

Abstract
We report the identities of the members of a group of proteins that associate with BRCA1 to form a large complex that we have named BASC (BRCA1-associated genome surveillance complex). This complex includes tumor suppressors and DNA damage repair proteins MSH2, MSH6, MLH1, ATM, BLM, and the RAD50–MRE11–NBS1 protein complex. In addition, DNA replication factor C (RFC), a protein complex that facilitates the loading of PCNA onto DNA, is also part of BASC. We find that BRCA1, the BLM helicase, and the RAD50–MRE11–NBS1 complex colocalize to large nuclear foci that contain PCNA when cells are treated with agents that interfere with DNA synthesis. The association of BRCA1 with MSH2 and MSH6, which are required for transcription-coupled repair, provides a possible explanation for the role of BRCA1 in this pathway. Strikingly, all members of this complex have roles in recognition of abnormal DNA structures or damaged DNA, suggesting that BASC may serve as a sensor for DNA damage. Several of these proteins also have roles in DNA replication-associated repair. Collectively, these results suggest that BRCA1 may function as a coordinator of multiple activities required for maintenance of genomic integrity during the process of DNA replication and point to a central role for BRCA1 in DNA repair.
Keywords: BASC, BRCA1, DNA repair, DNA structure, cancer
Two general classes of cancer genes have been identified (Kinzler and Vogelstein 1997). The first class consists of genes that control cell proliferation and tumor growth such as growth factors, cyclin-dependent kinase (Cdk) regulators such as cyclins, Cdk inhibitors (CKIs) and the retinoblastoma protein, apoptotic factors, and angiogenesis factors. These genes, when mutated or overproduced, promote the inappropriate accumulation of cells. The second class consists of genes that control the stability of the genome and prevent the accumulation of mutations in the first class of genes. These genes are called antimutators or caretaker genes and include DNA repair proteins, cell cycle checkpoint regulators, and genes that maintain the fidelity of chromosome segregation. Many genes of the second class have been identified, including the mismatch-repair genes, MSH2 and MLH1, which are linked to hereditary nonpolyposis colorectal cancer (Kinzler and Vogelstein 1996); the breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2; Futreal et al. 1994; Miki et al. 1994); the ATM gene, which is mutated in the cancer predisposition syndrome ataxia telangiectasia (AT; Savitsky et al. 1995); and the XP excision repair genes that are responsible for xeroderma pigmentosum. Other genetic disease genes that function in genome maintenance include NBS1, the gene mutated in Nijmegen breakage syndrome (NBS; Carney et al. 1998), BLM, which encodes a RecQ type DNA helicase and is mutated in Blooms' syndrome (Ellis et al. 1995), and MRE11, which is mutated in a variant of AT (Stewart et al. 1999). These proteins all function in DNA metabolism and repair. In addition, there is evidence that several of these proteins also participate in cell cycle checkpoint functions that halt cell cycle progression in the presence of damaged DNA (Shiloh and Rotman 1996; Jongmans et al. 1997).
BRCA1 contains an amino-terminal RING finger domain, a carboxy-terminal BRCT domain, and a SQ cluster domain (SCD) (Bork et al. 1997; Cortez et al. 1999). Disruption of the BRCA1 gene in mice causes embryonic lethality (Hakem et al. 1996; Gowen et al. 1996). Targeted deletion of exon 11 of BRCA1 in mouse mammary epithelial cells results in mammary tumor formation after long latency and genetic instability characterized by aneuploidy, chromosomal rearrangements, or alteration of p53 transcription (Xu et al. 1999a). The BRCA1 protein abundance is cell cycle regulated with low levels in G0 and G1 cells that increase as cells enter S phase (Chen et al. 1996; Ruffner and Verma 1997). BRCA1 localizes to nuclear foci during S phase that rapidly disperse when cells are treated with DNA damaging agents (Scully et al. 1997b). The BRCA1 protein is hyperphosphorylated in response to DNA damage and DNA replication blocks. Genetic evidence indicates that BRCA1 is required for transcription-coupled repair of oxidative DNA damage (Gowen et al. 1998) and homologous recombination in response to double-strand breaks (Moynahan et al. 1999). In addition, BRCA1 has been implicated in G2/M checkpoint control (Xu et al. 1999).
Biochemical evidence also supports a role for BRCA1 in DNA damage repair. BRCA1 is associated and colocalized with the DNA repair protein hRad51 (Scully et al. 1997c). BRCA1 associates with and is phosphorylated by the ATM protein kinase, a global regulator of the DNA damage response (Cortez et al. 1999). In addition, BRCA1 associates with the RAD50–MRE11–NBS1 complex, which functions in homologous recombination, nonhomologous end joining, meiotic recombination, and telomere maintenance (Zhong et al. 1999).
To further understand the function of BRCA1, we used immunoprecipitation and mass spectrometry to identify BRCA1-associated proteins. We found that BRCA1 resides in a large multisubunit protein complex of tumor suppressors, DNA damage sensors, and signal transducers that we have named BASC for BRCA1-associated genome surveillance complex.
Results
Biochemical purification and mass spectrometric identification of BRCA1-associated proteins
To facilitate the purification of the BRCA1 complex by antibody affinity, we raised two rabbit polyclonal antibodies against GST–BRCA1 1021–1552 (Ab80) and GST–BRCA1 1501–1861 (Ab81) produced in Escherichia coli. These antibodies were affinity purified using the respective antigens and shown to recognize a ∼220-kD BRCA1 protein by Western blotting and immunoprecipitation.
We purified BRCA1-associated proteins from unfractionated HeLa nuclear extracts by one-step immunoprecipitation with antibodies Ab80, Ab81, or the commercial carboxy-terminal epitope antibody C-20. After extensive washing in NETN buffer, the immunoprecipitates were eluted, separated by SDS-PAGE, and detected by Coomassie blue staining (Fig. 1A). Then, we sequenced all proteins that were just visible by Coomassie blue staining by mass spectrometry (Ogryzko et al. 1998).
Figure 1.


