

Abstract
In this article, progress in understanding proton coupled electron transfer (PCET) in photosystem II is reviewed. Changes in acidity/basicity may accompany oxidation/reduction reactions in biological catalysis. Alterations in the proton transfer pathway can then be used to alter the rates of the electron transfer reactions. Studies of the bioenergetic complexes have played a central role in advancing our understanding of PCET. Because oxidation of the tyrosine results in deprotonation of the phenolic oxygen, redox active tyrosines are involved in PCET reactions in several enzymes. This review focuses on PCET involving the redox active tyrosines in photosystem II. Photosystem II catalyzes the light-driven oxidation of water and reduction of plastoquinone. Photosystem II provides a paradigm for the study of redox active tyrosines, because this photosynthetic reaction center contains two tyrosines with different roles in catalysis. The tyrosines, YZ and YD, exhibit differences in kinetics and midpoint potentials, and these differences must be due to noncovalent interactions with the protein environment. Here, studies of YD and YZ and relevant model compounds are described.
Keywords: oxygen evolution, EPR spectroscopy, manganese cluster, midpoint potential, water oxidation
Redox-active tyrosine residues have been proposed to be mechanistically important in several enzymes, including ribonucleotide reductase (RNR) [1], photosystem II (PSII) [2, 3], prostaglandin H synthase [4], galactose oxidase [5], glyoxyl oxidase [6], and Mycobacterium tuberculosis catalase-peroxidase [7]. Redox-active tyrosine residues may mediate long range electron transfer (ET) reactions {reviewed in [8]}. Participation in proton transfer (PT) is possible because the oxidation of a protonated tyrosine at neutral pH values is coupled with the deprotonation of the phenolic oxygen [9]. Changes in the pK of the proton-accepting group and in the mechanism of proton/electron transfer can then alter the rate of the reaction [10-13]. In direct coupling reactions, the proton and electron may move simultaneously, sequentially, or their movement may be non-synchronous [14]. In this article, the term proton coupled electron transfer, PCET, will be used to refer to any reaction in which both an electron and a proton are transferred, regardless of mechanism [15, 16].
PSII
PSII contains two redox active tyrosines, YD and YZ, with different functional roles. PSII is the thylakoid membrane protein complex, which carries out the light-catalyzed oxidation of H2O and reduction of plastoquinone [17, 18]. PSII is found in plants, cyanobacteria, and algae. Electron transfer is initiated when light absorption and energy transfer in the pigment antenna lead to electronic excitation of the primary chlorophyll (chl) donor. Subsequent electron transfer reactions result in the production of a chl cation radical, P680+, and the sequential reduction of a pheophytin, a plastoquinone, QA (a single electron-acceptor), and a second plastoquinone, QB (a two electron-two proton acceptor). P680+ is very unstable and readily generates other oxidized species [19]. One of these oxidized species, YZ• [2, 3], is an electron transfer intermediate between P680+ and the catalytic site for water oxidation, the oxygen-evolving complex (OEC) [3, 20, 21].
PSII structural models, derived from X-ray diffraction on three-dimensional crystals, were presented at 3.8-2.9 Å resolution [22-27]. The issue of X-ray-induced Mn reduction has been raised as a potential concern in the interpretation of PSII electron density [25, 28, 29]. PSII is composed of multiple polypeptide components. The CP47 and CP43 subunits are intrinsic, chlorophyll binding proteins [30]. The Mn-stabilizing protein (MSP, psbO, or the 33 kDa protein) is an extrinsic subunit that modulates the properties of the catalytic site [31-33]. Cyanobacterial and plant PSII contain cytochrome b559 [34], as well as other small intrinsic and extrinsic subunits [35]. Two of the hydrophobic PSII subunits, called D1 and D2, bind most of the prosthetic groups that are involved in electron transfer [22-27, 36].
The OEC contains both calcium and manganese [22-27]. Chloride also plays an important role in oxygen evolution {reviewed in [37]} and has recently been identified 6.5 Å from the manganese cluster [27]. The OEC cycles among five oxidation states, called the Sn states, where n refers to the number of oxidizing equivalents stored at the active site [38]. Each S state advancement is driven by one light-driven charge separation in the reaction center ({for examples, see models in [39, 40]}. Oxygen release occurs during the S3 to S0 transition from an unstable intermediate, known as the S4 state. Four flashes are required to produce molecular oxygen from water, and the S1 state is the dark-adapted state [38]. Therefore, the oxygen-evolving reactions exhibit period four oscillations, with oxygen produced on the third flash and then on every fourth flash [38]. The S state transitions occur in the microsecond to millisecond time regime, with a rate that slows as oxidizing equivalents are stored in the OEC [20, 41].
Two redox active tyrosines, YZ and YD, in PSII
PSII is a prototypical system for the study of redox active amino acids, because two redox active tyrosine residues are involved in light-induced electron transfer reactions, but with different functional roles [19, 42, 43]. Site-directed mutagenesis showed that YZ is tyrosine 161 of the D1 polypeptide and that YD is tyrosine 160 of the D2 polypeptide [44-48]. Both redox active tyrosines are neutral radicals in the oxidized form, suggesting that PCET occurs [2, 3, 49]. YZ mediates electron transfer events between the OEC and the chl donor and is required for oxygen evolution [20, 21, 46, 47]. The other redox active tyrosine, YD, is also oxidized via P680+ and is in slow redox equilibrium with the OEC [2, 50-52]. YD may be involved in assembly of the OEC [53], but is not required for oxygen evolution [44, 45].
The PSII redox active tyrosines are in C2 symmetrically related positions in the D1 and D2 subunits (Figure 1). However, only one of the two side chains, YZ, is involved in the electron transfer events that lead to water oxidation. Such structural symmetry, but functional asymmetry, is also observed in the purple bacterial reaction center, which does not oxidize water. In this enzyme, there are two bacteriopheophytin acceptors, positioned with approximately C2 symmetry, but only one is used in functionally significant electron-transfer events [54].
Figure 1.

The environments of YZ (A) and YD (B) from the 2.9 Å X-ray structure of PSII [27]. In (A), the manganese ions are gray, and the calcium ion is orange.
Functional differences between YZ and YD
The tyrosyl radicals in PSII are distinguished by their different reduction kinetics and midpoint potentials {reviewed in [19, 42, 43]}. YZ and YD had midpoint potentials that differ by ~200 mV [47, 55]. YD• was stable after illumination for minutes to hours, depending on the PSII preparation, and its EPR signal (Figure 2) was observed as a dark stable signal {for examples, see [2, 43, 56]}. In oxygen-evolving samples, YZ• was reduced by the OEC on the microsecond time scale, with a rate that depends on S state [20]. In samples from which manganese, calcium, and chloride were removed (OEC-depleted), the EPR lifetime of YZ• increased to the hundreds of millisecond time scale [57, 58]. EPR signals from YZ• were detected in OEC-depleted preparations [59] (Figure 2) and in oxygen-evolving PSII [20, 60, 61]. Recent experiments were performed at temperatures >100 K and at high microwave powers, and the YZ• radical was shown to decay on the millisecond time scale after flash excitation, due to recombination with the PSII acceptor side [60, 61]. At these temperatures, the expected electron spin-spin interaction between the manganese cluster and YZ• collapsed, due to an increase in the relaxation rate of the metal cluster. It was proposed that YZ• may act as a hydrogen atom (H•) abstractor during the S state cycle [62], but in the current X-ray structures, YZ is too far from the OEC to directly abstract hydrogen from substrate water [22-27]. Currently, it is hypothesized that YZ is an intermediary in electron transfer on each S state transition [20, 21].
Figure 2.

Structures and spectra of tyrosyl radicals. (A) shows the dihedral angle at the methylene carbon, with the angles between the hydrogens and the pZ orbital defined as Θ1 and Θ2; (B) model of tyrosyl radical; (C) EPR spectra of the light induced signal from YZ• and YD• (or Z and D, solid line) and the dark stable YD• (or D, dashed line).
EPR studies of isotopically labeled tyrosinate model compounds revealed that tyrosine oxidation occurs from the aromatic ring (Figure 2). Oxidation generated a neutral radical with spin density located on the 1′, 3′, and 5′ carbon atoms and the phenolic oxygen [49, 63]. Rotation around the C1′-Cβ bond (Figure 2) altered the EPR line shape. The EPR lineshapes and g values of YZ• and YD• resembled the tyrosyl radical produced in tyrosinate powders at 77 K. The g values of the YZ• and YD• were consistent with the deprotonation of both residues (Figure 2) when oxidized [2, 3]. There was one spin of YD• per reaction center, and up to one spin of YZ• was generated in OEC-depleted preparations {reviewed in [19, 42, 43]}.
Because YD and YZ have different functions and midpoint potentials, each tyrosine must have a different interaction with its environment. Spectroscopic differences between the tyrosyl radicals were reported [3, 64-68]. Crystal structures of cyanobacterial PSII [24-27] showed that the environments of YZ and YD differ in the detailed placement of the calcium-manganese cluster and hydrophilic amino acids (Figure 1). YZ was 5 Å from the calcium in the OEC, while YD is 28 Å away from the metal cluster. YZ was predicted to hydrogen bond with H190D1 and is 5 Å from D170D1 and 8 Å from R357CP43. YZ may also interact electrostatically with a second histidine (H332D1) residue and a second aspartic acid (D342D1) residue, which coordinate manganese. YD was predicted to form a hydrogen bond with H189D2 and is 7 Å from an D333D2-R180D2 salt bridge. In addition, YD had potential pi-cation interactions with R272 of the CP47 subunit, at 8 Å, and with R294 of the D2 subunit, at 6.5 Å. The corresponding arginines are not found in the environment of YZ. Overall, the pockets are distinguished by increased hydrophobicity near YD.
Mechanism of PCET reactions
The mechanism of PCET may distinguish YD and YZ. Supporting this idea, differential pH effects on the oxidation kinetics of YD and YZ were observed [52]. Marcus electron transfer theory states that the electron transfer rate constant depends on the Gibbs free energy for the reaction, ΔGo, and on λ, the energy required to reorganize the nuclear configuration from that of the reactants to products [69]. The treatment is based on activated complex theory and simple harmonic potentials. The key parameters can be summarized in a plot of free energy versus reaction coordinate. Two overlapping parabolas represent potential energy surfaces for the reactants in the initial state and products in the final, electron transferred state. Changes in driving force, ΔGo (the free energy difference between reactants and products) cause vertical displacements of the parabolas; changes in λ cause horizontal displacements. Larger horizontal displacements (larger values of λ) produce larger activation barriers. At constant λ, increases in driving force increase and then decrease the rate, generating the well known inverted region. The maximum rate occurs when the absolute value of the driving force equals λ. The rate of electron transfer varied exponentially with distance, with an exponential factor, β, which is approximately 1.4 Å−1 [70]. However, the protein environment can alter the value of β, the reorganization energy and the driving force to fine-tune the reaction rate [71]. Theoretical treatments for PCET reactions, which are constrained to occur over short distances, have emerged {see for example [72, 73]. A Marcus-type treatment has also been applied to explain PCET [10, 74].
Three pathways for a PCET reaction, involving a redox active tyrosine, can be imagined (Figure 3) [10, 75, 76]. In the first pathway, proton transfer occurs first, followed by electron transfer (PTET). In a second pathway, electron transfer precedes proton transfer (ETPT). In a third pathway, electron and proton transfer occur simultaneously in a concerted manner (CPET). For the first and second pathways, either electron or proton transfer can be rate-limiting. CPET has the advantage of avoiding the production of high energy intermediates, such as the tyrosyl cation radical produced in ETPT.
