

Abstract
Under the conditions of transfer hydrogenation employing an ortho-cyclometallated iridium catalyst generated in situ from [Ir(cod)Cl]2, 4-cyano-3-nitrobenzoic acid and the chiral phosphine ligand (S)-SEGPHOS, α-methyl allyl acetate couples to alcohols 1a–1j with complete levels of branched regioselectivity to furnish products of carbonyl crotylation 3a–3j, which are formed with good levels of anti-diastereoselectivity and exceptional levels of enantioselectivity. An identical set of optically enriched carbonyl crotylation products 3a–3j is accessible from the corresponding aldehydes 2a–2j under the same conditions, but employing isopropanol as the terminal reductant. Experiments aimed at probing the origins of stereoselection establish a matched mode of ionization for the (R)-acetate and the iridium catalyst modified by (S)-SEGPHOS, as well as reversible ionization of the allylic acetate with rapid π-facial interconversion of the resulting π-crotyl intermediate in advance of C-C bond formation. Additionally, rapid alcohol-aldehyde redox equilibration in advance of carbonyl addition is demonstrated. Thus, anti-diastereo- and enantioselective carbonyl crotylation from the alcohol or aldehyde oxidation level is achieved in the absence of any stoichiometric metallic reagents or stoichiometric metallic byproducts.
Introduction
Carbonyl crotylation ranks among the foremost methods used for the construction of polypropionate natural products.1 The majority of enantioselective crotylation protocols exploit chirally modified crotylmetal reagents, such as the B-crotyl reagents developed by Hoffmann (1979),2a Brown (1986),2b,c Roush (1986),2d,e Masamune (1987),2f and Soderquist (2005),2g the Ti-crotyl reagents developed by Duthaler (1989),2h,i and the Si-crotyl reagents developed by Panek (1991)2j,k and Leighton (2004).2l Enantioselective Lewis acid catalyzed additions of crotylmetal reagents are reported by Yamamoto (1991),3a,b,c Mikami (1993),3d Nishiyama (2001),3e Evans (2006)3f and Hall (2006).3g,h Additionally, enantioselective Lewis base catalyzed carbonyl crotylations employing crotylmetal reagents are reported by Denmark (1994),4a,b,c Iseki (1997),4d Nakajima (1998),4e and Kočovsky (2003).4f Finally, a chiral diol catalyzed allylboration of ketones is reported by Schaus (2006).5
In these cases, the crotylating agent is a metal or metalloid that is itself prepared from a metallic precursor. For example, the crotylating agent developed by Brown2b,c is prepared through potassiation of butene employing Schlosser’s base6 followed by transmetallation to boron. Here, multiple manipulations and multiple preformed organometallics (n-BuLi, KC4H7, Ipc2BOMe) are required to prepare the desired crotylborane, which contributes to cost and waste generation.
An alternate approach to enantioselective carbonyl crotylation involves reductive generation of crotylmetal reagents from the corresponding halides, as in catalytic enantioselective variants of the Nozaki-Hiyama reaction.7,8 Here, metallic reductants are required for catalytic turnover and modest diastereoselectivities are typically observed. Related reductive couplings of allylic alcohols, acetates and carboxylates, which constitute an umpolung of π-allyl chemistry, also have been disclosed.9–13 However, catalytic enantioselective crotylations based on this approach are absent and, with one exception,12 metallic terminal reductants are again required.
Metal catalyzed reductive C-C coupling under the conditions of hydrogenation or transfer hydrogenation provides an alternative to the use of preformed organometallic reagents in an ever-increasing range of C=X (X = O, NR) addition processes.14 A powerful manifestation of these concepts resides in the coupling of unsaturates to carbonyl partners to furnish homoallylic alcohols,15–17 wherein allenes,15 dienes16 or allyl acetate17 serve as surrogates to preformed allylmetal reagents. The iridium catalyzed transfer hydrogenative couplings of allyl acetate17 represent an especially significant advance over established carbonyl allylation protocols,1 as highly enantioselective carbonyl allylation is achieved from the alcohol or aldehyde oxidation level in the absence of any stoichiometric metallic reagents. Inspired by these results, stereoselective carbonyl crotylations employing α-methyl allyl acetate as the crotyl donor were sought. Here, we report a second generation ortho-cyclometallated iridium catalyst modified by 4-cyano-3-nitrobenzoic acid and (S)-SEGPHOS that promotes highly regio- and enantioselective carbonyl crotylation from the alcohol or aldehyde oxidation level with good levels of anti-diastereoselectivity (Scheme 1).
Scheme 1.

Carbonyl crotylation from the alcohol or aldehyde oxidation level via transfer hydrogenative C-C coupling.