Immunoprecipitation of BRCA1-associated proteins and identification by mass spectrometry. (A) Immunoprecipitates of BRCA1-associated proteins using antibodies Ab80 and Ab81 were resolved on a 4%–20% gradient SDS–polyacrylamide gel and stained with Coomassie blue. Labeled protein bands were identified by mass spectrometry. (B) A representative MS/MS spectrum that identifies the 160-kD band from Ab80 immunoprecipitate as the RecQ DNA helicase BLM.
Nonspecific immunoprecipitating proteins were identified by immunoprecipitation with pre-immune serum and nonrelevant antibodies such as an anti-GST antibody followed by mass spectrometric sequencing. Proteins that are common to both anti-BRCA1 immunoprecipitates and negative controls are designated as nonspecific binding proteins. To identify proteins that may be cross-reacting to antibodies Ab80 and Ab81, we carried out immunoprecipitation in high detergent-containing buffers such as RIPA with the same antibodies. Under these conditions, most of the truly associated proteins will be completely or partially dissociated, whereas the cross-reacting proteins and BRCA1 will be immunoprecipitated.
In total, 40 proteins (excluding nonspecific-binding and cross-reacting proteins) were identified from Ab80, Ab81, and C-20 immunoprecipitates by mass spectrometry. As expected, we found several known BRCA1-interacting proteins such as BRCA1-associated RING domain protein (BARD1) and histone deacetylase 1 (HDAC1; Wu et al. 1996; Yarden and Brody 1999). Then, we focused our effort on the characterization of proteins that are involved in DNA damage repair (listed in Table 1 and described below) while the rest of the proteins await further characterization.
Table 1.
DNA repair proteins identified with mass spectrometry from immunoprecipitations of different BRCA1 antibodies
| SDS-PAGE (kD)
|
Ab80
|
Ab81
|
C20
|
|---|---|---|---|
| 350 | ATM | ||
| 220 | BRCA1 | BRCA1 | BRCA1 |
| 160 | MSH6, BLM | MSH6 | |
| 150 | RAD50 | ||
| 140 | RFC, p140 | RFC, p140 | |
| 100 | MSH2 | ||
| 90 | MLH1 | MLH1 | |
| 40 | RFC, p40 | RFC, p40 | |
| 37 | RFC, p37 | RFC, p37 |
BRCA1 associates with multiple DNA repair proteins to form BASC
One band that migrates at ∼150 kD from an anti-BRCA1 (C-20) immunoprecipitation was identified as RAD50. During the course of this study, a second group independently reported the association of RAD50 and BRCA1 (Zhong et al. 1999). A band in the Ab81 immunoprecipitate that migrates slightly slower on SDS-PAGE than BRCA1 was determined to be the protein kinase ATM, consistent with our previous observations (Cortez et al. 1999). In addition to the RAD50 complex and ATM, we also identified a 160-kD band from the Ab80 IP as BLM, the RecQ helicase (see Fig. 1B). Using a database search program PROWL (http://prowl.rockefeller.edu), the mass of the parent peptide (m/z 812, 2+ charged) and the masses of fragments we identified a sequence 41TSSDNNVSVTNNSVAK56, which is unique to BLM. Protein bands migrating at 160 kD from the Ab80 and C-20 immunoprecipitations were identified as MSH6. A 100 kD protein band from the Ab80 IP was identified as MSH2, and protein bands with molecular masses of 90 kD from Ab80 and Ab81 immunoprecipitates were identified as MLH1. Three bands that migrate at 140, 37, and 34 kD from both Ab80 and Ab81 immunoprecipitations were determined to be three subunits of the replication factor C (RFC) complex (p140, p37, and p34, respectively). These results are summarized in Table 1.
To confirm that the proteins identified by mass spectrometry did interact with BRCA1 and to determine the binding interrelationship among all the associated proteins, we carried out multiple reciprocal co-immunoprecipitations with antibodies against these proteins. As shown in Figure 2 (A,B), anti-BRCA1 antibodies co-immunoprecipitate RFC (p140), MSH6, BLM, ATM, and RAD50. Multiple independent BRCA1 antibodies (polyclonal Ab80, Ab81, and C-20, and monoclonal D-9) also co-immunoprecipitate MSH2, MLH1, MRE11 and NBS1 (data not shown). In addition, antibodies to ATM, RAD50, MSH2, and BLM reciprocally immunoprecipitate BRCA1 (Fig. 2C,D). The presence of MRE11 and NBS1 in the BRCA1 complex was confirmed by immunoblotting (Fig. 2D and data not shown). Moreover, we also found that antibodies to ATM, MSH6, MLH1, BLM, and RFC could immunoprecipitate RFC, MSH6, and BLM (Fig. 2A); antibodies to RAD50, MRE11, ATM, MSH2, MLH1, BLM, and RFC could immunoprecipitate ATM and RAD50 with the exception that RFC did not immunoprecipitate ATM (Fig. 2B), although antibodies to ATM did immunoprecipitate RFC (Fig. 2A); and antibodies to NBS1 could immunoprecipitate BLM, RAD50, MRE11, and ATM (Fig. 2D and data not shown). In summary, nearly all of these BRCA1-associated proteins can co-immunoprecipitate each other. Thus, BRCA1 and these DNA repair and checkpoint signaling proteins may reside in the same protein complex.
Figure 2.


BRCA1-associated proteins that form a BASC. (A–D) Components of the BASC coimmunoprecipitate. Immunoprecipitations were done with HeLa nuclear extracts (NE) (A–C) and 293T whole-cell extracts (TCL) (D). Antibodies and immunoprecipitation/Western conditions are described in the Materials and Methods section. (E) BRCA1 resides in a large complex of >2 MD. Components of the BASC complex cofractionate on DEAE and Superose 6 columns. HeLa nuclear extracts were fractionated and step eluted (0.2–0.4 m KCl) on a DEAE column. The majority of BRCA1 eluted in the 0.3 m fraction. The 0.3 m fraction was fractionated further on a Superose 6 gel filtration column. BRCA1 was detectable only in the void volume (>2 MD). Other components of BASC were detected as multiple peaks; all contain one peak in the void volume that co-elutes with BRCA1. Other peaks of smaller sizes exist independent of BRCA1. The majority of the RAD50 complex that is independent of BRCA1 elutes in the 0.2 m KCl on the DEAE column.
