

Abstract
Msh4 (MutS homolog 4) is a member of the mammalian mismatch repair gene family whose members are involved in postreplicative DNA mismatch repair as well as in the control of meiotic recombination. In this report we show that MSH4 has an essential role in the control of male and female meiosis. We demonstrate that MSH4 is present in the nuclei of spermatocytes early in prophase I and that it forms discrete foci along meiotic chromosomes during the zygotene and pachytene stages of meiosis. Disruption of the Msh4 gene in mice results in male and female sterility due to meiotic failure. Although meiosis is initiated in Msh4 mutant male and female mice, as indicated by the chromosomal localization of RAD51 and COR1 during leptonema/zygonema, the chromosomes fail to undergo normal pairing. Our results show that MSH4 localization on chromosomes during the early stages of meiosis is essential for normal chromosome synapsis in prophase I and that it acts in the same pathway as MSH5.
Keywords: Mismatch repair, meiosis, chromosome synapsis, recombination, germ cell
The DNA mismatch repair system (MMR) in eukaryotic cells is responsible for the repair of DNA mismatches that can result from a number of different mechanisms including DNA replication, genetic recombination, and chemical modification of DNA or nucleotide pools. Studies in yeast, and more recently in mice, have also revealed a role for MMR proteins in the control of meiotic recombination. The bacterial DNA mismatch repair system typified by the Escherichia coli Mut HLS system is the simplest and best understood. This system is capable of repairing both single nucleotide mismatches as well as small insertion/deletion mismatches (for reviews, see Kolodner 1996; Modrich and Lahue 1996). In E. coli, the MutS protein recognizes and binds to mismatched nucleotides. In a subsequent step a second protein, MutL, interacts with MutS and activates a third protein, MutH, which is an endonuclease. MutH nicks the unmethylated strand of hemimethylated DNA in the vicinity of a mismatch, thereby directing the repair of the newly synthesized strand.
Although the essential components of this MMR system have been conserved in eukaryotes, the repair system is more complex than in E. coli and involves several MutS and MutL homologs. In yeast Saccharomyces cerevisiae there are six homologs of the DNA-binding protein MutS designated MutS homolog (MSH) 1–6. There are also four known homologs of the MutL gene in yeast, designated MLH1, MLH2, PMS1, and MLH3 (for review, see Kolodner 1996; Crouse 1998). The mammalian genome has homologs for all of these genes except MSH1, which, if present, is yet to be discovered (Buermeyer et al. 1999; Kolodner and Marsischky 1999).
It is well established that in eukaryotes the products of the MSH2, MSH3, MSH6, as well as MLH1, PMS1, and MLH3 genes are involved in DNA mismatch repair. In eukaryotes, MMR requires a complex of MSH2–MSH6 for the repair of single base mispairs and either a complex of MSH2–MSH6 or MSH2–MSH3 for the repair of insertion/deletion mispairs (Acharya et al. 1996; Marsischky et al. 1996; Genschel et al. 1998; Guerrette et al. 1998; Umar et al. 1998). The two MSH complexes interact with the complexes of MLH1–PMS1 (PMS2 in human) or MLH1–MLH3 for the repair of the different mismatches (Prolla et al. 1994; Li and Modrich 1995; Habraken et al. 1997; Pang et al. 1997; Flores-Rozas and Kolodner 1998; Wang et al. 1999).
Germ-line mutations in some of the MMR genes in humans are associated with the cancer predisposition syndrome, hereditary nonpolyposis colon cancer (HNPCC). This syndrome is inherited in an autosomal dominant fashion and is characterized by a predispostion to develop colonic and extracolonic tumors where the tumors have a characteristic replication error (RER+) phenotype (Kinzler and Vogelstein 1996). Germ-line mutations in MSH2 and MLH1 account for a majority of HNPCC families (Peltomaki and Vasen 1997). Recently, it is was found that MSH6 germ-line mutations account for a small number of HNPCC families but appear to be also responsible for a larger number of late-onset familial colorectal cancer cases (Kolodner et al. 1999; Wu et al. 1999).
Studies in bacteria and yeast showed that the MMR system is also involved in the control of recombination. For example, genetic analysis in yeast showed that the complexes consisting of the MMR proteins MSH2–MSH6, MSH2–MSH3, and MLH1–PMS1 function in the prevention of recombination between divergent DNA sequences. This role in recombination is dependent on interactions with other proteins including RAD1–RAD10 and EXO1 (Nakagawa et al. 1999). Two other members of the yeast MSH family, MSH4 and MSH5, play a role specifically in meiotic recombination. Yeast strains carrying null mutations in either MSH4 or MSH5 show reduced rates of crossing over but not gene conversion, increased chromosomal nondisjunction, and reduced spore viability (Ross-Macdonald and Roeder 1994; Hollingsworth et al. 1995). The analysis of MSH4–MSH5 double mutant yeast strains indicates that MSH4 and MSH5 function in the same genetic pathway with MSH5 being epistatic to MSH4 (Hollingsworth et al. 1995). Yeast MSH4 and MSH5 are able to form heterodimeric complexes similar to the mitotic MSH proteins (Pochart et al. 1997). In a manner analogous to mitotic MMR, the analysis of MSH4–MLH1 double mutant yeast strains indicated that the meiosis-specific MutS homologs require the function of MLH1 for the promotion of meiotic crossing over (Hunter and Borts 1997).
To understand the role of the mammalian mismatch repair genes in DNA repair, cancer predisposition and meiosis, several mouse lines with targeted mutations in MMR genes have been generated. Mice that carry mutations in the mismatch repair genes Msh2 (de Wind et al. 1995; Reitmair et al. 1995), Msh3 (de Wind et al. 1999; Edelmann et al. 2000), Msh6 (Edelmann et al. 1997), Mlh1 (Baker et al. 1996; Edelmann et al. 1996), Pms2 (Baker et al. 1995), and Pms1 (Prolla et al. 1998) have been described. Msh2−/−, Mlh1−/−, Msh6−/−, and Pms2−/− mice display a predisposition to tumors, although the degree of this predisposition and the latency for tumor development differ. Mice lacking Msh3 and Pms1 are reported to be normal.
Mice that are homozygous for mutations in the somatic members of the MSH gene family (Msh2, Msh3, and Msh6), are viable and fully fertile (de Wind et al. 1995; Reitmair et al. 1995; Edelmann et al. 1997, 2000). However, mice that are mutant for the MutL homologs Pms2 and Mlh1 also exhibit a meiotic defect in addition to their cancer predisposition phenotypes. Male mice bearing a homozygous mutation in Pms2 show abnormal chromosome pairing during meiosis and are sterile whereas the females are fertile (Baker et al. 1995). Mice with mutations in the Mlh1 gene are viable but both sexes are sterile. Normal chromosome pairing was observed in pachynema of prophase I in spermatocytes from Mlh1 mutant males, but most of the cells fail to progress beyond pachynema (Baker et al. 1996; Edelmann et al. 1996).
