

Abstract
A series of analogues were synthesized by optimizing the structure of papaverine. The in vitro PDE10A binding affinity ( IC50) values for these new analogues were measured; for compounds that have IC50 value less than 60 nM for PDE10A, the binding affinities (IC50 value) for PDE3A and PDE3B were tested. Of these analogues, compounds 6a, 6b, 6n, 8b, 8c and 11 displayed relatively higher PDE10A potency with IC50 value in the range of 28–60 nM. The most potent compound 1-(4-(2-(2-fluoroethoxy)ethoxy)-3-methoxybenzyl)-6,7-dimethoxyisoquinoline (8c) has the IC50 value of 28 ± 1.2 nM for PDE10A, 2200 ± 437 nM for PDE3A and 2520 ± 210 nM for PDE3B. Compared to papavarine, compound 8c displayed similar PDE10A potency but improved selectivity to PDE10A versus PDE3A and PDE3B. To identify high potent PDE10A inhibitor, further optimization of the structures of these analogues is necessary.
Keywords: PDE10A, CNS, Schizophrenia
1. Introduction
Abnormal striatal output is caused by the dysfunction of striatopllaidal transmission, which is the major contributor to the pathophysiology of central nervous system (CNS) disorders including schizophrenia, Parkinson’s disease, Huntington’s disease, Torrette’s syndrome, and drug abuse [1–4]. The cyclic nucleotide phosphodiesterase enzymes have 11 gene families that contain 21 phosphodiesterase genes. The cyclic nucleotide phosphodiesterase 10A (PDE10A) is one of the 11 gene families and is highly expressed in the primary input neurons of the basal ganglia circuit, which are striatal medium-sized spiny neurons (MSNs) [5]. The inhibition of PDE10A alternates the basal ganglia circuit output and increases striatopallidal MSN activation, which represents a new therapeutic approach for the treatment of some of CNS diseases [5–9].
PDEs regulate levels of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) by hydrolysis of the cyclic nucleotide into their respective nucleotide monophosphates. PDE10A is a unique dual specificity phosphodiesterase that can convert both cAMP to adenosine monophosphate (AMP) and cGMP to guanosine monophosphate (GMP) [10–12].
Across species, compared to other PDE families, the PDE10A protein has a high degree of homology and is uniquely localized in mammals. PDE10 enzyme is expressed only in the testis and the brain [10–12]. Within the brain, the expression of the PDE10A enzyme is the highest in striatum (caudate and putamen), nucleus accumbens, and the olfactory tubercle of MSNs [10, 11, 13, 14]. Based on the fact that PDE10A expression parallels D2 localization in the brain, inhibition of PDE10A may have similar functional effects as D2 inhibition. The inhibition of PDE10A may mediate the clinical antipsychotic effects. However, there are distinct differences between PDE10A inhibition and classical antagonists of D2 receptors. The D2 receptor has signaling components besides cAMP [15] and the inhibition of PDE10A interferes with cAMP levels that may negatively modulate rather than directly antagonize dopamine signaling. The inhibition of PDE10A may avoid the limitations that are observed with strong D2 antagonism to treat schizophrenia and other CNS diseases. Furthermore, PDE10A is also expressed in D1 expressing striatal neurons [14]. The activation of the D1 receptor leads to the stimulation of adenylate cyclases, which results in the increase of cAMP levels. Therefore, the inhibition of PDE10A is likely to have effects that simulate D1 receptor agonism. Together, PDE10A inhibition will increase cAMP in cells and can be expected to increase cGMP levels. cGMP activates a number of target proteins in the cells like cAMP and it also interacts with the cAMP signaling pathways. Consequentially, inhibition of PDE10A to elevate cAMP and cGMP could be a novel therapeutic strategy for the treatment of associated neurological and psychiatric disorders and may avoid unwanted side effects such as extrapyramidal symptoms associated with conventional therapeutics.
Over the past 10 years, tremendous efforts have been made to develop PDE10A inhibitors for the treatment of schizophrenia and associated mental disorders [16–20]. The compounds in Figure 1 have attracted more attention [18, 19, 21]. Although papaverine has only moderate potency (IC50 = 36 nM) and poor selectivity (9 fold) over other PDE isoforms, papaverine was the first compound used to explore the role of PDE10A in the CNS. It was reported that papaverine potentiated the cataleptic effect of the D2 receptor antagonist haloperidol in rats, but did not cause catalepsy on its own. Papaverine as well as more potent and more selective PDE 10A inhibitors PQ-10, MP-10 and TP-10, were all able to reduce hyperactivity in rats induced by phenylcyclohexylpiperidine (PCP) and amphetamine [8]. Furthermore, PDE10A inhibition with papaverine reverses subchronic PCP-induced deficits in attention set-shifting in rats [22]. Papaverine has also shown efficacy in rat novel object recognition [23]. Together, these data suggest that PDE10A inhibition might alleviate cognitive deficits associated with schizophrenia and psychosis.
Figure 1.

Structures and in Vitro Assays of the Lead Compounds
In this paper, we will report the synthesis and biological binding affinity measurement of the new analogues as PD10A inhibitors based on the modification of the molecular structure of papaverine. Our investigation was inspired by (1) papaverine has PDE10A inhibition function with moderate potency for PDE10A and low selectivity; (2) the rapid in vivo metabolism of papaverine limits it serving as a PET ligand for imaging in PDE10A in vivo [24]; and (3) the demand of a suitable PET probe for imaging the PDE10A enzyme in vivo.
2. Results and discussion
2.1. Chemistry
Papaverine, 1-(3,4-dimethoxybenzyl-6,7-dimethoxy)isoquinoline, which possesses four methoxy groups in its molecular structure, is now known to be a potent inhibitor of phosphodiesterase 10A (PDE10A) and has been reported to increase cognitive performance in the rat model of schizophrenia. Radioactive [3H]papaverine, [14C]papaverine and [11C]papaverine have successfully been made to conduct pharmacologic studies. To develop the F-18 labeled PDE10A PET tracer, we synthesized a series of analogues by replacing the methoxy group(s) with fluoroethoxyl group or other groups.
The synthesis of the target analogues 6a–n, 8a–c and 11 was shown in Scheme 1–3. Substituted benzaldehydes 1a and 1b were condensed with nitromethane to generate 2-phenyl-1-nitroethene derivatives 2a and 2b [25], which reacted with sodium methoxide to give (1-methoxy-2-nitroethyl)benzene derivatives 3a and 3b [26]. It was found that the conversion of compounds 2a and 2b to 3a and 3b was complete in just 10 min when one equivalent of compound 2 was added into two equivalents of sodium methoxide solution in methanol. Reduction of the nitro group of 3a and 3b with LiAlH4 in THF gave amines 4a and 4b [26]. Due to the higher hydrophilicity of 4a and 4b, the extraction of 4a and 4b with organic solvent from their aqueous solution was very difficult. To resolve this problem, the excess lithium aluminum hydride was decomposed by carefully adding a very small amount of water followed by filtering the reaction mixture through celite to remove the precipitate, which was washed with THF. The THF was directly evaporated under vacuum without washing with water. When this procedure was followed, the yields of 4a and 4b were improved from 40% to 90%. Amines 4a and 4b were coupled with the corresponding substituted benzyl carboxylic acid using 1,1′-carbonyldiimidazole as a coupling agent to give amide 5a–m and 5o. Commercially available 2-amino-1-(3,4-dimethylphenyl)ethanol reacted with 2-substituted acetyl chloride to afford intermediate compounds 5n and 5p. Cyclization reaction of 5a–p with POCl3 in acetonitrile afforded compounds 6a–p. Compounds 6a–n were converted into oxalic salts for in vitro affinity measurements. Based on our experiments, the yield of the cyclization reaction was very good when the reaction time was limited to 30 min, otherwise the yield was much lower. Hydrogenolysis to remove the benzyl group in 6o and 6p afforded phenol intermediates 7a and 7b which were reacted with 2-bromo-1-fluoroethane or 1-chloro-2-(2-fluoroethoxy)ethane via O-alkylation to afford compounds 8a–c. Compounds 8a–c were converted into oxalic salts for determining their binding affinities. To test whether replacing the isoquinoline ring with a quinazoline ring increases the binding affinity of PDE10A, compound 7 was synthesized according to Scheme 3. Starting with 4-chloro-6,7-dimethoxyquinazoline, 9 was treated with (4-methoxybenzyl) magnesium chloride, 10 in the presence of Manganese (II) chloride in THF to afford compound 11 which was converted into its oxalate.
Scheme 1.

Reagents and conditions:
i) MeNO2, NH4OAc, HOAc; ii) MeONa/MeOH, THF; iii) LiAlH4, THF;
iv) CDI, CH2Cl2, 2-(substituted phenyl)acetic acid or 2-(substituted phenyl)acetic chloride;
v) POCl3, acetonitrile
Scheme 3.