To further confirm that these proteins reside in a complex with BRCA1 and to estimate the size of the complex, we performed a two-step fractionation of the nuclear extracts using a DEAE ion-exchange column and a Superose 6 gel-filtration column. BRCA1 elutes at 0.2, 0.3, and 0.4 m KCl from a step-eluted DEAE column. The 0.3 m KCl fraction that contains the majority of cellular BRCA1 as detected by Western blotting was further separated on a Superose 6 gel-filtration column. BRCA1 was eluted exclusively in the void volume. All the associated DNA repair proteins were detected in multiple fractions from the DEAE column and as multiple peaks from the gel-filtration column, but all of them could be detected co-eluting with BRCA1 in the void volume in the 0.3 m KCl fraction (Fig. 2E), thus establishing that this BRCA1-containing complex is >2 MD.
To exclude the possibility that this BRCA1 complex is organized by DNA or RNA instead of through protein–protein interactions, we treated the nuclear extracts with DNaseI, RNase A, or ethidium bromide before performing the co-immunoprecipitation experiments. We could not detect any significant difference in protein associations between treated and untreated nuclear extracts (data not shown).
Taken together, these results suggest that BRCA1 and these identified BRCA1-associated proteins are likely in the same protein complex and, therefore, may function in the same DNA damage response pathway.
Colocalization of BRCA1 and BLM before and after exposure to DNA damage and replication blocks
To examine when and where BASC may function, we analyzed the localization of these proteins in untreated cells and cells exposed to DNA-damaging agents and DNA replication inhibitors. We found that, in untreated cells, the BLM protein has several staining patterns that often occur within the same cell including a diffuse nucleoplasmic staining, brighter patches of staining, and bright discrete foci (Fig. 3B,E). BRCA1 foci partially overlap with the bright BLM foci but rarely with the BLM patches (Fig. 3A–F). The amount of BRCA1 and BLM colocalization is greatly increased in a subset of cells following treatment with hydroxyurea (HU) for 6–8 hr (Fig. 3G–L). The HU-induced relocalization of BLM and BRCA1 appears to be specific for cells that are in mid- to late S phase or G2 at the time of HU addition as the foci are rarely observed following synchronization of the cells in early S phase or G1 (data not shown). This result may also explain why a relatively small percentage of asynchronous populations of cells (∼10%) show this relocalization following HU treatment. Exposure of cells to ionizing radiation also causes a more subtle redistribution of BLM into an increased number of nuclear foci in some cells that partially overlap with BRCA1 foci (Fig. 3M–O).
Figure 3.

Colocalization of BRCA1 with BLM before and after exposure to genotoxic agents. (A–F) Asynchronous, logarithmically growing MCF7 cells were fixed with methanol/acetone and stained with antibodies to BRCA1 (Ab-1, Calbiochem) and BLM followed by the appropriate FITC and Cy3-conjugated secondary antibodies. (G–L) Cells were treated with 1 mm HU for 6–8 hr followed by fixation and staining. (M–O) Cells were exposed to 12 Gy of γ-irradiation and incubated for 8 hr prior to fixation and staining. Confocal images were captured at 1260× magnification.
Two distinct localization patterns of the RAD50– MRE11–NBS1 complex in response to DNA damage
BRCA1 partially colocalizes with the RAD50–MRE11–NBS1 complex in a subset of cells that are exposed to ionizing radiation and then allowed to recover for various times (2–8 hr) (Fig. 4; data not shown). However, we often found cells that displayed little if any detectable BRCA1 signal but strong RAD50–MRE11–NBS1 foci. Further examination revealed two distinct localization patterns for these proteins after treatment with ionizing radiation. Cells that formed bright, discrete RAD50–MRE11–NBS1 foci often expressed too little BRCA1 to be visualized by immunofluorescence (Fig. 4, D–F and P–R). Cells that formed bright BRCA1 foci also exhibited RAD50–MRE11 foci but also showed a more diffuse nucleoplasmic RAD50–MRE11 staining as well. In many cases, the RAD50–MRE11 foci in these cells did overlap significantly with BRCA1 foci.
Figure 4.

Cell cycle dependent colocalization of BRCA1 with the MRE11–RAD50–NBS1 complex after ionizing radiation. (A–C,M–O) Asynchronous MCF7 cells were fixed and stained with (A–C) anti-RAD50 and anti-BRCA1 or (M–O) anti-MRE11 and anti-BRCA1 antibodies. Asynchronous (D–F,P–R), G0/G1 arrested (G–I,S–U), or S-phase synchronized (J–L,V–X) MCF7 cells were exposed to 12 Gy of γ-irradiation then incubated for 5 hr prior to fixation and immunostaining. Appropriate FITC or Cy3-conjugated secondary antibodies were used for indirect visualization of the epitopes. Confocal images were captured on a Bio-Rad confocal microscope at 1260× magnification.
Because BRCA1 expression peaks in S phase, we tested whether these two staining patterns reflected cell cycle regulation. MCF-7 cells were synchronized by serum starvation and re-addition of serum. Then, we irradiated the cells immediately (G0/G1 cells) or waited 24 hr to allow the cells to progress into S phase and then irradiated them. The cells were fixed and stained 5 hr after irradiation. This procedure causes the cells to either arrest at the G1 DNA damage checkpoint or the G2/M DNA damage checkpoint. We observed that bright RAD50–MRE11 foci and low BRCA1 staining were specific to the G1-arrested cells whereas diffuse nucleoplasmic and focal RAD50–MRE11 staining accompanied by the bright BRCA1 foci were specific to cells irradiated in S phase (Fig. 4, G–L and S–X). The highest level of colocalization between BRCA1 and RAD50–MRE11 was seen in the bright foci among the S phase-irradiated cells.
