

Abstract
The chemokine receptor CXCR3 has been proposed to play a critical role in host anti-tumor responses. In this study, we defined CXCR3-expressing immune cell infiltration in human skin squamous cell carcinomas and then used CXCR3 deficient mice to assess the contribution of CXCR3 to skin tumorigenesis. Our studies employed two established protocols for chemical skin carcinogenesis (MCA or DMBA/TPA models). CXCR3 deletion did not affect tumor development in the MCA model; however, CXCR3 was important in the DMBA/TPA model where gene deletion reduced the incidence of skin tumors. This decreased incidence of skin tumors did not reflect differences in epidermal development but rather was associated with reduced epidermal thickness and proliferation in CXCR3−/− mice implicating the CXCR3 pathway in DMBA/TPA-induced epidermal inflammation and proliferation. Notably, CXCR3 expressed in CD4+ and CD8+ T cells was found to be important for enhanced epidermal proliferation. Specifically, CXCR3-deficient mice reconstituted with T cells isolated from wild-type mice treated with DMBA/TPA restored wild-type levels of epidermal proliferation in the mutant mice. Taken together, our findings establish that CXCR3 promotes epidermal tumorigenesis likely through a T cell-dependent induction of keratinocyte proliferation.
Keywords: CXCR3, epidermal proliferation, skin tumorigenesis, chemical carcinogenesis
Introduction
The chemokine receptor CXCR3 and its three ligands CXCL9, 10 and 11 are part of a large family of chemotactic cytokines whose major function is the chemo-attraction of specific hematopoietic cellular subsets during homeostatic or inflammatory conditions. In addition, chemokines function in diverse processes such as T cell proliferation, tissue remodeling and angiogenesis (1–3). The majority of studies assessing the role of CXCR3 in tumor growth and rejection have relied on enforced expression of ligands in syngeneic tumors, xenograft or therapeutic models. For example, enforced CXCL10 expression in melanoma xenografts or CXCL9/10 in lymphoma xenografts reduced but did not abrogate growth of tumors in immunodeficient mice (4–6). CXCL10 was also implicated as the critical mediator of IL-12’s actions in a therapeutic model of tumor rejection via its effects on angiogenesis (7). Thus, limited studies to date have explored the role of endogenously produced CXCR3 ligands or their receptor in the host anti-tumor response in models of primary tumor development.
To address the role of CXCR3 in skin tumorigenesis, we first analyzed human cutaneous squamous cell carcinomas (CSCC) and identified infiltration of CXCR3 expressing cells. We then utilized CXCR3 null mice in two of the most well characterized chemical carcinogen models of primary tumor formation—the methylcholanthrene (MCA) fibrosarcoma and the 7,12-dimethylbenz(a)anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA) skin tumor systems. Over the last 15 years, Schreiber, Smyth and other groups used the MCA model to show that the impact of the immune system on nascently transformed cells results in successful control of neoplastic growth consistent with the predictions of Burnet and Thomas and which is now more broadly described as cancer immunoediting (8). Dissection of this dynamic process has revealed the critical role of lymphocytes and immunologically relevant cytokines in protecting the murine host from primary tumor development. For example, mice deficient in the interferon-γ (IFNγ) receptor α-chain (IFNGR1) display an increased MCA induced tumor burden compared to WT counterparts (9). Importantly, the downstream mediators of IFNγ’s actions have been incompletely defined but may include CXCR3 ligands, which are directly induced by this cytokine. Although MCA carcinogenesis has been utilized predominantly to reveal the host protective actions of the immune system, it is surprisingly also influenced by inflammation (10). A second commonly used model, based on DMBA/TPA application, revealed the essential role of epithelial restricted γδ T cells in immune surveillance (11). However, this model has also been utilized to demonstrate the contribution of cancer-promoting inflammation. Mice deficient in tumor promoting CD8+ T cells (12), inflammatory molecules, such as cycloxygenase-2 (COX-2) (13), or inflammatory environment regulators such as RAGE (14), all show a decreased incidence of tumors compared to WT mice. Thus, depending on the model, the specific polarization of the immune response to one that is either inflammatory or host protective leads to one of two diametrically opposed outcomes for the host (15).
Herein, we detail our findings using both of these classical carcinogenesis models in CXCR3−/− mice where (1) DMBA/TPA treatment revealed decreased tumor development suggesting that CXCR3 contributes to the inflammatory environment and (2) no role was found for CXCR3 in protecting mice from carcinogenesis in the MCA system. Although there were no developmental differences between WT and CXCR3−/− mice in epidermal differentiation markers, analysis of epidermal proliferation in response to DMBA/TPA demonstrated a decreased proliferative response in CXCR3−/− mice. We then determined that both CD4+ and CD8+ T cells, known CXCR3 expressing cells, were the critical immune cells regulating epidermal proliferation in WT mice and were able to reconstitute the epidermal proliferation deficit in CXCR3−/− mice. Whereas CXCR3 does not affect immune surveillance in MCA carcinogenesis, it does promote DMBA/TPA tumor development and enhances DMBA/TPA T cell dependent keratinocyte proliferation.
Materials and Methods
Animals
Studies were performed under approved protocols of the Animal Studies Committees of Washington University and the Department of Veteran’s Affairs. C57BL/6, RAG2−/− C57BL/6 (both from Taconic Farms, NY) and CXCR3−/− mice were used (16). Mice were maintained in a specific pathogen-free environment and were sex and age-matched for experiments.
Antibodies
Antibodies to mouse CD3 (145-2C11), CD4 (RM4-5), CD8 (Ly-2, 53-6.7), CD16/CD32 (2.4G2), CD45 (30-F11), Gr-1 (RB6/8C5), CD11b (M1/70), γδ TCR (GL3), Vγ5 (536), human CXCR3 (1C6) and isotype controls were from BioLegend (San Diego, CA) and BD Biosciences (San Diego, CA). For in vivo studies, control and antibodies targeting CD4, CD8 and NK1.1 were from Bio X Cell (West Lebanon, NH). Anti-keratin antibodies were from Covance (Emeryville, CA).