Figure 3.

Three possible PCET reactions for tyrosyl radical reduction, with an imidazole as the proton donor. Reprinted with permission from [122]. Copyright year 2007 American Chemical Society.
These three mechanisms can be distinguished by the magnitude of the solvent isotope effect {reviewed in [10]}. ETPT shows only a small (<1.3) kinetic isotope effect, while a more significant kinetic isotope effect (1.6-81.0) is expected for CPET [10, 77-89]. PTET will give only a small equilibrium isotope effect (~0.5 ΔpK) in 2H2O buffers [10, 90, 91].
The pH dependence of the rate constant also helps to distinguish PCET pathways. PTET predicts a pH dependent rate with a rate constant that changes by a factor of 10 per pH unit [10, 89, 92, 93]. ETPT will exhibit no significant pH dependence, if electron transfer is the rate limiting step [89]. Under some conditions, CPET reactions have been reported to be pH dependent [75, 89, 91, 94], although the origin of this pH dependence is controversial {reviewed in [95]}.
Mechanism can also be investigated using temperature dependent studies. These experiments establish the reorganization energy for the PCET reaction, assuming an adiabatic Marcus-type treatment [10, 74]. Substitution of modified electron transfer cofactors with altered potentials and pK is helpful in deducing mechanism. Examples are the use of 3-fluorotyrosine to investigate YZ electron transfer and the use of altered quinones to investigate electron transfer in the bacterial reaction center (see discussions below).
Models for PCET reactions
For tyrosyl radicals, model systems were used to investigate the effect of specific intramolecular interactions in hydrogen bonded, modified phenols [10]. Studies involving phenol derivatives with pendant bases showed that hydrogen bonding decreases the reduction potential of tyrosine [10]. Tyrosine and tryptophan residues were linked to ruthenium photosensitizers [75, 91, 96]. Studies of tyrosine appended to rhenium complexes [76] investigated the pH and buffer dependence of the PCET reactions [91]. Electrochemistry and other techniques have been used to examine PCET reactions for the oxidation of phenols in water [89, 97].
A CPET, rather than the stepwise PTET or ETPT, mechanism was inferred for tyrosine/phenol model compounds under some conditions [10, 75, 76, 91, 97, 98]. For example, in the work of [10, 99], PCET in phenols with pendant, hydrogen bonded bases was studied. The bases were an amine, an imidazole, and a pyridine. The phenol was oxidized with an accompanying intramolecular proton transfer to the hydrogen bonded base. From studies of the dependence of the rate on driving force and temperature, it was concluded that adiabatic Marcus theory can be applied to the PCET reaction. Further, evaluation of the kinetic data led to the suggestion that proton transfer occurs by a CPET mechanism. It was pointed out that the reactions proceed with a large reorganization energy (~2.4 eV or 56 kcal/mole, for one example). The suggested criteria for CPET were threefold. First, the magnitudes of the isotope effects (1.6-2.8) were considered to be consistent with CPET, but not with an ETPT or PTET mechanism. Second, it was concluded that the intermediates produced in ETPT and PTET (the phenoxyl cation radical and the phenolate species) were too high in energy to be accessible during the reactions. Third, the activation energy was observed to depend on the driving force for the reaction [10, 99]. For the reaction rates measured, this was judged to be consistent with CPET, but inconsistent with ETPT and PTET.
A CPET mechanism was also proposed for linked tyrosine-histidine-ruthenium model compounds [75, 84, 100]. For example, in the model compound experiments of [75], photoexcitation of ruthenium reduces an exogenous acceptor. The tyrosine then transfers an electron to the oxidized ruthenium. The experiment was conducted by monitoring the ground state recovery of Ru(II) at 450 nm. For pH values below the pK of the tyrosine (~10), the rate constant increased with pH and had a solvent isotope effect of ~2.0 [75, 82]. Under these conditions, a large reorganization energy, 2 eV, was deduced from studies of the temperature dependence. For pH values above the pK of the tyrosine, the tyrosine was deprotonated, and the rate increased and became pH independent. The reorganization energy decreased to 0.9 eV. The interpretation of these results was that mechanism is CPET below pH 10, but is simple ET, from the unprotonated tyrosine, above pH 10 [75]. It was suggested that a large reorganization energy is a characteristic of the CPET transfer, and that the observed pH dependence arises from the entropy term in the driving force {but see discussion in [95]}.
Calculations were conducted on a model of this system using multistate continuum theory [84]. The solute was described by a multistate valence bond model, the transferring hydrogen nucleus was treated quantum mechanically, and the solvent was represented as a dielectric continuum. The free energy surfaces for ET as a function of a single solvent coordinate or for PCET as a function of two solvent coordinates, corresponding to PT and ET, were calculated. Rate expressions for the ET and CPET reactions were also provided. The pH dependence of the CPET reaction was attributed to the decrease in driving force with pH {[84], but see [89] and [95] references therein). The larger rate for the pure ET reaction was attributed to a smaller solvent reorganization energy for ET and a vibrational overlap factor in the CPET rate expression. The predicted temperature dependence, the solvent isotope effects, and the solvent reorganization energy were reproduced in the calculation [84]. The extension of this approach to more complex systems, which model the PSII reaction center, would be welcome.
Protein and peptide models for PCET
The complexity of enzyme structures, such as PSII, can make it difficult to isolate and manipulate specific variables that influence rate and mechanism. Therefore, the development of simpler peptide/protein electron transfer model systems of defined structure is of value. In one previous approach, small electron transfer proteins such as cytochrome c or azurin, were modified to contain a bound ruthenium ion, with which intermolecular or intramolecular electron transfer was photoinitiated {see, for example, [13, 101] and references therein}.
Another approach is to use a designed, de novo sequence as a framework to investigate electron transfer. The effect of noncovalent interactions on the potential of redox active cofactors, such as hemes, iron sulfur clusters, and other metal centers, has been systematically explored in these peptide models or maquettes {for examples, see refs [102-108]}. For redox active tyrosines, previous work has reported a shift in tyrosine reduction potential in designed helical proteins that contain tyrosine residues [109, 110]. The redox potential of tyrosine was modulated by placement in a non-polar environment and by shielding from proton acceptors [109, 110]. Cyclodecapeptides have also been used to model redox active tyrosine residues in proteins [111].
A beta hairpin maquette exhibits a PCET reaction
Another peptide system in which specific intermolecular and covalent interactions can be probed was recently described. Peptide A consisted of 18 amino acids, IMDRYRVRNGDRIHIRLR, and folded into a beta hairpin [112] (Figure 4). For previous examples of designed beta hairpin peptides, see refs [113, 114]. Peptide A contained a single tyrosine residue (Y5), which was oxidized by UV photolysis or by electrochemical techniques. The experimental results suggested that this peptide exhibits an interstrand PCET reaction between a histidine (H14) and Y5, which are involved in a pi-pi stacking interaction. Substitutions at the histidine were observed to eliminate inflection points in the Pourbaix diagram, which plots peak potential versus pH [112, 115]. This result implied that the inflection points are due to oxidation-induced pK shifts in the histidine. Fits to the data gave pK values for the histidine in the oxidized and reduced forms of the peptide [115]. The extracted pK values suggested a shift from histidine pK 6 in the reduced form of the peptide to histidine pK 9 in the oxidized form. This analysis implied that oxidation of the tyrosine is accompanied by protonation of the histidine in the mid pH region. Although histidine and tyrosine are not directly hydrogen bonded, proton transfer may occur through a bridge of one or more hydrogen bonded water molecules [91, 97, 112, 116]. This peptide exhibits an example of a PCET reaction in which the electron and proton acceptors are two distinct species. Electrochemical data also provided evidence that histidine protonation decreases tyrosine midpoint potential. This effect was attributed to electrostatic stabilization of electron density on the tyrosyl radical phenolic oxygen [112].
Figure 4.
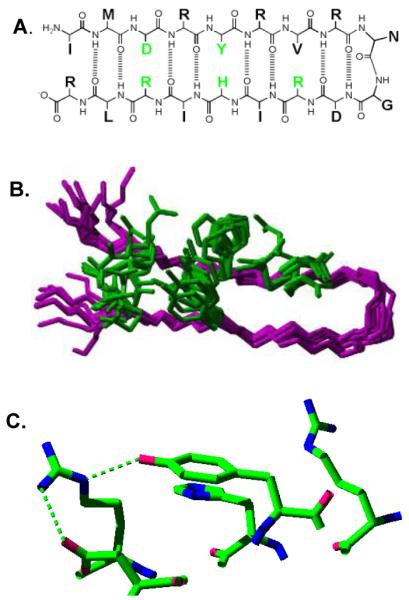
Structure of a beta hairpin peptide, IMDRYRVRNGDRIHIRLR, in which a tyrosine-histidine pi-pi interaction lowers the tyrosine redox potential and a PCET reaction occurs between tyrosine and histidine. (A) shows the primary sequence and some of the predicted hydrogen bonds and cross strand interactions; (B) is the overlap of the 20 lowest energy 2-D NMR structures, with only five amino acid side chains shown (see part A, green); and (C) is the redox active tyrosine and its immediate environment in the averaged, minimized NMR structure. Predicted hydrogen bonds are shown with dashed lines. Reprinted with permission from [112]. Copyright year 2007 American Chemical Society.
YD has a lower midpoint potential than YZ [47, 55], and its protein environment is distinguished by potential pi-cation interactions with arginine residues. In peptide A, the single tyrosine is hydrogen bonded to an arginine residue, R16, and a second arginine, R12, has a pi-cation interaction with Y5. Beta hairpin maquettes were used to evaluate the effect of these hydrogen bonding and electrostatic interactions on the PCET reactions [115]. Optical titrations showed that R16 substitution changes the pK of Y5. Removal of R16 or R12 increased the midpoint potential for tyrosine oxidation. The effects of R12 substitution were consistent with the midpoint potential difference, which is observed for the PSII redox active tyrosine residues. These results demonstrated that a pi-cation interaction, hydrogen bonding, and changes in the nature of PCET reactions can alter redox active tyrosine function. Each interaction may contribute equally to the control of midpoint potential [115].
PCET and YD in PSII
In PSII structures (Figure 1), YD was predicted to hydrogen bond to H189D2 [24-27]. Spectroscopic evidence suggested that proton transfer occurs from a protonated YD to the nearby histidine, H189D2 [117], and that the neutral radical, YD•, is hydrogen bonded to this histidine [118]. In site directed mutants at H189D2, reconstitution of imidazole accelerated the rate of YD• reduction. This result implied that YD proton transfer is rate limiting and that proton and electron transfer are kinetically coupled [117]. Mutations at H189D2 substantially changed the EPR lineshape of YD [119-121]. At pH 7.5, reaction-induced FT-IR spectroscopy showed a perturbation of the H189D2 NH stretching vibrations. The perturbation was attributed to a proton transfer reaction from the protonated imidazole side chain of H189D2 to YD• [117].