The RAD50 complex properly localizes in BRCA1-deficient HCC1937 cells
We also examined how RAD50 foci relocalize in BRCA1-deficient HCC1937 cells. Before treatment of the cells, we observed RAD50–MRE11–NBS1 foci in ∼11% of the cells using antibodies to each of these components (Fig. 5A,D). After treatment of the cells with 12 Gy of ionizing radiation, we observed an increase in the number of RAD50–MRE11–NBS1 foci-containing cells to ∼60%, which is consistent with the response in BRCA1-proficient cell lines (Fig. 5B,E; Maser et al. 1997). Transient transfection of wild-type BRCA1 driven by either a CMV promoter or a LTR promoter had no effect on the percentage of cells that displayed the focal staining pattern within the transfected population (data not shown). As this result appears to be in direct contradiction to previously published data (Zhong et al. 1999), we obtained HCC1937 cells from three different sources to confirm its reproducibility. Two of the sources behaved virtually identically as reported above. The cells from the third source showed a reduced increase in foci formation after ionizing radiation, but in no case did transfection of wild-type BRCA1 have any effect. Similar results have been obtained by others (D. Livingston, pers. comm.).
Figure 5.
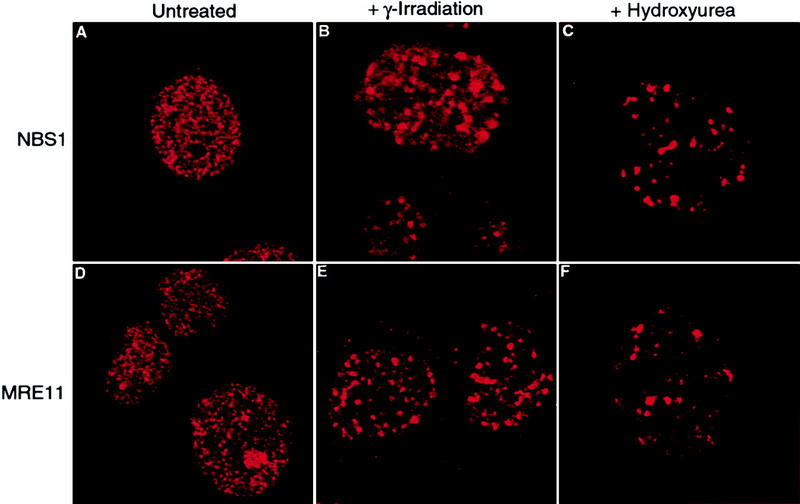
Relocalization of the RAD50–MRE11–NBS1 complex following exposure to ionizing radiation or HU is independent of BRCA1. Exponentially growing HCC1937 cells were either left untreated (A,D), exposed to 12 Gy of ionizing radiation followed by an 8-hr incubation (B,E), or treated with 1 mm HU for 8 hr (C,F). Cells were fixed and stained with antibodies to NBS1 (Novus) or MRE11 (Novus) as indicated. (A,D) Cells without foci; (B,C,E,F) cells that contain foci. Confocal images were captured at 1260× magnification. The percentage of cells with foci in each condition was determined by scoring cells as positive if they contained >10 foci. In each experiment, >200 cells were counted.
BLM colocalizes with BRCA1 and the RAD50–MRE11–NBS1 complex to replication forks in cells treated with inhibitors of replication
Significantly, the RAD50 complex also redistributes to large foci in a subset of asynchronous cells exposed to HU (Fig. 6). We confirmed that this redistribution is to the same location as the BRCA1 and BLM proteins by staining cells simultaneously for RAD50 and BRCA1 (Fig. 6A–C), MRE11 and BRCA1 (Fig. 6D–F), MRE11 and BLM (Fig. 6G–I), or RAD50 and BLM (Fig. 6J–L). The relocalization of the RAD50–MRE11–NBS1 complex after HU treatment does not appear to require BRCA1 function as we observed an increase (from 11% to 23%) in the percentage of HCC1937 cells displaying a focal RAD50–MRE11–NBS1 staining after HU treatment for 8 hr (Fig. 5C,F). This increase is consistent with the ∼10% increase observed in HU-treated cells containing wild-type BRCA1.
Figure 6.

Colocalization of RAD50, MRE11, BRCA1, and BLM following treatment of cells with HU. Asynchronous MCF7 cells were treated with 1 mm HU for 4–8 hr. Cells were fixed and stained with the following antibodies: (A–C) anti-RAD50 (13B3, GeneTex) and anti-BRCA1 (Ab-2, Neomarkers); (D–F) anti-MRE11 (12D, GeneTex) and anti-BRCA1 (Ab-2 NeoMarkers); (G–I) anti-MRE11 (12D7 GeneTex); and affinity-purified polyclonal anti-BLM; (J–L) anti-RAD50 (13B3, GeneTex) and affinity-purified polyclonal anti-BLM. Confocal images were captured at 1260× magnification. It should be noted that the majority of cells after HU treatment did not appear significantly different from the untreated cells. However, ∼10% showed redistribution into nuclear foci as represented in these images.
BRCA1 has been shown previously to colocalize with PCNA at replication forks in a small subset of late S phase cells exposed to ionizing radiation (Scully et al. 1997a). Therefore, we examined whether the BLM–BRCA1–MRE11–RAD50 nuclear domains that formed in response to HU treatment were also at replication forks. Staining with PCNA and BRCA1, PCNA and BLM, or PCNA and RAD50 confirmed that this BRCA1 complex does redistribute to PCNA-positive replication forks after HU treatment (Fig. 7).
Figure 7.