Cutaneous carcinogenesis
The protocol of Girardi (17) was followed with 100 μg DMBA for initiation and 25μg TPA for promotion (both from Sigma, St. Louis, MO). Tumor development was monitored weekly and lesions ≥ 2mm were counted as positive. A short-term experiment was also adapted where back skin was shaved, treated with a single dose of DMBA followed by 3 doses of TPA, and harvested 16–20 hours after the final TPA application. For MCA carcinogenesis, tumors were induced and monitored as described (9).
Immunohistochemistry
Skin samples were fixed in formalin, paraffin embedded and sectioned. After antigen retrieval using an IHC-Tek Epitope Retrieval Set (IHC World, MD), incubations were carried out with the primary (4°C, overnight) and secondary biotinylated IgG (room temperature, 30 min). The Vectastain Elite ABC and Peroxidase Substrate Kit were used for detection (Vector Laboratories, Burlingame, CA). IHC sections were counterstained with hematoxylin (Fisher Scientific, Pittsburgh, PA). Human CSCC samples were obtained under a Washington University IRB approved protocol and were immunostained after citrate based antigen retrieval (Santa Cruz Biotechnology, Santa Cruz, CA) with an antibody to human CXCR3 (1C6, BD Pharmingen, San Diego, CA). They were evaluated by a single study pathologist (M.E.P.) for the presence of staining in both lymphocytes and tumor cells. Staining was subjectively graded on a scale of 1 to 3 with 1 representing scattered cell staining, 2 was clusters and 3 was large aggregates or bands.
ELISA
CXCL9 and CXCL10 kits (R&D Systems, Minneapolis, MN) were used to detect protein in total lysates from mouse skin.
Proliferation Analysis
DMBA/TPA treated mice (16–20 hours after last TPA dosing) were injected with 100 mg/kg of BrdU (Sigma, St. Louis, MO) either 1 hour or 24 hours before harvest. BrdU staining on fixed sections was carried out using the Labeling and Detection Kit II (Roche, Indianapolis, IN) according to the manufacturer’s protocol with eosin counterstain (Fisher Scientific, Pittsburgh, PA). All BrdU+ cells and total basal keratinocytes were counted for each section. A proliferation index was calculated using the formula ((total number BrdU+ cells/total number of basal keratinocytes) × 1000) to compare between sections.
Fetal liver chimeric mice
Fetal liver cells from E13.5 embryos were harvested and transferred into sublethally radiated (1100 rads) recipient mice. Fetal liver cells have been successfully used to reconstitute epidermal T cell responses (18). Mice were allowed to reconstitute for 12 weeks and analyzed for appropriate reconstitution by FACS of splenocytes at the end of the DMBA/TPA experiments.
Skin infiltrating lymphocyte analysis
Epidermal preparations of DMBA/TPA treated skin were made by treating with Dispase II (2.4 units/ml, Roche, Indianapolis, IN) at 37°C for 2–4 hours. Single cell suspensions were generated by treatment with 0.25% trypsin (Hyclone, Rockford, IL), filtered, washed and plated overnight in complete RPMI medium to allow antigen re-expression (17). Staining and FACS were performed as described (19, 20).
Hyperplasia
H&E stained DMBA/TPA treated mouse skin was analyzed for the thickness of the epidermis using a calibrated ocular micrometer (at 40×).
Adoptive T cell transfers
WT or CXCR3−/− spleen and lymph nodes were used to isolate CD3+ T cells using magnetic bead based negative selection (Miltenyi Biotec, Auburn, CA). Purity of cells was between 85–95% by FACS and 15 × 106 purified CD3+ cells were transferred into the indicated recipients one week after DMBA application, followed by short course TPA (3 doses) treatment and analysis for BrdU incorporation.
Statistics
All statistical analysis was performed in Prism (GraphPad Software, La Jolla, CA). Tumor incidence in the MCA groups was analyzed by survival curve analysis and the Log Rank (Mantel-Cox) test. The paired, two-tailed T test was used for the DMBA/TPA experiments and an unpaired, two-tailed T test was used to analyze all other data. A p<0.05 was considered significant and all error bars represent S.E.M.
Results
CXCR3 is expressed on infiltrating cells in human CSCC
To assess the association of CXCR3 with human CSCC, we examined 24 individual patient samples by IHC for CXCR3 expressing cells (Figure 1A). The key findings of these analyses were that (1) CXCR3 expression was limited to infiltrating cells and was not present on tumor cells, (2) the majority of tumors (20/24) had strong (grade 3) peritumoral CXCR3+ cells, (3) intratumoral CXCR3+ staining was more variable (grade 1=9, 2=8 and 3=6) and (4) no CXCR3 was detected on adjacent normal epidermis (Figure 1B). This analysis demonstrated that infiltrating cells expressing CXCR3 were present in human CSCC and that neither tumor nor other adjacent normal cells express CXCR3. Due to a limited number of samples, patient outcomes relative to CXCR3 expression could not be determined.
Figure 1.

CXCR3 is associated with human cutaneous squamous cell carcinomas (CSCC), and has contrasting roles in mouse tumorigenesis where it promotes DMBA/TPA induced tumor development and has no role in MCA tumorigenesis. (A) Representative IHC analysis of human CSCC with infiltrating (IT, block arrow) or peritumoral CXCR3 expressing cells (PT, bracketed cells). Note there is no expression of CXCR3 by tumor cells (40X). (B) Adjacent tumor free epidermis shows no CXCR3 staining in normal epidermal layer (NE, bracketed layer, 10X). (C) Grading of 24 separate tumors is shown at right. (D) DMBA/TPA tumorigenesis in WT (triangles) and CXCR3−/− (squares) male mice shows increased tumor formation in WT mice (***p<0.001, n=15 for WT and 8 for CXCR3−/−). (E) MCA tumorigenesis in WT (triangles) and CXCR3−/− (squares) mice shows no difference in tumor formation (n=29 for WT and 34 for CXCR3−/−).