2.2. In vitro assessment of affinities
The binding affinities of PDE10A for the newly synthesized analogues were first determined by measuring IC50 values. It was reported that papaverine was not only a relatively potent inhibitor of PDE10A, but also exhibited moderate cross-reactivity versus PDE3A and PDE3B (Ki as 279, and 417 nM, respectively) [19]. The use of PDE3A/B inhibitor could lead to arrhythmia and increased mortality [27, 28]. Therefore, compounds that had IC50 value less than 60 nM for PDE10A, were further determined by their binding affinities for PDE3A/B. The PDE activities were measured according to the procedure described in the experimental section. The binding affinities of the analogues were shown in Table 1.
Table 1.
Structure and Affinities of New Analoguesa
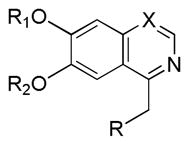 | ||||||||
|---|---|---|---|---|---|---|---|---|
| X | R1 | R2 | R | IC50 (nM) | Log Pb | |||
| PDE10A | PDE3A | PDE3B | ||||||
| Papaverine | C | Me | Me |
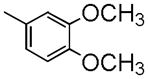
|
21±1.2 | 917 ± 100 | 1030 ± 88 | 3.71 |
| 6a | C | -CH2CH2F | Me |
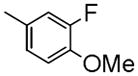
|
49±1.1 | 1290 ± 97 | 1250 ± 220 | 4.13 |
| 6b | C | -CH2CH2F | Me |
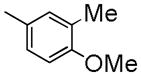
|
58.5±1.1 | 1190 ± 130 | 1670 ± 240 | 4.57 |
| 6c | C | -CH2CH2F | Me |
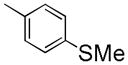
|
112±1.2 | NDc | NDc | 4.69 |
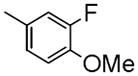
| 6d | C | Me | -CH2CH2F |
|
198±1.1 | NDc | NDc | 4.13 |
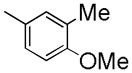
| 6e | C | Me | -CH2CH2F |
|
244±1.1 | NDc | NDc | 4.57 |
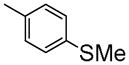
| 6f | C | Me | -CH2CH2F |
|
237±1.1 | NDc | NDc | 4.68 |
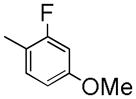
| 6g | C | Me | -CH2CH2F |
|
500±1.2 | NDc | NDc | 4.23 |
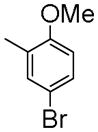
| 6h | C | Me | -CH2CH2F |
|
872±1.3 | NDc | NDc | 5.15 |
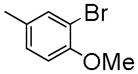
| 6i | C | Me | -CH2CH2F |
|
248±1.1 | NDc | NDc | 4.69 |
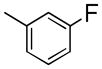
| 6j | C | Me | -CH2CH2F |
|
469±1.3 | NDc | NDc | 4.26 |
| 6k | C | Me | -CH2CH2F |
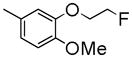
|
200±1.1 | NDc | NDc | 4.17 |
| 6l | C | Me | -CH2CH2F |
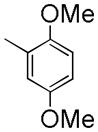
|
262±1.1 | NDc | NDc | 3.92 |
| 6m | C | Me | -CH2CH2F |
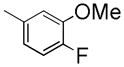
|
303±1.1 | NDc | NDc | 4.13 |
| 6n | C | Me | Me |
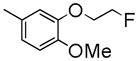
|
55±1.0 | 1940 ± 490 | 2510 ± 140 | 3.93 |
| 8a | C | Me | -CH2CH2F |
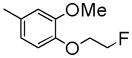
|
246±1.1 | NDc | NDc | 4.17 |
| 8b | C | Me | Me |
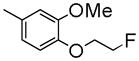
|
55±1.2 | 1740 ± 390 | 2170 ± 260 | 3.93 |
| 8c | C | Me | Me |
|
28±1.2 | 2200 ± 440 | 2520 ± 210 | 3.54 |
| 11 | C | Me | Me |
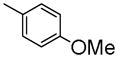
|
48.8±1.07 | 3980 ± 510 | 3100 ± 270 | 2.53 |
IC50 is defined as the concentration of the inhibitor required to reduce the [3H]cAMP hydrolysis activity of recombinant human PED10A by 50% with scintillation proximity assay.
Calculated value at pH = 7.4 by ACD/I-Lab ver. 7.0 (Advanced Chemistry Development, Inc., Canada).
Not determined.
To develop fluorine analogues, we took the following approach. First, we replaced the 6-methoxy in papaverine with a 6-fluoroethoxy group and replaced the 3-methoxy with a fluorine or methyl group. It was found that compounds 6a and 6b have a modest affinity for PDE10A (IC50 value for 6a was 49 nM and for 6b was 58 nM). However, after replacing the 4-methoxy with a 4-methylthio group (6c), the IC50 value for PDE10A dropped to 112 nM. Second, when the 6-methoxy was retained and the 7-methoxy was replaced with a 7-fluroethoxy, compounds 6d–m and 8a were generated. Compared to 6a–c and 8b, the PDE10A binding affinities for 6d vs. 6a, 6e vs. 6b, 8a vs. 8b were reduced by 5 fold; for 6f vs. 6c, it was reduced by approximately 2 fold. The binding affinities were 49 ± 1.1, 58.5 ± 1.1, 112 ± 1.2 and 55 ± 1.2 nM for 6a, 6b, 6c and 8b, 198 ± 1.1, 244 ± 1.1, 237 ± 1.1 and 246 ± 1.1 nM for 6d, 6e, 6f and 8a respectively. Similar or even greater reduction in the PDE10A binding affinities was found for analogues 6g–m and 8a. Third, we retained both the 6 and 7-methoxy groups and replaced the 3-methoxy or 4-methoxy on the substituted benzyl group and found that both the 6n and 8b displayed modest affinities for PDE10A (IC50 value as 55 ± 1.0 nM for 6n and 55 ± 1.2 nM for 8b). Extending the 4-fluroethoxy group to a 4-(2-(2-fluoroethoxy))ethoxy group increased PDE10A affinity by 2-fold, from 55 ± 1.2 nM for 8b to 28 ± 1.2 nM for 8c). Fourth, we replaced 6,7-dimethoxyisoquinoline with 6,7-dimethoxy quinazoline, while removing the 3-methoxy group, which generated 11 that had a modest PDE10A affinity of IC50 = 48.8 ± 1.07 nM.
3. Conclusions
In the current study, the papaverine structure was modified by incorporating fluorine containing substituted group(s) and replacing the isoquinoline ring with a quinazoline ring. The in vitro PDE10A binding affinity results of these compounds indicated that the compound 8c, 1-(4-(2-(2-fluoroethoxy)ethoxy)-3-methoxybenzyl)-6,7-dimethoxyisoquinoline has a comparable binding affinity for PDE10A to that of papaverine. Compared to papaverine, compound 8c has a much lower binding affinity for PDE3A (2200 nM for 8c vs 279 nM for papaverine) and a lower binding affinity for PDE3B (2517 nM for 8c vs 417 nM for papaverine) than that of papaverine [19]. Compound 8c is a fluorine containing compound that can take advantage of the isotopic properties of F-18 (half-life is 110 min, widely commercial availability) compared to that of C-11 (half-live of 20 min; C-11 tracers have to be made in-house at a cyclotron facility). Fluorine-18 labeled probes are more suitable for conducting the imaging studies of PDE10A in living animals. The radioactive [18F]8c will be radiosynthesized for in vivo evaluation to test its potential as a PET tracer for imaging the PDE10A in vivo.
4. Experimental
4.1 Chemistry
1H NMR spectra were recorded on a Varian 300 MHz NMR spectrometer. Chemical shifts are reported in δ values with respect to tetramethylsilane (TMS) as an internal reference standard. The following abbreviations are used for multiplicity of NMR signals: br s = broad singlet, s = singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplet, t = triplet, m = multiplet, q = quartet. Melting points were determined on an electrothermal melting point apparatus and are uncorrected. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA, and were within ±0.4% of the calculated values. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant No. P41RR0954). All reactions were carried out under an inert atmosphere of nitrogen.
Procedure A: synthesis of compounds 1a and 1b
4-(2-Fluoroethoxy)-3-methoxybenzaldehyde (1a)
Potassium carbonate (30.0 g, 0.22 mol) was added to a solution of 4-hydroxy-3-methoxybenzaldehyde (15.2 g, 0.1 mol) and 2-bromo-1-fluoroethane (25.4 g, 0.2 mol) in acetone (100 mL). The reaction mixture was refluxed overnight until the reaction was complete as determined by thin layer chromatography (TLC). After the solvent was removed, 200 mL of water was added to the flask, and the mixture was extracted with ethyl acetate (50 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate. Filtered and concentrated, the crude product was purified by silica gel column chromatography with EtOAc/MeOH (95/5, v/v) as mobile phase to give 1a (15.3 g, 77%) as a pale yellow solid. 1H NMR (300 MHz, CDCl3) δ 3.94 (s, 3 H), 4.37 (dt, J = 27.3, 4.2 Hz, 2 H), 4.83 (dt, J = 47.4, 4.2 Hz, 2 H), 7.01 (d, J = 8.1 Hz, 1 H), 7.44–7.47 (m, 2 H), 9.87 (s, 1 H).