The observation that mutations in the MutL homologous genes result in a different meiotic phenotype compared to mutations in the MutS homologous genes with which they interact during mitotic DNA mismatch repair indicates that the MLH proteins employ different members of the MSH family as partners during meiosis. Recently, the human homologs of the yeast MSH4 and MSH5 genes have been isolated and their expression in human germ cells (Paquis-Flucklinger et al. 1997; Her and Doggett 1998; Winand et al. 1998) suggests that one or both of these gene products may be partners for MLH1 during meiosis. Indeed, Msh5 mutant mice are viable, but both males and females are sterile. Meiosis in these mice cannot progress normally because chromosome pairing is severely affected during prophase I (de Vries et al. 1999; Edelmann et al. 1999).
To study the meiotic function of mammalian MSH4 we analyzed its expression during the different stages of meiosis I. We also assessed the role of MSH4 in meiosis by generating mice that carry a null mutation in this gene. Our results show that MSH4 is required for normal chromosome pairing during prophase I. The combination of the Msh4 mutation with a mutation in Msh5 further showed that MSH4 and MSH5 are both essential for proper chromosome pairing during mammalian meiosis and that they act in the same pathway.
Results
Association of MSH4 with meiotic chromosomes
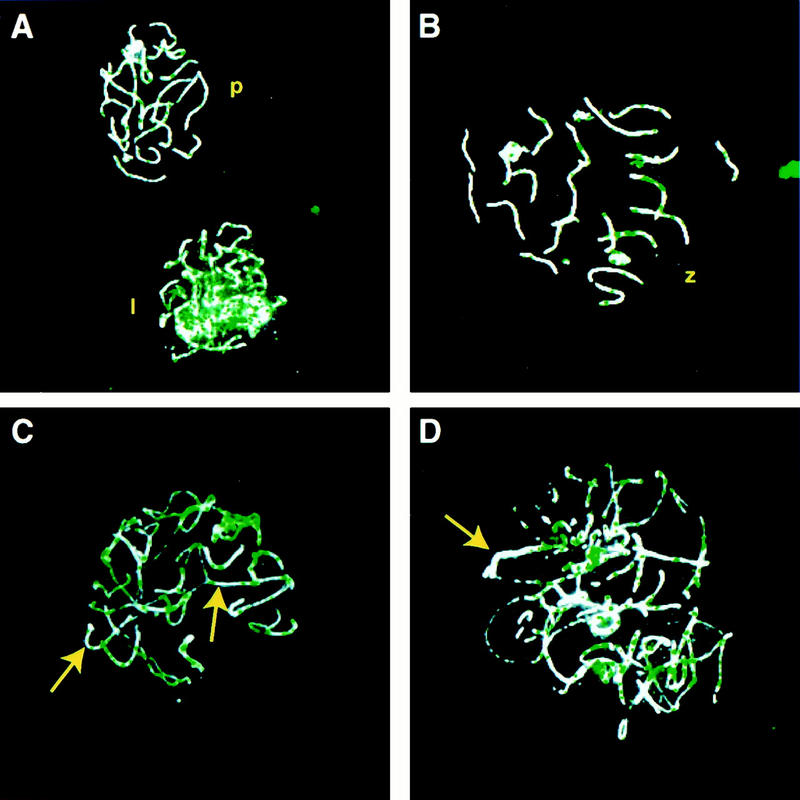
To determine the tissue-specific expression pattern of Msh4 Northern blot analysis was performed using a cDNA probe spanning the entire coding region of the human MSH4 gene (Paquis-Flucklinger et al. 1997). A 3.2-kb mRNA transcript was detected in testis but was virtually absent in all other tissues tested including skin, lung, liver, thymus, spleen, brain, heart, kidney, stomach, small intestine, and skeletal muscle (data not shown). The testis-specific expression of Msh4 suggested a role similar to its yeast counterpart in the control of meiotic processes. To further investigate this possibility, the distribution of MSH4 protein along meiotic chromosomes was analyzed by immunofluorescent methods. The MSH4 protein colocalized with the synaptonemal complex (SC) and axial element protein COR1 on chromosome spreads prepared from wild-type testes at day 17 postpartum (pp). MSH4 foci were found to be colocalized with the SC from leptonema up until pachynema (Fig. 1). At leptonema, MSH4 was localized throughout the nuclear region, although not intimately associated with the axial element backbone of unsynapsed chromosomes (Fig. 1A,E). By zygonema, MSH4 staining was associated directly with the axial elements themselves, being distributed along much of the length of the meiotic chromosomes. The foci at this stage were of variable size but were still quantifiable (142 ± 24.7 per nucleus; Fig. 1B,E). Early in pachynema, MSH4 was still present in discrete foci along the SC of synapsed chromosomes, with the number of foci per bivalent remaining high (90 ± 4.5 per nucleus; Fig. 1C,E). By midpachynema the number of MSH4 foci declined further, with an average of 47 ± 4.5 foci per nucleus (Fig. 1D,E).
Figure 1.