Results and Discussion
Using our first generation catalytic system,17a which employs an iridium complex generated in situ from [Ir(cod)Cl]2, m-nitrobenzoic acid and chelating chiral phosphine ligand, carbonyl couplings employing α-methyl allyl acetate occur with complete branch regioselectivity. However, poor anti-diastereoselectivities were observed and the level of diastereoselection was insensitive to changes in the character of the phosphine ligand. Our subsequent discovery that the active catalyst is an ortho-cyclometallated iridium C,O-benzoate17b unveiled new opportunities to direct diastereoselectivity involving modification of the cyclometallating agent.
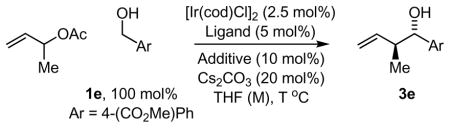
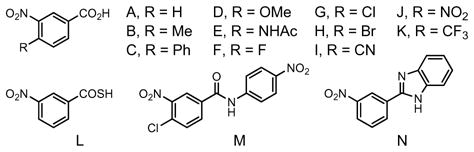
Accordingly, a range of substituted m-nitrobenzoic acids were assayed in the transfer hydrogenative coupling of α-methyl allyl acetate and alcohol 1e to furnish homoallyl alcohol 3e (Table 1). This assay focused primarily on 4-substituted-3-nitrobenzoic acids, as substitution at the 5- and 6-positions substantially diminished the reactivity of the resulting catalytic complex. Iridium catalysts modified by 4-cyano-3-nitrobenzoic acid I and 3,4-dinitrobenzoic acid J displayed the highest levels of diastereocontrol, providing the homoallyl alcohol 3e in 3.0:1 and 3.5:1 anti:syn ratios, respectively (Table 1, entries 9 and 10). Alternative cyclometallating agents, as represented by compounds L–M, were explored
Table 1.
Optimizing relative and absolute stereocontrol in transfer hydrogenative carbonyl crotylation from the alcohol oxidation level.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ligand | Acid | OAc (eq) | THF (M) | T °C | Y (%) | dr (ee%) |
| 1 | BIPHEP | A | 10 | 0.2 | 100 | 85 | 2.0:1 |
| 2 | BIPHEP | B | 10 | 0.2 | 100 | 72 | 2.7:1 |
| 3 | BIPHEP | C | 10 | 0.2 | 100 | 10 | 2.0:1 |
| 4 | BIPHEP | D | 10 | 0.2 | 100 | 68 | 2.2:1 |
| 5 | BIPHEP | E | 10 | 0.2 | 100 | 50 | 1.5:1 |
| 6 | BIPHEP | F | 10 | 0.2 | 100 | 78 | 2.3:1 |
| 7 | BIPHEP | G | 10 | 0.2 | 100 | 93 | 2.6:1 |
| 8 | BIPHEP | H | 10 | 0.2 | 100 | 80 | 2.4:1 |
| 9 | BIPHEP | I | 10 | 0.2 | 100 | 70 | 3.0:1 |
| 10 | BIPHEP | J | 10 | 0.2 | 100 | 65 | 3.5:1 |
| 11 | BIPHEP | K | 10 | 0.2 | 100 | 86 | 2.4:1 |
| 12 | BIPHEP | L | 10 | 0.2 | 100 | 38 | 1.9:1 |
| 13 | BIPHEP | M | 10 | 0.2 | 100 | 7 | 1.9:1 |
| 14 | BIPHEP | N | 10 | 0.2 | 100 | 5 | 2.1:1 |
| 15 | BIPHEP | I | 5 | 0.2 | 100 | 57 | 3.7:1 |
| 16 | BIPHEP | I | 2 | 0.2 | 100 | 55 | 4.3:1 |
| 17 | BIPHEP | I | 2 | 0.5 | 100 | 77 | 4.8:1 |
| 18 | BIPHEP | I | 2 | 1.0 | 100 | 75 | 7.1:1 |
| 19 | BIPHEP | I | 2 | 1.0 | 90 | 78 | 7.5:1 |
| 20 | BIPHEP | J | 2 | 1.0 | 90 | 42 | 7.6:1 |
| 21 | (S)-BINAP | I | 2 | 1.0 | 90 | 75 | 3.5:1 (95) |
| 22 | (S)-MeO-BIPHEP | I | 2 | 1.0 | 90 | 63 | 5.8:1 (94) |
| 23 | S)-Cl,MeO-BIPHEP | I | 2 | 1.0 | 90 | 67 | 3.0:1 (96) |
| ⇨ 24 | (S)-SEGPHOS | I | 2 | 1.0 | 90 | 70 | 7.4:1 (95) |
| 25 | (S)-C2-TUNEPHOS | I | 2 | 1.0 | 90 | 68 | 7.7:1 (91) |
| ⇨ 26 | (S)-C3-TUNEPHOS | I | 2 | 1.0 | 90 | 77 | 8.0:1 (97) |
| 27 | (S)-C4-TUNEPHOS | I | 2 | 1.0 | 90 | 71 | 6.4:1 (92) |
|
| |||||||
| |||||||
| |||||||
All reactions were performed in 13 × 100 mm pressure tubes. The cited yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis via comparison to racemic diastereomeric mixtures. For entries 1–18, the reaction was allowed to run for 20 hours. For entries 19–27, the reaction was allowed to run for 48 hours. See experimental section for further details.