CXCR3 promotes DMBA/TPA tumorigenesis
To assess the role of CXCR3 in tumor development, we first compared the susceptibilities of WT and CXCR3−/− mice in DMBA/TPA tumorigenesis. Groups of male WT C57BL/6 and CXCR3−/− mice were treated with DMBA (100 μg) followed by twice-weekly TPA (25 μg) and monitored for tumor development (Figure 1D). Note, the background of the CXCR3−/− mice was confirmed via genome wide analysis of informative polymorphic markers between the 129Sv/Ev and C57BL/6 strains, which demonstrated that the gene deficient mice were 99.3% (145/146 markers) C57BL/6 strain (data not shown). These experiments showed that, as a group, the CXCR3−/− mice developed two-fold fewer tumors/mouse compared to WT mice. Similar results were obtained in independent, repeat experiments with male and female mice (combined cohort totals for all experiments was 59 WT and 45 CXCR3−/− mice). The difference in tumor incidence between WT and CXCR3−/− mice was more robust in males than in females, which led us to use males for the remainder of this study (Supplementary Figure 1A and B). In separate experiments, directly comparing male and female cohorts, we noted a significant gender difference in that WT males were more sensitive than females to DMBA/TPA whereas male and female CXCR3−/− mice were uniformly resistant to tumor development (data not shown). C57BL/6 mice develop mostly papillomas in response to DMBA/TPA with rare progression to squamous cell carcinomas (21). In these experiments, there were no differences in tumor sizes or progression to carcinomas between WT and CXCR3−/− mice (data not shown). Thus, CXCR3 has a tumor-promoting role in the inflammation induced DMBA/TPA model of cutaneous tumorigenesis.
Similar incidence of MCA induced tumors in CXCR3−/− and WT mice
Although the MCA model has been critical for delineating the central tenets of cancer immunoediting, it also relies on an inflammatory contribution for tumor development (10). To assess the role of CXCR3 deficiency in fibrosarcoma development, large cohorts of WT (n=60), RAG2−/− (n=29) and CXCR3−/− (n=67) mice were injected with 2 different doses of MCA and monitored. Consistent with the known role for lymphocytes in immunosurveillance, all 14 RAG2−/− mice developed tumors when treated with 25 μg of MCA compared to 20/29 WT mice (p=0.0067, data not shown). In contrast, CXCR3−/− mice displayed tumor incidences that were similar to that of WT mice both at the 25μg dose where 24/34 mice developed tumors (Figure 1E) and at a 6.25 μg dose (Supplementary Figure 1C). In addition, CXCR3−/− mice were transplanted with 21 different RAG regressor tumors, which are RAG2−/− derived tumor cell lines that are rejected in WT mice and grow progressively in RAG2−/− mice (19, 20). These experiments revealed that the vast majority (20/21) of transplanted tumor lines underwent rejection in CXCR3−/− mice similar to WT mice (Supplementary Figure 1D). Thus, CXCR3 did not contribute to immune surveillance or tumor promotion in MCA carcinogenesis and was very rarely required to reject transplanted tumors.
No differences in epidermal differentiation in CXCR3−/− versus WT mice
Two possible explanations for the effects of CXCR3 on skin tumor development were that there was an inherent developmental defect in CXCR3−/− skin or that there was dysregulated chemokine biology influencing the developing tumor or microenvironment. To address the former possibility, we first examined standard H&E stained WT and CXCR3−/− skin and saw no differences (data not shown). To compare developmental markers in the epidermis, IHC was used to assess keratin expression in WT and CXCR3−/− mice. In untreated mice, the epidermis is thin and no keratin differences were observed. To better examine keratin distribution, DMBA/TPA treated skin was stained and showed no differences in the distribution of keratin 5, which is expressed in all layers of epidermal keratinocytes (data not shown) and of keratin 1, which is expressed only in developing but not basal keratinocytes (Figure 2A and B). Therefore, CXCR3 deficiency has no effect on epidermal development and thus, the decreased tumor development must be due to altered chemokine biology.
Figure 2.

Epidermal differentiation is unaffected in CXCR3−/− mice relative to WT mice and CXCR3 ligands are induced in DMBA/TPA treated skin. Analysis of keratin 1 (A, B) and keratin 5 (data not shown) in DMBA/TPA treated skin shows no difference in WT (A) and CXCR3−/− mice (B). The arrowhead highlights a basal keratinocyte with minimal keratin 1 staining. Representative sections shown from n=3 mice for each genotype. (C) CXCL9 and (D) CXCL10 ELISA of total skin lysates from indicated mice shows induction of both chemokines with DMBA and 3 dose TPA treatment (D/T) and shows increased levels in WT vs. CXCR3−/−mice (**p<0.01 and ***p<0.001, n=3 independent treated mice each, representative of 2 independent experiments).
DMBA/TPA treatment induces CXCL9 and CXCL10
To establish that CXCR3 ligands are induced in DMBA/TPA treated mouse skin thus serving as a target for CXCR3 expressing cells, we assessed ligand levels by ELISA. Importantly, we focused only on CXCL9 and CXCL10 because C57BL/6 mice do not express CXCL11 due to a 2-bp insertion in the coding sequence that leads to a premature stop codon (22). Untreated or vehicle treated skin (no differences between these two were noted) had low levels of both CXCL9 and CXCL10 protein in both WT and CXCR3−/− mice (Figure 2C and D). Upon DMBA/TPA treatment, significant induction of CXCL9 and CXCL10 was detected in both WT and CXCR3−/− mice. Interestingly, compared to WT mice, CXCR3−/− mice had significantly lower levels of both CXCL9 and CXCL10, suggesting that CXCR3-expressing cells potentially induce expression of CXCR3 ligands in a feed-forward pathway. Thus, consistent with the effects of CXCR3 in tumor development, DMBA/TPA lead to induction of CXCR3 ligands in the skin.