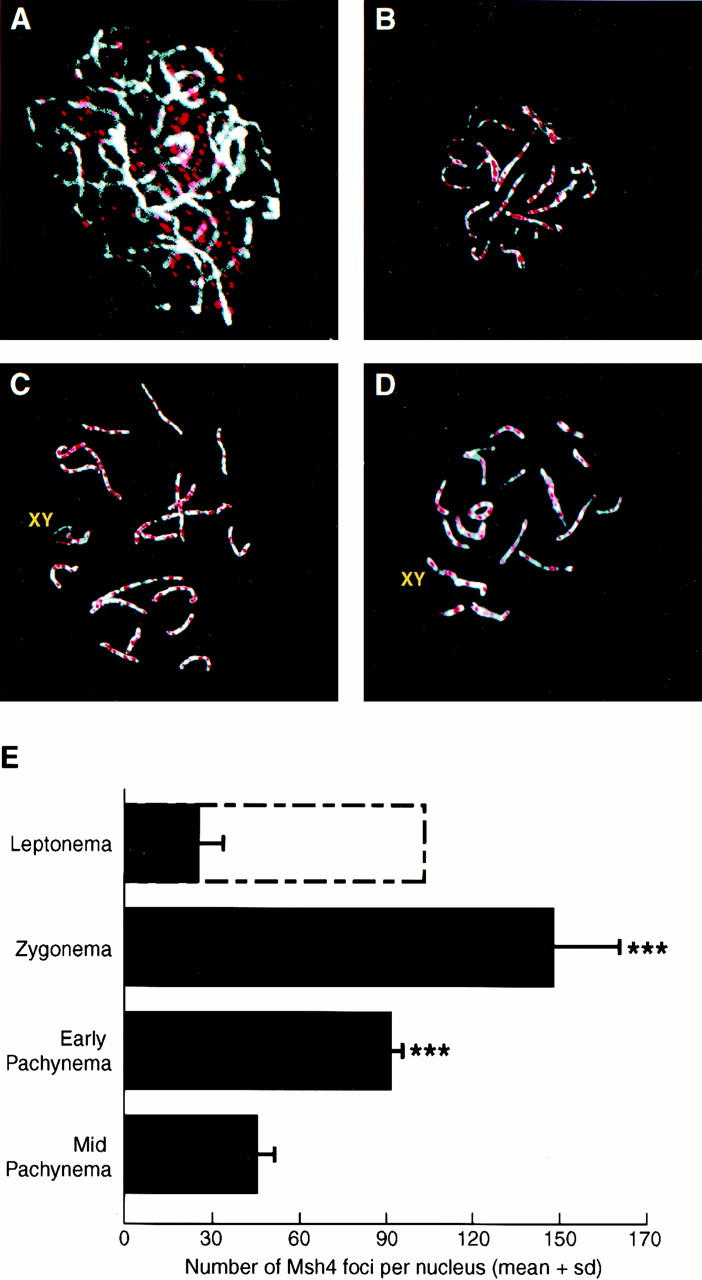
MSH4 localization on meiotic chromosomes during prophase I. (A–D) Immunofluorescent colocalization of MSH4 (red) and the synaptonemal complex protein COR1 (white) on chromosome spreads from wild-type spermatocytes at day 17 pp. (A) Leptonema; (B) zygonema; (C) early pachynema; (D) mid-pachynema. (E) Quantitation of foci associated with meiotic chromosomes during prophase I (mean ± s.d.). Solid bars represent mean number of foci associated with the COR1 protein at each stage of prophase I; the open bar represents the number of foci observed throughout the nucleus. One-way ANOVA reveals a high degree of significance across the stages (P < 0.0001). Asterisk indicate statistically significant differences from both leptotene foci (on chromosomes) and mid-pachytene foci (Dunn's multiple post-test, P < 0.001, n = 12 nuclei per genotype).
Generation of Msh4 mutant mice
The localization of MSH4 on meiotic chromosomes supported a role for this protein in meiosis. To determine the importance of MSH4 for mammalian meiosis we generated a mouse line that carries an inactivating mutation in the germ line. The gene targeting vector pMsh4ex4 was designed to introduce a PGK hygromycin resistance cassette into exon 4 corresponding to codon 252 of the human MSH4 cDNA (Fig. 2A). This modification introduces multiple stop codons into the Msh4 reading frame as verified by sequencing and is predicted to result in an inactivating mutation. A truncated protein, if produced by the modified Msh4 locus, would lack the nucleotide-binding domain and the helix–loop–helix domain located at the COOH-end that are essential for the function of the MutS family of proteins (Ross-Macdonald and Roeder 1994; Paquis-Flucklinger et al. 1997). The targeting vector pMsh4ex4 was linearized and electroporated into embryonic stem (ES) cells. One hundred ninety-six hygromycin-resistant clones were isolated and screened for the homologous recombination event by PCR (Fig. 2A). Forty-three (22%) of the analyzed clones tested positive for the correct targeting event. The appropriate modification was verified by Southern blot analysis (Fig. 2B). Three independently derived Msh4 ES clones were injected into C57/Bl6 blastocysts. Chimeric animals from all three cell lines transmitted the disrupted allele through the germ line. Heterozygous F1 animals were interbred to obtain homozygous mutant mice. We obtained 518 F2 offspring animals from 11 mating pairs. Genotyping of the F2 mice revealed that 139 animals were wild type, 263 animals were heterozygous, and 116 animals were homozygous for the mutant allele. This result is consistent with a normal Mendelian pattern of inheritance and indicates that MSH4 is not essential for normal development.
Figure 2.

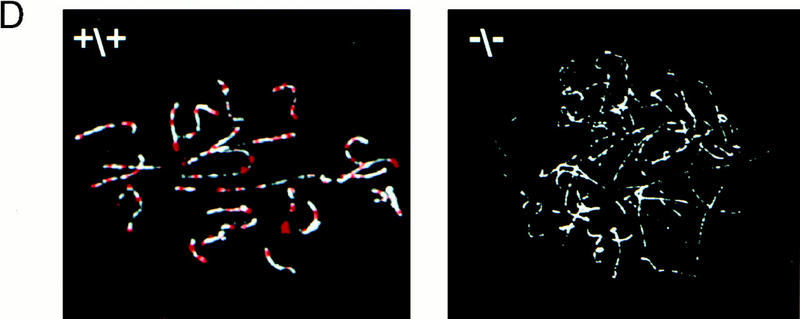
Generation of Msh4 mutant mice. (A) Gene-targeting strategy. Schematic representation of the Msh4 wild-type gene locus, the pMsh4Ex4 targeting construct and the targeted Msh4 locus. The exons are shown as black boxes. The PCR primers located in the PGK hygromycin cassette and in exon 6 that were used for detecting the gene-targeting events are indicated by arrows connected by a dotted line. The diagnostic BglII digestion products for the wild-type Msh4 locus and the modified Msh4 locus that are recognized by the hybridization probe are shown. (B) Southern blot hybridization of DNA from mice of the F2 generation. Tail DNA was digested with BglII and hybridized with the probe shown in A. The 5.2-kb band corresponds to the wild type and the 3.6-kb band corresponds to the targeted allele. (+/+) Wild type; (+/−) heterozygous; (−/−) homozygous. (C) Detection of Msh4 by Northern blot analysis. Testis poly(A) RNA of the different genotypes was analyzed with a cDNA probe corresponding to the entire Msh4-coding sequence. A human β-actin-specific probe was used as a control. (+/+) Wild type; (+/−) heterozygous; (−/−) homozygous. (D) Detection of MSH4 protein on meiotic chromosomes. Immunofluorescent colocalization of MSH4 (red) and the synaptonemal complex protein COR1 (white) on chromosome spreads of spermatocytes from 20-day-old males. (+/+) Chromosome spread from wild-type spermatocytes; (−/−) chromosome spread from homozygous mutant spermatocytes.