Using the catalyst modified by 4-cyano-3-nitrobenzoic acid I, it was found that the level of diastereoselection was dependant upon the loading of α-methyl allyl acetate. Specifically, a reduction in the loading of α-methyl allyl acetate from 1000 mol% to 500 mol% to 200 mol% was found to increase the level of anti-diastereoselection from 3.0:1 to 3.7:1 to 4.3:1, respectively (Table 1, entries 9, 15 and 16). Remarkably, at 200 mol% loadings of α-methyl allyl acetate, both diastereoselectivity and isolated yield were found to improve substantially with increasing concentration. At 0.2, 0.5 and 1.0 M concentrations, anti-diastereoselectivities increased from 4.3:1 to 4.8:1 to 7.1:1, respectively (Table 1, entries 16–18). Finally, by decreasing reaction temperature from 100 °C to 90 °C, homoallyl alcohol 3e was formed in 78% yield with 7.5:1 anti-diastereoselectivity (Table 1, entry 19). Notably, the catalyst modified by 3,4-dinitrobenzoic acid J was highly sensitive to reaction temperature, and under identical conditions at 90 °C, homoallyl alcohol 3e was produced in only 42% yield with 7.6:1 anti-diastereoselectivity (Table 1, entry 20).
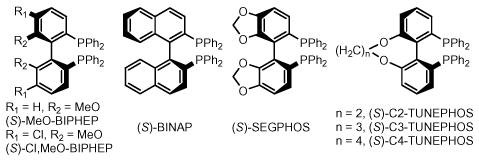
At this point, chirally modified catalysts were assayed. Although catalysts incorporating the ligands (S)-BINAP, (S)-MeO-BIPHEP, and (S)-Cl,MeO-BIPHEP were found to enforce high levels of absolute stereocontrol, significant erosion of diastereoselectivity was observed (Table 1, entries 21–23). Using a catalyst modified by (S)-SEGPHOS, the carbonyl crotylation product 3e is formed in 70% isolated yield with 7.4:1 anti-diastereoselectivity and 95% enantiomeric excess (Table 1, entries 24). The TUNEPHOS ligands of Zhang18 also were assayed (Table 1, entries 25–27). Here, (S)-C3-TUNEPHOS was found to provide the carbonyl crotylation product 3e in 77% isolated yield with 8:1 anti-diastereoselectivity and 97% enantiomeric excess (Table 1, entry 26).
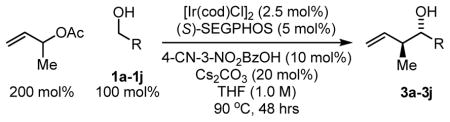
Using the aforementioned coupling conditions employing (S)-SEGPHOS and (S)-C3-TUNEPHOS, a range of alcohols were subjected to carbonyl crotylation (Table 2). In most cases, the catalyst modified by (S)-SEGPHOS gave superior results. In terms of scope, diverse substituted benzylic alcohols 1a–1f are converted to the corresponding carbonyl crotylation products 3a–3f in good to excellent yield, very good levels of anti-diastereoselectivity and with exceptional levels of absolute stereocontrol (Table 2, entries 1–6). As demonstrated by the conversion of cinnamyl alcohol 1g to homoallyl alcohol 3g, allylic alcohols participate in the coupling (Table 2, entry 7). Finally, unactivated aliphatic alcohols 1h–1j are transformed to the corresponding carbonyl crotylation products 3h–3j in good yield with equally high levels of relative and absolute stereocontrol (Table 2, entries 8–10).
Table 2.