3-(2-Fluoroethoxy)-4-methoxybenzaldehyde (1b)
Starting with 3-hydroxy-4-methoxybenzaldehyde, Procedure A was followed to afford compound 1b (5.9 g, 80%) as a pale yellow solid. 1H NMR (300 MHz, CDCl3) δ 3.97 (s, 3 H), 4.34 (dt, J = 27.3, 4.2 Hz, 2 H), 4.83 (dt, J = 47.4, 4.2 Hz, 2 H), 7.01 (d, J = 8.4 Hz, 1 H), 7.43 (d, J = 2.1 Hz, 1 H), 7.51 (dd, J = 8.4, 2.1 Hz, 1 H), 9.86 (s, 1 H).
Procedure B: synthesis of compounds 2a and 2b
(E)-1-(2-Fluoroethoxy)-2-methoxy-4-(2-nitrovinyl)benzene (2a)
A stirred mixture of 1a (12.5 g, 63.1 mmol), nitromethane (12.0 mL, 221.7 mmol) and anhydrous ammonium acetate (12.0 g, 155.8 mmol) in glacial acetic acid (28 mL) was refluxed for 30 min. After cooling to room temperature, the reaction mixture was poured into water (200 mL). The solid was collected by filtration to give 2a (13.0 g, 85%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 3.93 (s, 3 H), 4.34 (dt, J = 27.3, 4.2 Hz, 2 H), 4.82 (dt, J = 47.4, 4.2 Hz, 2 H), 6.94 (d, J = 8.1 Hz, 1 H), 7.03 (d, J = 2.1 Hz, 1 H), 7.15 (dd, J = 8.1, 2.1 Hz, 1 H), 7.53 (d, J = 13.8 Hz, 1 H), 7.96 (d, J = 13.8 Hz, 1 H).
(E)-2-(2-fluoroethoxy)-1-methoxy-4-(2-nitrovinyl)benzene (2b)
Starting with compound 1b, procedure B was followed to afford compound 2b (7.8 g, 78%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 3.97 (s, 3 H), 4.34 (dt, J = 27.3, 4.2 Hz, 2 H), 4.83 (dt, J = 47.4, 4.2 Hz, 2 H), 7.01–7.13 (m, 2 H), 7.51 (d, J = 8.4 Hz, 1 H), 7.56 (d, J = 13.5 Hz, 1 H), 7.90 (d, J = 13.5 Hz, 1 H).
Procedure C: synthesis of compounds 3a and 3b
1-(2-Fluoroethoxy)-2-methoxy-4-(1-methoxy-2-nitroethyl)benzene (3a)
A solution of 2a (3.3 g, 13.7 mmol) in dry THF (50 mL) was added in one portion to a stirred solution of sodium methoxide (25% in MeOH, 9 mL) at room temperature. The resulting mixture was stirred for 5 min and poured into water. The crude product was collected by filtration to give 3a (2.6 g, 70%) as a pale yellow solid. 1H NMR (300 MHz, CDCl3) δ 3.27 (s, 3H ), 3.89 (s, 3H ), 4.29 (dt, J = 27.3, 3.9 Hz, 2 H), 4.38 (dd, J = 12.6, 3.6 Hz, 1 H), 4.61 (dd, J = 12.3, 9.9 Hz, 1 H), 4.70–4.92 (m, 3 H), 6,88–6.95 (m, 3 H).
2-(2-Fluoroethoxy)-1-methoxy-4-(1-methoxy-2-nitroethyl)benzene (3b)
Starting with 2b, procedure C was followed to afford compound 3b (7.3 g, 78%) as a pale yellow solid. 1H NMR (300 MHz, CDCl3) δ 3.26 (s, 3 H), 3.88 (s, 3 H), 4.24–4.40 (m, 3 H), 4.60 (dd, J = 10.8, 9.9 Hz, 1 H), 4.71–4.91 (m, 3 H), 6.90–6.97 (m, 3 H).
Procedure D: synthesis of compounds 4a and 4b
2-(4-(2-Fluoroethoxy)-3-methoxyphenyl)-2-methoxyethanamine (4a)
A solution of 3a (2.6 g, 9.5 mmol) in THF (45 mL) was added to lithium aluminum hydride (1.1 g, 29.1 mmol) in dry THF (15 mL) dropwise. The reaction mixture was refluxed for 6–8 h. The excess lithium aluminum hydride was decomposed by carefully adding a small amount of water. The suspension was filtered through (Celite) and washed with THF. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography with CH2Cl2/MeOH/Et3N (95/5/1, v/v/v) as mobile phase to give 4a (1.53 g, 66%) as a pale yellow oil. 1H NMR (300 MHz, CDCl3) δ 2.81(dd, J = 13.6, 4.8 Hz, 1 H), 2.92 (dd, J = 13.2, 7.5 Hz, 1 H), 3.27 (s, 3 H), 4.08 (dd, J = 7.5, 4.2 Hz, 1 H), 3.88 (s, 3H), 4.28 (dt, J = 27.3, 4.5 Hz, 2 H), 4.78 (dt, J = 47.4, 4.5 Hz, 2 H), 6,83–6.91 (m, 3 H).
2-(3-(2-Fluoroethoxy)-4-methoxyphenyl)-2-methoxyethanamine (4b)
Starting with 3b, procedure D was followed to afford compound 4b (1.90 g, 82%). 1H NMR (300 MHz, CDCl3) δ 2.81(dd, J = 13.2, 4.2 Hz, 1 H), 2.90 (dd, J = 13.2, 7.5 Hz, 1 H), 3.25 (s, 3 H), 3.88 (s, 3 H), 4.06 (dd, J = 7.5, 4.2 Hz, 1 H), 4.28 (dt, J = 27.9, 3.9 Hz, 2 H), 4.78 (dt, J = 47.7, 3.9 Hz, 2 H), 6.87 (s, 3 H).
Procedure E: synthesis of compounds 5a–o
2-(3-Fluoro-4-methoxyphenyl)-N-(2-(3-(2-fluoroethoxy)-4-methoxyphenyl)-2-methoxyethyl)acetamide (5a)
In a typical conversion, 1,1′-Carbonyldiimidazole (CDI, 0.32 g, 2 mmol) was added to a solution of 2-(4-fluoro-3-methoxyphenyl)acetic acid (0.37 g, 2 mmol) in 20 mL of dichloromethane, and the mixture was stirred at room temperautre for 2 h, at which point, 2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethanamine 4a (0.48 g, 2.0 mmol) was added and the mixture was stirred overnight. The reaction mixture was washed with saturated sodium carbonate solution, dried over anhydrous sodium sulfate. After concentrating, the residue 5a was used in the next step without further purification. 1H NMR (300 MHz, CDCl3) δ 3.17–3.26 (m, 4 H), 3.47 (s, 2 H), 3.59 (m, 1 H), 3.87 (s, 3 H), 3.89 (s, 3 H), 4.13 (dd, J = 8.4, 4.5 Hz, 1 H), 4.26 (dt, J = 27.9, 4.2 Hz, 2 H), 4.77 (dt, J = 47.4, 4.2 Hz, 2 H), 5.74 (br, 1 H), 6.78–6.99 (m, 6 H).
N-(2-(3-(2-Fluoroethoxy)-4-methoxyphenyl)-2-methoxyethyl)-2-(4-methoxy-3-methylphenyl)acetamide (5b)
Procedure E was followed to prepare compound 5b. 1H NMR (300 MHz, CDCl3) δ 2.20 (s, 3 H), 3.1–3.22 (m, 4 H), 3.47 (s, 2 H), 3.58 (m, 1 H), 3.83 (s, 3 H), 3.86 (s, 3 H), 4.12 (dd, J = 8.4, 4.5 Hz, 1 H), 4.24 (dt, J = 27.3, 4.5 Hz, 2 H), 4.77 (dd, J = 47.4, 4.5 Hz, 2 H), 5.77 (br, 1 H), 6.70–7.01 (m, 6 H).