The effect of solvent isotope exchange on the reduction kinetics of YD• was examined. Experiments were conducted as a function of pL (the negative logarithm of the lyonium ion concentration). The kinetic isotope effect (KIE) on YD• reduction was measured by EPR spectroscopy [122]. YD• was detected independently of YZ•, because YZ• decayed in an interval that was not used in kinetic analysis. The rate of YD• reduction was slow, and so it was assumed that PT, which occurs over short distances {discussed in [10] and references therein}, does not limit the reaction rate. These studies showed that YD proton transfer reactions proceed with different mechanisms at high and low pH [122, 123]. YD• reduction exhibited a pL-dependent rate constant, with a minimum at ~pL 7.0-7.5 (Figure 5). At high pL (>7.5), the values of the KIE were greater than 2.0 and were consistent with a CPET mechanism. At low pL, smaller KIE’s were observed, and this result was attributed to a PTET mechanism in which the protonation of YD• occurs first. The rate acceleration observed at low pH was consistent with PTET.
Figure 5.
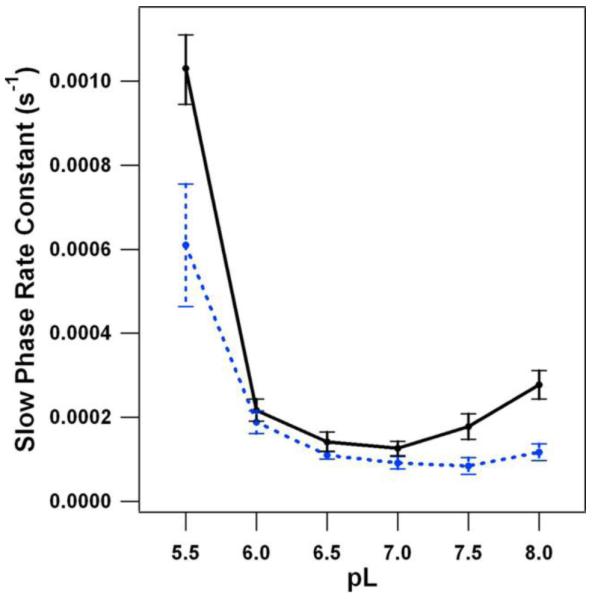
pL dependence of the YD• reduction rate, as determined by EPR spectroscopy, in 1H2O (black) and 2H2O (blue) buffers. This slow phase corresponds to ≥85% of the signal amplitude. Reprinted with permission from [122]. Copyright year 2007 American Chemical Society.
A proton inventory was recorded to determine the number of protons transferred at high pH [123]. This technique uses changes in the mole fraction of 2H2O to determine the number of protons transferred in the rate-limiting step of a reaction [124]. The decay of YD• was monitored as a function of 2H2O mole fraction at pL 8.0, where there is a significant isotope effect. Again, EPR transients were recorded on a time scale in which YZ• does not contribute to the data. When the rate was plotted versus 2H2O content, hypercurvature was observed. This hypercurvature provided evidence for the existence of multiple, proton-donation pathways to YD• (Figure 6). For comparison, a rocking model [125] in which histidine donates and then reaccepts a proton would give a linear proton inventory, corresponding to the transfer of one proton in the transition state. Simulation of the proton inventory data suggested that at least one of the proton transfer pathways involves the transfer of more than one proton (Figure 6). Modeling of the data supported the conclusion that there are two competing proton transfer pathways, one involving one proton and the other a multiproton (≥3 proton) pathway (Figure 6). It was proposed that one pathway involves H189D2 as proton donor and that the other pathway involves water as the proton donor [123].
Figure 6.

Two possible models for proton transfer involving YD•, based on the observation of hypercurvature in the proton inventory. The arrows indicate proton, not electron, movement. A. In pathway 1, histidine (blue) is involved in the multiproton pathway, which must involve three or more protons, of which two are shown. In pathway 2, a water molecule (red) is proposed to act as a single proton donor. B. In pathway 1, histidine (blue) is proposed to act as a single proton donor. In pathway 2, a chain of water molecules (red) are involved in the multiproton pathway, which must involve three or more protons, of which only two are shown. Reprinted with permission from [123]. Copyright year 2009 American Chemical Society.
H190D1 and YZ in PSII
In PSII structures, YZ was predicted to hydrogen bond to H190D1 [24-27]. YZ was suggested to be protonated at high temperatures [61] and will therefore lose a proton when oxidized. From chemical complementation studies, it was proposed that H190D1 is the proton acceptor for YZ and that the hydroxyl proton of YZ• remains bound to H190D1 during the lifetime of the radical [126, 127]. In these studies, the rate of YZ oxidation was monitored by optical studies of P680+ reduction at 432.5 and at 810 nm. The rate of YZ• reduction was monitored by optical studies at 242 nm. Mutants at H190D1 did not bind the OEC and are therefore OEC-depleted [126, 128, 129]. The rates of YZ oxidation and reduction slowed in site directed mutations at H190D1, but accelerated when imidazole and other small bases were reconstituted. An imidazole effect was observed both in wildtype and mutant PSII [126]. The mutant result suggested that YZ proton transfer is rate limiting and that electron and proton transfer are coupled for the oxidation and the reduction reaction. The fact that imidazole addition gave increased rates both for YZ oxidation and reduction even in wildtype PSII, which contains H190D1, was attributed to a suboptimal interaction between YZ and H190D1, in the absence of the manganese cluster [126, 127]. High concentrations of imidazole also accelerated P680+ reduction in the Y161D1F mutant, which lacks tyrosine Z. The etiology of this effect is unclear.
Contrasting kinetic results were obtained by room and low temperature EPR studies in a H190D1Q mutant [129]. In that work, the EPR lineshape of YZ• was only slightly altered by the mutation {see also [130]}. This was in contrast to the situation in the H189D2 mutant, in which substantial YD• lineshape changes were observed (see above). The reason for this difference between the D1 and D2 mutants is not yet clear. At −10°C, , the yield of YZ• in His190D1Q was found to be lower than that of wildtype, but only by a factor of 1.6, and the reduction kinetics were not significantly altered by the mutation. At room temperature, the YZ• yield was decreased by a factor of 2.6 in this mutant, but again, the kinetics of YZ• reduction appeared similar to wildtype [129]. These results suggested that an alternant proton donor, other than H190, is active in YZ• reduction reactions under these conditions. One possible explanation is that the identity of the proton donor/acceptor is different for the oxidation and reduction reaction [129]. This could occur if YZ• oxidation induces a local structural change, which changes its proximity to local bases and acids.
If oxidation of YZ triggers a conformational change, temperature dependence might be expected in the redox reactions. Significant temperature dependence was observed in the yield of the YZ• EPR signal in the His190D1Q mutant [129]. The yield at 4 °C was 70% of the −10 °C yield, and the yield at 21 °C was 40% of the −10 °C yield. However, there was no significant change in the overall kinetics of YZ• decay at the three temperatures. This observed temperature dependence in the His190D1Q mutant was a reversible phenomenon, with a recovery of 80% of the yield when the 21°C sample was reequilibrated at −10°C. The field-swept EPR spectra acquired under continuous illumination were indistinguishable at all three temperatures (data not shown). In summary, EPR analysis of the H190D1Q mutant suggests complexity in the YZ proton transfer reactions, which is congruent with some of the interpretations described below.
PCET reactions and YZ
Based on EPR studies, it was suggested that the mechanism of YZ PCET is different in oxygen-evolving and OEC-depleted PSII [61]. OEC removal may cause changes in the environment of YZ•, including more disordered hydrogen bonding to the tyrosyl radical [61, 65, 67, 68, 131]. Also, in the presence of the OEC, YZ• is in a more electropositive environment, compared to YZ• in an OEC-depleted preparations [61]. A synopsis of EPR and optical studies on PCET reactions in oxygen-evolving and OEC-depleted PSII is given below. Investigations have focused both on the oxidation and reduction reaction.
EPR spectroscopy investigates solvent isotope effects in OEC-depleted and oxygen-evolving PSII
Solvent isotope and pH effects on YZ redox reactions were reported using EPR spectroscopy to study spinach PSII. In [132], an EPR study was reported on oxygen-evolving and OEC-depleted PSII at room temperature. The kinetics of YZ• reduction were monitored. No significant solvent isotope effect was observed on YZ• reduction at pL 6.0 in OEC-depleted PSII, but KIE values of 2.9, 1.3, and 1.6 were reported for the S1 to YZ•, S2 to YZ•, and S3 to YZ• reduction reactions. These isotope effects could be due to PCET events at the OEC or at YZ. The value for the S1 to YZ• reduction reaction was larger than the KIE measured by optical spectroscopy, which gave values of 1.3-1.4 for each S state transition in the same type of PSII preparation [133]. This discrepancy could be caused by a difference in the extent of solvent isotope exchange in the two cases. In [134], another room temperature EPR study of YZ• reduction from the OEC was reported. In this case, PSII retained the manganese cluster, but Sr+2 was substituted for Ca+2 in order to slow the rate of YZ• reduction. A KIE of 1.4 was reported at pL 6.5, suggesting that metal ion substitution did not change the mechanism.
An EPR study reported a proton inventory for the YZ oxidation reaction in OEC-depleted spinach PSII [135]. The isotope and pH effects were assessed indirectly by measurement of YZ• yield under illumination. The findings were that one proton transferred during the rate-limiting step of the reaction and that the oxidation rate of YZ was pH dependent. The derived values were reported as KIE=1.4 at pL≤5 and pL>7 and as KIE=5 at pL 6.0. Thus, this experiment supports the conclusion that there is a small isotope effect on the YZ oxidation reaction. A more detailed interpretation of the mechanism awaits further studies.
Optical spectroscopy suggests two different proton acceptors for YZ in OEC-depleted PSII and shows no significant solvent isotope effect for YZ oxidation in oxygen-evolving PSII
Optical spectroscopy was used to study YZ oxidation in oxygen-evolving and OEC-depleted PSII from pea [136]. The reduction of P680+ was monitored. In OEC-depleted PSII, the overall decay time was pH dependent and slowed as the pH decreased. The data were fit with three exponentials. The two fastest phases were attributed to electron transfer between YZ and P680+. The fast phase (1 microsecond half decay time) was largely pH independent, and the slower kinetic component (35 microsecond half decay time at pH 4) exhibited only a five-fold decrease in rate from pH 8.0 to pH 4.0. However, the amplitudes were pH dependent, with the fast phase dominating at high pH. The fast phase was insensitive to solvent isotope exchange; the slower phase showed an isotope effect of 2.5. In oxygen-evolving centers, the rate of YZ to P680+ electron transfer occurred in the nanosecond time regime and showed no significant isotope effect. These results concerning the nanosecond rates agree with previous reports {see [133, 137, 138] and references therein}. In oxygen-evolving centers below pH 5, the nanosecond components were replaced by slower components, attributed to a pH-induced inactivation of PSII centers {[136], but see [138]}.
The experiments of ref [136] on OEC-depleted pea PSII were reinterpreted in [75], based on comparison to new model studies. Similarities in reorganization energy and in the pH dependence of the rates were observed when OEC-depleted PSII was compared to ruthenium-tyrosine containing compounds. In the model compounds, it was concluded that a CPET mechanism applies at low pH and that at pH values above the pK of tyrosine, pure ET occurs. Comparison to data from OEC-depleted PSII showed a similarity in pH dependence and activation energy in the low pH regime. This similarity led to the suggestion that a CPET mechanism applies at pH <7.6 and a pure ET mechanism applies at pH >7.6 in PSII. At pH<7.6, proton release was proposed to occur directly into bulk water. At pH>7.6, it was suggested that a hydrogen bond forms between H190 and YZ; this hydrogen bond was suggested to facilitate electron transfer by decreasing the reorganization energy {see also [84]}. The hydrogen bond between H190 and YZ was suggested to be broken at lower pH [75]. Thus, this interpretation suggests two different proton acceptors for YZ in OEC-depleted PSII. The nature of proton transfer in oxygen-evolving PSII is not clear, because no significant isotope effect was observed. More experiments are necessary to elucidate this question.