CXCR3−/− mice have decreased hyperplasia in response to DMBA/TPA
As there were no differences in epidermal cell differentiation, we then asked whether there was a differential response to DMBA/TPA between WT and CXCR3−/− mice. Epidermal hyperplasia was analyzed in mouse skin treated with DMBA and then 3 doses of TPA (Figure 3A, short-term) or DMBA and then bi-weekly TPA for 30 weeks (Figure 3B, long-term) by measuring the thickness from the basement membrane to the stratum corneum. In the short-term protocol, there was a decrease in the mean measured thickness from 73.7 ± 4.9μm in WT mice to 53.9 ± 1.3μm in CXCR3−/−mice and in the long term protocol the mean measured thickness decreased from 71.2 ± 3μm in WT mice to 51.8 ± 2.7μm in CXCR3−/− mice. These data suggest that WT mice have a CXCR3 dependent epidermal proliferative response to DMBA/TPA. In addition, the short-term assay replicates the findings of the long-term assay at the level of epidermal hyperplasia and provides a surrogate assay to dissect the components contributing to CXCR3 related epidermal proliferation.
Figure 3.

Reduced epidermal hyperplasia and proliferation in response to DMBA/TPA in CXCR3−/− mice relative to WT mice. CXCR3−/− mice exhibit decreased epidermal hyperplasia upon (A) short term (**p<0.01, n=5 for WT and n=4 for CXCR3−/− mice) or (B) long term DMBA/TPA application (** p<0.01, n=3 each). To assess proliferation, WT or CXCR3−/− mice were treated with DMBA/TPA and then injected with BrdU (100 mg/kg). (C) Representative sections of skin after short term DMBA/TPA with 1 hour BrdU incorporation, and staining for BrdU (dark cells). (D, E) The calculated proliferation index with (D) short (**p<0.01, n=2 mice each, representative of 3–5 independent experiments) or (E) long term (20 weeks) DMBA/TPA (**p<0.01, n=3 mice each). (F) Representative section of 24 hour BrdU uptake shows decreased proliferation and migration in CXCR3−/− mice (from 4 mice per group of each genotype).
Decreased epidermal proliferation in CXCR3−/− mice
As the epidermal hyperplasia assessment provides a “snapshot” end result view of DMBA/TPA treatment, we next asked whether 5-bromo-2-deoxyuridine (BrdU) incorporation in proliferating keratinocytes would also reveal differences between WT and CXCR3−/− mice, as this is a more dynamic assay revealing specific cellular behavior. Using both DMBA/TPA application protocols (short and long), mice were pulsed for 1 hour with BrdU followed by immediate harvest of skin. Incorporated BrdU was detected after antigen recovery and a proliferation index was calculated for each stained section by counting BrdU positive cells in the basal layer and normalizing this number relative to total numbers of basal keratinocytes. As expected keratinocyte proliferation was increased relative to untreated mice (data not shown). However, there was a marked contrast in the proliferative response upon DMBA/TPA application between WT and CXCR3−/− mice (Figure 3C, D and E). In WT mice, more basal keratinocytes incorporated BrdU both in the short-term protocol (proliferation index of 522.6 ± 27 WT vs. 165.7 ± 9.5 in CXCR3−/− mice) and in the long-term protocols (proliferation index of 482 ± 19.8 WT vs. 310.1 ± 30.3 in CXCR3−/− mice). When BrdU was injected 24 hours prior to harvest of skin, the differences between WT and CXCR3−/−mice continued and extended into the upper layers of the epidermis (Figure 3F). Whereas WT mice showed incorporation of BrdU into proliferating basal keratinocytes and migration of these cells, CXCR3−/− mice continued to have both poor basal proliferation and decreased migration. We also examined these sections by TUNEL staining and saw no differences between WT and CXCR3−/− mice (data not shown). These data demonstrate that CXCR3 deficiency leads to impaired DMBA/TPA induced proliferation in the epidermis.
CXCR3 expressing hematopoietic cells drive proliferation
Having demonstrated a quantitative difference in the response of WT versus CXCR3−/− mice to DMBA/TPA treatment, the specific compartment (hematopoietic or non-hematopoietic) where CXCR3 acts to promote this phenotype was next addressed. Although most data show that CXCR3 expression is limited to immune cells and we found no CXCR3 expression in human epidermis (Figure 1B) some groups suggest that it is also expressed in keratinocytes (for example see (23)). To address this issue, we generated chimeric mice by sublethal radiation of recipient mice and subsequent transfer of fetal liver cells to reconstitute the hematopoietic system. Using this technique, we generated four different groups of mice (1) WT→WT, (2) WT→CXCR3−/−, (3) WT→RAG2−/− and (4) CXCR3−/−→RAG2−/−. RAG2−/− mice were used as recipients to avoid any contribution of radio resistant CXCR3 expressing B or T cells. At the end of the experiments, all mice were analyzed by counting total splenocytes and FACS for CXCR3 expressing lymphocyte populations (Supplementary Figure 2). Reconstitution restored splenocyte populations to levels seen in WT mice and CXCR3 expression in T and NK cells was similar to our previous work (24).
Using these mice, we then assessed epidermal proliferation after DMBA/TPA using the BrdU incorporation assay. As shown in Figure 4, we found that all mice expressing CXCR3 in the hematopoietic compartment displayed proliferation rates similar to WT mice. These groups and proliferation indexes were (1) WT→WT, 481.6 ± 27.9 (2) WT→RAG2−/−, 495.2 ± 35.4 and (3) WT→ CXCR3−/−, 478.7 ± 10.4. In contrast, mice that lacked CXCR3 in the hematopoietic compartment (the CXCR3−/−→RAG2−/− mice) had significantly decreased epidermal proliferation (index=249.3 ± 28.2). The proliferation index from CXCR3−/−→RAG2−/− was similar to that seen in DMBA/TPA treated CXCR3−/− mice (data not shown). These findings demonstrate that CXCR3 is necessary and sufficient in hematopoietic cells to induce epidermal proliferation by DMBA/TPA treatment.
Figure 4.

Fetal liver chimeric (FLC) mice reveal that CXCR3 expression in the hematopoietic system promotes DMBA/TPA induced epidermal proliferation. (A) The indicated mice were generated with fetal liver transplantation of irradiated recipients and treated with short course DMBA/TPA (**p<0.01, n=3 for WT→WT, n=3 for WT→RAG2−/−, n=4 for WT→ CXCR3−/− and n=4 for CXCR3−/−→RAG2−/−, combined from 2 different experiments). BrdU incorporation was then determined and all mice (representative section in B of WT→CXCR3−/−) displayed WT levels of proliferation, except the CXCR3−/−→RAG2−/− (representative section in C).
