Alami M, Marsaud V, Stella B, Renoir J-M. New novobiocin analogues as antiproliferative agents in breast cancer cells and potential inhibitors of heat shock protein 90. J. Med. Chem. 2007;50:6189–6200. doi: 10.1021/jm0707774. [DOI] [PubMed] [Google Scholar]
- 35.Radanyi C, Le Bras G, Messaoudi S, Bouclier C, Peyrat J-F, Brion J-D, Marsaud V, Renoir J-M, Alami M. Synthesis and biological activity of simplified denoviose-coumarins related to novobiocin as potent inhibitors of heat-shock protein 90 (hsp90) Bioorg. Med. Chem. Lett. 2008;18:2495–2498. doi: 10.1016/j.bmcl.2008.01.128. [DOI] [PubMed] [Google Scholar]
- 36.Yu XM, Shen G, Blagg BSJ. Synthesis of (−)-noviose from 2,3-O-isopropylidene-D-erythronolactol. J. Org. Chem. 2004;69:7375. doi: 10.1021/jo048953t. [DOI] [PubMed] [Google Scholar]
- 37.Hosmane RS, Hong M. How important is the N-3 sugar moiety in the tight-binding interaction of coformycin with adenosine deaminase? Biochim. Biophys. Res. Commun. 1997;236:88–93. doi: 10.1006/bbrc.1997.6920. [DOI] [PubMed] [Google Scholar]
- 38.Eis C, Nidetzky B. Substrate-binding recognition and specificity of trehalose phosphorylase from Schizophyllum commune examined in steady-state kinetic studies with deoxy and deoxyfluoro substrate analogues and inhibitors. Biochem. J. 2002;363:335–340. doi: 10.1042/0264-6021:3630335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Werz DB, Seeberger PH. Carbohydrates as the next frontier in pharmaceutical research. Chemistry. 2005;11:3194–3206. doi: 10.1002/chem.200500025. [DOI] [PubMed] [Google Scholar]
- 40.Yu YM, Han H, Blagg BSJ. Synthesis of mono- and dihydroxylated furanoses, pyranoses, and an oxepananose for the preparation of natural product analogue libraries. J. Org. Chem. 2005;70:5599. doi: 10.1021/jo050558v. [DOI] [PubMed] [Google Scholar]
- 41.Donnelly A, Zhao H, Kusuma BR, Blagg BSJ. Cytotoxic sugar analogues of an optimized novobiocin scaffold. MedChemComm. 2010;1:165–170. [Google Scholar]
- 42.Zhao H, Kusuma BR, Blagg BSJ. Synthesis and evaluation of noviose replacements on novobiocin that manifest antiproliferative activity. ACS Med. Chem. Lett. 2010;1:311–315. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.