Optical spectroscopy on cyanobacterial PSII shows significant solvent isotope effects and pH dependence for YZ redox reactions
The cyanobacterial Y160D2F mutant lacks YD, and thus the YZ redox reactions can be studied in a background that lacks the stable tyrosyl radical signal from YD. However, the mutation may alter proton transfer pathways on the donor side of PSII [139]. In OEC-depleted PSII from the Y160D2F mutant, the oxidation of YZ was monitored by decreases in the P680+ signal [131]. The KIE value was 3.6 at pL 7.0 (maximal) and was pH dependent. The oxidation rate also depended on pL, with the reaction rate increasing at higher pL values. The rate constants in 1H2O varied from 30.6 ms−1 at pH 5.5 to 1260 ms−1 at pH 9.5. There results were interpreted with a mechanism, which is electron transfer limited at high pH values and conformationally gated. 0at low pH values. However, results from fluorotyrosine labeling (described below) suggest a reinterpretation of these results. The rate of YZ reduction was also measured, by monitoring its recombination with QA− at 325 nm. The KIE for the reduction reaction was 2.0 and was smaller than the KIE for the oxidation reaction. The rate of the reduction reaction showed less dependence on pL. It was concluded that there was no rate limitation on proton transfer in the YZ reduction reaction [131]. However, in this case, the origin of the isotope effect is unclear.
In a study of OEC-depleted PSII from wildtype cyanobacteria (retaining YD), the rate of YZ oxidation and reduction were also monitored by optical spectroscopy. The kinetics of P680+ decay, as monitored at 810 nm, were fit with four exponentials. Again, pH dependence was observed. While the individual rate constants did not vary extensively with pH, the relative amplitudes showed variation [127]. At pH 7.5, the k−1 values were 340 ns, 3.4 μs, 21 μs, and 120 μs. Details of the observed pH dependence were different from the measurements of [131]. The isotope effects were also smaller and pH independent (1.7-2.6). Differences between ref [127] and [131] may be due to the use of the YF160D2 mutant in [131]. However, ref [127] attributed the difference to lower time resolution in the measurements reported in [131]. In ref [127], addition of imidazole and other bases accelerated the rate of YZ oxidation and reduction, even in these wildtype PSII preparations. The experiments were interpreted as supporting either a CPET mechanism or a mechanism in which proton transfer is rate limiting [127]. The observed influence of exogenous bases on electron transfer in wildtype PSII is interesting. The explanation was that when manganese is removed, the hydrogen bond between YZ and H190 is broken. The suggestion was that the rate acceleration in wildtype is due to access of exogenous proton donors to YZ•.
To summarize, studies of mutant and wildtype cyanobacterial PSII agree that the rate of YZ oxidation accelerates with increasing pH. Isotope effects have been reported on the oxidation and reduction reactions [127, 131]. Small differences in rates and isotope effects, when pea [136] and cyanobacteria [127, 131] were compared, may reflect biochemical differences between the preparations. Alterations in the PCET pathway may complicate interpretation of studies on the Y160D2 mutant [131].
Fluorinated tyrosine labeling suggests a YZ oxidation mechanism in OEC-depleted PSII
A study of PCET has been reported for YZ using fluorinated tyrosine [140]. Fluorinated tyrosine has been used previously to investigate the mechanism of PCET in RNR [141, 142]. In PSII, the extent of fluorination was reported as 75%, based on analysis of the altered electron spin echo spectrum. Fluorinated tyrosine has both an altered pK and midpoint potential [141]; therefore, global incorporation of fluorinated tyrosine is expected to change the pK and midpoint potential of YZ. In some PSII experiments, a site directed mutant was employed in which a histidine mutation changes the midpoint potential of P680+. The reduction of P680+ was monitored by optical spectroscopy, and the samples were OEC-depleted cyanobacterial PSII. The pH dependence of the rate was not altered in the fluorotyrosine-containing samples. Because the pK should decrease by 1.5 units in the fluorotyrosine derivative, it was concluded that the pH dependence reflects a base other than YZ. The rate at pH 9.2 was observed to increase in the fluorotyrosine samples, consistent with the pK change altering a rate-limiting step in the reaction. The activation energy, as assessed by the temperature dependence of the rate, decreased by 110 meV, which corresponds to the expected change in ΔG for the proton transfer reaction in fluorotyrosine. This was distinct from observations with the histidine mutant, which was assumed to change only the driving force for the reaction. From this, it was concluded that the change in activation energy is due to the pK change in fluorotyrosine, not from a difference in driving force. To explain these data, a CPET mechanism was proposed for YZ at low pH, and a sequential PTET mechanism was proposed at high pH [140]. It was concluded that the mechanism of PTET is limited by a proton transfer reaction. To explain the pH dependence, the authors proposed that the network of hydrogen bonds around YZ changes as a function of pH. It was also suggested that an alternate proton acceptor may be available, other than H190, at low pH.
To summarize, at low pH values, both [140] and [75] predict a CPET mechanism for YZ oxidation in OEC-depleted PSII. Fluorotyrosine labeling suggested a PTET mechanism at high pH and complexity in the proton transfer pathway [140]. The effect of OEC removal on YZ PCET remains to be evaluated.
Are redox reactions at YZ coupled with a conformational change?
Reaction-induced FT-IR and EPR spectroscopies were used to study tyrosine oxidation in tyrosinate powders and in tyrosine-peptides [143, 144]. This model compound work suggested that Y• reduction is accompanied by a conformational change. In model compounds, Y• can be generated by UV photolysis at liquid nitrogen temperatures (77 K). The difference or reaction-induced FT-IR spectra reflect the changes in CO and ring stretching vibrations, which are associated with formation of the radical. When the reaction-induced FT-IR spectra acquired on tyrosinate and on peptides were compared, the peptide spectra exhibited unique bands in the amide I (1650 cm−1) region of the spectrum [143-145]. The frequencies of the bands were found depend on amino acid composition and/or sequence. Further, an EPR transient, with a lifetime on the seconds time scale, was observed at 85 K in tyrosinate powders [144].
The EPR transient was attributed to a conformational change, which is expected from DFT studies. At room temperature, DFT calculations predict two stable conformers at the beta methylene bond for tyrosinate, but only one stable conformer for the tyrosyl radical [145, 146]. For tyrosine, conformer A had a ring dihedral angle of +168°, and conformer B had a ring dihedral angle of −69°. ESEEM and FT-IR studies suggested that the B conformer is favored by solvent interactions [143, 146]. For tyrosyl radical, DFT calculations predicted one stable conformer, with a ring dihedral angle of +165°. Therefore, when the radical is generated at low temperature, the initial, populated radical states have ring dihedral angles representative of the tyrosine ground state. The initially populated B state will be unstable at higher temperatures [147] and its relaxation could give rise to the EPR transient [144].
What is the origin of the amide I bands in the model Y• FT-IR spectra? Interestingly, amide I bands have also been detected in FT-IR studies of the tyrosyl radical in RNR. Reaction-induced FT-IR spectroscopy was used to study the hydroxurea-induced reduction of tyrosyl radical, Y122•, in the beta subunit [148]. EPR spectroscopy had previously provided evidence for a conformational change at the beta methylene position [149]. Reaction-induced FT-IR spectroscopy showed that reduction of the Y122• radical was accompanied by amide I contributions, which had similar frequencies to those observed in a pentapeptide, RSTYH [148]. RSTYH is the primary sequence surrounding Y122 [144].
One possible origin for the perturbation of the amide bond frequencies could be spin delocalization to the amide nitrogen [150]. However, this was not consistent with subsequent ESEEM studies of model compounds [146]. An alternate origin for the changes in amide I frequencies is an alteration in amide bond ionic character. Rotation around the amide bond changes the electronegativity of the amide nitrogen, the ionic character of the CN bond, and the force constants for the CN stretching modes [151]. Such a change in amide bond planarity could be the conformational change detected and could be driven by the change in electron density, which makes the phenolic oxygen atom more negative in the radical state [112].
In PSII, reaction-induced FT-IR spectra of YZ have also detected amide I frequencies, perturbed by YZ oxidation and reduction [150, 152, 153]. Structural changes preceding the S state transitions have been reported [154, 155]; these changes may correspond to alterations in amino acid protonation state and may be driven by YZ redox or conformational reactions. If oxidation of YZ causes a conformational change, such an alteration could expose the YZ phenolic oxygen to a different acid/base environment. For example, after oxidation of YZ and protonation of H190D1, such a conformational change could rotate YZ• towards positively charged species, such as Arg357 or the calcium ion (Figure 1). For recent suggestions concerning the role of calcium and Arg357 in the water oxidation cycle see refs [156, 157]. After YZ• reduction occurs, a decreased distance from a positive charge would stabilize a negative charge on YZ−, decreasing its pK. Because YZ− is now in a new microenvironment with a potentially different pK, a different species could protonate the tyrosyl radical. Thus, a local conformational change could control PCET reactions involving the redox active tyrosine and could provide a mechanism to translocate protons [156]. If YZ is conformationally flexible, then variations in experimental results could be due to small changes in biochemical preparations, pH, manganese content, or measurement temperatures. The idea that YZ is conformationally flexible in oxygen-evolving PSII has been proposed previously [67].
Do redox reactions at the OEC alter YZ PCET reactions?
As exhibited in the discussion above, YZ PCET reactions are complex, and there is no consensus yet concerning mechanism, at least in oxygen-evolving PSII. Redox-linked changes in the OEC may influence the protonation state of YZ and PCET reactions [61]. A speculative model giving an example of such a redox-linked reaction is shown in Figure 7. PCET to YZ• is depicted as ETPT, i.e., two sequential steps, in which a proton transfer follows a rate-limiting electron transfer. However, as discussed above, other possible mechanisms cannot be excluded at the present time. In Figure 7, transfer of protons from the oxidized OEC to a nearby base, B1, modulates the pK of B2, the immediate proton donor for YZ•. Redox-linked changes in the pK of B2 ensure that YZ• reprotonates and therefore has the high midpoint potential [158] necessary to drive S state advancement {see discussion in [61]}. This idea implies that the microenvironment of YZ and pK of proton donor/s acceptors may be S state dependent. More experiments on oxygen-evolving PSII will be necessary to evaluate YZ PCET reactions.
Figure 7.

Speculative two base model for YZ PCET reactions in the presence of the OEC, adapted from [61]. The reduction and protonation of YZ• may occur in these two sequential steps (ETPT) as shown, or by a PTET or CPET mechanism. In the first step (YZH oxidation), YZ is oxidized and loses an electron and a proton to form a neutral tyrosyl radical. B2 is the amino acid side chain that accepts the proton, forming H+B2. In the second step (S state advancement), the OEC is oxidized by YZ•, generating YZ−, and the pK of a second amino acid side chain, B1, is altered so that B1 protonates, forming H+B1. In the third step (YZ− protonation), generation of the positive charge on H+B1 changes the pK of H+B2 so that YZ− protonates to form YZH.
Comparison to PCET involving the quinone acceptors
Comparison can be made to PCET involving quinone acceptors in the bacterial reaction center, in which the mechanism of PCET has been elucidated. PCET events on the PSII acceptor side resemble acceptor side transfer in the bacterial reaction center.