N-(2-(3-(2-Fluoroethoxy)-4-methoxyphenyl)-2-methoxyethyl)-2-(4-(methylthio)phenyl)acetamide (5c)
Procedure E was followed to prepare compound 5c. 1H NMR (300 MHz, CDCl3) δ 2.49 (s, 3 H), 3.15–3.23 (m, 4 H), 3.51 (s, 2 H), 3.59 (m, 1 H), 3.87 (s, 3 H), 4.11 (m, 1 H), 4.24 (dt, J = 27.6, 4.5 Hz, 2 H), 4.77 (dt, J = 47.1, 4.5 Hz, 2 H), 5.74 (br, 1 H), 6.73–6.85 (m,3 H), 7.13–7.25 (m, 4 H).
2-(3-Fluoro-4-methoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)acetamide (5d)
Procedure E was followed to prepare compound 5d. 1H NMR (300 MHz, CDCl3) δ 3.20–3.28 (m, 4 H), 3.49 (s, 2 H), 3.62 (m, 1 H), 3.85 (s, 3 H), 3.89 (s, 3 H), 4.13 (m, 1 H), 4.27 (dt, J = 27.3, 4.2 Hz, 2 H), 4.79 (dt, J = 47.4, 4.2 Hz, 2 H), 5.77 (br, 1 H), 6.72–6.94 (m, 6 H).
N-(2-(4-(2-Fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)-2-(4-methoxy-3-methylphenyl)acetamide (5e)
Procedure E was followed to prepare compound 5e. 1H NMR (300 MHz, CDCl3) δ 2.21 (s, 3 H), 3.15–3.23 (m, 4 H), 3.48 (s, 2 H), 3.59 (m, 1 H), 3.83 (s, 3 H), 3.84 (s, 3 H), 4.13 (m, 1 H), 4.26 (dt, J = 27.3, 4.2 Hz, 2 H), 4.78 (dt, J = 47.4, 4.2 Hz, 2 H), 5.78 (br, 1 H), 6.71–6.86 (m, 4 H), 6.98–7.01 (m, 2 H).
N-(2-(4-(2-Fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)-2-(4-(methylthio)phenyl)acetamide (5f)
Procedure E was followed to afford compound 5f. 1H NMR (300 MHz, CDCl3) δ 2.52 (s, 3 H), 3.21–3.27 (m, 4 H), 3.55 (s, 2 H), 3.63 (m, 1 H), 3.87 (s, 3 H), 4.13 (m, 1 H), 4.30 (dt, J = 27.3, 4.2 Hz, 2 H), 4.71 (dt, J = 47.4, 4.2 Hz, 2 H), 5.77 (br, 1 H), 6.73–6.86 (m, 3 H), 6.89–7.26 (m, 4 H).
2-(2-Fluoro-4-methoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)acetamide (5g)
Procedure E was followed to prepare compound 5g. 1H NMR (300 MHz, CDCl3) δ 3.14–3.26 (m, 4 H), 3.51 (s, 2 H), 3.62 (m, 1 H), 3.80 (s, 3 H), 3.85 (s, 3 H), 4.15 (m, 1 H), 4.26 (dt, J = 27.6, 4.2 Hz, 2 H), 4.78 (dt, J = 47.1, 4.2 Hz, 2 H), 5.85 (br, 1 H), 6.62–6.87 (m, 5 H), 7.15 (m, 1 H).
2-(5-Bromo-2-methoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)acetamide (5h)
Procedure E was followed to prepare compound 5h. 1H NMR (300 MHz, CDCl3) δ 3.04–3.13 (m, 4 H), 3.43 (s, 2 H), 3.55 (m, 1 H), 3.74 (s, 3 H), 3.76 (s, 3 H), 4.03 (m, 1 H), 4.24 (dt, J = 27.9, 4.2 Hz, 2 H), 4.70 (dt, J = 47.4, 4.2 Hz, 2 H), 6.06 (br, 1 H), 6.63–6.79 (m, 4 H), 7.19 (s, 1 H), 7.30 (m, 1 H).
2-(3-Bromo-4-methoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)acetamide (5i)
Procedure E was followed to prepare compound 5i. 1H NMR (300 MHz, CDCl3) δ 3.16–3.27 (m, 4 H), 3.47 (s, 2 H), 3.61 (m, 1 H), 3.85 (s, 3 H), 3.89 (s, 3 H), 4.15 (m, 1 H), 4.27 (dd, J = 27.3, 4.2 Hz, 2 H), 4.78 (dd, J = 47.1, 4.2 Hz, 2 H), 5.75 (br, 1 H), 6.70–6.88 (m, 4 H), 7.15 (m, 1 H), 7.43 (s, 1 H).
N-(2-(4-(2-Fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)-2-(3-fluorophenyl)acetamide (5j)
Procedure E was followed to prepare compound 5j. 1H NMR (300 MHz, CDCl3) δ 3.14–3.26 (m, 4 H), 3.56 (s, 2 H), 3.64 (m, 1 H), 3.85 (s, 3 H), 4.15 (m, 1 H), 4.27 (dt, J = 27.6, 4.5 Hz, 2 H), 4.70 (dt, J = 47.1, 4.5 Hz, 2 H), 5.80 (br, 1 H), 6.73–6.80 (m, 3 H), 6.96–7.04 (m, 3 H), 7.27–7.35 (m, 1 H).
N-(2-(4-(2-Fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)-2-(3-(2-fluoroethoxy)-4-methoxyphenyl)acetamide (5k)
Procedure E was followed to prepare compound 5k. 1H NMR (300 MHz, CDCl3) δ 3.17–3.25 (m, 4 H), 3.49 (s, 2 H), 3.58 (m, 1 H), 3.85 (s, 3 H), 3.88 (s, 3 H), 4.13 (m, 1 H), 4.19–4.33 (m, 4 H), 4.86–4.88 (m, 4 H), 5.76 (br, 1 H), 6.70–6.87 (m, 6 H).
2-(3,5-Dimethoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)acetamide (5l)
Procedure E was followed to prepare compound 5l. 1H NMR (300 MHz, CDCl3) δ 3.18–3.24 (m, 4 H), 3.50 (s, 2 H), 3.60 (m, 1 H), 3.78 (s, 6 H), 3.85 (s, 3 H), 4.12 (m, 1 H), 4.26 (dt, J = 27.3, 4.5 Hz, 2 H), 4.78 (dt, J = 47.4, 4.5 Hz, 2 H), 5.84 (br, 1 H), 6.38 (m, 3 H), 6.70–6.86 (m, 3 H).
2-(4-Fluoro-3-methoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-methoxyethyl)acetamide (5m)
Procedure E was followed to prepare compound 5m. 1H NMR (300 MHz, CDCl3) δ 3.18–3.25 (m, 4 H), 3.51 (s, 2 H), 3.61 (m, 1 H), 3.85 (s, 3 H), 3.88 (s, 3 H), 4.12 (m, 1 H), 4.27 (dt, J = 27.3, 4.5 Hz, 2 H), 4.78 (dt, J = 47.4, 4.5 Hz, 2 H), 5.79 (br, 1H), 6.69–6.87 (m, 4 H), 7.00–7.11 (m, 2 H).
N-(2-(3,4-dimethoxyphenyl)-2-hydroxyethyl)-2-(3-(2-fluoroethoxy)-4-methoxyphenyl)acetamide (5n)
Procedure E was followed to afford compound 5n. The 1H NMR (300 MHz, CDCl3) is δ 3.33 (m, 1 H), 3.51 (s, 2 H), 3.59 (m, 1 H), 3.89 (m, 9 H), 4.24 (dt, J = 28.8, 4.5 Hz, 2 H), 4.74 (dd, J = 8.1, 3.6 Hz, 1 H), 4.78 (dt, J = 47.4, 4.5 Hz, 2 H), 5.81 (br, 1 H), 6.78–6.86 (m, 6 H).
2-(4-(Benzyloxy)-3-methoxyphenyl)-N-(2-(4-(2-fluoroethoxy)-3-methoxyphenyl)-2-ethoxyethyl)acetamide (5o)
Procedure E was followed to prepare compound 5o. The 1H NMR (300 MHz, CDCl3) is δ 3.14–3.20 (m, 3 H), 3.49 (s, 2 H), 3.60 (m, 1 H), 3.84 (s, 3 H), 3.87 (s, 3 H), 4.15 (m, 1 H), 4.22 (dt, J = 27.3, 4.5 Hz, 2 H), 4.82 (dt, J = 47.4, 4.5 Hz, 2 H), 5.16 (s, 2 H), 5.79 (br, 1 H), 6.67–6.87 (m, 4 H), 7.20–7.50 (m, 7 H).