Colocalization of BLM, BRCA1, and RAD50 with PCNA following treatment with HU. Cells were left either untreated or exposed to 1 mm HU for 7 hr. Then, cells were fixed by methanol/acetone treatment and stained by indirect immunofluorescence with the following primary antibodies: (A–F) anti-PCNA (PC10, Santa Cruz) and anti-BRCA1 (Ab-2, Neomarkers); (G–L) anti-PCNA (PC10, Santa Cruz) and anti-BLM; (M–R) anti-PCNA (FL261, Santa Cruz) and anti-RAD50 (GeneTex). Appropriate Cy3- or FITC-conjugated secondary antibodies were used and confocal images were captured at 1260× magnification. It should be noted that the majority of cells after HU treatment did not appear significantly different from the untreated cells. However, ∼10% showed redistribution into nuclear foci as represented in these images.
Discussion
In this study we have partially purified the BRCA1 protein complex and identified its components using mass spectrometry. We found that all cellular BRCA1 protein resides in a large protein complex(s) (>2 MD) consistent with previous studies using sedimentation analysis (Scully et al. 1997a). We characterized a group of BRCA1-associated proteins that form what we refer to as a BASC. This work not only reinforces the current view of the involvement of BRCA1 in DNA damage repair but also allows us to propose two testable models for the function of BASC and the function of BRCA1 within BASC (see below). Furthermore, the fact that many proteins in BASC are themselves tumor suppressors suggests that interference with the function of BASC may be a central event in tumorigenesis.
BASC, a super complex of DNA repair protein complexes
The BASC characterized in this paper contains at least 15 subunits. In addition to BRCA1, ATM, and BLM, BASC contains four subprotein complexes including the RAD50–MRE11–NBS1 complex, the MSH2–MSH6 heterodimer, the MLH1–PMS2 heterodimer (the presence of PMS2 is inferred) and the RFC complex (five subunits total, of which three were detected by mass spectrometric analysis, and two are hypothesized to be present on the basis of known interactions with the first three). All of the BRCA1-associated proteins reported here also form smaller, stable subcomplexes independent of BRCA1 as evidenced by column fractionation. The relatively low abundance of the BRCA1 protein suggests that these other subcomplexes may have functions independent of BRCA1. BRCA1 may regulate the functions of these subcomplexes for a specialized repair function, or perhaps these complexes confer special properties to BRCA1.
The assembly of BRCA1 with different proteins may be a dynamic process changing throughout the cell cycle and within subnuclear domains. Thus, the associated proteins that we have identified may represent multiple distinct complexes that assemble and disassemble at various sites of BRCA1 function such as DNA double-strand breaks and stalled replication forks. However, because we were able to co-immunoprecipitate many of these DNA repair proteins in BASC with each other, it is likely that this BASC functions coordinately on DNA. In our analysis, we have not found BRCA2 and RAD51, two proteins that were previously reported to interact with BRCA1 (Chen et al. 1998; Scully et al. 1997c). The reason for this result is not clear at present. These proteins may be displaced by the three antibodies that we used, which were all raised against different segments of the carboxyl-terminus of BRCA1 (amino acids 1021–1861). As shown in Table 1 and Figure 1, BRCA1-associated proteins are immunoprecipitated to different extents by different antibodies, possibly due to competition for binding-site accessibility. Alternatively, these proteins may be substoichiometric components of BASC whose abundance is below the level detectable by Coomassie blue and mass spectrometry.
Is BASC a DNA structure surveillance machine?
An intriguing feature of these BRCA1-associated DNA repair proteins is that they each possess the ability to bind abnormal DNA structures, such as double-strand breaks, base-pair mismatches, Holliday junctions, cruciform DNA, template–primer junctions, and telomere repeat sequences (Uchiumi et al. 1996; Alani et al. 1997; Bennett et al. 1999; Marsischky et al. 1999). Therefore, these proteins have the potential to act as sensors of these structures. The RAD50–MRE11–NBS1 complex and the checkpoint kinase ATM may be sensors for DNA double-strand breaks as they interact with these breaks and are regulated by them (Maser et al. 1997; Smith et al. 1999; Stewart et al. 1999). The mismatch repair proteins may act as sensors of abnormal DNA structures caused both by distortions of the helix by mismatches and chemical alterations of the helix by cisplatin, DNA-methylating agents, and other chemicals. The tyrosine kinase c-Abl regulates p73 in the apoptotic response to cisplatin-induced DNA damage in a MLH1-dependent manner (Gong et al. 1999). Furthermore, treatment of cells with methylating agents results in p53 phosphorylation that is dependent on the presence of functional MSH2–MSH6 and MLH1 proteins (Duckett et al. 1999). These observations implicate the mismatch repair system in the initial step of a damage-signaling cascade that leads to activation of the DNA damage response. In addition, the MSH2–MSH6 heterodimer binds not only to mismatched DNA, but also has affinity for Holliday junctions (Alani et al. 1997; Marsischky et al. 1999) thereby acting as potential sensors of recombination and replication fork damage. The BLM DNA helicase may be a sensor of abnormal double-stranded DNA structures during replication. Its yeast ortholog Sgs1 binds a variety of abnormal DNA structures including forked DNA, synthetic cruciforms, and telomeric G4 DNA in vitro (Bennett et al. 1999). In addition, the Sgs1 helicase has been shown to act upstream of Rad53 in the DNA replication checkpoint (Frei and Gasser 2000). The RFC complex is known to bind the 3′ end of an elongating DNA primer and to recruit PCNA onto DNA polymerase δ, serving the role of the clamp loader in replication and a potential sensor of gapped DNA. Rfc3 mutant Schizosaccharomyces pombe cells are sensitive to HU, methanesulfonate, gamma irradiation, and UV irradiation. Phosphorylation of Chk1 and the replication checkpoint are deficient in Rfc3 mutant cells (Shimada et al. 1999). Work in budding yeast has also revealed a role for RFC in DNA repair and S-phase checkpoint regulation, suggesting that the RFC complex plays a direct role in sensing the state of DNA (Sugimoto et al. 1996; Noskov et al. 1998).