Lack of Msh4 transcripts or MSH4 protein in testis of Msh4−/− mice
Two lines of evidence indicated that the insertion of the PGK hygromycin cassette into exon 4 resulted in an inactivating mutation. First, to confirm that Msh4−/− mice did not produce normal transcripts we subjected testis poly(A) RNA to Northern blot analysis (Fig. 2C). The RNA from wild-type mice contained a 3.2-kb transcript that was also present at reduced level in heterozygous mice. No Msh4 transcript was detectable in the testis RNA of homozygous mutant mice. Second, the inactivation of MSH4 in homozygous mutant mice was further confirmed by immunolocalization experiments. Although MSH4 foci are readily detectable in maximal numbers on wild-type spermatocyte chromosomes at zygonema, no MSH4 protein was present on or around the meiotic chromosomes of Msh4−/− mice at the comparable stage of meiosis (Fig. 2D).
Fertility of Msh4−/− male mice
Msh4 mutant animals up to 12 months of age appeared to develop normally without any discernable disease phenotype. However, whereas Msh4+/+ and Msh4+/− males were fertile, matings between Msh4−/− males and wild-type females did not produce any offspring. Msh4−/− males exhibited normal sexual behavior and aggression, but were infertile. The testis weights of Msh4−/− adult males was only ∼50% of wild type and closer analysis revealed no spermatozoa within the epididymides of Msh4−/− adult males (data not shown) or within the seminiferous tubular lumen of their testes (Fig. 3B,D). In contrast, Msh4+/+ adult male littermates had normal numbers of epididymal spermatozoa and numerous spermatozoa within the seminiferous tubules, as identified by the sperm tails protruding into the tubular lumen (Fig. 3A,C, arrowheads). To investigate the progression of the first meiotic wave in Msh4+/+ and Msh4−/− males, the appearance and progression of germ cells through meiosis was assessed morphologically at day 23 pp, representing the time when the first meiosis I is completed. The seminiferous tubules of Msh4+/+ males contained an abundance of meiotic germ cells, ranging from early spermatogonia, flattened against the basement membrane of the tubule, to spermatocytes entering and progressing through prophase I (Fig. 3E). These meiosis I cells were readily identified by their enlarged size, their gradual loss of the signal for GCNA1, a germ cell-specific marker during mitosis and meiosis whose loss is indicative of progression to pachynema (Enders and May 1994), and their position further toward the lumen of the seminiferous tubules (Fig. 3E). Some differentiating spermatids were also apparent within tubules of Msh4+/+ males, indicating the completion of meiosis II in these cells (Fig. 3E). In contrast, even during the first wave of meiosis between day 13 and 26 pp, seminiferous tubules of Msh4−/− males exhibited a severe depletion of spermatocytes, but not of primary spermatogonia (Fig. 3F). Cells further in toward the lumen of the seminiferous tubules were densely stained with GCNA1 and remained small compared to those cells seen within the tubules of wild-type males. No luminal cells appeared to be at meiotic stages beyond zygonema. In addition, many cells appeared to be apoptotic as assessed by routine morphological criteria (Fig. 3B). Thus, by adulthood, seminiferous tubules of Msh4+/+ males contained a cellular profile representative of all stages of the spermatogenic wave (Fig. 3A), whereas the tubules of Msh4−/− males were devoid of many spermatogenic cells, having lost most of the resident type A and B spermatogonia and all of the spermocytes (Fig. 3B). Many seminiferous tubules of adult Msh4−/− males contained only a single layer of spermatogonia and Sertoli cells (Fig. 3B). Interestingly, the interstitial areas of the testes of Msh4−/− males appeared to contain many more Leydig cells (LC, Fig. 3B,D) than those of wild-type males (Fig. 3A,C).
Figure 3.
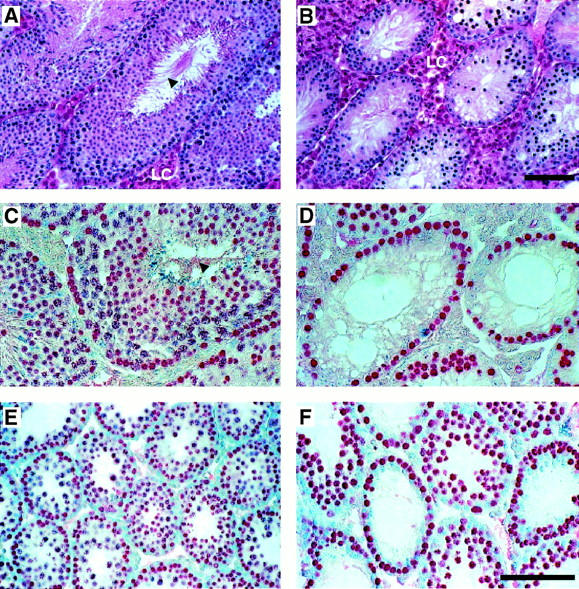
Testis morphology in Msh4+/+ and Msh4−/− males. (A,C,E) Wild-type males. (B,D,F) Msh4−/− mutant males. Hematoxylin and eosin staining (A,B) and immunohistochemical localization of GCNA1-positive spermatogonia and spermatocytes (C,D) in sections of adult testis. (E,F) GCNA1 localization of spermatogonia and spermatocytes at the end of the first wave of prophase I at day 23 pp. (LC) Leydig cells. Arrowheads show mature spermatozoa within the lumen of testes from wild-type adult males. (A–F) Bar, 200 μm.
Chromosome pairing analysis in male germ cells
Analysis of meiotic chromosomes in Msh4−/− males revealed severe abnormalities in pairing at the zygotene stage of prophase I, with most chromosomes failing to undergo any degree of pairing or synapsis. However, most of the nuclei showed at least some signs of chromosomal interactions. At day 23 pp, when most (>90%) spermatocyte nuclei from wild-type males contained bivalent chromosomes in late zygonema or pachynema (Fig. 4A; Table 1), <70% of spermatocyte nuclei from Msh4−/− males contained any paired chromosomes (Fig. 4B,C,D; Table 1). Of the Msh4−/− cells that did show chromosome pairing, between 2 to 3 (mean = 2.74 ± 0.23) chromosomes per cell showed some degree of pairing. However, most pairing was between nonhomologous chromosomes as revealed by interactions between chromosomes of different lengths (Fig. 4C,D, arrows). The small degree of homologous pairing that does occur is limited and rarely apparent across the entire length of the chromosomes (Fig. 4C,D, arrowheads). Occasionally, homologous pairing was evident at one or both ends of a chromosome (Fig. 4D, arrows), whereas some chromosomes were paired intermittently along their entire length (Fig. 4D, arrowhead). Chromosomes from Msh4−/− males were never condensed, whereas pairing in wild-type spermatocytes was always accompanied by chromosome condensation. The degree of chromosomal interactions in Msh4−/− spermatocytes was significantly more advanced than that seen in spermatocytes taken from either Msh5−/− or Msh4−/−/Msh5−/− males (Fig. 4E,F; Edelmann et al. 1999), but remained significantly disrupted when compared to wild-type littermates (Table 1).