T cells promote epidermal proliferation
Having demonstrated that CXCR3 is important in the hematopoietic compartment for DMBA/TPA induced responses, we next addressed which subset(s) of the CXCR3 expressing immune cell(s) were involved. CXCR3 was originally associated with CD4+Th1 differentiation (25, 26) and in our previous work, we established that the major CXCR3 expressing cells in naïve mice included populations of CD4+ and CD8+ T cells, a subset of NK and all NK-T cells (24). To compare possible epidermal infiltration differences between WT and CXCR3−/− mice, we used FACS to characterize the immune infiltrate in epidermal preparations of untreated or DMBA/TPA treated mice. We found minimal infiltration of NK or NK-T cells into treated skin in either set of mice (data not shown). As expected there was an overall increase in inflammatory cell infiltrate consisting of CD11b+/Gr1+ and T cell subsets in both WT and CXCR3−/− mice after DMBA/TPA treatment. However, there were no differences in infiltration of CD11b+, CD11b+/Gr1+ or importantly epidermal Vγ5+γδ T cells in WT mice compared to CXCR3−/− mice (either untreated or DMBA/TPA treated, Supplementary Figure 3). The most significant difference noted was a modest but statistically significant decrease of both CD4+ and CD8+ T cells in DMBA/TPA treated CXCR3−/− mice compared to WT mice (Figure 5A).
Figure 5.

T cell infiltration is reduced in CXCR3−/− compared to WT mice and T cell depletion in WT mice attenuates epidermal proliferation. (A) FACS analysis of DMBA/TPA treated skin reveals reduced CD4+ and CD8+ T cells in CXCR3−/− compared to WT mice. Epidermal preparations were generated from untreated WT and CXCR3−/− mice or short course DMBA/TPA treated skin (designated as D/T) and FACS analysis was performed for CD4+ and CD8+ T cells (data shown) and CD11b+ cells, Gr1+CD11b+cells and γδ/vγ5+ cells (data in Supplementary Figure 4). Each point represents an individual mouse and data are expressed as percentage of cells relative to total CD45+/PI− cells and revealed significant reductions in CD4+ and CD8+ T cells (*p<0.05). (B) WT C57BL/6 mice were treated with the indicated mAbs and DMBA/TPA treated skin was assessed for proliferation (***p<0.001 for WT (n=4) versus CXCR3−/− (n=3), ***p<0.001 for control IgG (n=2) versus anti-CD8 (n=7) and **p<0.01 for control IgG (n=2) versus anti-CD4 (n=6), combined data from 3 experiments).
Having shown a decreased infiltration of T cells into treated CXCR3−/− mouse epidermis, we next assessed their contribution to CXCR3 dependent epidermal proliferation. WT mice were treated with control or depleting monoclonal antibodies (mAb) that ablated CD4, CD8 or NK1.1 expressing populations. By FACS analysis of mouse spleens at the end of the experiment, the specific cell populations were reduced by >95% (data not shown). After mAb treatment, mice were treated with DMBA/TPA and analyzed with the BrdU proliferation assay. In these experiments, the proliferation index for WT and CXCR3−/− mice was 550 ± 37 and 187 ± 15, respectively. Treatment of WT mice with a rat IgG control resulted in a proliferation index of 517 ± 20, which was significantly reduced in mice treated with αCD8 mAb (255 ± 19) or αCD4 mAb (374 ± 14.9). When we depleted mice with αNk1.1 mAb, no decrease in the proliferation index was apparent compared to mice treated with control mouse mAb (Figure 5B). These experiments revealed that CD4+or CD8+ T cells had the capacity to induce epidermal proliferation in response to short course DMBA/TPA treatment.
CXCR3+ T cells reconstitute epidermal proliferation in CXCR3−/− mice
Finally, having identified that T cells could promote DMBA/TPA induced epidermal proliferation in WT mice, we next asked whether CXCR3−/− mice could be reconstituted with WT total CD3+ T cells to enhance epidermal proliferation. This was tested using the short term DMBA/TPA protocol and by transferring purified CD3+ T cells from WT mice into CXCR3−/− mice one week after DMBA treatment. T cells were transferred after DMBA treatment due to previous reports of similar successful reconstitution (17) and as the pro-inflammatory effects in this model are due to TPA application (14). Additionally, separate CXCR3−/− mice receiving cells from CXCR3−/− donors served as controls. When tested in these experiments, CXCR3−/− mice again displayed a reduction in BrdU incorporation with the proliferation index falling from 487 ± 15 to 255 ± 54.3 (Figure 6A). When CXCR3−/− mice were reconstituted with WT CD3+ T cells, the proliferation index increased to 428 ± 29 (representative image Figure 6B). In contrast, control CXCR3−/−mice that received T cells purified from CXCR3−/− mice displayed a proliferation index of 299 ± 38, which was significantly lower than mice that received WT T cells (representative image Figure 6C). Thus, WT T cells, but not CXCR3−/− T cells, are able to complement the DMBA/TPA induced epidermal proliferation deficiency in CXCR3−/− mice.
Figure 6.