In PSII and the bacterial reaction center, light induced electron transfer leads to the sequential reduction of two quinone molecules, QA and QB. Two sequential photo-induced reductions occur to generate a reduced quinol, QBH2, in the QB site. While QB is a two electron, two proton acceptor, QA is a single electron acceptor and is not protonated {reviewed in [159]}. Unlike PSII, the bacterial reaction center does not split water and lacks the redox active tyrosines. Instead, a cytochrome acts as an electron donor. Substitutions of quinones at the QA and QB sites in the bacterial reaction center led to the elucidation of PCET reactions between these electron acceptors. The quinone substitutions changed midpoint potential and pK.
For the first electron transfer from QA− to QB, it was observed that the rate constant did not depend on driving force, although a substantial activation barrier had been deduced for this reaction {see [160] and references therein}. These results suggested that electron transfer occurs alone, with no proton transfer, and that a protein conformation change controls the rate [160]. This conformation change was attributed to an alteration in the position of QB, which was observed in crystals under illumination [161]. For the second electron transfer from QA− to QB−, which involves proton transfer, it was concluded that reversible proton transfer occurs, followed by rate limiting electron transfer [92]. Thus, a two step PTET mechanism applies. The second protonation reaction, to form QBH2, follows the production of QBH−. The semiquinone anion radical, QBH−, was detected in the QB site of reaction centers, which contained a rhodoquinone with an altered pK value [93]. Comparison with donor side electron transfer in PSII suggests that quinone cofactors catalyze step-wise transfers, while YD and YZ involve CPET mechanisms.
Conclusions
Control of catalysis can occur through modulation of PCET reactions. Both the electron and proton tunnel, but the proton transfer is constrained to occur over short distances. When the distance is short, the proton and electron may be transferred together. However, when the distance is long, separate or orthogonal electron and proton acceptors may be used [11]. Modification of the proton transfer reaction can be a key feature in determining the electron transfer rate. In PSII, much remains to be determined about how the responsive protein matrix participates in PCET reactions. Model compounds and model peptides provide an arena, in which hypotheses generated from PSII can be tested. For example, a linked tyrosine-histidine-ruthenium model compounds mimic PCET reactions in OEC-depleted PSII [75, 84, 100]. A beta hairpin peptide [112, 115] exhibits a PCET reaction between tyrosine and histidine and shows a functional difference induced by a pi-cation interaction, which is consistent with expectations from the PSII structure. The mechanism of PCET may help to distinguish YD and YZ, but more information is necessary concerning PCET in oxygen-evolving PSII samples.
In this article, progress in understanding proton coupled electron transfer (PCET) in photosystem II, the photosynthetic water oxidizing complex, is reviewed. PCET may be catalyzed by redox active tyrosines. Photosystem II provides a paradigm for the study of redox active tyrosines and PCET, because this photosynthetic reaction center contains two tyrosines with different roles in catalysis. Here, studies of YD and YZ and relevant model compounds are described.
Acknowledgments
The PCET studies from the author’s group, described in this review, were supported by the National Institute of General Medical Sciences and the National Eye Institute at the National Institutes of Health (GM43272). The author acknowledges the many important contributions of her former students and postdoctoral fellows and thanks her current group, Dr. Cynthia Pagba, Dr. Robin Sibert, Tina Dreaden, Dr. Adam Offenbacher, Brandon Polander, and James Keough.
Abbreviations
- ET
electron transfer
- KIE
kinetic isotope effect
- L
lyonium ion
- OEC
oxygen-evolving complex
- PCET
proton coupled electron transfer
- PSII
photosystem II
- PT
proton transfer
- RNR
ribonucleotide reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sjoberg B-M, Reichard P, Graslund A, Ehrenberg A. The tyrosine free radical in ribonucleotide reductase from Escherichia Coli. J. Biol. Chem. 1978;253:6863–6865. [PubMed] [Google Scholar]
- [2].Barry BA, Babcock GT. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc. Nat. Acad. Sci. 1987;84:7099–7103. doi: 10.1073/pnas.84.20.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Boerner RJ, Barry BA. Isotopic labeling and EPR spectroscopy show that a tyrosine residue is the terminal electron donor, Z, in manganese-depleted photosystem II preparations. J. Biol. Chem. 1993;268:17151–17154. [PubMed] [Google Scholar]
- [4].Kulmacz RJ, Ren Y, Tsai A-L, Palmer G. Prostaglandin H synthase: Spectroscopic studies of the interaction with hydroperoxides and with indomethacin. Biochemistry. 1990;29:8760–8771. doi: 10.1021/bi00489a037. [DOI] [PubMed] [Google Scholar]
- [5].Whittaker MM, Whittaker JW. Tyrosine-derived free radical in apogalactose oxidase. J. Biol. Chem. 1990;265:9610–9613. [PubMed] [Google Scholar]
- [6].Whittaker MM, Kersten PJ, Nakamura N, Sanders-Loehr J, Schweizer ES, Whittaker JW. Glyoxal oxidase from Phanerochaete chrysosporium is a new radical-copper oxidase. J. Biol. Chem. 1996;271:681–687. doi: 10.1074/jbc.271.2.681. [DOI] [PubMed] [Google Scholar]
- [7].Chouchane S, Girotto S, Yu S, Magliozzo RS. Identification and characterization of tyrosyl radical formation in Mycobacterium tuberculosis catalase-peroxidase (KatG) J. Biol. Chem. 2002;277:42633–42638. doi: 10.1074/jbc.M207916200. [DOI] [PubMed] [Google Scholar]
- [8].Stubbe J, van der Donk WA. Protein radicals in enzyme catalysis. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- [9].Dixon WT, Murphy D. Determination of the acidity constants of some phenol radical cations by means of electron spin resonance. J. Chem. Soc. London, Faraday Trans. II. 1976;72:1221–1230. [Google Scholar]
- [10].Rhile IJ, Markle TF, Nagao H, DiPasquale AG, Lam OP, Lockwood MA, Rotter K, Mayer JM. Concerted proton-electron transfer in the oxidation of hydrogen bonded phenols. J. Am. Chem. Soc. 2006;128:6075–6088. doi: 10.1021/ja054167+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology. Phil. Trans. Royal Soc. B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moser C, Page C, Farid R, Dutton P. Biological electron transfer. J. Bioenerg. Biomembr. 1995;27:263–274. doi: 10.1007/BF02110096. [DOI] [PubMed] [Google Scholar]
- [13].Gray HB, Winkler JR. Long-range electron transfer. Proc. Nat. Acad. Sci. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chang CJ, Chang MCY, Damrauer NH, Nocera DG. Proton-coupled electron transfer: a unifying mechanism for biological charge transport, amino acid radical initiation and propagation, and bond making/bond breaking reactions of water and oxygen. Biochim. Biophys. Acta. 2004;1655:13–28. doi: 10.1016/j.bbabio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- [15].Huynh MHV, Meyer TJ. Proton-coupled electron transfer. Chem. Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Costentin C. Electrochemical approach to the mechanistic study of proton-coupled electron transfer. Chem. Rev. 2008;108:2145–2179. doi: 10.1021/cr068065t. [DOI] [PubMed] [Google Scholar]
- [17].Nelson N, Yocum CF. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 2006;57:521–565. doi: 10.1146/annurev.arplant.57.032905.105350. [DOI] [PubMed] [Google Scholar]
- [18].Barber J. Photosystem II: an enzyme of global significance. Biochem. Soc. Trans. 2006;34:619–631. doi: 10.1042/BST0340619. [DOI] [PubMed] [Google Scholar]
- [19].Barry BA. Tyrosyl radicals in photosystem II. Methods Enzymol. 1995;258:303–319. doi: 10.1016/0076-6879(95)58053-0. [DOI] [PubMed] [Google Scholar]
- [20].Babcock GT, Blankenship RE, Sauer K. Reaction kinetics for positive charge accumulation on the water side of chloroplast photosystem II. FEBS Lett. 1976;61:286–289. doi: 10.1016/0014-5793(76)81058-7. [DOI] [PubMed] [Google Scholar]
- [21].Gerken S, Brettel K, Schlodder E, Witt HT. Optical characterization of the immediate donor to Chlorophyll aII+ in O2-evolving photosystem II complexes. FEBS Lett. 1988;237:69–75. [Google Scholar]
- [22].Zouni A, Witt HT, Kern J, Fromme P, Krauß N, Saenger W, Orth P. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- [23].Kamiya N, Shen J-R. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 A resolution. Proc. Nat. Acad. Sci. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- [25].Biesiadka J, Loll B, Kern J, Irrgang K-D, Zouni A. Crystal structure of cyanobacterial photosystem II at 3.2 Å resolution: a closer look at the Mn-cluster. Phys. Chem. Chem. Phys. 2004;20:4733–4736. [Google Scholar]
- [26].Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- [27].Guskov A, Kern J, Gabdulkhakov A, Boser M, Zouni A, Saenger W. Nature Struc. Mol. Biol. 2009;19:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- [28].Yano J, Kern J, Irrgang K-D, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Nat. Acad. Sci. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grabolle M, Haumann M, Muller C, Liebisch P, Dau H. Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by X-ray irradiation at 10-300 K. J. Biol. Chem. 2006;281:4580–4588. doi: 10.1074/jbc.M509724200. [DOI] [PubMed] [Google Scholar]
- [30].Bricker TM. The structure and function of CPa-1 and CPa-2 in Photosystem II. Photosynth. Res. 1990;24:1–13. doi: 10.1007/BF00032639. [DOI] [PubMed] [Google Scholar]
- [31].Bricker T, Frankel L. The structure and function of the 33 kDa extrinsic protein of Photosystem II: A critical assessment. Photosynth. Res. 1998;56:157–173. [Google Scholar]
- [32].Hutchison RS, Steenhuis JJ, Yocum CF, Razeghifard RM, Barry BA. Deprotonation of the 33 kDa extrinsic, manganese stabilizing protein accompanies photooxidation of manganese in photosystem II. J. Biol. Chem. 1999;274:31987–31995. doi: 10.1074/jbc.274.45.31987. [DOI] [PubMed] [Google Scholar]
- [33].Sachs R, Halverson KM, Barry BA. Specific isotopic labeling and photooxidation-linked structural changes in the manganese-stabilizing subunit of photosystem II. J. Biol. Chem. 2003;278:44222–44229. doi: 10.1074/jbc.M307148200. [DOI] [PubMed] [Google Scholar]