Procedure F: synthesis of compound 5p
2-(4-(Benzyloxy)-3-methoxyphenyl)-N-(2-(3,4-dimethoxyphenyl)-2-hydroxyethyl)acetamide (5p)
Saturated sodium carbonate solution (5 mL) was added to a mixture of 2-amino-1-(3,4-dimethoxyphenyl)ethanol hydrochloride (0.41 g, 1.75 mmol) in ether (10 mL) and the mixture was stirred for 5 min. To the mixture, 2-(4-(benzyloxy)-3-methoxyphenyl)acetyl chloride (0.57 g, 1.96 mmol) in ether was added dropwise. The mixture was stirred for 2 h. The solid was collected by filtration to give 5p (0.70 g, 88%). 1H NMR (300 MHz, CDCl3) δ 3.13 (br, 1 H), 3.28–3.38 (m, 1 H), 3.52 (s, 2 H), 3.53–3.62 (m, 1 H), 3.86 (s, 9 H), 4.75 (m, 1 H), 5.15 (s, 2 H), 5.80 (br, 1 H), 6.66–6.87 (m, 6 H), 7.25–7.46 (m, 5 H).
Procedure G: synthesis of compounds 6a–p
1-(3-Fluoro-4-methoxybenzyl)-6-(2-fluoroethoxy)-7-methoxyisoquinoline oxalate (6a)
A mixture of 5a and POCl3 (0.5 mL) in acetonitrile (20 mL) was stirred under reflux for 30 min. After being cooled to room temperature and quenched with 25 mL of saturated sodium bicarbonate, the mixture was extracted with methylene chloride (10 mL × 3) and dried over MgSO4. After evaporation of the solvent, the residue was purified by column chromatography with CH2Cl2/MeOH (20/1, v/v) as mobile phase to give the isoquinoline 6a (0.41 g, 66%) as a white solid. To a solution of the 6a in ethyl acetate/methanol (5 mL) was added 1 equivalent of oxalic acid in MeOH (5 mL), and the resulting mixture was stirred for 1 h at room temperature to give a crude oxalate salt. The white solid was filtered, washed thoroughly with ethyl acetate and methanol, separately. The free base was converted into oxalate salt for in vitro affinity measurements and elemental analysis. Mp (oxalate salt): 179.7–180.9 °C; 1H NMR(300 MHz, free base, CDCl3) δ 3.83 (s, 3 H), 3.89 (s, 3 H), 4.39 (dt, J = 27.0, 4.2 Hz, 2 H), 4.51 (s, 2 H), 4.87 (dt, J = 47.7, 4.2 Hz, 2 H), 6.81–7.00 (m, 3 H), 7.08 (s, 1 H), 7.28 (s, 1 H), 7.42 (d, J = 5.7 Hz, 1 H), 8.35 (d, J = 5.7 Hz, 1 H). 13C NMR (75 MHz, free base, CDCl3) δ 40.4, 55.1 (d, J = 33 Hz), 66.9 (d, J = 20.5 Hz), 76.2, 80.4 (d, J = 168.6 Hz), 103.5, 105.9, 112.6, 115.3 (d, J = 18.2 Hz), 117.8, 122.2, 123.0 (d, J = 3.4 Hz), 131.7 (d, J = 5.6 Hz), 132.2, 140.1, 145.1 (d, J = 11.4 Hz), 149.2, 150.5, 151.4 (d, J = 244.8 Hz), 156.3. Anal. Calcd for C20H19F2NO3•H2C2O4: C, 58.80; H, 4.71; N, 3.12. Found: C, 58.75; H, 4.64; N, 3.09.
6-(2-Fluoroethoxy)-7-methoxy-1-(4-methoxy-3-methylbenzyl)isoquinoline oxalate (6b)
Starting with 5b, procedure G was followed to prepare compound 6b as a white solid. Mp (oxalate salt): 172.9–175.0 °C; 1H NMR (300 MHz, free base, CDCl3) δ 2.14 (s, 3 H), 3.76 (s, 3 H), 3.89 (s, 3 H), 4.38 (dt, J = 27.0, 4.2 Hz, 2 H), 4.50 (s, 2 H), 4.86 (dt, J = 47.4, 4.2 Hz, 2 H), 6.71 (d, J = 8.7 Hz, 1 H), 7.04–7.07 (m, 3 H), 7.38 (s, 1 H), 7.39 (d, J = 5.7 Hz, 1 H), 8.36 (d, J = 6.0 Hz, 1 H). 13C NMR (75 MHz, CDCl3) δ 31.1, 41.0, 55.8 (d, J = 47.8 Hz), 68.2 (d, J = 20.5 Hz), 77.4, 80.4 (d, J = 168.6 Hz), 105.3, 107.0, 110.4, 119.4, 126.9, 127.0, 130.3, 130.6, 131.1, 134.0, 135.3, 139.1, 150.8, 154.0, 157.8. Anal. Calcd for (C21H22FNO3•H2C2O4: C, 62.02; H, 5.43; N, 3.14. Found: C, 6175; H, 5.52; N, 3.12.
6-(2-Fluoroethoxy)-7-methoxy-1-(4-(methylthio)benzyl)isoquinoline oxalate (6c)
Starting with 5c, procedure G was followed to prepare compound 6c (0.23 g, 72%) as a white solid. Mp (oxalate salt): 138.5–140.0 °C; 1H NMR (300 MHz, free base, CDCl3) δ 2.42 (s, 3 H), 3.88 (s, 3 H), 4.38 (dt, J = 27.0, 4.2 Hz, 2 H), 4.55 (s, 2 H), 4.86 (dt, J = 47.4, 4.2 Hz, 2 H), 7.06 (s, 1 H), 7.14–7.21 (m, 4 H), 7.38 (s, 1H), 7.41 (d, J = 6.0 Hz, 1 H), 8.36 (d, J = 6.0 Hz, 1 H). Anal. Calcd for C20H20FNO2S•H2C2O4•0.5H2O: C, 57.88; H, 5.08; N, 3.07. Found: C, 57.89; H, 5.03; N, 2.93.
1-(4-Fluoro-3-methoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6d)
Starting with 5d, procedure G was followed to prepare 6d (0.21 g, 66%) as a white solid. Mp (oxalate salt): 207.7–208.7 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.83 (s, 3 H), 4.00 (s, 3 H), 4.27 (dt, J = 27.3, 3.9 Hz, 2 H), 4.50 (s, 2 H), 4.81 (dt, J = 47.1, 3.9 Hz, 2 H), 6.82–7.00 (m, 3 H), 7.07 (s, 1 H), 7.31 (s, 1 H), 7.44 (d, J = 5.7 Hz, 1 H), 8.37 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C20H19F2NO3•H2C2O4: C, 58.80; H, 4.71; N, 3.12. Found: C, 58.78; H, 4.73; N, 3.23.
7-(2-Fluoroethoxy)-6-methoxy-1-(4-methoxy-3-methylbenzyl)isoquinoline oxalate (6e)
Starting with 5e, procedure G was followed to prepare compound 6e (0.13 g, 61%) as a white solid. Mp (oxalate salt): 179.4–180.2 °C; 1H NMR (300 MHz, free base, CDCl3) δ 2.14 (s, 3 H), 3.76 (s, 3 H), 3.99 (s, 3 H), 4.26 (dt, J = 27.3, 4.5 Hz, 2 H), 4.48 (s, 2 H), 4.80 (dt, J = 47.4, 4.5 Hz, 2 H), 6.70 (d, J = 7.8 Hz, 1 H), 7.00–7.10 (m, 3 H), 7.39 (s, 1 H), 7.41 (d, J = 5.7 Hz, 1 H), 8.37 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C21H22FNO3•H2C2O4: C, 62.02; H, 5.43; N, 3.14. Found: C, 62.09; H, 5.51; N, 3.26.
7-(2-Fluoroethoxy)-6-methoxy-1-(4-(methylthio)benzyl)isoquinoline oxalate (6f)
Starting with 5f, procedure G was followed to prepare compound 6f (0.15 g, 70%) as a white solid. Mp (oxalate salt): 199.5–200.5 °C; 1H NMR(300 MHz, free base, CDCl3) δ 2.43 (s, 3 H), 3.99 (s, 3 H), 4.26 (dt, J = 27.0, 4.2 Hz, 2 H), 4.53 (s, 2 H), 4.80 (dt, J = 47.4, 4.2 Hz, 2 H), 7.07 ( s, 1 H), 7.16 (m, 4 H), 7.32 (s, 1 H), 7.43 (d, J = 5.7 Hz, 1 H), 8.37 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C20H20FNO2S•H2C2O4: C, 59.05; H, 4.96; N, 3.13. Found: C, 59.06; H, 4.93; N, 3.10.
1-(2-Fluoro-4-methoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6g)
Starting with 5g, procedure G was followed to prepare compound 6g (0.13 g, 67%) as a pale solid. Mp (oxalate salt): 203.5–204.2 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.74 (s, 3 H), 3.99 (s, 3 H), 4.35 (dt, J = 27.3, 4.2 Hz, 2 H), 4.49 (s, 2 H), 4.85 (dt, J = 47.4, 4.2 Hz, 2 H), 6.55 (dd, J = 8.7, 1.8 Hz, 1 H), 6.64 (dd, J = 12.0, 2.7 Hz, 1 H), 7.00–7.13 (m, 2 H), 7.40–7.43 (m, 2 H), 8.36 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C20H19F2NO3•H2C2O4: C, 58.80; H, 4.71; N, 3.12. Found: C, 58.90; H, 4.75; N, 3.11.