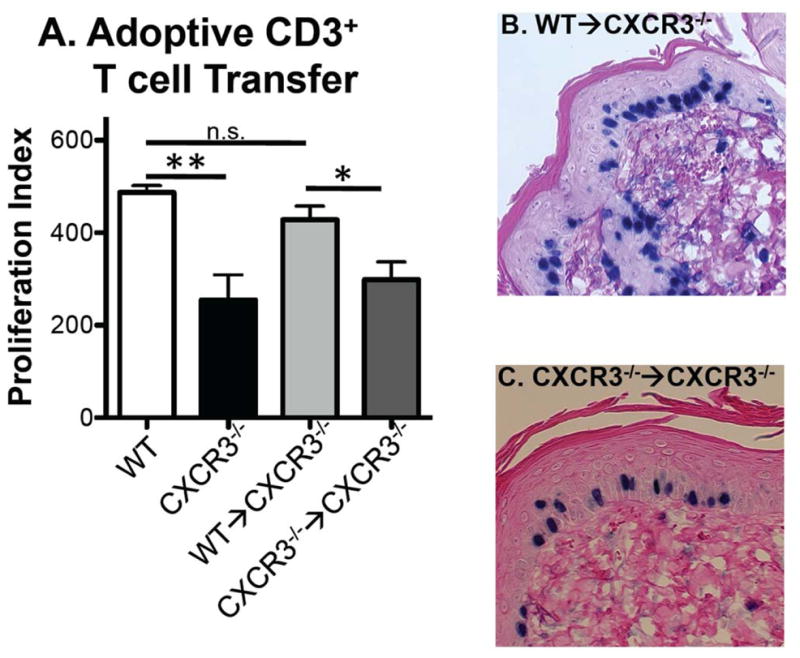
CXCR3 expressing CD4+ and CD8+ cells are critical for DMBA/TPA induced epidermal proliferation. (A) Control WT and CXCR3−/− mice showed significant proliferation differences with DMBA/TPA application (**p<0.01, n=3 for each genotype). CXCR3−/− mice were reconstituted with purified WT or CXCR3−/− CD3+ T cells and assessed for BrdU incorporation revealing that WT but not CXCR3−/− cells are able to enhance epidermal proliferation (*p<0.05, n=8 for WT→CXCR3−/− and n=6 for CXCR3−/−→CXCR3−/−, combined from 3 separate adoptive transfers). Representative images of (B) WT→CXCR3−/− and (C) CXCR3−/−→CXCR3−/−. Comparison of WT and WT→CXCR3−/− groups showed no statistical significance demonstrating reconstitution to WT levels (n.s.= not significant).
Discussion
Previous studies have implicated CXCR3 and its ligands as mediators of tumor growth inhibition via effects both on protective host immune infiltration and tumor vasculature. All of these studies relied on therapeutic manipulation of transplanted tumors (for example see (7, 27–29)) and many of these studies were xenografts in the setting of immunodeficient mice, which ignores the actions of CXCR3 and its ligands on the immune system (for example see (4–6)). One study demonstrated that CXCR3 deficiency in the TRAMP prostate cancer model enhanced tumor growth; however, the genetic background of these mice and CXCR3 expression on prostate cancer cells themselves makes the interpretation of the immune impact on tumor development unclear (30, 31). Importantly, all these studies shed light on the capacity of CXCR3 and its ligands to contribute to angiostasis and immune infiltration but the question of the role of endogenously produced CXCR3 chemokines in primary tumor development in immune competent hosts remained unanswered. We chose to address this question by assessing the contribution of CXCR3 in two commonly utilized chemical carcinogen induced tumor models. Both the MCA and DMBA/TPA models have been used to dissect the immune system’s contribution to tumor development in host protective and tumor promoting modes. We found that CXCR3 promoted tumor development in the DMBA/TPA model whereas it had no role in MCA carcinogenesis or in rejection of transplantable, immunogenic tumor cell lines. CXCR3 was important for epidermal proliferation in response to DMBA/TPA and this effect was likely due to CXCR3 activity in the CD3+ T cells of the hematopoietic system. As has been documented in a number of studies, we used epidermal proliferation and thickness as surrogate markers for tumor development (reviewed in (21)) and we will focus our future studies on translating this finding to the causation of tumorigenesis by specific subsets of T cells. These studies support a model where CXCR3 chemokines are induced in response to DMBA/TPA treatment and recruit CXCR3 expressing CD4+ and CD8+ cells that induce inflammatory epidermal proliferation, which, in the context of DMBA induced RAS mutations, then promotes tumor development. Consistent with this, CXCR3−/− mice would recruit fewer CD4+ and CD8+ T cells relative to WT mice thus attenuating the inflammatory environment resulting in decreased tumorigenesis. WT T cell transfers into CXCR3−/− mice followed by tumorigenesis will delineate the specific T cell requirements and further clarify this model—these experiments will be the subject of a follow-up study. In contrast, CXCR3 is not involved in promoting or preventing MCA-induced sarcoma development. This minimal effect may reflect a redundancy in the chemokine system that allows lymphocyte recruitment in the absence of CXCR3. In FACS analysis of some of the RAG regressor tumors transplanted into WT or CXCR3−/− mice, where the vast majority of tumors transplanted into CXCR3−/− mice were rejected at the same kinetics as WT mice (20/21 tumors, Supplementary Figure 1D), minimal differences were noted in lymphocyte recruitment suggesting that CXCR3 did not contribute to recruiting the critical lymphocytes needed for tumor rejection (data not shown). These results, along with our previous work definitively showing that IFNγ is required for MCA tumor surveillance, suggests that IFNγ mediated surveillance of MCA sarcomas is not dependent on CXCR3.
Our initial analysis of human CSCC showed an association of CXCR3 expressing infiltrating cells especially in peritumoral regions. The impact of this infiltrate in clinically evident tumors is unclear, as opposed to our findings in the mouse system where CXCR3 expressing cells enhance tumor development. Several studies on inflammatory lesions such as those found in psoriasis and in CSCC have repeatedly shown an association with CXCR3+ lymphocytes. For example, CXCR3 expressing CD8+ T cells have been found at the basal epidermal layer in lichenoid graft-versus-host disease and in lichen planus lesions (32) and dermal CXCR3+ expressing CD3+ T cells were strongly associated with psoriatic plaques (33). In CSCC, a strong interferon associated transcriptional signature, including CXCL9, and IHC evidence of CXCR3+ cells, CD3+ cells and granzyme B+ cells correlating with the signature have been described (34). No studies have correlated patient outcomes with CXCR3 receptor or ligand expression. Clearly, the association of CXCR3-expresing cells in human CSCC—either correlatively or causally—deserves further study.