In addition to these built-in sensors, BASC also has signal transducers including the ATM kinase, the ATR kinase (B. Abraham, pers. comm.), and possibly other proteins that have not yet been identified. The initial effector may be BRCA1 itself, which is hyperphosphorylated in response to various types of DNA damage by multiple kinases including ATM (Cortez et al. 1999). Other proteins within BASC, such as NBS1 and MRE11, may also be targets for the ATM kinase (Kim et al. 1999). Thus, it is conceivable that the function of BRCA1 in the context of BASC is as a scaffold protein that organizes different types of DNA damage sensors, then serves as an effector in response to DNA damage to coordinate repair. A role for BASC as a surveillance machine is consistent with the observation that BRCA1 is associated constitutively with these DNA damage sensors and signal transducers. When aberrant DNA structures occur, in theory, BASC could respond rapidly to signal the DNA damage response. Phosphorylation of components of BASC by signal transducers such as ATM may regulate the repair functions of these proteins. In addition, the signal transducers could activate cell cycle checkpoints through phosphorylation and activation of the p53 and Chk proteins (Banin et al. 1998; Canman et al. 1998; Matsuoka et al. 1998).
A role for BASC in postreplicational repair
Another common feature of the repair proteins associated with BRCA1 is their roles in DNA postreplicational repair. Many of these proteins function directly in DNA replication or repair of damage that can occur at replication forks. The RAD50–MRE11–NBS1 complex is involved in repair of double-strand breaks generated at stalled replication forks (Haber 1998). We have previously identified mutants in the Saccharomyces cerevisiae homolog of RAD50 and NBS1 (Xrs2) as mutants sensitive to the replication inhibitor HU (Allen et al. 1994). HU treatment induces sister chromatid exchange (SCE) and mutants in BLM have high levels of spontaneous SCE. Furthermore, the S. pombe homolog of BLM was initially identified on the basis of its extreme sensitivity to HU (Stewart et al. 1997) further supporting a role in repair of replication errors. RFC complexes are also clearly involved in replicational repair of DNA damage and mutants in yeast that are sensitive to HU have been identified (Sugimoto et al. 1996; Noskov et al. 1998; Shimomura et al. 1998; Shimada et al. 1999). Mismatch repair proteins would also be required not only to check the fidelity of newly synthesized DNA during repair processes, but also to recognize and initiate repair of abnormal structures generated at the site of collapsed replication forks. Given the roles of these proteins in replicational repair and the fact that BRCA1 expression, phosphorylation, and localization all peak or change during S phase, it is tempting to speculate that BRCA1 might act to coordinate the repair and surveillance functions of these proteins at sites of DNA replicational stress. The resolution of aberrant DNA structures that occur during DNA replication likely occurs through a tightly regulated process that is linked to the actions of the replication machinery. BRCA1 might act to funnel certain types of damage through particular pathways. For example, it could identify a broken replication fork, allow BLM and mismatch repair proteins to unwind and remove inappropriate DNA helical conformations, then allow the RAD50–MRE11–NBS1 complex to initiate homologous recombinational repair, and finally load the RFC complex to recruit DNA polymerase to complete repair. Although speculative, this model is consistent with the observation that BRCA1-deficient cells are hypersensitive to DNA-damaging and replication blocking agents (Abbott et al. 1999; Scully et al. 1999). It should be noted that a role in directing repair does not rule out a role for this complex as a sensor and signal transducer.
The colocalization of BLM and BRCA1 with PCNA further suggests a role in replicational repair. The relocalization of BRCA1, BLM, and RAD50–MRE11–NBS1 after HU treatment appears to be specific to mid to late S phase cells. This observation could indicate a specific requirement for these proteins in the replication/repair of late replicating DNA. Alternatively, it may be that the number of active replication forks and the size of DNA replication factories early in S phase simply are not large enough to produce the large immunostaining domains of these proteins. The association may occur throughout S phase to resolve problems associated with DNA metabolism, and HU may simply amplify the number of problems sufficiently to observe an accumulation of the proteins at intranuclear domains of replication.
The identification of the RAD50 complex in BASC provides further support for the role of BRCA1 in control of homologous recombination (Moynahan et al. 1999). The role of BRCA1 in this process is currently unknown. Although it was previously published that cells deficient for BRCA1 fail to properly regulate and localize the RAD50 complex in response to DNA damage (Zhong et al. 1999), we could not find such a role for BRCA1 in our studies. Analysis of three different sources of HCC1937 cells defective for BRCA1, including the cells analyzed by Zhong and colleagues showed BRCA1-independent foci formation. Our results suggest that BRCA1 is not required to localize the RAD50 complex in response to DNA damage and may carry out a different role with respect to this complex. The cell cycle-regulated colocalization of BRCA1 with the RAD50 complex suggests that it may regulate the type of repair activity that functions to repair double-strand break lesions.
Association of mismatch repair proteins with BRCA1 may explain the role of BRCA1 in transcription-coupled repair
The identification of multiple mismatch repair proteins associated with BRCA1 provides support for genetic observations that BRCA1 is required for transcription-coupled repair of oxidation induced damage (Gowen et al. 1998). Previously it was noted that MSH2-deficient cell lines have a defect in transcription-coupled repair for both UV-induced and oxidation-induced DNA damage (Mellon et al. 1996). The association of BRCA1 with MSH2–MSH6 and MLH1–PMS2 suggests that the defective transcription-coupled repair in BRCA1−/− cells may arise from deregulation of mismatch repair genes or an inability of the mismatch repair genes to signal the presence of damage to BRCA1. We also examined colocalization of mismatch repair proteins (MSH2 and MLH1) with BRCA1 by immunofluorescence. The mismatch repair proteins stain the nucleus uniformly before DNA damage and do not show a detectable change upon DNA damage (ionizing radiation and HU treatment). We reason that even if there are foci formed in response to DNA damage, which is not known at present, the intense uniform nucleoplasm staining might mask the foci formed for these mismatch repair proteins. Therefore, it is not presently possible to judge colocalization of mismatch repair proteins with BRCA1 in nuclear foci. It is possible that the mismatch repair proteins use their association with BRCA1, which is known to interact with RNA polymerase (Scully et al. 1997a), to identify the transcribed strand for preferential repair. Alternatively, this association may instruct BRCA1 to prevent active transcription complexes from disrupting repair of the transcribed strand. This connection may provide insight for the initiation of mechanistic studies aimed at understanding the mechanism of transcription-coupled repair.