1-(5-Bromo-2-methoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6h)
Starting with 5h, procedure G was followed to prepare compound 6h (0.14 g, 49%) as a white solid. Mp (oxalate salt): 195.7–197.3 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.90 (s, 3 H), 4.00 (s, 3 H), 4.27 (dt, J = 27.1, 4.5 Hz, 2 H), 4.52 (s, 2 H), 4.81 (dt, J = 47.4, 4.5 Hz, 2 H), 6.78 (d, J = 9.0 Hz, 1 H), 7.07 (s, 1 H), 7.16–7.43 (m, 4 H), 8.37 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C20H19BrFNO3•H2C2O4: C, 51.78; H, 4.15; N, 2.74. Found: C, 51.90; H, 4.21; N, 2.80.
1-(3-Bromo-4-methoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6i)
Starting with 5i, procedure G was followed to prepare compound 6i (0.16 g, 47%) as a white solid. Mp (oxalate salt): 208.4–209.2°C; 1H NMR (300 MHz, free base, CDCl3) δ 3.83 (s, 3 H), 4.00 (s, 3 H), 4.29 (dt, J = 27.3, 4.5 Hz, 2 H), 4.49 (s, 2 H), 4.83 (dt, J = 47.4, 4.5 Hz, 2 H), 6.78 (d, J = 8.1 Hz, 1 H), 7.07 (s, 1H), 7.13 (dd, J = 8.1, 1.8 Hz, 1 H), 7.32 (s, 1 H), 7.43 (d, J = 5.7 Hz, 1 H), 7.49 (d, J = 1.8 Hz, 1 H), 8.37 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C20H19BrFNO3•H2C2O4: C, 51.78; H, 4.15; N, 2.74. Found: C, 51.92; H, 4.21; N, 2.80.
1-(3-Fluorobenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6j)
Starting with 5j, procedure G was followed to prepare compound 6j (0.12 g, 35%) as a white solid. Mp (oxalate salt): 182.4–183.4 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.92 (s, 3 H), 4.18 (dt, J = 27.3, 4.2 Hz, 2 H), 4.49 (s, 2 H), 4.73 (dt, J = 47.4, 4.2 Hz, 2 H), 6.76–6.87 (m, 2 H), 6.95–7.00 (m, 2 H), 7.10–7.21 (m, 2 H), 7.37 (d, J = 5.7 Hz, 1 H), 8.36 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C19H17F2NO2•H2C2O4: C, 60.14; H, 4.57; N, 3.34. Found: C, 59.98; H, 4.65; N, 3.42.
7-(2-Fluoroethoxy)-1-(3-(2-fluoroethoxy)-4-methoxybenzyl)-6-methoxyisoquinoline oxalate (6k)
Starting with 5k, procedure G was followed to prepare compound 6k (0.14 g, 58%) as a white solid. Mp (oxalate salt): 171.2–172.0 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.74 (s, 3 H), 3.99 (s, 3 H), 4.16–4.35 (m, 4 H), 4.49 (s, 2 H), 4.70–4.80 (m, 4 H), 6.55–7.13 (m, 4 H), 7.40–7.43 (m, 2 H), 8.36 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C22H23F2NO4•H2C2O4: C, 58.42; H, 5.11; N, 2.84. Found: C, 58.58; H, 5.22; N, 2.94.
1-(3,5-Dimethoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6l)
Starting with 5l, procedure G was followed to prepare compound 6l (75 mg, 22%) as a white solid. Mp (oxalate salt): 176.2–176.7 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.70 (s, 6 H), 3.97 (s, 3 H), 4.26 (dt, J = 27.3, 4.5 Hz, 2 H), 4.50 (s, 2 H), 4.78(dt, J = 47.4, 4.5 Hz, 2 H), 6.28 (t, J = 2.4 Hz, 1 H), 6.41 (m, 2 H), 7.04 (s, 1 H), 7.36 (s, 1 H), 7.41 (d, J = 5.7 Hz, 1 H), 8.36 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C21H22FNO4•H2C2O4: C, 59.87; H, 5.24; N, 3.04. Found: C, 59.91; H, 5.22; N, 3.07.
1-(4-Fluoro-3-methoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline oxalate (6m)
Starting with 5m, procedure G was followed to prepare compound 6m (65 mg, 36%) as a white solid. Mp (oxalate salt): 154.8–155.6 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.76 (s, 3 H), 3.99 (s, 3 H), 4.26 (dt, J = 27.3, 4.5 Hz, 2 H), 4.48 (s, 2 H), 4.80 (dt, J = 47.4, 4.5 Hz, 2 H), 6.70 (d, J = 7.8 Hz, 1 H), 7.00–7.10 (m, 3 H), 7.39 (s, 1 H), 7.41 (d, J = 5.7 H z, 1 H), 8.37 (d, J = 5.7 Hz, 1 H). Anal. Calcd for C20H19F2NO3•H2C2O4: C, 58.80; H, 4.71; N, 3.12. Found: C, 58.82; H, 4.81; N, 3.21.
1-(3-(2-Fluoroethoxy)-4-methoxybenzyl)-6,7-dimethoxyisoquinoline (6n)
Starting with 5n, procedure G was followed to prepare compound 6n (125 mg, 50%) as a pale solid. Mp (oxalate salt): 149.7–150.6 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.81 (s, 3 H), 3.90 (s, 3 H), 4.01 (s, 3 H), 4.16 (dt, J = 27.9, 4.5 Hz, 2 H), 4.52 (s, 2 H), 4.70 (dt, J = 47.1, 4.5 Hz, 2 H), 6.78–6.89 (m, 3 H), 7.05 (s, 1 H), 7.32 (s, 1 H), 7.43 (d, J = 5.7, 1 H), 8.36 (d, J = 5.7 Hz, 1 H). 13C NMR (75 MHz, CDCl3) δ 37.2, 56.3, 56.6, 56.9, 69.3 (d, J = 21.6 Hz), 80.9 (d, J = 169.6 Hz), 105.0, 106.0, 112.6, 115.5, 121.6, 122.1, 122.9, 128.5, 131.5, 136.9, 152.7, 154.7, 157.0, 160.7, 163.3. Anal. Calcd for C21H22FNO4•H2C2O4: C, 59.87; H, 5.24; N, 3.04. Found: C, 59.78; H, 5.26; N, 3.04.
1-(4-(Benzyloxy)-3-methoxybenzyl)-7-(2-fluoroethoxy)-6-methoxyisoquinoline (6o)
Starting with 5o, procedure G was followed to prepare compound 6o (0.16 g, 40%) as a white solid. 1H NMR (300 MHz, free base, CDCl3) δ 3.77 (s, 3 H), 3.99 (s, 3 H), 4.25 (dt, J = 27.3, 4.5 Hz, 2 H), 4.50 (s, 2 H), 4.85 (dt, J = 47.4, 4.5 Hz, 2 H), 5.09 (s, 2 H), 6.70–6.80 (d, J = 7.8 Hz, 4 H), 7.06 (s, 1 H), 7.29–7.44 (m, 6 H), 8.37 (d, J = 5.7 Hz, 1 H).
1-(4-(Benzyloxy)-3-methoxybenzyl)-7-(2-methoxy)-6-methoxyisoquinoline (6p)
Starting with 5p, procedure G was followed to prepare compound 6p (0.23 g, 29%) as a white solid. 1H NMR (300 MHz, free base, CDCl3) δ 3.77 (s, 3 H), 3.86 (s, 3 H), 4.00 (s, 3 H), 4.44 (d, J = 5.7 Hz, 2 H), 4.52 (s, 2 H), 5.09 (s, 2 H), 6.75 (s, 1 H), 6.76 (s, 1 H), 6.83 (s, 1 H), 7.05 (s, 1 H), 7.30–7.44 (m, 8 H), 8.70 (d, J = 7.5 Hz, 1 H).
Procedure H: synthesis of compounds 7a and 7b
4-((7-(2-Fluoroethoxy)-6-methoxyisoquinolin-1-yl)methyl)-2-methoxyphenol (7a)
A mixture of 6o (0.45 g, 1.0 mmol), hydrochloric acid (4.0 M, 3 mL), and ethanol (5 mL) was refluxed for 2h. After diluting the reaction mixture with ethyl acetate (20 mL), saturated NaHCO3 solution was added and then extracted with ethyl acetate (30 mL × 3). The organic layer was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified on a silica gel column with CH2Cl2/MeOH (20/1, v/v/) as mobile phase to give 7a (180 mg, 50%) as a white solid. 1H NMR (300 MHz, CDCl3) is δ 3.75 (s, 3 H), 3.99 (s, 3 H), 4.26 (dt, J = 27.3, 4.5 Hz, 2 H), 4.50 (s, 1 H), 4.81 (dt, J = 47.4, 4.5 Hz, 2 H), 5.58 (br, 1 H), 6.7–6.82 (m, 3 H), 7.06 (s, 1 H), 7.39 (s, 1 H), 7.43 (d, J = 5.7 Hz, 1 H), 8.37 (d, J = 5.7 Hz, 1 H).