Ir-catalyzed transfer hydrogenative crotylation of alcohols 1a–1j.a
 | |||||
|---|---|---|---|---|---|
| 1a, R = Ph | 1f, R = 2-(N-Me-indole) | ||||
| 1b, R = 3-MeOPh | 1g, R = CH=CHPh | ||||
| 1c, R = 4-MeOPh | 1h, R = 2-phenyl-1-ethyl | ||||
| 1d, R = 4-BrPh | 1i, R = 3-(benzyloxy)propyl | ||||
| 1e, R = 4-(CO2Me)Ph | 1j, R = (CH2)7CH3 | ||||
|
| |||||
| Entry | Alcohol | Product | Yield (%) | ee (%) | anti:syn |
|
| |||||
| 1 | 1a |
 3a |
65 | 95 | 6:1 |
| 2 | 1b |
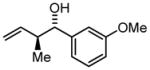 3b |
70 | 95 | 6:1 |
| 3b | 1c |
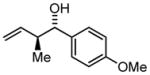 3c |
67 73 |
90 92 |
5:1 4:1c |
| 4 | 1d |
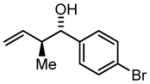 3d |
73 | 95 | 8:1 |
| 5 | 1e |
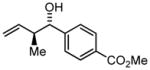 3e |
70 77 |
95 97 |
7:1 8:1c |
| 6b | 1f |
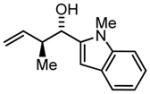 3f |
73 | 95 | 5:1 |
| 7 | 1g |
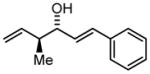 3g |
61 63 |
86 90 |
7:1 6:1c |
| 8 | 1h |
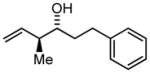 3h |
69 | 97 | 7:1 |
| 9 | 1i |
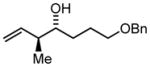 3i |
68 | 97 | 7:1 |
| 10 | 1j |
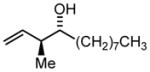 3j |
69 | 97 | 7:1 |
Reactions were performed in 13 × 100 mm pressure tubes. Cited yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis via comparison to racemic diastereomeric mixtures. See experimental section for further details.
80 °C, 72 h.
(S)-C3-TUNEPHOS was used as ligand.
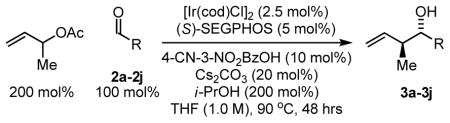
Carbonyl crotylation from the aldehyde oxidation level employing isopropanol as the terminal reductant also was explored (Table 3). To our delight, under conditions identical to those cited in Table 2, but in the presence of isopropanol (200 mol%), aryl aldehydes 2a–2f, cinnamaldehyde 2g, and unactivated aliphatic aldehydes 2h–2j are converted to the corresponding carbonyl crotylation products 3a–3f with complete levels of regioselectivity, very good levels of anti-diastereoselectivity and with exceptional levels of enantioselectivity. Thus, carbonyl crotylation is achieved from the aldehyde or alcohol oxidation level in the absence of preformed crotylmetal reagents.
Table 3.
Ir-catalyzed transfer hydrogenative crotylation of aldehydes 2a-2j.a
 | |||||
|---|---|---|---|---|---|
| 2a, R = Ph | 2f, R = 2-(N-Me-indole) | ||||
| 2b, R = 3-MeOPh | 2g, R = CH=CHPh | ||||
| 2c, R = 4-MeOPh | 2h, R = 2-phenyl-1-ethyl | ||||
| 2d, R = 4-BrPh | 2i, R = 3-(benzyloxy)propyl | ||||
| 2e, R = 4-(CO2Me)Ph | 2j, R = (CH2)7CH3 | ||||
|
| |||||
| Entry | Aldehyde | Product | Yield (%) | ee (%) | anti:syn |
|
| |||||
| 1 | 2a |
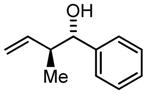 3a |
77 | 98 | 9:1 |
| 2 | 2b |
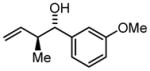 3b |
74 | 98 | 9:1 |
| 3b | 2c |
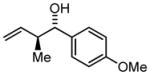 3c |
75 77 |
97 98 |
7:1 6:1c |
| 4 | 2d |
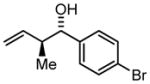 3d |
78 | 97 | 11:1 |
| 5 | 2e |
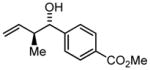 3e |
80 82 |
96 97 |
11:1 13:1c |
| 6b | 2f |
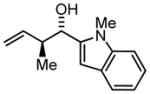 3f |
78 | 97 | 6:1 |
| 7 | 2g |
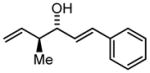 3g |
66 68 |
98 98 |
7:1 8:1c |
| 8 | 2h |
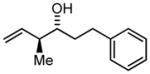 3h |
71 | 97 | 11:1 |
| 9 | 2i |
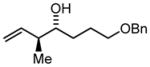 3i |
68 | 97 | 11:1 |
| 10 | 2j |
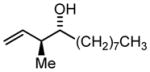 3j |
75 | 97 | 11:1 |
Reactions were performed in 13 × 100 mm pressure tubes. Cited yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis via comparison to racemic diastereomeric mixtures. See experimental section for further details.
80 °C, 72 h.
(S)-C3-TUNEPHOS was used as ligand.