Figure 4.
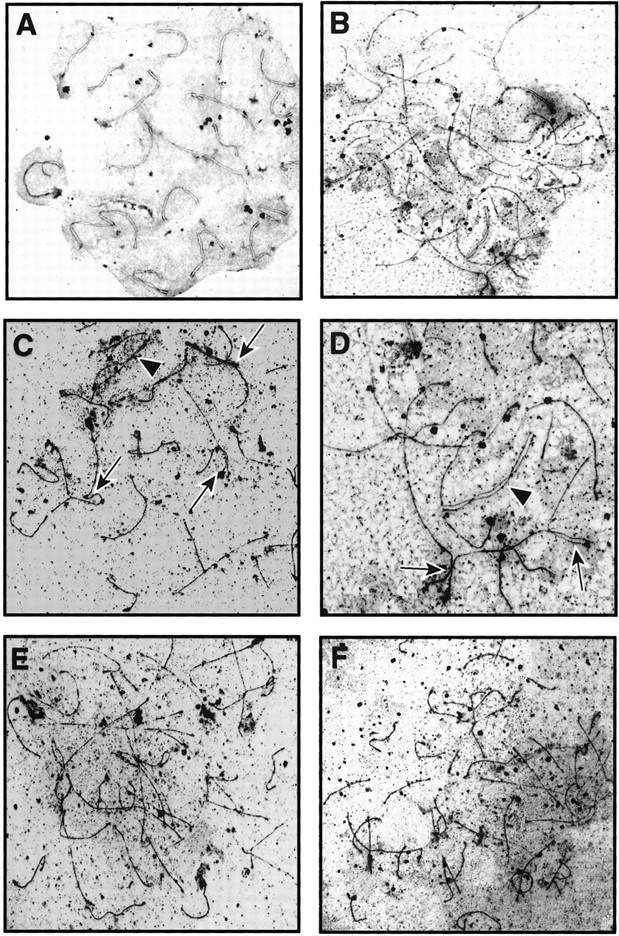
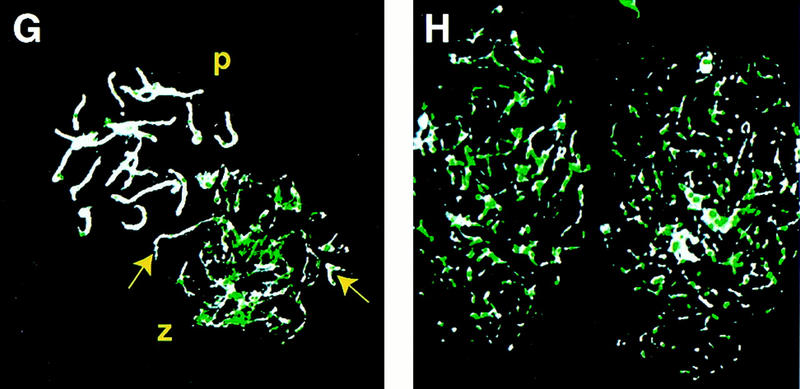
Analysis of chromosome spreads from spermatocytes of Msh4+/+ and Msh4−/− males. (A–D) Electron micrographs of silver-stained chromosome spreads from spermatocytes of wild-type (A) and Msh4−/− homozygous mutant (C,D) males, showing small degree of pairing in the Msh4−/− males compared to complete synapsis of chromsomes from spermatocytes of Msh4+/+ males. Arrows indicate nonhomologous pairing between two (C) or more (D) chromosomes; arrowheads indicate regions of apparent homologous pairing (C,D). (E,F) Silver-stained chromosome spreads from spermatocytes taken from Msh5−/− (E) and Msh4−/−/Msh5−/− (F) males are shown for comparison, with less pairing than in chromosome spreads from Msh4−/− spermatocytes. (G,H) Immunofluorescent colocalization of RAD51 (green) and the axial element/synaptonemal complex protein COR1 (white) on chromosome spreads from day 23 pp spermatocytes. (P) Pachynema; (Z) zygonema. Yellow arrows indicate regions of pairing, and coincident loss of RAD51 foci at zygonema in Msh4+/+ spermatocytes (G), whereas RAD51 localization on chromosomes of Msh4−/− spermatocytes remains high (H).
Table 1.
Quantitation of pairing between meiotic chromosomes in Msh4−/−, Msh5−/−, and Msh4−/−/Msh5−/−spermatocytes
|
|
Msh4+/+
|
Msh4−/−
|
Msh5−/−
|
Msh4−/−/Msh5−/−
|
|---|---|---|---|---|
| Percent nuclei with 100% pairing | 64.9a | 0 | 0 | 0 |
| (48/74) | ||||
| Percent nuclei with no pairing | 9.4b | 31 | 90 | 92 |
| (7/74) | (18/58) | (54/60) | (46/50) | |
| Percent nuclei with partial pairing | 25.7c | 69 | 10 | 8 |
| (19/74) | (40/58) | (6/60) | (4/50) | |
| Analysis of partially paired chromosomesd | ||||
| Percent nonpaired chromosomes | 0 | 86.0 | 89.6 | 90.6 |
| (0/760) | (1376/1600) | (251/240) | (145/160) | |
| Percent homologous partial pairing | 100 | 7.1 | 6.25 | 0 |
| (760/760) | (114/1600) | (15/240) | (0/160) | |
| Percent homologous pairing | 0 | 6.9 | 4.2 | 9.4 |
| (110/1600) | (10/240) | (15/160) | ||
| Statistical analysis of pairing trends between genotypese | ||||
| χ2 comparison with Msh4−/− | P < 0.0001 | — | P < 0.0001 | P < 0.0001 |
| χ2 comparison with Msh5−/− | P < 0.0001 | P < 0.0001 | — | N.S. (P = 0.09) |
Values are percent, with number values in parentheses.
Nuclei at pachynema.
Nuclei at leptonema.
Nuclei at zygonema (with all chromosomes being partially paired).
Total numbers of chromosomes are obtained by multiplying 40 (total number of chromosomes per cell) by the number of nuclei showing some pairing.
Chi-square analyses were performed between different pairs of groups. (N.S.) Not significant.