Relevant to our findings are three other investigations on the role of other chemokines and receptors in DMBA/TPA skin tumorigenesis. In their findings on the contribution of TNFα to DMBA/TPA tumorigenesis, Balkwill and colleagues also examined the role of CCL2 (MCP-1 or monocyte chemoattractant protein-1) and found a 50% reduction in tumor development in CCL2−/− mice (35). Although these investigators did not pursue the associated mechanisms in these mice, further work by Graham and colleagues on the decoy chemokine receptor D6 revealed a critical role for chemokine mediated inflammation in tumor development (36). D6 acts as a sink to remove the inflammatory CC (iCC) class of chemokines, which includes CCL2, 3, 4, 5, 7, 8, 11, 17 and 22. Thus, D6−/− mice, which have a reduced clearance of iCC, develop a T cell dependent exaggerated inflammatory epidermal response which is associated with enhanced tumor development in response to DMBA/TPA application. Finally, CXCR2 expression on keratinocytes was found to be critical for epidermal migration and tumorigenesis (37).
The broad range of chemokines addressed by the decoy receptor D6 did not reveal the specific contributing molecules for controlling tumorigenesis but highlighted the effects of T cell trafficking to epidermal inflammation. Other skin conditions, such as allergic contact dermatitis, psoriasis and atopic dermatitis, are mediated by an inflammatory infiltrate that includes neutrophils and T cells and is in a large part dependent on specific chemokine production in the epidermal compartment (reviewed in (38)). CXCR3’s involvement in skin wound healing has been the focus of several studies and the specific mechanism is under investigation (23, 39, 40). In skin tumorigenesis induced by DMBA/TPA, T cell contributions are variable depending on the genetic background of the mouse and the dose of TPA used to promote cutaneous lesions. For example, in the FVB/n strain, CD4+ and CD8+ T cells have a protective role when promoted with low dose TPA. However, with high dose TPA, a population of IL17 producing, RORγt+ CD8+ T cells promoted malignant degeneration of benign lesions (12, 17). By contrast, in C3H/HeN, where allergic contact hypersensitivity to DMBA plays a key role in tumor development, CD8+ T cells are host protective and CD4+ T cells promoted tumor development (41). Our data on epidermal hyperplasia in the C57BL/6 background revealed a contribution of both CD8+ and CD4+ T cells—the characterization of how CXCR3 and these cells work together in this model constitutes our current focus. Candidates for the cytokines emanating from these T cells to promote DMBA/TPA tumorigenesis include IL-17 (42), and IFNγ (43). Additionally, we noted a gender difference in tumor development with WT male mice developing more tumors (data not shown). To our knowledge this has not been described previously for the DMBA/TPA model but gender differences in UVB induced skin tumorigenesis (44) and DEN induced hepatocellular carcinomas have been reported (45). Interestingly, the latter study involves differential secretion of IL-6, which has been shown to be a growth factor for keratinocytes and may potentially promote tumor formation (46). Finally, the CXCR3 ligand(s) regulating lymphocyte recruitment to the skin are undefined. Notably, C57BL/6 mice do not express CXCL11 (22), thus identifying either CXCL9 or CXCL10 (or both) as the chemoattractant signal. Analysis of DMBA/TPA treated skin showed induction of both chemokines but the levels were reduced in CXCR3−/− mice suggesting the lack of an inducing signal. Although speculative, one possible scenario is that the reduced immune infiltrate in CXCR3−/− mice results in a decrease of a chemokine inducing cytokine. Our current work is aimed at defining the specific cytokine(s) and other chemokines contributing to tumor development. In conclusion, our work highlights the influence of CXCR3 expressing T cells in epidermal proliferation and likely tumorigenesis and lays the foundation for further delineation of specific chemoattractant pathways in cutaneous tumorigenesis.
Supplementary Material
Acknowledgments
RU was supported by grants from the NCI (K08CA090403), the American Academy of Otolaryngology/Head and Neck Surgery and the Veteran’s Affairs Research Service. RDS was supported by NIH grants CA43059 and CA107527.
We thank Jack D. Bui, Gavin P. Dunn, S. Ruby Chan, and Charles G. Rickert for comments on the manuscript and helpful discussions. We also thank Jessica Archambault, J. Michael White and Mark Diamond for advice and help with fetal liver chimeric mice generation and Brian Faddis and Pat Keller for instrumental assistance in histology processing. Experimental support was provided by the RCAVS Histology Core (NIH/NIDCD Grant P30DC04665) and the Speed Congenics Facility of the Rheumatic Diseases Core.
Footnotes
Disclosure: No conflict of interest
References
- 1.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 2.Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–80. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- 3.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–15. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sgadari C, Angiolillo AL, Cherney BW, et al. Interferon-inducible protein-10 identified as a mediator of tumor necrosis in vivo. Proc Natl Acad Sci U S A. 1996;93:13791–6. doi: 10.1073/pnas.93.24.13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Richmond A. The angiostatic activity of interferon-inducible protein-10/CXCL10 in human melanoma depends on binding to CXCR3 but not to glycosaminoglycan. Mol Ther. 2004;9:846–55. doi: 10.1016/j.ymthe.2004.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sgadari C, Farber JM, Angiolillo AL, et al. Mig, the monokine induced by interferon-gamma, promotes tumor necrosis in vivo. Blood. 1997;89:2635–43. [PubMed] [Google Scholar]
- 7.Coughlin CM, Salhany KE, Gee MS, et al. Tumor cell responses to IFNgamma affect tumorigenicity and response to IL-12 therapy and antiangiogenesis. Immunity. 1998;9:25–34. doi: 10.1016/s1074-7613(00)80585-3. [DOI] [PubMed] [Google Scholar]
- 8.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–61. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swann JB, Vesely MD, Silva A, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105:652–6. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girardi M, Glusac E, Filler RB, et al. The distinct contributions of murine T cell receptor (TCR)gammadelta+ and TCRalphabeta+ T cells to different stages of chemically induced skin cancer. J Exp Med. 2003;198:747–55. doi: 10.1084/jem.20021282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwong BY, Roberts SJ, Silberzahn T, et al. Molecular analysis of tumor-promoting CD8+ T cells in two-stage cutaneous chemical carcinogenesis. J Invest Dermatol. 130:1726–36. doi: 10.1038/jid.2009.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiano HF, Loftin CD, Akunda J, et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002;62:3395–401. [PubMed] [Google Scholar]