In prior studies of the parent allylation reaction,17b intervention of symmetric iridium π-allyl intermediates or rapid interconversion of σ-allyl haptomers through the agency of a symmetric π-allyl was inferred on the basis of isotopic labeling studies. To evaluate the nature of the purported π-crotyl iridium intermediate, optically enriched allylic acetate 4 (98% ee) was coupled to alcohol 1e under standard conditions employing the achiral ligand BIPHEP. The product, homoallyl alcohol 5, is produced in 48% isolated yield with 9:1 anti-diastereoselectivity and 14% enantiomeric excess (Scheme 2, eqn. 2). These data suggest racemization via π-facial interconversion of the kinetic π-crotyl iridium complex occurs at a rate only marginally slower than the rate of carbonyl addition.
Scheme 2.

Experiments aimed at probing the origins of stereoselection in Ir-catalyzed transfer hydrogenative crotylation (Ar = (4-(CO2Me)Ph).
aReactions were performed in 13 × 100 mm pressure tubes. Cited yields are of material isolated by silica gel chromatography. Enantiomeric excess was determined by chiral stationary phase HPLC analysis via comparison to racemic diastereomeric mixtures. See experimental section for further details.
To further probe the origins of stereoselection, optically enriched allylic acetate 4 (98% ee) was coupled to alcohol 1e under standard conditions employing iridium catalysts modified by (S)-SEGPHOS and (R)-SEGPHOS (Scheme 2, eqns. 3 and 4, respectively). In the former case, excellent levels of relative and absolute stereocontrol are observed. In the later case, diminished efficiencies and reduced levels of diastereo- and enantioselectivity are evident. In both cases, recovered allylic acetate 4 exhibits significant erosion of optical purity. These experiments suggest that ionization of the (R)-allylic acetate 4 by the (S)-SEGPHOS modified iridium catalyst represents the lower energy diastereomeric pathway, that is, a stereochemically matched ionization mode. Partial racemization of recovered allylic acetate 4 suggests that ionization occurs reversibly with incomplete kinetic stereoselectivity.
Finally, in reactions employing racemic allylic acetate 4 and the iridium catalyst modified by (S)-SEGPHOS, recovered 4 exhibits a substantial degree of optical enrichment favoring the (S)-enantiomer (Scheme 2, eqn. 5). This result is consistent with the notion that consumption of the (R)-allylic acetate 4 by the (S)-SEGPHOS modified iridium catalyst represents a more rapid stereochemically matched reaction pathway. Notably, the degree of optical enrichment of recovered (S)-4 does not increase as a function of conversion due to erosion of the optical purity of unreacted allylic acetate 4 via reversible ionization, as established in a preceding experiment (Scheme 2, eqn. 2).
The observed anti-diastereoselectivity presumably arises via kinetic formation of the cis-π-crotyl complex,19 which engages the carbonyl partner by way of an (E)-crotyl iridium intermediate (Scheme 3). Erosion of diastereoselectivity may result via isomerization to the trans-π-crotyl complex, which engages the carbonyl partner by way of the (Z)-crotyl iridium complex to form the syn-diastereomer. This interpretation may account for the fact that couplings to aldehydes exhibit uniformly higher levels of diastereoselectivity. For the aldehyde couplings, carbonyl addition is anticipated to be faster as higher concentrations of aldehyde are present throughout the course of the reaction, promoting rapid capture of the kinetic allyliridium intermediate. The observation that diastereoselectivity increases with increasing concentration in couplings of alcohols further supports the veracity of this interpretation (Table 1, entries 16–18). The favorable influence of the 4-cyano-3-nitro-C,O-benzoate moiety on anti-diastereoselection is the subject of computational study.
Scheme 3.

Stereochemical features associated with formation and isomerization of the purported crotyl iridium intermediates (*Ln = (S)-SEGPHOS and C,O-benzoate of 4-cyano-3-nitrobenzoic acid I).
Exposure of α-methyl allyl acetate to equimolar quantities of 1a and 2e under standard conditions employing BIPHEP as ligand provides 3a and 3e in 83% yield in a 1:2.3 ratio, respectively. Exposure of α-methyl allyl acetate to equimolar quantities of 2a and 1e under otherwise identical conditions provides 3a and 3e in 74% yield in a 1:1.1 ratio, respectively. These experiments demonstrate rapid redox equilibration in advance of carbonyl addition, which is relevant to crotylations conducted from the alcohol oxidation level (Scheme 4).
Scheme 4.

Experiments establishing rapid redox equilibration in advance of carbonyl addition (Ar = (4-(CO2Me)Ph).a
aReactions were performed in 13 × 100 mm pressure tubes. Cited yields are of material isolated by silica gel chromatography. See experimental section for further details.