The association of the RecA homolog RAD51 on unpaired chromosomes at leptonema and zygonema, and loss of such association upon homologous pairing, is now well established (Rockmill et al. 1995; Barlow et al. 1998). To assess the localization of this recombination protein on meiotic chromosomes in the absence of MSH4, we performed coimmunofluorescence using rabbit anti-mouse RAD51 and a mouse monoclonal antibody raised against the synaptonemal complex protein COR1. As expected, RAD51 foci were readily detectable on chromosomes from wild-type spermatocytes at zygonema (Fig. 4G), but were lost as pairing proceeded (Fig. 4G, arrow) and completely absent by pachynema, a time when synapsis is complete. In contrast, the failure of chromosomal pairing in spermatocytes from Msh4−/− males was associated with increased localization of RAD51 foci on the meiotic chromosomes (Fig. 4H).
Analysis of female meiosis and ovarian development in Msh4−/− females
Meiosis I occurs synchronously in female oogonia from embryonic day (E) 16 until birth in mice, after which time the oocytes enter a period of dictyate arrest just after pachynema. To analyze the progression of this first stage of meiosis I, ovaries from Msh4+/+ and Msh4−/− females were removed during the neonatal period (E18 through to day 4 pp). Staining of germ cells with an antibody against GCNA1 revealed a steady loss of germ cells in Msh4−/− females soon after birth (Fig. 5A–F). At E18, the number of GCNA1-positive cells in Msh4+/+ and Msh4−/− ovaries was similar (Fig. 5A,B). By day 2 pp, the earliest signs of follicular development as part of ovarian reorganization were apparent in Msh4+/+ ovaries (Fig. 5C, arrows), whereas many of the oogonia in Msh4−/− cells had already been lost (Fig. 5D). By day 4 pp, when all of the remaining oocytes in Msh4+/+ ovaries were enclosed by readily identifiable primordial follicles (Fig. 5E, arrows), the vast majority of oocytes in Msh4−/− ovaries had been lost (Fig. 5F), indicating a loss of oocytes before dictyate arrest in the Msh4−/− females.
Figure 5.
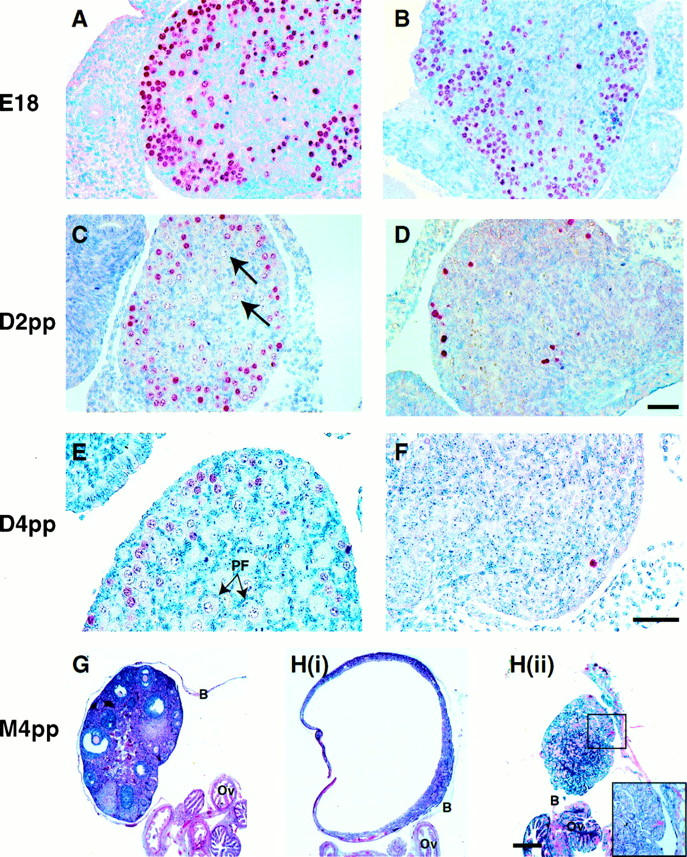
Histological analysis of neonatal and adult ovaries from Msh4+/+ and Msh4−/− females. Neonatal ovaries at E18 (A,B), day 2 pp (C,D), and day 4 pp (E,F) were stained with anti-GCNA1 antibody (red precipitate). GCNA1 is localized to oogonia and oocytes entering the initial stages of meiosis I, and is then lost as oocytes enter dictyate arrest before metaphase I. Oocytes in dictyate arrest are indicated by arrows in C and E. (PF) Primordial follicles. Bar, 100 μm. (G,H) Adult ovaries at 4 months pp were stained with hematoxylin and eosin. Adult Msh4−/− ovaries from the same animal frequently showed strikingly different phenotypes [H(i), H(ii)]. (B) Ovarian bursa; (Ov) oviduct.
To assess the consequences of oocyte loss in Msh4−/− females, ovaries were taken at 4, 16, and 28 weeks of life. In contrast to the oocyte-rich ovary in 4-week-old Msh4+/+ females (Fig. 5G), Msh4−/− females had very small ovaries containing few if any oocytes at this age (Fig. 5H). Often, many divergent morphologies were present within the same female, as is the case for the pair of ovaries shown in Figure 5H (i, ii). Immunohistochemical analysis using an antibody directed against P450 side chain cleavage enzyme revealed that these structures were not producing steroids, but were epithelial in origin, as demonstrated by immunohistochemical staining with an anticytokeratin antibody (data not shown). These ovarian structures were completely encased within their ovarian bursas and were attached to apparently normal oviductal and uterine structures (data not shown). By 7 months of age, the ovaries of Msh4−/− females were virtually nonexistent, or consisted of massively convoluted tissue structures (data not shown), with no resemblance to the wild-type ovaries at the same age. These structures occasionally, but not always, contained a single or a number of large fluid and blood-filled cysts (see Fig. 5H, below).
Chromosomal pairing in Msh4−/− oocytes
Analysis of chromosomal pairing in oocytes from Msh4+/+ and Msh4−/− females was performed at E19 by immunofluorescence because the extremely small amount of tissue precluded the use of the silver staining technique. However, these analyses demonstrated clearly that the oocytes from both Msh4+/+ and Msh4−/− ovaries enter leptonema, and acquire both their initial synaptonemal complex proteins and at least one key recombination nodule protein, RAD51 (Fig. 6). As in the males, the number of RAD51 foci on meiotic chromosomes remained higher in the Msh4−/− oocytes, compared to wild-type oocytes (Fig. 6A,B), indicative of a failure to undergo complete pairing and to enter pachynema (Fig. 6C,D). In those regions of chromosomes that did undergo pairing, RAD51 foci were lost (Fig.6 C,D, arrows), indicating that the pairing failure itself might be responsible for the persistence of RAD51 foci. Pairing of homologous chromosomes appeared to be a more frequent occurrence in Msh4 mutant oocytes than in Msh4 mutant spermatocytes, although the small amount of tissue available in the embryonic ovaries precluded the possibility of a more quantitative assessment of pairing in the female germ cells.