The molecular dissection of BASC presented here has provided significant insights into the role of BRCA1 in the process of DNA repair. Knowledge of the composition of this complex has allowed us to propose two models for the role of this complex in the cell, as a sensor of abnormal DNA structures and/or as a regulator of the post-replicational repair process. This knowledge sets the stage for the dissection of the significance of the presence of the individual members of this complex and the elucidation of their roles in the preservation of genomic stability and tumor suppression.
Materials and methods
Nuclear extract fractionation
HeLa-S3 nuclear extracts prepared according to the standard Dignam protocol were first loaded on a DEAE column equilibrated with 20 mm Tris-HCl (pH 8.0), 100 mm KCl, and 0.5 mm DTT and step-eluted in 0.2 m KCl, 0.3 m KCl and 0.4 m KCl, all in the equilibration buffer. The 0.3 m fraction was then applied to a Superose 6 (Pharmacia) column equilibrated with 20 mm HEPES (pH 7.5), 0.2 m KCl and 0.5 mm DTT. Fractions were collected and an equal volume from each fraction was loaded onto a 4%–12% SDS–polyacrylamide gel.
Immunoprecipitation, Western blotting, and antibodies
Large scale immunoprecipitation was carried out with unfractionated HeLa nuclear extracts. From 50 to 100 μg of affinity-purified antibodies as indicated was added to 5 ml of HeLa nuclear extracts (∼10 mg/ml) and rotated for 2 hr at 4°C. Two hundred microliters of protein A–Sepharose beads (50% slurry) was added to the mixture and rotated for an additional 2 hr. The immunoprecipitates were then washed in 10 ml of NETN [20 mm Tris-HCl (pH 8.0), 0.1 m NaCl, 1 mm EDTA, 0.5% NP-40] three times. The precipitated proteins were eluted into Laemmli buffer and separated on a 4%–20% SDS–polyacrylamide gel.
For immunoprecipitation/Western analysis, immunoprecipitation was done in essentially the same manner, except in a smaller scale. Briefly, 100 μl of nuclear extracts, 5 μg of antibody, and 15 μl of protein A beads were used in each reaction. The wash was done with 1 ml of NETN three times. Samples were separated on 4%–12% SDS–polyacrylamide gels, transferred to nitrocellulose membranes by semi-dry method, and probed with the appropriate antibodies.
The primary antibodies used in this work were as follows: rabbit polyclonal Ab80 and Ab81 were raised in rabbits against GST–BRCA1 1021–1552 and GST–BRCA1 1501–1861, respectively. GST–BRCA1 fusion proteins were produced in E. coli. Ab80 and Ab81 antibodies were affinity purified using the respective antigens following a conventional affinity-purification protocol. Affinity-purified rabbit polyclonal anti-NBS1C′ antibody was raised against a carboxy-terminal NBS1 peptide: CDDLFRYNPYLKRRR conjugated to KLH. Rabbit anti-BLM polyclonal antibody was prepared as described (Neff et al. 1999). Commercial anti-BRCA1 antibodies were D-9, C-20 (mouse monoclonal and rabbit polyclonal, respectively, Santa Cruz), Ab-2 (rabbit polyclonal, NeoMarker), and Ab-1 (monoclonal, CalBiochem). Other antibodies were rabbit polyclonal anti-ATM (Novus), mouse monoclonal anti-ATM-2C1 (GeneTex), rabbit polyclonal anti-ATM (H248, Santa Cruz, Ab-3, CalBiochem), mouse monoclonal anti-RAD50-13B3 (GeneTex), rabbit polyclonal anti-NBS1 (Novus), rabbit polyclonal anti-MRE11 (Novus), mouse monoclonal anti-MRE11-12D7 (GeneTex), goat polyclonal anti-MSH6 (GTBP, N-20, Santa Cruz), rabbit polyclonal anti-MSH2 (N-20, Santa Cruz), rabbit polyclonal anti-hMLH1 (C-20, Santa Cruz), mouse monoclonal anti-PCNA (PC10 and FL261, Santa Cruz), and mouse monoclonal anti-RFC p140 (kind gift from Dr. Bruce Stillman, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
Identification of proteins by mass spectrometry
Protein sequencing using mass spectrometry was carried out as described with the exception that O18-labeled H2O was omitted (Ogryzko et al. 1998). Briefly, the Coomassie blue stained protein band was in-gel digested with trypsin, and the recovered peptides were analyzed using an electrospray ion trap mass spectrometer (LCQ, Finnigan MAT, San Jose, CA) coupled on-line with a capillary HPLC (Magic 2002, Michrom BioResources, Auburn, CA) to acquire mass spectrometry/mass spectrometry (MS/MS) spectra. A 0.1 × 50 mm-MAGICMS C18 column (5-μm particle diameter, 200 Å pore size) with mobile phases of A (methanol:water:acetic acid, 5 : 94 : 1) and B (methanol:water:acetic acid, 85 : 14 : 1) was used. Data derived from the MS/MS spectra were used to search a compiled protein database that was composed of the protein database NR and a six-reading frame translated EST database to identify the protein by use of the program PROWL, which is publicly available (http://prowl.rockefeller.edu).
Immunofluorescence
Indirect immunofluorescence was performed by growing cells on glass coverslips in 35-mm dishes. Fixation and permeabilization were performed with either 100% methanol at −20°C for 20 min followed by 100% acetone at −20°C for 20 seconds or 3% paraformaldehyde followed by 0.5% triton X-100. Some samples were blocked with BSA or normal donkey serum. Staining was performed with antibodies diluted in PBS at 37°C for 20–30 min. Cy3-, Texas Red-, or FITC-conjugated secondary antibodies were obtained from Jackson Immunoresearch Laboratories and Amersham. Images were captured on a Zeiss microscope with a Bio-Rad confocal imaging system.