A simplified catalytic mechanism consistent with our collective results is depicted in Scheme 5. The ortho-cyclometallated iridium hydride I undergoes deprotonation in the presence of Cs2CO3 to furnish the anionic iridium(I) C,O-benzoate II.20 Oxidative addition to α-methyl allyl acetate delivers the iridium π-crotyl complex III. The parent BINAP-ligated iridium π-allyl C,O-benzoate complex has been characterized by single crystal x-ray diffraction and has been demonstrated to be catalytically relevant.17b Aldehyde addition by way of the (E)-σ-crotyliridium complex IV through a closed chair-like transition structure delivers the anti-homo-allyl iridium alkoxide V. This intermediate is stable with respect to β-hydride elimination of the carbinol C-H due to occupation of the remaining coordination site at iridium(III) by the olefin moiety of the homo-allylic alcohol. Exchange of the homo-allyl alcohol for isopropanol or a reactant alcohol provides VI, which has an open coordination site and, consequently, β-hydride eliminates to regenerate the ortho-cyclometallated complex I. A stereochemical model accounting for the observed stereochemistry is analogous to that previously proposed (Scheme 5, right).
Scheme 5.

Left: Simplified catalytic mechanism proposed for the iridium catalyzed transfer hydrogenative crotylation. Right: Model accounting for the observed sense of relative and absolute stereocontrol.
Summary
We report a protocol for anti-diastereo- and enantioselective carbonyl crotylation from the alcohol or aldehyde oxidation level under the conditions of iridium catalyzed transfer hydrogenation. This method circumvents the use of chirally modified crotylmetal reagents or metallic terminal reductants and, consequently, avoids generation of stoichiometric metallic byproducts. Furthermore, the ability to conduct carbonyl crotylation directly from the alcohol oxidation level allows one to bypass the oxidation chemistry typically required to convert alcohols to aldehydes.
Mechanistic studies establish a matched mode of ionization for the (R)-acetate and the iridium catalyst modified by (S)-SEGPHOS, as well as reversible ionization of the allylic acetate with rapid π-facial interconversion of the resulting π-crotyl intermediate in advance of C-C bond formation. Additionally, rapid alcohol-aldehyde redox equilibration in advance of carbonyl addition is demonstrated. The 4-cyano-3-nitro-C,O-benzoate moiety appears to facilitate partitioning of the (E)- and (Z)-σ-crotyliridium intermediates and, hence, anti- and syn-diastereoselectivity. However, the specific interactions that mediate partitioning remain unclear and are currently the subject of computational analysis.
Our collective studies on C-C bond forming hydrogenation and transfer hydrogenation define a departure from the use of preformed organometallic reagents in carbonyl addition chemistry. Future studies will focus on the development of related transformations, including imine additions from the amine oxidation level.
Supplementary Material
Acknowledgments
Acknowledgment is made to Merck, the Robert A. Welch Foundation, the ACS-GCI Pharmaceutical Roundtable, the NIH-NIGMS (RO1-GM069445) and the Korea Research Foundation (KRF-2007-356-E00037) for partial support of this research. Dr. Oliver Briel of Umicore is thanked for the generous donation of [Ir(cod)Cl]2. Dr. Hideo Shimizu and Dr. Wataru Kuriyama of Takasago are thanked for the generous donation of (S)-SEGPHOS. Professor Xumu Zhang is thanked for the generous donation of (S)-C2-TUNEPHOS, (S)-C3-TUNEPHOS and (S)-C4-TUNEPHOS.
Footnotes
Supporting Information Available: Experimental details and spectroscopic data. For unknown compounds (3f and 5), 1H-NMR, 13C-NMR, IR, HRMS and HPLC data are provided. For known compounds (3a–3e and 3g–3j), 1H-NMR, 13C-NMR and HPLC data are provided. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on enantioselective carbonyl allylation and crotylation, see: Hoffmann RW. Angew Chem, Int Ed Engl. 1982;21:555.Yamamoto Y, Asao N. Chem Rev. 1993;93:2207.Ramachandran PV. Aldrichim Acta. 2002;35:23.Kennedy JWJ, Hall DG. Angew Chem, Int Ed. 2003;42:4732. doi: 10.1002/anie.200301632.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.
- 2.For enantioselective carbonyl additions of chirally modified crotylmetal reagents, see: Hoffmann RW, Ladner W. Tetrahedron Lett. 1979;20:4653.Brown HC, Bhat KS. J Am Chem Soc. 1986;108:293. doi: 10.1021/ja00279a042.Brown HC, Bhat KS. J Am Chem Soc. 1986;108:5919. doi: 10.1021/ja00279a042.Roush WR, Halterman RL. J Am Chem Soc. 1986;108:294.Roush WR, Ando K, Powers DB, Palkowitz AD, Halterman RL. J Am Chem Soc. 1990;112:6339.Garcia J, Kim BM, Masamune S. J Org Chem. 1987;52:4831.Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044. doi: 10.1021/ja043612i.Riediker M, Duthaler RO. Angew Chem, Int Ed Engl. 1989;28:494.Hafner A, Duthaler RO, Marti R, Rihs G, Rothe-Streit P, Schwarzenbach F. J Am Chem Soc. 1992;114:2321.Panek JS, Yang M. J Am Chem Soc. 1991;113:6594.Hu T, Takenaka N, Panek JS. J Am Chem Soc. 2002;124:12806. doi: 10.1021/ja020853m.Hackman BM, Lombardi PJ, Leighton JL. Org Lett. 2004;6:4375. doi: 10.1021/ol0480731.