Figure 6.
Immunofluorescent localization of COR1 and RAD51 on meiotic chromosomes from Msh4+/+ and Msh4−/− females. The SC protein COR1 and the RecA homolog RAD51 were immunolocalized on chromosome spreads from oocytes at E19. COR1 immunofluorescence is shown in white, and RAD51 foci are localized in green. (A,B) chromosome spreads from wild-type oocytes at early zygonema (A, lower nucleus), late zygonema (B) and pachynema (A, top nucleus). (C,D) chromosome spreads from Msh4−/− oocytes at zygonema, showing at least partial synapsis of chromosomes (C,D, arrows) and the coincident loss of RAD51 colocalization.
MSH4 and MSH5 function in the same genetic pathway
The comparison of the meiotic phenotype between Msh4−/− and Msh5−/− mice revealed that both MutS homologs are required in the early stages of meiosis I and are essential for normal chromosome synapsis during zygonema. In yeast, genetic analysis has shown that MSH4 and MSH5 function in the same epistasis group. To investigate whether mammalian MSH4 and MSH5 behave in a similar manner double mutant Msh4−/−/Msh5−/− mice were generated. As with the single mutant mice, no adverse phenotype was observed with regard to the viability and survival of Msh4−/−/Msh5−/− mice and no notable somatic phenotype was apparent. Both male and female double homozygous mutant mice were infertile. The degree of chromosomal pairing during meiosis I in spermatocytes was similar to that seen in Msh5−/− mice, in that <10% of nuclei contained chromosomes with any degree of pairing, and no chromosomal condensation was apparent (see Fig. 4E,F; Table 1). The similarity in meiotic phenotype between Msh5−/− and Msh4−/−/Msh5−/− mice indicates that mammalian MSH5 functions upstream of MSH4 within the same epistasis group and both are required at the same time in meiosis to ensure proper chromosome synapsis.
Discussion
The generation of mouse lines with inactivating mutations in the genes encoding homologs of the MutS and MutL proteins that function in the MutHLS mismatch repair system has revealed roles for many of these proteins in mitotic mismatch repair and in meiotic recombination. The phenotypes observed in these mutant mouse lines have shown that some of these genes are required for both processes, whereas others are required only for mitotic mismatch repair or only for meiotic recombination. Our results show that MSH4 is essential for male and female meiosis and acts in the same pathway as MSH5.
MSH4 is expressed in meiotic tissues and forms discrete foci on chromosomes during the progression from leptonema to pachynema
In yeast, MSH4 expression is restricted to meiotic cells undergoing sporulation. Similarly, expression analysis of multiple tissues revealed that Msh4 mRNA is only detectable in testis and is absent in all somatic tissues. This result is consistent with that observed in human tissues and suggests a role for mammalian MSH4 in meiotic recombination (Paquis-Flucklinger et al. 1997). Our analysis of MSH4 expression in meiotic nuclei revealed that it is present at the various stages of male prophase I. Early in meiosis during the leptotene stage of prophase I, MSH4 protein accumulated within the nuclear matrix. Subsequently, in zygonema, MSH4 colocalized with the synaptonemal complex protein COR1 and was found at multiple sites along unpaired meiotic chromosomes. The number of these MSH4 foci decreased until midpachynema to ∼47 foci per nucleus, a figure that represents almost twice the number of estimated recombination sites found in mouse spermatocytes at diplonema (27 per nucleus; Polani and Jagiello 1976). This numerical and temporal correlation between MSH4 foci and chiasmata frequency could indicate a specific functional relationship between the two phenomena, a suggestion that could be addressed in future studies. The formation of MSH4 foci on zygotene chromosomes in mice differs from that seen in yeast where MSH4 only localizes at discrete locations along synapsed chromosomes during pachynema (Ross-Macdonald and Roeder 1994). This temporal difference in binding might indicate different roles of MSH4 during recombination in yeast and mammals.
Meiotic defects in the Msh4−/− male mice
Msh4−/− mutant mice developed normally and the males showed normal reproductive behavior. Histological analysis of the testis of Msh4−/− mutant males indicated the presence of primary spermatogonia A and B. However, none of the spermatocytes progressed beyond zygonema because most of the cells that were present at this stage became apoptotic. The failure of spermatogonial maturation appears to be related to abnormal chromosome pairing during the zygotene phase of meiotic prophase I. Analysis of chromosome spreads revealed that most chromosomes at this stage failed to undergo pairing, indicative of a role for MSH4 in chromosome synapsis that is supported by the presence of a large number of MSH4 foci on meiotic chromosomes during zygonema as described above. The presence of MSH4 foci during pachynema in wild-type mice suggests an additional role for MSH4 at later stages of meiosis. However, the analysis of such a later meiotic role was precluded in Msh4−/− males because of the induction of apoptosis in the spermatocytes following the failure of chromosome pairing at zygonema.
The phenotype in yeast is consistent with a role for yeast MSH4 in the resolution of recombination intermediates. As mentioned above, yeast MSH4 only localizes on chromosomes at pachynema, and MSH4 null mutant yeast strains do not show abnormal chromosome synapsis but display a reduction in crossing over as well as an increase in chromosomal nondisjunction resulting in reduced spore viability (Ross-Macdonald and Roeder 1994). The comparison of the distribution of MSH4 foci along the meiotic chromosomes at the various stages of prophase I as well as the phenotypes of MSH4 mutant yeast and mouse strains indicate a functional difference. Interestingly, the Caenorhabditis elegans gene him-14, an ortholog of yeast MSH4, is also not required for pairing or synapsis. Similar to its yeast counterpart, him-14 appears to be required during pachynema and to have a role in crossover formation (Zalevsky et al. 1999). This functional difference of MSH4 in yeast and C. elegans versus mouse may indicate a divergence of function during evolution.