4-((6,7-Dimethoxyisoquinolin-1-yl)methyl)-2-methoxyphenol (7b)
Starting with 6p, following the above procedure H to afford 7b (0.12 g, 49%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 3.75 (s, 3 H), 3.90 (s, 3 H), 4.00 (s, 3 H), 4.52 (s, 2 H), 5.65 (br, 1 H), 6.76 (s, 1 H), 6.82 (s, 2 H), 7.05 (s, 1 H), 7.35 (s, 1 H), 7.42 (d, J = 5.7 Hz, 1 H), 8.36 (d, J = 5.7 Hz, 1 H).
Synthesis of compounds 8a and 8b
7-(2-Fluoroethoxy)-1-(4-(2-fluoroethoxy)-3-methoxybenzyl)-6-methoxyisoquinoline oxalate (8a)
Starting with 7a, procedure A was followed to prepare 8a (0.15 g, 42%) as a pale solid. The oxalate salt of 8a was prepared from 8a according to the procedure described for the oxalate slat of 6a. Mp (oxalate salt): 167.2–168.0 °C; 1H NMR (300 MHz, free base, CDCl3) is δ 3.76 (s, 3 H), 3.99 (s, 3 H), 4.20 (dt, J = 27.3, 4.5 Hz, 2 H), 4.26 (dt, J = 27.3, 4.2 Hz, 2 H), 4.52 (s, 2H), 4.73 (dt, J = 47.4, 4.2 Hz, 2 H), 4.80 (dt, J = 47.7, 4.5 Hz, 2 H), 6.75–6.83 (m, 3 H), 7.07 (s, 1H), 7.38 (s, 1 H), 7.43 (d, J = 5.4 Hz, 1 H), 8.38 (d, J = 5.4 Hz, 1 H). Anal. Calcd for C22H23F2NO4•H2C2O4: C, 58.42; H, 5.11; N, 2.84. Found: C, 58.45; H, 5.14; N, 2.75.
1-(4-(2-Fluoroethoxy)-3-methoxybenzyl)-6,7-dimethoxyisoquinoline oxalate (8b)
Starting with 7b, procedure A was followed to prepare 8b (0.17 g, 55%) as a white sold. The oxalate salt of 8b was prepared from 8b according to the procedure described for the oxalate slat of 6a. Mp (oxalate salt): 157.3–158.2 °C. 1H NMR (300 MHz, CDCl3) δ 3.76 (s, 3 H), 3.90 (s, 3 H), 4.01 (s, 3 H), 4.21 (dt, J = 27.6, 3.9 Hz, 2 H), 4.54 (s, 2 H), 4.74 (dt, J = 47.7, 3.9 Hz, 2 H), 6.80–6.86 (m, 3 H), 7.05 (s, 1 H), 7.33 (s, 1 H), 7.43 (d, J = 5.4 Hz, 1 H), 8.37 (d, J = 5.4 Hz, 1 H). 13C NMR (75 MHz, CDCl3) δ 37.4, 56.4, 56.6, 56.9, 69.0 (d, J = 21.6 Hz), 82.2 (d, J = 169.6 Hz), 105.0, 106.0, 113.2, 115.3, 120.9, 122.6, 122.9, 130.0, 131.6, 136.9, 147.6, 150.8, 154.6, 157.0, 163.3. Anal. Calcd for C21H22FNO4•H2C2O4: C, 59.87; H, 5.24; N, 3.04. Found: C, 59.70; H, 5.21; N, 2.96.
Procedure I: synthesis of compound 8c
1-(4-(2-(2-Fluoroethoxy)ethoxy)-3-methoxybenzyl)-6,7-dimethoxyisoquinoline oxalate (8c)
A mixture of 7b (0.2 g, 0.62 mmol), 1-chloro-2-(2-fluoroethoxy)ethane (0.2 g, 1.59 mmol), and K2CO3 (0.25 g, 1.81 mmol) in DMF (10 mL) was stirred at room temperature overnight. The mixture was poured into water (50 mL) and extracted with ethyl acetate (10 mL × 3). The organic layer was dried over MgSO4. After evaporation of the solvent, the residue was purified by silica gel column chromatography with CH2Cl2/MeOH (20/1, v/v/) as mobile phase to give 8c (0.14 g, 54%) as a white solid. The oxalate salt of 8c was prepared from 8c according to the procedure described for the oxalate slat of 6a. Mp (oxalate salt):152.2–153.0 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.74 (s, 3 H), 3.83–3.90 (m, 7 H), 4.00 (s, 3 H), 4.14 (t, J = 5.4 Hz, 2 H), 4.53 (s, 2 H), 4.55 (dt, J = 47.7, 4.2 Hz, 2 H), 6.80–6.84 (m, 3 H), 7.05 (s, 1 H), 7.33 (s, 1 H), 7.43 (d, J = 5.7 Hz, 1 H), 8.37 (d, J = 5.7 Hz, 1 H). 13C NMR (75 MHz, CDCl3) δ 37.5, 56.4, 56.6, 56.9, 69.1, 70.0, 70.5 (d, J = 21.8 Hz), 83.3 (d, J = 168.4 Hz), 105.1, 106.0, 113.0, 114.7, 120.9, 121.6, 123.0, 139.4, 131.6, 136.9, 148.0, 152.6, 154.7, 157.0, 163.3. Anal. Calcd for C23H26FNO5•H2C2O4: C, 59.40; H, 5.58; N, 2.77. Found: C, 59.26; H, 5.60; N, 2.78.
Procedure J: synthesis of compound 11
6,7-Dimethoxy-4-(4-methoxybenzyl)quinazoline oxalate (11)
To a solution of 4-chloro-6,7-dimethoxyquinazoline, 9 (0.25 g, 1.1 mmol) and manganese (II) chloride (12.6 mg, 0.1 mmol) in THF (20 mL) was added 4-methoxybenzyl magnesium chloride, 10 (3.3 mmol). After the reaction mixture was stirred at room temperature for 6 h, water (30 mL) was added, and the mixture was extracted with ethyl acetate (20 mL × 3). The organic layer was dried over anhydrous sodium sulfate. After concentrating, the residue was purified by silica gel column with CH2Cl2/MeOH (20/1, v/v/) as mobile phase to give 11 (0.21 g, 58%) as a white solid. The oxalate salt of 11 was prepared from 11 according to the procedure described for the oxalate slat of 6a. Mp (oxalate salt): 127.6–128.4 °C; 1H NMR (300 MHz, free base, CDCl3) δ 3.76 (s, 3 H), 3.93 (s, 3 H), 4.03 (s, 3 H), 4.48 (s, 2 H), 6.81–6.85 (m, 2 H), 7.19–7.30 (m, 4 H), 9.09 (s, 1 H). 13C NMR (75 MHz, CDCl3) δ 40.9, 55.3, 56.1, 56.4, 102.6, 107.3, 114.2, 119.5, 129.8, 130.1, 148.5, 150.2, 153.7, 155.6, 158.5, 166.3. Anal. Calcd for C18H18N2O3: C, 69.66; H, 5.85; N, 9.03. Found: C, 69.53; H, 5.81; N, 8.99.
4.2. In vitro Assessment of affinities
To determine the potency of the new synthesized analogues, first, compounds were screened in vitro for their affinities toward PDE10A to determine IC50 values. Compounds having a high affinity for PDE10A (IC50 values < 60 nM), were further assessed for their selectivity for PDE10A versus other PDE isoforms, PDE3A and PDE3B. All compounds were independently assayed at least two times.
4.2.1. PDE10A enzyme assay protocol
The screening method followed published procedures [10–12, 19, 29]. PDE activity was measured using the Phosphodiesterase [3H]cAMP Scintillation Proximity Assay (SPA) (Cat. #TRKQ7090, Perkin Elmer, Waltham, MA) with minor modifications to the manufacturer’s protocol. Briefly, the effect of PDE inhibitors is determined by assaying a fixed amount of enzyme in the presence of varying compound concentrations and a low [3H]cAMP substrate concentration; the substrate concentration used in the assay is 1/3 of the Km concentration, allowing for comparisons of lC50 values across a panel of different PDE enzymes. Reactions are initiated with enzyme, incubated to give ~30% substrate turnover, and terminated with yttrium silicate SPA beads. Plates are sealed, allowed to settle, and counted on a Trilux Micro-Beta Counter (PerkinElmer, Waltham, MA). Radioactivity units can be converted to percent activity of an uninhibited control (100%), plotted against inhibitor concentration and inhibitor IC50 values were calculated.