- 3.For enantioselective Lewis acid catalyzed carbonyl additions of crotylmetal reagents, see: Furuta K, Mouri M, Yamamoto H. Synlett. 1991:561.Yanagasawa A, Ishiba A, Nakashima H, Yamamoto H. Synlett. 1997:88.Yanagasawa A, Kageyama H, Nakatsuka Y, Asakawa K, Matsumoto Y, Yamamoto H. Angew Chem, Int Ed. 1999;38:3701. doi: 10.1002/(sici)1521-3773(19991216)38:24<3701::aid-anie3701>3.0.co;2-d.Aoki S, Mikami K, Terada M, Nakai T. Tetrahedron Lett. 1993;49:1783.Motoyama Y, Okano M, Narusawa H, Makihara N, Aoki K, Nishiyama H. Organometallics. 2001;20:1580.Evans DA, Aye Y, Wu J. Org Lett. 2006;8:2071. doi: 10.1021/ol0604771.Rauniyar V, Hall DG. Angew Chem, Int Ed. 2006;45:2426. doi: 10.1002/anie.200504432.Rauniyar V, Zhai H, Hall DG. J Am Chem Soc. 2008;130:8481. doi: 10.1021/ja8016076.
- 4.For enantioselective Lewis base catalyzed carbonyl additions of crotylmetal reagents, see: Denmark SE, Coe DM, Pratt NE, Griedel BD. J Org Chem. 1994;59:6161. doi: 10.1021/jo052202p.Denmark SE, Coe DM, Pratt NE, Griedel BD. J Org Chem. 2001;59:6161. doi: 10.1021/jo052202p.Denmark SE, Fu J. J Am Chem Soc. 2001;123:9488. doi: 10.1021/ja016552e.Iseki K, Kuroki Y, Takahashi M, Kishimoto S, Kobayashi Y. Tetrahedron. 1997;53:3513.Nakajima M, Saito M, Shiro M, Hashimoto SI. J Am Chem Soc. 1998;120:6419.Malkov AV, Dufková L, Farrugia L, Kočovsky P. Angew Chem, Int Ed. 2003;42:3674. doi: 10.1002/anie.200351737.
- 5.For enantioselective chiral diol catalyzed carbonyl additions of crotylmetal reagents, see: Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308.
- 6.(a) Schlosser M, Rauchschwalbe G. J Am Chem Soc. 1978;100:3258. [Google Scholar]; (b) Schlosser M, Stähle M. Angew Chem, Int Ed. 1980;19:487. [Google Scholar]
- 7.For catalytic enantioselective carbonyl crotylation via Nozaki-Hiyama coupling, see: Bandini M, Cozzi PG, Umani-Ronchi A. Polyhedron. 2000;19:537.Bandini M, Cozzi PG, Umani-Ronchi A. Tetrahedron. 2001;57:835.Bandini M, Cozzi PG, Umani-Ronchi A. Angew Chem, Int Ed. 2000;39:2327. doi: 10.1002/1521-3773(20000703)39:13<2327::aid-anie2327>3.0.co;2-9.Inoue M, Suzuki T, Nakada M. J Am Chem Soc. 2003;125:1140. doi: 10.1021/ja021243p.Lee JY, Miller JJ, Hamilton SS, SIgman MS. Org Lett. 2005;7:1837. doi: 10.1021/ol050528e.McManus HA, Cozzi PG, Guiry PJ. Adv Synth Catal. 2006;348:551.Xia G, Yamamoto H. J Am Chem Soc. 2006;128:2554. doi: 10.1021/ja058454p.
- 8.For recent reviews of catalytic Nozaki-Hiyama coupling, see: Avalos M, Babiano R, Cintas P, Jiménez JL, Palacios JC. Chem Soc Rev. 1999;28:169.Bandini M, Cozzi PG, Umani-Ronchi A. Chem Commun. 2002:919. doi: 10.1039/b109945k.Hargaden GC, Guiry PJ. Adv Synth Catal. 2007;349:2407.