- [34].MacDonald GM, Boerner RJ, Everly RM, Cramer WA, Debus RJ, Barry BA. Comparison of cytochrome b-559 content in photosystem II complexes from spinach and Synechocystis sp. PCC 6803. Biochemistry. 1994;33:4393–4400. doi: 10.1021/bi00180a037. [DOI] [PubMed] [Google Scholar]
- [35].Kashino Y, Lauber WM, Carroll JA, Wang Q, Whitmarsh J, Satoh K, Pakrasi H. Proteome analysis of a highly active photosystem II preparation from the cyanobacterium Synechocystis sp. PCC 6803 reveals the presence of novel polypeptides. Biochemistry. 2002;41:8004–8012. doi: 10.1021/bi026012+. [DOI] [PubMed] [Google Scholar]
- [36].Nanba O, Satoh K. Isolation of a photosystem II reaction center consisting of D-1 and D-2 polypeptides and cytochrome b-559. Proc. Nat. Acad. Sci. 1987;84:109–112. doi: 10.1073/pnas.84.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Popelkova H, Yocum CF. Current status of the role of Cl− ion in the oxygen-evolving complex. Photosynth. Res. 2007;93:111–121. doi: 10.1007/s11120-006-9121-5. [DOI] [PubMed] [Google Scholar]
- [38].Joliot P, Kok B. Oxygen evolution in photosynthesis. In: Govindjee, editor. Bioenergetics of Photosynthesis. Academic Press; New York: 1975. pp. 388–412. [Google Scholar]
- [39].McEvoy JP, Brudvig GW. Water-splitting chemistry of photosystem II. Chem. Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- [40].Meyer TJ, Huynh MHV, Thorp HH. The possible role of proton-coupled electron transfer (PCET) in water oxidation by photosystem II. Angew. Chem. 2007;46:5284–5304. doi: 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]
- [41].Dekker JP, Plijter JJ, Ouwehand L, van Gorkom HJ. Kinetics of manganese redox transitions in the oxygen-evolving apparatus of photosynthesis. Biochim. Biophys. Acta. 1984;767:176–179. [Google Scholar]
- [42].Pujols-Ayala I, Barry BA. Tyrosyl radicals in Photosystem II. Biochim. Biophys. Acta. 2004;1655:205–216. doi: 10.1016/j.bbabio.2003.07.010. [DOI] [PubMed] [Google Scholar]
- [43].Rutherford AW, Boussac A, Faller P. The stable tyrosyl radical in Photosystem II: why D? Biochim. Biophys. Acta. 2004;1655:222–230. doi: 10.1016/j.bbabio.2003.10.016. [DOI] [PubMed] [Google Scholar]
- [44].Debus RJ, Barry BA, Babcock GT, McIntosh L. Site-specific mutagenesis identifies a tyrosine radical involved in the photosynthetic oxygen-evolving complex. Proc. Nat. Acad. Sci. 1988;85:427–430. doi: 10.1073/pnas.85.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vermaas WFJ, Rutherford AW, Hansson Ö. Site-directed mutagenesis in photosystem II of the cyanobacterium Synechocystis sp. PCC 6803: Donor D is a tyrosine residue in the D2 protein. Proc. Nat. Acad. Sci. 1988;85:8477–8481. doi: 10.1073/pnas.85.22.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Debus RJ, Barry BA, Sithole I, Babcock GT, McIntosh L. Directed mutagenesis indicates that the donor to P680+ in photosystem II is Tyr-161 of the D1 polypeptide. Biochemistry. 1988;27:9071–9074. doi: 10.1021/bi00426a001. [DOI] [PubMed] [Google Scholar]
- [47].Metz JG, Nixon PJ, Rögner M, Brudvig GW, Diner BA. Directed alteration of the D1 polypeptide of photosystem II: evidence that tyrosine-161 is the redox component, Z, connecting the oxygen-evolving complex to the primary electron donor, P680. Biochemistry. 1989;28:6960–6969. doi: 10.1021/bi00443a028. [DOI] [PubMed] [Google Scholar]
- [48].Noren GH, Barry BA. The YF161D1 mutant of Synechocystis 6803 exhibits an EPR signal from a light-induced photosystem II radical. Biochemistry. 1992;31:3335–3342. doi: 10.1021/bi00128a005. [DOI] [PubMed] [Google Scholar]
- [49].Barry BA, El-Deeb MK, Sandusky PO, Babcock GT. Tyrosine radicals in photosystem II and related model compounds. J. Biol. Chem. 1990;265:20139–20143. [PubMed] [Google Scholar]
- [50].Babcock GT, Sauer K. Electron paramagnetic resonance Signal II in spinach chloroplasts. Biochim. Biophys. Acta. 1973;325:483–503. doi: 10.1016/0005-2728(73)90209-0. [DOI] [PubMed] [Google Scholar]
- [51].Babcock GT, Sauer K. Electron paramagnetic resonance signal in spinach chloroplasts II. Alternative spectral forms and inhibitor effects on the kinetics of Signal II in flashing light. Biochim. Biophys. Acta. 1973;325:504–519. doi: 10.1016/0005-2728(73)90210-7. [DOI] [PubMed] [Google Scholar]
- [52].Faller P, Debus RJ, Brettel K, Sugiura M, Rutherford AW, Boussac A. Rapid formation of the stable tyrosyl radical in photosystem II. Proc. Nat. Acad. Sci. 2001;98:14368–14373. doi: 10.1073/pnas.251382598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ananyev GM, Sakiyan I, Diner BA, Dismukes GC. A functional role for tyrosine-D in assembly of the inorganic core of the water oxidase complex of photosystem II and the kinetics of water oxidation. Biochemistry. 2002;41:974–980. doi: 10.1021/bi011528z. [DOI] [PubMed] [Google Scholar]
- [54].Deisenhofer J, Michel H. The photosynthetic reaction center from the purple bacterium Rhodopseudomonas viridis. Science. 1989;245:1463–1473. doi: 10.1126/science.245.4925.1463. [DOI] [PubMed] [Google Scholar]
- [55].Boussac A, Etienne AL. Midpoint potential of Signal II (Slow) in Tris-washed photosystem-II particles. Biochim. Biophys. Acta. 1984;766:576–581. [Google Scholar]
- [56].Kim S, Ayala I, Steenhuis JJ, Gonzalez ET, Razeghifard MR, Barry BA. Infrared spectroscopic identification of the C-O stretching vibration associated with the tyrosyl Z• and D• radicals in photosystem II. Biochim. Biophys. Acta. 1998;1366:330–354. doi: 10.1016/s0005-2728(98)00016-4. [DOI] [PubMed] [Google Scholar]
- [57].Boska M, Sauer K. Kinetics of EPR Signal IIvf in chloroplast photosystem II. Biochim. Biophys. Acta. 1984;765:84–87. doi: 10.1016/0005-2728(77)90062-7. [DOI] [PubMed] [Google Scholar]
- [58].Dekker JP, van Gorkom HJ, Brok M, Ouwehand L. Optical characterization of photosystem II electron donors. Biochim. Biophys. Acta. 1984;764:301–309. [Google Scholar]
- [59].Babcock GT, Sauer K. A rapid, light-induced transient in electron paramagnetic resonance Signal II activated upon inhibition of photosynthetic oxygen evolution. Biochim. Biophys. Acta. 1975;376:315–328. doi: 10.1016/0005-2728(75)90024-9. [DOI] [PubMed] [Google Scholar]
- [60].Zahariou G, Ioannidis N, Sioros G, Petrouleas V. The collapse of the tyrosine Z•-Mn spin-spin interaction above approximately 100 K reveals the spectrum of tyrosine Z•. An application of rapid-scan EPR to the study of intermediates of the water splitting mechanism of photosystem II. Biochemistry. 2007;46:14335–14341. doi: 10.1021/bi7018767. [DOI] [PubMed] [Google Scholar]
- [61].Ioannidis N, Zahariou G, Petrouleas V. The EPR spectrum of tyrosine Z• and its decay kinetics in O2-evolving photosystem II preparations. Biochemistry. 2008;47:6292–6300. doi: 10.1021/bi800390r. [DOI] [PubMed] [Google Scholar]
- [62].Hoganson C, Babcock GT. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- [63].Hulsebosch RJ, van der Brink JS, Niewenhuis SAM, Gast P, Raap J, Lugtenburg J, Hoff AJ. Electronic structure of the neutral tyrosine radical in frozen solution. Selective 2H-, 13C-, and 17O-isotope labeling and EPR spectroscopy at 9 and 35 GHz. J. Am. Chem. Soc. 1997;119:8685–8694. [Google Scholar]
- [64].MacDonald GM, Bixby KA, Barry BA. A difference FT-IR study of two redox-active tyrosine residues in photosystem II. Proc. Nat. Acad. Sci. 1993;90:11024–11028. doi: 10.1073/pnas.90.23.11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Warnke K, Babcock GT, McCracken J. Structure of the YD tyrosine radical in photosystem II as revealed by 2H electron spin echo envelope modulation (ESEEM) spectroscopic analysis of hydrogen hyperfine interactions. J. Am. Chem. Soc. 1994;116:7332–7340. [Google Scholar]
- [66].Bernard MT, MacDonald GM, Nguyen AP, Debus RJ, Barry BA. A difference infrared study of hydrogen bonding to the Z• tyrosyl radical of photosystem II. J. Biol. Chem. 1995;270:1589–1594. doi: 10.1074/jbc.270.4.1589. [DOI] [PubMed] [Google Scholar]
- [67].Tommos C, Tang X-S, Warncke K, Hoganson CW, Styring S, McCracken J, Diner BA, Babcock GT. Spin-density distribution, conformation, and hydrogen bonding of the redox-active tyrosine Yz in photosystem II from multiple electron magnetic resonance spectroscopies: Implications for photosynthetic oxygen evolution. J. Am. Chem. Soc. 1995;117:10325–10335. [Google Scholar]
- [68].Tang XS, Zheng M, Chisholm DA, Dismukes GC, Diner BA. Investigation of the differences in the local protein environments surrounding tyrosine radicals YZ and YD in photosystem II using wild-type and the D2-Tyr160Phe mutant of Synechocystis 6803. Biochemistry. 1996;35:1475–1484. doi: 10.1021/bi951489p. [DOI] [PubMed] [Google Scholar]
- [69].Marcus RA. Electron transfer reactions in chemistry: theory and experiment. Pure Appl. Chem. 1997;69:13–29. [Google Scholar]
- [70].Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature of biological electron transfer. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- [71].Beratan DN, Onuchic JN, Winkler JR, Gray HB. Electron-tunneling pathways in proteins. Science. 1992;258:1740–1741. doi: 10.1126/science.1334572. [DOI] [PubMed] [Google Scholar]
- [72].Cukier RI. Theory and simulation of proton-coupled electron transfer, hydrogen atom transfer, and proton translocation in proteins. Biochim. Biophys. Acta. 2004;1655:37–44. doi: 10.1016/j.bbabio.2003.06.011. [DOI] [PubMed] [Google Scholar]
- [73].Hammes-Schiffer S, Iordanova N. Theoretical studies of proton-coupled electron transfer reactions. Biochim. Biophys. Acta. 2004;1655:29–36. doi: 10.1016/j.bbabio.2003.07.009. [DOI] [PubMed] [Google Scholar]
- [74].Klinman JP. The role of tunneling in enzyme catalysis of C-H activation. Biochim. Biophys. Acta. 2006;1757:981–987. doi: 10.1016/j.bbabio.2005.12.004. [DOI] [PubMed] [Google Scholar]
- [75].Sjodin M, Styring S, Akermark B, Sun L, Hammarstrom L. Proton-coupled electron transfer from tyrosine in a tyrosine-ruthenium-tris-bipyridine complex: comparison with tyrosine Z oxidation in photosystem II. J. Am. Chem. Soc. 2000;122:3932–3936. [Google Scholar]
- [76].Reece SY, Nocera DG. Direct tyrosine oxidation using the MLCT excited states of rhenium polypyridyl complexes. J. Am. Chem. Soc. 2005;127:9448–9458. doi: 10.1021/ja0510360. [DOI] [PubMed] [Google Scholar]