Acknowledgments
We are grateful to Drs. Robert Roeder and Yoshihiro Nakatani for HeLa nuclear extracts, Dr. John Petrini for sharing Nbs1 antibodies, Dr. Bruce Stillman for providing anti-RFC140 antibodies, and Dr. David Livingston for sharing unpublished results. We also thank Tim H. Lee for technical assistance. D.C. is a Fellow of the Jane Coffin Childs Memorial Fund for Medical Research. This work was supported by grants GM44664 and Q1187 (Welch) to S.J.E. S.J.E. is an investigator with the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jqin@bcm.tmc.edu; FAX (713) 798-1625.
References
- Abbott DW, Thompson ME, Robinson-Benion C, Tomlinson G, Jensen RA, Holt JT. BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. J Biol Chem. 1999;274:18808–18812. doi: 10.1074/jbc.274.26.18808. [DOI] [PubMed] [Google Scholar]
- Alani E, Lee S, Kane MF, Griffith J, Kolodner RD. Saccharomyces cerevisiae MSH2, a mispaired base recognition protein, also recognizes Holliday junctions in DNA. J Mol Biol. 1997;265:289–301. doi: 10.1006/jmbi.1996.0743. [DOI] [PubMed] [Google Scholar]
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage- induced transcription in yeast. Genes & Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Keck JL, Wang JC. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J Mol Biol. 1999;289:235–248. doi: 10.1006/jmbi.1999.2739. [DOI] [PubMed] [Google Scholar]
- Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- Chen Y, Farmer AA, Chen CF, Jones DC, Chen PL, Lee WH. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer Res. 1996;56:3168–3172. [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of BRCA1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Duckett DR, Bronstein SM, Taya Y, Modrich P. hMutSα- and hMutLα-dependent phosphorylation of p53 in response to DNA methylator damage. Proc Natl Acad Sci. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes & Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WGJ, Levrero M, Wang JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Avrutskaya AV, Latour AM, Koller BH, Leadon SA. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- Haber JE. The many interfaces of Mre11. Cell. 1998;95:583–586. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, Firpo E, Hui CC, Roberts J, Rossant J, Mak TW. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- Jongmans W, Vuillaume M, Chrzanowska K, Smeets D, Sperling K, Hall J. Nijmegen breakage syndrome cells fail to induce the p53-mediated DNA damage response following exposure to ionizing radiation. Mol Cell Biol. 1997;17:5016–5022. doi: 10.1128/mcb.17.9.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- ————— Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Lee S, Griffith J, Kolodner RD. Saccharomyces cerevisiae MSH2/6 complex interacts with Holliday junctions and facilitates their cleavage by phage resolution enzymes. J Biol Chem. 1999;274:7200–7206. doi: 10.1074/jbc.274.11.7200. [DOI] [PubMed] [Google Scholar]
- Maser RS, Monsen KJ, Nelms BE, Petrini JH. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6087–6096. doi: 10.1128/mcb.17.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Mellon I, Rajpal DK, Koi M, Boland CR, Champe GN. Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science. 1996;272:557–560. doi: 10.1126/science.272.5261.557. [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Neff NF, Ellis NA, Ye TZ, Noonan J, Huang K, Sanz M, Proytcheva M. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of bloom syndrome cells. Mol Biol Cell. 1999;10:665–676. doi: 10.1091/mbc.10.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov VN, Araki H, Sugino A. The RFC2 gene, encoding the third-largest subunit of the replication factor C complex, is required for an S-phase checkpoint in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:4914–4923. doi: 10.1128/mcb.18.8.4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, Kotani T, Zhang X, Schlitz RL, Howard T, Yang XJ, Howard BH, Qin J, Nakatani Y. Histone-like TAFs within the PCAF histone acetylase complex. Cell. 1998;94:35–44. doi: 10.1016/s0092-8674(00)81219-2. [DOI] [PubMed] [Google Scholar]
- Ruffner H, Verma IM. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc Natl Acad Sci. 1997;94:7138–7143. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Scully R, Anderson SF, Chao DM, Wei W, Ye L, Young RA, Livingston DM, Parvin JD. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci. 1997a;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997b;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997c;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Rotman G. Ataxia-telangiectasia and the ATM gene: Linking neurodegeneration, immunodeficiency, and cancer to cell cycle checkpoints. J Clin Immunol. 1996;16:254–260. doi: 10.1007/BF01541389. [DOI] [PubMed] [Google Scholar]
- Shimada M, Okuzaki D, Tanaka S, Tougan T, Tamai KK, Shimoda C, Nojima H. Replication factor C3 of Schizosaccharomyces pombe, a small subunit of replication factor C complex, plays a role in both replication and damage checkpoints. Mol Biol Cell. 1999;10:3991–4003. doi: 10.1091/mbc.10.12.3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T, Ando S, Matsumoto K, Sugimoto K. Functional and physical interaction between Rad24 and Rfc5 in the yeast checkpoint pathways. Mol Cell Biol. 1998;18:5485–5491. doi: 10.1128/mcb.18.9.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GC, Divecha N, Lakin ND, Jackson SP. DNA-dependent protein kinase and related proteins. Biochem Soc Symp. 1999;64:91–104. [PubMed] [Google Scholar]
- Stewart E, Chapman CR, Al-Khodairy F, Carr AM, Enoch T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 1997;16:2682–2692. doi: 10.1093/emboj/16.10.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Shimomura T, Hashimoto K, Araki H, Sugino A, Matsumoto K. Rfc5, a small subunit of replication factor C complex, couples DNA replication and mitosis in budding yeast. Proc Natl Acad Sci. 1996;93:7048–7052. doi: 10.1073/pnas.93.14.7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiumi F, Ohta T, Tanuma S. Replication factor C recognizes 5′-phosphate ends of telomeres. Biochem Biophys Res Commun. 1996;229:310–315. doi: 10.1006/bbrc.1996.1798. [DOI] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999a;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999b;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci. 1999;96:4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, Chen PL, Sharp ZD, Lee WH. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]