- 9.For catalytic carbonyl allylation via reductive coupling of π-allyls based on palladium, see: Tabuchi T, Inanaga J, Yamaguchi M. Tetrahedron Lett. 1986;27:1195.Takahara JP, Masuyama Y, Kurusu Y. J Am Chem Soc. 1992;114:2577.Kimura M, Ogawa Y, Shimizu M, Sueishi M, Tanaka S, Tamaru Y. Tetrahedron Lett. 1998;39:6903.Kimura M, Tomizawa T, Horino Y, Tanaka S, Tamaru Y. Tetrahedron Lett. 2000;41:3627.Kimura M, Shimizu M, Shibata K, Tazoe M, Tamaru Y. Angew Chem, Int Ed. 2003;42:3392. doi: 10.1002/anie.200351182.Zanoni G, Gladiali S, Marchetti A, Piccinini P, Tredici I, Vidari G. Angew Chem, Int Ed. 2004;43:846. doi: 10.1002/anie.200352743.Kimura M, Shimizu M, Tanaka S, Tamaru Y. Tetrahedron. 2005;61:3709.Howell GP, Minnaard AJ, Feringa BL. Org Biomol Chem. 2006;4:1278. doi: 10.1039/b518311a.Barczak NT, Grote RE, Jarvo ER. Organometallics. 2007;26:4863.
- 10.For catalytic carbonyl allylation via reductive coupling of π-allyls based on rhodium, see: Masuyama Y, Kaneko Y, Kurusu Y. Tetrahedron Lett. 2004;45:8969.
- 11.For catalytic carbonyl allylation via reductive coupling of π-allyls based on iridium, see: Masuyama Y, Chiyo T, Kurusu Y. Synlett. 2005:2251.Banerjee M, Roy S. J Mol Catal A. 2006;246:231.Masuyama Y, Marukawa M. Tetrahedron Lett. 2007;48:5963.
- 12.For catalytic carbonyl allylation via reductive coupling of π-allyls based on ruthenium, see: Tsuji Y, Mukai T, Kondo T, Watanabe Y. J Organomet Chem. 1989;369:C51.Kondo T, Ono H, Satake N, Mitsudo T-a, Watanabe Y. Organometallics. 1995;14:1945.
- 13.For selected reviews covering carbonyl allylation via umpolung of π-allyls, see: Masuyama Y. In: Advances in Metal-Organic Chemistry. Liebeskind LS, editor. Vol. 3. JAI Press; Greenwich: 1994. p. 255.Tamaru Y. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i, de Meijere A., editors. Vol. 2. Wiley; New York: 2002. p. 1917.Tamaru Y. In: Perspectives in Organopalladium Chemistry for the XXI Century. Tsuji J, editor. Elsevier; Amsterdam: 1999. p. 215.Kondo T, Mitsudo T-a. Curr Org Chem. 2002;6:1163.Tamaru Y. Eur J Org Chem. 2005:2647.Zanoni G, Pontiroli A, Marchetti A, Vidari G. Eur J Org Chem. 2007:3599.
- 14.For selected reviews of hydrogenative C-C coupling, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Iida H, Krische MJ. Top Curr Chem. 2007;279:77.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102. doi: 10.1246/cl.2008.1102.
- 15.For hydrogenative and transfer hydrogenative carbonyl allylations employing allenes as allyl donors, see: Skucas E, Bower JF, Krische MJ. J Am Chem Soc. 2007;129:12678. doi: 10.1021/ja075971u.Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134. doi: 10.1021/ja077389b.Ngai MY, Skucas E, Krische MJ. Org Lett. 2008;10:2705. doi: 10.1021/ol800836v.
- 16.For hydrogenative carbonyl allylations employing dienes as allyl donors, see: Bower JF, Patman RL, Krische MJ. Org Lett. 2008;10:1033. doi: 10.1021/ol800159w.Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338. doi: 10.1021/ja801213x.
- 17.For hydrogenative carbonyl allylations employing allyl acetate as allyl donor, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.
- 18.(a) Zhang X. 6521769. US Patent. 2003 (filed 1999); (b) Zhang Z, Qian H, Longmire J, Zhang X. J Org Chem. 2000;65:6223. doi: 10.1021/jo000462v. [DOI] [PubMed] [Google Scholar]; (c) Sun X, Zhou L, Li W, Zhang X. J Org Chem. 2008;73:1143. doi: 10.1021/jo702068w. [DOI] [PubMed] [Google Scholar]
- 19.The designation of “cis” is made in reference to the substituent of the central carbon atom of the π-allyl complex.
- 20.As described in reference 17b, we report a crystal structure of the BINAP-ligated iridium C,O-benzoate derived from m-nitrobenzoic acid. Such ortho-cyclometallation onto m-nitrobenzoate were described previously for C5Me5-iridium complexes: Kisenyi JM, Sunley GJ, Cabeza JA, Smith AJ, Adams H, Salt NJ, Maitlis PMJ. Chem Soc, Dalton Trans. 1987:2459.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.