- 14.Gebhardt C, Riehl A, Durchdewald M, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205:275–85. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johansson M, Denardo DG, Coussens LM. Polarized immune responses differentially regulate cancer development. Immunol Rev. 2008;222:145–54. doi: 10.1111/j.1600-065X.2008.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hancock WW, Lu B, Gao W, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–20. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts SJ, Ng BY, Filler RB, et al. Characterizing tumor-promoting T cells in chemically induced cutaneous carcinogenesis. Proc Natl Acad Sci U S A. 2007;104:6770–5. doi: 10.1073/pnas.0604982104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y, Yang W, Pan M, et al. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med. 2003;198:433–42. doi: 10.1084/jem.20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bui JD, Uppaluri R, Hsieh CS, Schreiber RD. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res. 2006;66:7301–9. doi: 10.1158/0008-5472.CAN-06-0556. [DOI] [PubMed] [Google Scholar]
- 20.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 21.Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4:1350–62. doi: 10.1038/nprot.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sierro F, Biben C, Martinez-Munoz L, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104:14759–64. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yates CC, Whaley D, Kulasekeran P, et al. Delayed and deficient dermal maturation in mice lacking the CXCR3 ELR-negative CXC chemokine receptor. Am J Pathol. 2007;171:484–95. doi: 10.2353/ajpath.2007.061092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uppaluri R, Sheehan KC, Wang L, et al. Prolongation of cardiac and islet allograft survival by a blocking hamster anti-mouse CXCR3 monoclonal antibody. Transplantation. 2008;86:137–47. doi: 10.1097/TP.0b013e31817b8e4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loetscher P, Uguccioni M, Bordoli L, et al. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391:344–5. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 26.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–83. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conforti R, Ma Y, Morel Y, et al. Opposing effects of toll-like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res. 2010;70:490–500. doi: 10.1158/0008-5472.CAN-09-1890. [DOI] [PubMed] [Google Scholar]
- 28.Andersson A, Yang SC, Huang M, et al. IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J Immunol. 2009;182:6951–8. doi: 10.4049/jimmunol.0803340. [DOI] [PubMed] [Google Scholar]
- 29.Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008;68:8437–45. doi: 10.1158/0008-5472.CAN-08-1440. [DOI] [PubMed] [Google Scholar]
- 30.Shen H, Schuster R, Lu B, Waltz SE, Lentsch AB. Critical and opposing roles of the chemokine receptors CXCR2 and CXCR3 in prostate tumor growth. Prostate. 2006;66:1721–8. doi: 10.1002/pros.20476. [DOI] [PubMed] [Google Scholar]
- 31.Nagpal ML, Davis J, Lin T. Overexpression of CXCL10 in human prostate LNCaP cells activates its receptor (CXCR3) expression and inhibits cell proliferation. Biochim Biophys Acta. 2006;1762:811–8. doi: 10.1016/j.bbadis.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 32.Wenzel J, Lucas S, Zahn S, et al. CXCR3 <-> ligand-mediated skin inflammation in cutaneous lichenoid graft-versus-host disease. J Am Acad Dermatol. 2008;58:437–42. doi: 10.1016/j.jaad.2007.10.647. [DOI] [PubMed] [Google Scholar]
- 33.Chen SC, de Groot M, Kinsley D, et al. Expression of chemokine receptor CXCR3 by lymphocytes and plasmacytoid dendritic cells in human psoriatic lesions. Arch Dermatol Res. 2010;302:113–23. doi: 10.1007/s00403-009-0966-2. [DOI] [PubMed] [Google Scholar]
- 34.Wenzel J, Tomiuk S, Zahn S, et al. Transcriptional profiling identifies an interferon-associated host immune response in invasive squamous cell carcinoma of the skin. Int J Cancer. 2008;123:2605–15. doi: 10.1002/ijc.23799. [DOI] [PubMed] [Google Scholar]
- 35.Moore RJ, Owens DM, Stamp G, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- 36.Nibbs RJ, Gilchrist DS, King V, et al. The atypical chemokine receptor D6 suppresses the development of chemically induced skin tumors. J Clin Invest. 2007;117:1884–92. doi: 10.1172/JCI30068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cataisson C, Ohman R, Patel G, et al. Inducible cutaneous inflammation reveals a protumorigenic role for keratinocyte CXCR2 in skin carcinogenesis. Cancer Res. 2009;69:319–28. doi: 10.1158/0008-5472.CAN-08-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lonsdorf AS, Hwang ST, Enk AH. Chemokine receptors in T-cell-mediated diseases of the skin. J Invest Dermatol. 2009;129:2552–66. doi: 10.1038/jid.2009.122. [DOI] [PubMed] [Google Scholar]
- 39.Luster AD, Cardiff RD, MacLean JA, Crowe K, Granstein RD. Delayed wound healing and disorganized neovascularization in transgenic mice expressing the IP-10 chemokine. Proc Assoc Am Physicians. 1998;110:183–96. [PubMed] [Google Scholar]
- 40.Yates CC, Krishna P, Whaley D, Bodnar R, Turner T, Wells A. Lack of CXC chemokine receptor 3 signaling leads to hypertrophic and hypercellular scarring. American Journal Of Pathology. 2010;176:1743–55. doi: 10.2353/ajpath.2010.090564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yusuf N, Nasti TH, Katiyar SK, et al. Antagonistic roles of CD4+ and CD8+ T-cells in 7,12-dimethylbenz(a)anthracene cutaneous carcinogenesis. Cancer Res. 2008;68:3924–30. doi: 10.1158/0008-5472.CAN-07-3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Yi T, Zhang W, Pardoll DM, Yu H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res. 2010;70:10112–20. doi: 10.1158/0008-5472.CAN-10-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao M, Wang C, Zhang J, Li Z, Zhao X, Qin Z. IFNgamma promotes papilloma development by up-regulating Th17-associated inflammation. Cancer Res. 2009;69:2010–7. doi: 10.1158/0008-5472.CAN-08-3479. [DOI] [PubMed] [Google Scholar]
- 44.Thomas-Ahner JM, Wulff BC, Tober KL, Kusewitt DF, Riggenbach JA, Oberyszyn TM. Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res. 2007;67:3468–74. doi: 10.1158/0008-5472.CAN-06-3798. [DOI] [PubMed] [Google Scholar]
- 45.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 46.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–9. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.