4.2.2. Measure the in vitro affinities for PDE 3A and PDE 3B to determine selectivity of PDE10A vs PDE3A and PDE3B for analogues with high PDE10A affinity
Compounds with high affinity for PDE10A (IC50 < 60 nM) were further assessed for their PDE10A selectivity over other PDE3A and PDE3B. The screening method is the same as the protocol for PDE10A except the assays will use other PDE isoforms and their concentrations are adjusted based on the Ki value for the different PDE isoforms.
Scheme 2.

Reagents and conditions:
i) 1:1 HCl(aq), EtOH; ii) 2-bromo-1-fluoroethane or 1-chloro-2-(2-fluoroethoxy)ethane, K2CO3, DMF
Research highlights.
Successfully synthesized and evaluated a series of analogues as PDE10A inhibitors.
Identified six compounds which have PDE10 binding affinities ranging between 28–60 nM.
Compound 8c has high affinity for PDE10A and high selectivity for PDE10A vs. PDE3A/B.
Compound 8c can be radiosynthesized with fluorine-18 serving as PDE10A PET probe.
Acknowledgments
Financial support for these studies was provided by the National Institute of Health under MH081281-04 (PI: Mach), NS061025 (PI: Tu) and MH092797 (PI: Tu).
Abbreviations
- AMP
Adenosine monophosphate
- Anal
Analysis
- Calcd
Calculated
- cAMP
cyclic Adenosine monophosphate
- CDI
1,1′-Carbonyldiimidazole
- CIMS
Chemical ionization mass spectrometry
- CNS
Central nervous system
- cGMP
cyclic Guanosine monophosphate
- DCC
N,N′-Dicyclohexylcarbodiimide
- DMF
N,N-Dimethylformamide
- DMSO
Dimethyl sulfoxide
- GMP
Guanosine monophosphate
- MSNs
Medium-sized spiny neurons
- ND
Not determined
- PA
Papaverine
- PCP
Phenylcyclohexylpiperidine
- PDE10A
Phosphodiesterase 10A
- PDE3A
Phosphodiesterase 3A
- PDE3B
Phosphodiesterase 3B
- PET
Positron emission tomography
- PLD
Phospholipase D
- THF
Tetrahydrofuran
- TLC
Thin layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Groenewegen HJ, van den Heuvel OA, Cath DC, Voorn P, Veltman DJ. Brain Dev. 2003;25(Suppl 1):S3–S14. doi: 10.1016/s0387-7604(03)90001-5. [DOI] [PubMed] [Google Scholar]
- 2.Winterer G, Weinberger DR. Trends Neurosci. 2004;27:683–90. doi: 10.1016/j.tins.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 3.DeLong MR, Wichmann T. Arch Neurol. 2007;64:20–4. doi: 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- 4.Winterer G, Carver FW, Musso F, Mattay V, Weinberger DR, Coppola R. Hum Brain Mapp. 2007;28:805–16. doi: 10.1002/hbm.20322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menniti FS, Chappie TA, Humphrey JM, Schmidt CJ. Curr Opin Investig Drugs. 2007;8:54–9. [PubMed] [Google Scholar]
- 6.Menniti FS, Faraci WS, Schmidt CJ. Nat Rev Drug Discov. 2006;5:660–70. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 7.Siuciak JA, McCarthy SA, Chapin DS, Fujiwara RA, James LC, Williams RD, Stock JL, McNeish JD, Strick CA, Menniti FS, Schmidt CJ. Neuropharmacology. 2006;51:374–85. doi: 10.1016/j.neuropharm.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Siuciak JA, Chapin DS, Harms JF, Lebel LA, McCarthy SA, Chambers L, Shrikhande A, Wong S, Menniti FS, Schmidt CJ. Neuropharmacology. 2006;51:386–96. doi: 10.1016/j.neuropharm.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms JF, Hoffman WE, Lebel LA, McCarthy SA, Nelson FR, Proulx-LaFrance C, Majchrzak MJ, Ramirez AD, Schmidt K, Seymour PA, Siuciak JA, Tingley FD, 3rd, Williams RD, Verhoest PR, Menniti FS. J Pharmacol Exp Ther. 2008;325:681–90. doi: 10.1124/jpet.107.132910. [DOI] [PubMed] [Google Scholar]
- 10.Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA. Gene. 1999;234:109–17. doi: 10.1016/s0378-1119(99)00171-7. [DOI] [PubMed] [Google Scholar]
- 11.Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K, Omori K. J Biol Chem. 1999;274:18438–45. doi: 10.1074/jbc.274.26.18438. [DOI] [PubMed] [Google Scholar]
- 12.Soderling SH, Bayuga SJ, Beavo JA. Proc Natl Acad Sci U S A. 1999;96:7071–6. doi: 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotera J, Fujishige K, Yuasa K, Omori K. Biochem Biophys Res Commun. 1999;261:551–7. doi: 10.1006/bbrc.1999.1013. [DOI] [PubMed] [Google Scholar]
- 14.Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, Lanfear J, Ryan AM, Schmidt CJ, Strick CA, Varghese AH, Williams RD, Wylie PG, Menniti FS. Brain Res. 2003;985:113–26. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- 15.Neve KA, Seamans JK, Trantham-Davidson H. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- 16.Chappie T, Humphrey J, Menniti F, Schmidt C. Current Opinion in Drug Discovery and Development. 2009;12:458–67. [PubMed] [Google Scholar]
- 17.Kehler J, Kilburn JP. Expert Opinion on Therapeutic Patents. 2009;19:1715–25. doi: 10.1517/13543770903431050. [DOI] [PubMed] [Google Scholar]
- 18.Verhoest PR, Chapin DS, Corman M, Fonseca K, Harms JF, Hou X, Marr ES, Menniti FS, Nelson F, O’Connor R, Pandit J, Proulx-LaFrance C, Schmidt AW, Schmidt CJ, Suiciak JA, Liras S. Journal of Medicinal Chemistry. 2009;52:5188–96. doi: 10.1021/jm900521k. [DOI] [PubMed] [Google Scholar]
- 19.Chappie TA, Humphrey JM, Allen MP, Estep KG, Fox CB, Lebel LA, Liras S, Marr ES, Menniti FS, Pandit J, Schmidt CJ, Tu M, Williams RD, Yang FV. J Med Chem. 2007;50:182–5. doi: 10.1021/jm060653b. [DOI] [PubMed] [Google Scholar]
- 20.Hofgen N, Stange H, Schindler R, Lankau HJ, Grunwald C, Langen B, Egerland U, Tremmel P, Pangalos MN, Marquis KL, Hage T, Harrison BL, Malamas MS, Brandon NJ, Kronbach T. J Med Chem. 2010;53:4399–411. doi: 10.1021/jm1002793. [DOI] [PubMed] [Google Scholar]
- 21.Tu Z, Xu J, Li S, Jones LA, Mach RH. Annual meeting of Society of Nuclear Medicine. Supplement 2. Vol. 50. Toronto, ON, Canada: 2009. p. 618. [Google Scholar]
- 22.Rodefer JS, Murphy ER, Baxter MG. Eur J Neurosci. 2005;21:1070–6. doi: 10.1111/j.1460-9568.2005.03937.x. [DOI] [PubMed] [Google Scholar]
- 23.Liu F, Zhang G, Kelley C. Society for Neuroscience. Atlanta (GA): Oct 11–18, 2006. [Google Scholar]
- 24.Tu Z, Xu J, Jones LA, Li S, Mach RH. Nucl Med Biol. 2010;37:509–16. doi: 10.1016/j.nucmedbio.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grigg R, Inman M, Kilner C, Köppen I, Marchbank J, Selby P, Sridharan V. Tetrahedron. 2007;63:6152–69. [Google Scholar]
- 26.Rao KV, Beach JW. Journal of Medicinal Chemistry. 1991;34:1871–9. doi: 10.1021/jm00110a018. [DOI] [PubMed] [Google Scholar]
- 27.Mager G, Klocke RK, Kux A, Hopp HW, Hilger HH. Am Heart J. 1991;121:1974–83. doi: 10.1016/0002-8703(91)90834-5. [DOI] [PubMed] [Google Scholar]
- 28.Movsesian M, Stehlik J, Vandeput F, Bristow MR. Heart Fail Rev. 2009;14:255–63. doi: 10.1007/s10741-008-9130-x. [DOI] [PubMed] [Google Scholar]
- 29.Fawcett L, Baxendale R, Stacey P, McGrouther C, Harrow I, Soderling S, Hetman J, Beavo JA, Phillips SC. Proc Natl Acad Sci U S A. 2000;97:3702–7. doi: 10.1073/pnas.050585197. [DOI] [PMC free article] [PubMed] [Google Scholar]