- [77].Buhks E, Bixon M, Jortner J. Deuterium isotope effects on outer-sphere electron-transfer reactions. J. Phys. Chem. 1981;85:3763–3766. [Google Scholar]
- [78].Gould IR, Farid S. Specific deuterium isotope effects on the rates of electron transfer within geminate radical-ion pairs. J. Am. Chem. Soc. 1988;110:7883–7885. [Google Scholar]
- [79].Hille R. Electron transfer within xanthine oxidase: a solvent kinetic isotope effect study. Biochemistry. 1991;30:8522–8529. doi: 10.1021/bi00099a004. [DOI] [PubMed] [Google Scholar]
- [80].Huynh MHV, Meyer TJ, White PS. Proton-coupled electron transfer from nitrogen. A N-H/N-D kinetic isotope effect of 41.4. J. Am. Chem. Soc. 1999;121:4530–4531. [Google Scholar]
- [81].Shafirovich V, Dourandin A, Luneva NP, Geacintov NE. The kinetic deuterium isotope effect as a probe of a proton coupled electron fransfer mechanism in the oxidation of guanine by 2-aminopurine radicals. J. Phys. Chem. B. 2000;104:137–139. [Google Scholar]
- [82].Sjodin M, Styring S, Akermark B, Sun L, Hammarstrom L. The mechanism for proton-coupled electron transfer from tyrosine in a model complex and comparisons with YZ oxidation in photosystem II. Phil. Trans. Royal Soc. B. 2002;357:1471–1478. doi: 10.1098/rstb.2002.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Knapp MJ, Rickert K, Klinman JP. Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- [84].Carra C, Iordanova N, Hammes-Schiffer S. Proton-coupled electron transfer in a model for tyrosine oxidation in Photosystem II. J. Am. Chem. Soc. 2003;125:10429–10436. doi: 10.1021/ja035588z. [DOI] [PubMed] [Google Scholar]
- [85].Lehnert N, Solomon EI. Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J. Biol. Inorg. Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]
- [86].Weatherly SC, Yang IV, Armistead PA, Thorp HH. Proton-coupled electron transfer in guanine oxidation: effects of isotope, solvent, and chemical modification. J. Phys. Chem. B. 2003;107:372–378. [Google Scholar]
- [87].Costentin C, Robert M, Saveant J-M. Carboxylates as proton-accepting groups in concerted proton-electron transfers. electrochemistry of the 2,5-dicarboxylate 1,4-hydrobenzoquinone/2,5-dicarboxy 1,4-benzoquinone couple. J. Am. Chem. Soc. 2006;128:8726–8727. doi: 10.1021/ja0621750. [DOI] [PubMed] [Google Scholar]
- [88].Hatcher E, Soudackov AV, Hammes-Schiffer S. Proton-coupled electron transfer in soybean lipoxygenase: dynamical behavior and temperature dependence of kinetic isotope effects. J. Am. Chem. Soc. 2007;129:187–196. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- [89].Bonin J, Costentin C, Louault C, Robert M, Routier M, Savéant J-M. Intrinsic reactivity and driving force dependence in concerted proton-electron transfers to water illustrated by phenol oxidation. Proc. Nat. Acad. Sci. 2010;107:3367–3372. doi: 10.1073/pnas.0914693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Schowen KB, Schowen RL. Solvent isotope effects of enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- [91].Irebo T, Reece SY, Sjödin M, Nocera DG, Hammarström L. Proton-coupled electron transfer of tyrosine oxidation: buffer dependence and parallel mechanisms. J. Am. Chem. Soc. 2007;129:15462–15464. doi: 10.1021/ja073012u. [DOI] [PubMed] [Google Scholar]
- [92].Graige MS, Paddock ML, Bruce JM, Feher G, Okamura MY. Mechanism of proton-coupled electron transfer for quinone (QB) reduction in reaction centers of Rb. sphaeroides. J. Am. Chem. Soc. 1996;118:9005–9016. [Google Scholar]
- [93].Graige MS, Paddock ML, Feher G, Okamura MY. Observation of the protonated semiquinone intermediate in isolated reaction centers from Rhodobacter sphaeroides: Implications for the mechanism of electron and proton transfer in proteins. Biochemistry. 1999;38:11465–11473. doi: 10.1021/bi990708u. [DOI] [PubMed] [Google Scholar]
- [94].Costentin C, Robert M, Saveant J-M. Concerted proton-electron transfer reactions in water. Are the driving force and rate constant depending on pH when water acts as a proton donor or acceptor? J. Am. Chem. Soc. 2007;129:5870–5879. doi: 10.1021/ja067950q. [DOI] [PubMed] [Google Scholar]
- [95].Hammes-Schiffer S, Soudackov AV. Proton-coupled electron transfer in solution, proteins, and electrochemistry. J. Phys. Chem. B. 2008;112:14108–14123. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lomoth R, Magnuson A, Sjodin M, Huang P, Styring S, Hammarstrom L. Mimicking the electron donor side of photosystem II in artificial photosynthesis. Photosynth. Res. 2006;87:25–40. doi: 10.1007/s11120-005-9005-0. [DOI] [PubMed] [Google Scholar]
- [97].Costentin C, Louault C, Robert M, Savéant J-M. The electrochemical approach to concerted proton-electron transfers in the oxidation of phenols in water. Proc. Nat. Acad. Sci. 2009;106:18143–18148. doi: 10.1073/pnas.0910065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Biczók L, Gupta N, Linschitz H. Coupled electron-proton transfer in interactions of triplet C60 with hydrogen-bonded phenols: effects of solvation, deuteration, and redox potentials. J. Amer. Chem. Soc. 1997;119:12601–12609. [Google Scholar]
- [99].Rhile IJ, Mayer JM. One-electron oxidation of a hydrogen-bonded phenol occurs by concerted proton-coupled electron transfer. J. Amer. Chem. Soc. 2004;126:12718–12719. doi: 10.1021/ja031583q. [DOI] [PubMed] [Google Scholar]
- [100].Magnuson A, Berglund H, Korall P, Hammarstrom L, Akermark B, Styring S, Sun L. Mimicking electron transfer reactions in photosystem II: synthesis and photochemical characterization of a ruthenium(II) tris(bipyridyl) complex with a covalently linked tyrosine. J. Am. Chem. Soc. 1997;119:10720–10725. [Google Scholar]
- [101].Engstrom G, Rajagukguk R, Saunders AJ, Patel CN, Rajagukguk S, Merbitz-Zahradnik T, Xiao K, Pielak GJ, Trumpower B, Yu CA, Yu L, Durham B, Millett F. Design of a ruthenium-labeled cytochrome c derivative to study electron transfer with the cytochrome bc1 complex. Biochemistry. 2003;42:2816–2824. doi: 10.1021/bi027213g. [DOI] [PubMed] [Google Scholar]
- [102].Jones G, II, Vullev V, Braswell EH, Zhu D. Multistep photoinduced electron transfer in a de novo helix bundle: multimer self-assembly of peptide chains including a chromophore special pair. J. Am. Chem. Soc. 2000;122:388–389. [Google Scholar]
- [103].Lombardi A, Nastri F, Pavone V. Peptide-based heme-protein models. Chem. Rev. 2001;101:3165–3190. doi: 10.1021/cr000055j. [DOI] [PubMed] [Google Scholar]
- [104].Gibney BR, Huang SS, Skalicky JJ, Fuentes EJ, Wand AJ, Dutton PL. Hydrophobic modulation of heme properties in heme protein maquettes. Biochemistry. 2001;40:10550–10561. doi: 10.1021/bi002806h. [DOI] [PubMed] [Google Scholar]
- [105].Kennedy ML, Gibney BR. Proton coupling to [4Fe-4S]2+/+ and [4Fe-4Se]2+/+ oxidation and reduction in a designed protein. J. Am. Chem. Soc. 2002;124:6826–6827. doi: 10.1021/ja0171613. [DOI] [PubMed] [Google Scholar]
- [106].Discher BM, Noy D, Strzalka J, Ye S, Moser CC, Lear JD, Blasie JK, Dutton PL. Design of amphiphilic protein maquettes: Controlling assembly, membrane insertion, and cofactor interactions. Biochemistry. 2005;44:12329–12343. doi: 10.1021/bi050695m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J. Am. Chem. Soc. 2005;127:1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- [108].Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Evaluation of electron-withdrawing group effects on heme binding in designed proteins: Implications for heme a in cytochrome c oxidase. Inorg. Chem. 2006;45:4685–4694. doi: 10.1021/ic060072c. [DOI] [PubMed] [Google Scholar]
- [109].Tommos C, Skalicky J, Pilloud DL, Wand AJ, Dutton LP. De novo proteins as models of radical enzymes. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- [110].Di Bilio AJ, Crane BR, Wehbi WA, Kiser CN, Abu-Omar MM, Carlos RM, Richards JH, Winkler JR, Gray HB. Properties of photogenerated tryptophan and tyrosyl radicals in structurally characterized proteins containing rhenium(I) tricarbonyl diimines. J. Am. Chem. Soc. 2001;123:3181–3182. doi: 10.1021/ja0043183. [DOI] [PubMed] [Google Scholar]
- [111].Hossain MA, Thomas F, Hamman S, Saint-Aman E, Boturyn D, Dumy P, Pierre JL. Cyclodecapeptides to mimic the radical site of tyrosyl-containing proteins. J. Pept. Sci. 2006;12:612–619. doi: 10.1002/psc.769. [DOI] [PubMed] [Google Scholar]
- [112].Sibert RS, Josowicz M, Porcelli F, Veglia G, Range K, Barry BA. Proton-coupled electron transfer in biomimetic peptide as a model of enzyme regulatory mechanism. J. Am. Chem. Soc. 2007;129:4393–4400. doi: 10.1021/ja068805f. [DOI] [PubMed] [Google Scholar]
- [113].Syud FA, Stanger HE, Gelman SH. Interstrand side chain-side chain interactions in a designed beta hairpin: Significance of both lateral and diagonal pairings. J. Am. Chem. Soc. 2001;123:8667–8677. doi: 10.1021/ja0109803. [DOI] [PubMed] [Google Scholar]
- [114].Tatko C, Waters ML. The geometry and efficacy of cation-pi interactions in a diagonal position of a designed beta hairpin. Protein Sci. 2003;12:2443–2452. doi: 10.1110/ps.03284003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sibert RS, Josowicz M, Barry BA. Control of proton and electron transfer in de novo designed, biomimetic β hairpins. ACS Chem. Biol. 2010;5:1157–1168. doi: 10.1021/cb100138m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Siwick BJ, Bakker HJ. On the role of water in intermolecular proton-transfer reactions. J. Am. Chem. Soc. 2007;129:13412–13420. doi: 10.1021/ja069265p. [DOI] [PubMed] [Google Scholar]
- [117].Kim S, Liang J, Barry BA. Chemical complementation identifies a proton acceptor for redox-active tyrosine D in photosystem II. Proc. Nat. Acad. Sci. 1997;94:14406–14411. doi: 10.1073/pnas.94.26.14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Campbell KA, Peloquin JM, Diner BA, Tang X-S, Chisholm DA, Britt RD. The τ-Nitrogen of D2 histidine 189 is the hydrogen bond donor to the tyrosine radical YD• of photosystem II. J. Am. Chem. Soc. 1997;119:4787–4788. [Google Scholar]
- [119].Tang X-S, Chisholm DA, Dismukes GC, Brudvig GW, Diner BA. Spectroscopic evidence from site-directed mutants of Synechocystis PCC6803 in favor of a close interaction between histidine 189 and redox-active tyrosine 160, both of the polypeptide D2 of the photosystem II reaction center. Biochemistry. 1993;32:13742–13748. doi: 10.1021/bi00212a045. [DOI] [PubMed] [Google Scholar]
- [120].Tommos C, Davidsson L, Svensson B, Madsen C, Vermaas W, Styring S. Modified EPR spectra of the tyrosineD radical in photosystem II in site-directed mutants of Synechocystis on the D2 sp. PCC 6803: Identification of side chains in the immediate vicinity of tyrosineD protein. Biochemistry. 1993;32:5436–5441. doi: 10.1021/bi00071a020. [DOI] [PubMed] [Google Scholar]
- [121].Un S, Tang X-S, Diner BA. 245 GHz high-field EPR study of tyrosine-Do and tyrosine-Zo in mutants of photosystem II. Biochemistry. 1996;35:679–684. doi: 10.1021/bi9523769. [DOI] [PubMed] [Google Scholar]
- [122].Jenson D, Evans A, Barry BA. Proton-coupled electron transfer and tyrosine D of photosystem II. J. Phys. Chem. B. 2007;111:12599–12604. doi: 10.1021/jp075726x. [DOI] [PubMed] [Google Scholar]
- [123].Jenson D, Barry BA. Proton-coupled electron transfer in Photosystem II: Proton inventory of a redox active tyrosine. J. Am. Chem. Soc. 2009;131:10567–10573. doi: 10.1021/ja902896e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Venkatasubban KS, Schowen RL. The proton inventory technique. In: Fasman GD, editor. Critical Reviews in Biochemistry. CRC Press; Boca Raton: 2000. pp. 1–44. [DOI] [PubMed] [Google Scholar]
- [125].Babcock GT, Barry BA, Debus RJ, Hoganson CW, Atamian M, McIntosh L, Sithole I, Yocum CF. Water oxidation in photosystem II: from radical chemistry to multielectron chemistry. Biochemistry. 1989;28:9557–9565. doi: 10.1021/bi00451a001. [DOI] [PubMed] [Google Scholar]
- [126].Hays A-MA, Vassiliev IR, Golbeck JH, Debus RJ. Role of D1-His190 in proton-coupled electron transfer reactions in photosystem II: a chemical complementation study. Biochemistry. 1998;37:11352–11365. doi: 10.1021/bi980510u. [DOI] [PubMed] [Google Scholar]
- [127].Hays AMA, Vassiliev IR, Golbeck JH, Debus RJ. Role of D1-His190 in the proton-coupled oxidation of tyrosine Y-Z in manganese-depleted photosystem II. Biochemistry. 1999;38:11851–11865. doi: 10.1021/bi990716a. [DOI] [PubMed] [Google Scholar]
- [128].Mamedov F, Sayre RT, Styring S. Involvement of histidine 190 on the D1 protein in electron/proton transfer reactions on the donor side of Photosystem II. Biochemistry. 1998;37:14245–14256. doi: 10.1021/bi980194j. [DOI] [PubMed] [Google Scholar]
- [129].Pujols-Ayala I, Barry BA. His 190-D1 and Glu 189-D1 provide structural stabilization in photosystem II. Biochemistry. 2002;41:11456–11465. doi: 10.1021/bi026172l. [DOI] [PubMed] [Google Scholar]
- [130].Roffey RA, Wijk K.-J.van, Sayre RT, Styring S. Spectroscopic characterization of TyrZ in histidine 190 mutants of the D1-protein in photosystem II in Chlamydomonas reinhardtii: Implications for the structural model of the donor side of PSII. J. Biol. Chem. 1994;269:5115–5121. [PubMed] [Google Scholar]
- [131].Diner BA, Force DA, Randall DW, Britt RD. Hydrogen bonding, solvent exchange, and coupled proton and electron transfer in the oxidation and reduction of redox-active tyrosine YZ in Mn-depleted core complexes of Photosystem II. Biochemistry. 1998;37:17931–17943. doi: 10.1021/bi981894r. [DOI] [PubMed] [Google Scholar]
- [132].Lydakis-Simantiris N, Ghanotakis DF, Babcock GT. Kinetic isotope effects on the reduction of the YZ radical in oxygen evolving and tris-washed photosystem II membranes by time-resolved EPR. Biochim. Biophys. Acta. 1997;1322:129–140. [Google Scholar]
- [133].Karge M, Irrgang K-D, Renger G. Analysis of the reaction coordinate of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O. Biochemistry. 1997;36:8904–8913. doi: 10.1021/bi962342g. [DOI] [PubMed] [Google Scholar]
- [134].Westphal KL, Lydakis-Simantris N, Cukier RI, Babcock GT. Effects of Sr+2 substitution on the reduction rates of Yz• in PSII membranes-Evidence for concerted hydrogen-atom transfer in oxygen evolution. Biochemistry. 2000;39:16220–16229. doi: 10.1021/bi0018077. [DOI] [PubMed] [Google Scholar]
- [135].Kühne H, Brudvig GW. Proton-coupled electron transfer involving tyrosine Z in Photosystem II. J. Phys. Chem. B. 2002;106:8189–8196. [Google Scholar]
- [136].Ahlbrink R, Haumann M, Cherepanov D, Bogershausen O, Mulkidjanian A, Junge W. Function of tyrosine Z in water oxidation by photosystem II: Electrostatical potential instead of hydrogen abstractor. Biochemistry. 1998;37:1131–1142. doi: 10.1021/bi9719152. [DOI] [PubMed] [Google Scholar]
- [137].Haumann M, Bogershausen O, Cherepanov D, Ahlbrink R, Junge W. Photosynthetic oxygen evolution: H/D isotope effects and the coupling between electron and proton transfer during redox reactions at the oxidizing side of Photosystem II. Photosynth. Res. 1997;51:193–208. [Google Scholar]
- [138].Schilstra MJ, Rappaport F, Nugent JHA, Barnett CJ, Klug DR. Proton/hydrogen transfer affects the S-state-dependent microsecond phases of P680+ reduction during water splitting. Biochemistry. 1998;37:3974–3981. doi: 10.1021/bi9713815. [DOI] [PubMed] [Google Scholar]
- [139].Jeans C, Schilstra MJ, Ray N, Husain S, Minagawa J, Nugent JHA, Klug DR. Replacement of tyrosine D with phenylalanine affects the normal proton transfer pathways for the reduction of P680+ in oxygen-evolving photosystem II particles from Chlamydomonas. Biochemistry. 2002;41:15754–15761. doi: 10.1021/bi020558e. [DOI] [PubMed] [Google Scholar]
- [140].Rappaport F, Boussac A, Force D, Peloquin J, Bryndal M, Sugiura M, Un S, Britt D, Diner B. Probing the coupling between proton and electron transfer in photosystem II core complexes containing a 3-fluorotyrosine. J. Am. Chem. Soc. 2009;131:4425–4433. doi: 10.1021/ja808604h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Seyedsayamdost MR, Reece SY, Nocera DG, Stubbe J. Mono-, di-, tri-, and tetra-substituted fluorotyrosines: New probes for enzymes that use tyrosyl radicals in catalysis. J. Am. Chem. Soc. 2006;128:1569–1579. doi: 10.1021/ja055926r. [DOI] [PubMed] [Google Scholar]
- [142].Seyedsayamdost MR, Yee CS, Reece SY, Nocera DG, Stubbe J. pH rate profiles of FnY356-R2s (n=2, 3, 4) in Escherichia coli ribonucleotide reductase: Evidence that Y-356 is a redox-active amino acid along the radical propagation pathway. J. Am. Chem. Soc. 2006;128:1562–1568. doi: 10.1021/ja055927j. [DOI] [PubMed] [Google Scholar]
- [143].Ayala I, Range K, York D, Barry BA. Spectroscopic properties of tyrosyl radicals in dipeptides. J. Am. Chem. Soc. 2002;124:5496–5505. doi: 10.1021/ja0164327. [DOI] [PubMed] [Google Scholar]
- [144].Vassiliev IR, Offenbacher AR, Barry BA. Redox-active tyrosine residues in pentapeptides. J. Phys. Chem. B. 2005;109:23077–23085. doi: 10.1021/jp054159f. [DOI] [PubMed] [Google Scholar]
- [145].Range K, Ayala I, York D, Barry BA. Normal modes of redox-active tyrosine: conformation dependence and comparison to experiment. J. Phys. Chem. 2006;110:10970–10981. doi: 10.1021/jp061503f. [DOI] [PubMed] [Google Scholar]
- [146].McCracken J, Vassiliev IR, Yang EC, Range K, Barry BA. ESEEM studies of peptide nitrogen hyperfine coupling in tyrosyl radicals and model peptides. J. Phys. Chem. B. 2007;111:6586–6592. doi: 10.1021/jp071402x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Warncke K, Perry MS. Redox state dependence of rotamer distributions in tyrosine and neutral tyrosyl radical. Biochim. Biophys. Acta. 2001;1545:1–5. doi: 10.1016/s0167-4838(00)00289-2. [DOI] [PubMed] [Google Scholar]
- [148].Offenbacher AR, Vassiliev IR, Seyedsayamdost MR, Stubbe J, Barry BA. Redox-linked structural changes in ribonucleotide reductase. J. Am. Chem. Soc. 2009;131:7496–7497. doi: 10.1021/ja901908j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Högbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzian F. Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-Å x-ray data. Proc. Nat. Acad. Sci. 2003;100:3209–3214. doi: 10.1073/pnas.0536684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Pujols-Ayala I, Sacksteder CA, Barry BA. Redox-active tyrosine residues: Role for the peptide bond in electron transfer. J. Am. Chem. Soc. 2003;125:7536–7538. doi: 10.1021/ja035005l. [DOI] [PubMed] [Google Scholar]
- [151].Wiberg KB, Brenemain CM. Resonance interactions in acyclic systems. 3. Formamide internal-rotation revisted-charge and energy redistribution along the C-N bond rotational pathway. J. Am. Chem. Soc. 1992;114:831–840. [Google Scholar]
- [152].Kim S, Barry BA. The protein environment surrounding tyrosyl radicals D• and Z• in photosystem II: a difference Fourier-transform infrared spectroscopic study. Biophys. J. 1998;74:2588–2600. doi: 10.1016/S0006-3495(98)77965-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ayala I, Kim S, Barry BA. A difference Fourier transform infrared study of tyrosyl radical Z• decay in photosystem II. Biophys. J. 1999;77:2137–2144. doi: 10.1016/S0006-3495(99)77054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- [155].Barry BA, Cooper IB, DeRiso A, Brewer SH, Vu D, Dyer RB. Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle. Proc. Nat. Acad. Sci. 2006;103:7288–7291. doi: 10.1073/pnas.0600216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Miqyass M, van Gorkom HJ, Yocum CF. The PSII calcium site revisited. Photosynth. Res. 2007;92:275–287. doi: 10.1007/s11120-006-9124-2. [DOI] [PubMed] [Google Scholar]
- [157].Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Quantum mechanics/molecular mechanics study of the catalytic cycle of water splitting in photosystem II. J. Am. Chem. Soc. 2008;130:3428–3442. doi: 10.1021/ja076130q. [DOI] [PubMed] [Google Scholar]
- [158].Harriman A. Further comments on the redox potentials of trypthophan and tyrosine. J. Phys. Chem. 1987;91:6102–6104. [Google Scholar]
- [159].Okamura MY, Paddock ML, Graige MS, Feher G. Proton and electron transfer in bacterial reaction centers. Biochim. Biophys. Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- [160].Graige MS, Feher G, Okamura MY. Conformational gating of the electron transfer reaction QA−.QB --> QAQB−. in bacterial reaction centers of Rhodobacter sphaeroides determined by a driving force assay. Proc. Nat. Acad. Sci. 1998;95:11679–11684. doi: 10.1073/pnas.95.20.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Stowell MHB, McPhillips TM, Rees DC, Soltis SM, Abresch E, Feher G. Light-induced structural changes in photosynthetic reaction center: Implications for mechanism of electron-proton transfer. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]