Ovarian meiosis and development in Msh4−/− female mice
As in males, meiosis was severely disrupted in Msh4 mutant females. Analysis of chromosomes in germ cells from embryonic and neonatal ovaries indicated that meiosis I was disrupted in Msh4−/− females at a similar stage compared to that of Msh4−/− males. Although some chromosome pairing was evident in many Msh4 mutant oocytes, the majority of chromosomes remained unpaired. The failure of pairing in these oocytes correlated with an induction of apoptosis, resulting in the almost complete obliteration of germ cell numbers within the ovaries of Msh4−/− females by day 4 pp (Fig. 5). As in the case of Msh5−/− females, the loss of the oocyte pool resulted in the disruption of ovarian development and the complete loss of ovarian structures in the adult Msh4−/− female. Such ovarian dysgenesis after germ cell loss is specific to mice lacking MSH5 and MSH4, as other mutations that result in germ cell depletion do not give rise to such a severe alteration in ovarian development. For example, female mice carrying various allelic mutations in the c-kit ligand Steel factor do not exhibit a loss of ovarian structures, despite a reduction in oocyte numbers to ∼10% of wild type at birth (Bedell et al. 1995). Similarly, mice lacking the RecA homolog DMC1 also exhibit a loss of germ cells at birth, but their ovarian structures remain intact, albeit considerably reduced in size (Pittman et al. 1998; Yoshida et al. 1998). These data clearly indicate that the progression of meiosis in the embryonic ovary is monitored by a developmental checkpoint that regulates the survival of meiosis I oocytes and the subsequent signals that trigger ovarian development and folliculogenic reorganization.
MSH complexes during meiosis
In yeast the two meiotic MutS homologs MSH4 and MSH5 form heterodimeric complexes resembling the complexes formed by the mitotic MutS homologs MSH2–MSH3 and MSH2–MSH6 (Marsischky et al. 1996). It was also observed that MSH5 deficiency causes a more severe phenotype than MSH4 deficiency indicating that MSH5 is epistatic to MSH4 (Hollingsworth et al. 1995). Recently it was reported that mammalian MSH4 and MSH5 are also capable of forming heterodimeric complexes (Winand et al. 1998; Bocker et al. 1999). The meiotic phenotypes of the Msh4−/− and Msh5−/− mice generated by our laboratory show that both proteins are required for chromosome synapsis. The comparison of the phenotypes in both mouse lines further shows that Msh5 inactivation causes a more severe defect in pairing than Msh4 inactivation. Although only very few of the nuclei in Msh5−/− spermatocytes contained chromosomes that underwent homologous or nonhomologous pairing (10%), the majority of the MSH4 nuclei (69%) contained a small number of paired chromosomes. In contrast, by the same age 64.9% of the wild-type spermatocytes were at pachynema with all chromosomes being fully synapsed. Our results suggest that mammalian MSH4 and MSH5 act within the same genetic pathway and that MSH5 might be epistatic to MSH4 similar to the situation in yeast. This notion is also supported by the phenotype of the Msh4−/−/Msh5−/− mice, which is similar to that of the MSH5 mutant mice with almost no detectable chromosome pairing (in only 10% of the nuclei). It should be mentioned that in an Msh5 mutant mouse line generated in another laboratory, chromosome synapsis was also affected but a higher degree of chromosome pairing was reported (de Vries et al. 1999). The difference in the degree of chromosome pairing observed in the two lines might be caused by differences in the genetic background. Another possibility is that the two Msh5−/− lines represent different Msh5 alleles. The Msh5−/− mouse line generated by our laboratory carries a null mutation as no Msh5 transcript or protein is expressed in testis. A direct comparison to the Msh5 mutant line generated by de Vries et al. (1999) is difficult to assess because no RNA or protein data were reported from meiotic tissue.
The role of MMR genes in mammalian meiosis
Msh4 is the last known MutS homolog that has been inactivated in the mouse germ line, and it is now possible to compare the biological function of all the known members of the MMR family of genes. Currently, there is no evidence for a role of three of the five MutS homologs MSH2, MSH3, and MSH6 in the control of meiosis. As expected from their germ cell-specific expression pattern and similarity to their yeast counterparts, mutations in the other two MutS homologs MSH4 and MSH5 cause a meiotic phenotype. The MSH2–MSH3 and MSH2–MSH6 complexes function in conjunction with MLH1–PMS2 and MLH1–MLH3 in mitotic mismatch repair (Wang et al. 1999; Lipkin et al. 2000). Although the identity of the proteins that interact with MSH4–MSH5 during meiotic recombination is not known, it is possible that MSH4–MSH5 functions in meiosis along with MLH1–PMS2 and/or MLH1–MLH3. The possibility of such an interaction is supported by the fact that MLH1 as well as PMS2 have a meiotic function in mouse. The phenotype in Pms2−/− mutant males corresponds to Msh4−/− and Msh5−/− mutant males in that they show abnormalities in zygonema with incomplete chromosome pairing. The Pms2−/− phenotype, however, is less severe in that apoptotic cell death was not reported and abnormal spermatozoa were produced. In addition, Pms2 mutant females appear to be able to produce functional oocytes. In contrast, in both males and females, MLH1 deficiency results in sterility that in males is caused by apoptotic cell death of the spermatocytes immediately after pachynema. However, unlike the Msh4, Msh5, and Pms2 mutants, chromosome synapsis in Mlh1 mutant males and females is not affected (Baker et al. 1996; Edelmann et al. 1996; Woods et al. 1999). The defect in Mlh1 mutants appears to be at a later stage in pachynema/diplonema and the absence of chiasmata in diplonema suggests that MLH1 is required for the formation and/or resolution of meiotic crossover sites. At present it is not clear at what steps MSH4 and MSH5 participate in during meiotic recombination, nor what structures in the DNA are recognized for binding. It is possible that both proteins are involved in the early and/or late events of meiotic recombination. An interesting possibility is that MSH4–MSH5 may be required for multiple steps of the recombination process and that at different stages their involvement requires the interaction with different protein complexes. Many of the proteins that mediate meiotic recombination have been identified in yeast (for review, see Roeder 1997). In mammals several homologs of these genes have been described and it will be very interesting to determine whether interactions exist between these homologs and MSH4 or MSH5.
Materials and methods
Inactivation of Msh4 in embryonic stem cells
A 4.1-kb Sac genomic fragment containing Msh4 exons 4 and 5 derived from a 129Ola λ phage library was subcloned into pUC19. Comparison of the mouse and human sequences spanning exons 4 and 5 revealed 90% homology between the two species. A single BglII site was inserted into exon 4 corresponding to human codon 252 by site-directed mutagenesis. A 2.0-kb BglII fragment containing PGKhygromycin was cloned into exon 4 in the same transcriptional orientation as the Msh4 gene and the resulting targeting vector was designated pMsh4Ex4. The targeting vector (40 μg) was linearized at the single BamHI side and electroporated into 2.0 × 107 WW6 ES cells as described previously (Edelmann et al. 1997). The ES cells were selected in hygromycinB (150 μg/ml) and resistant colonies were isolated after 10 days of selection. Genomic DNA from individual colonies was subjected to Long Range PCR analysis (Boehringer Mannheim) and positive ES cell colonies were identified by a 4.3-kb PCR fragment using forward primer 5′-TGGAAGGATTGGAGCTACGG-3′ and reverse primer 5′-GAAAGCAGCTGCTCCGTATC-3′. The PCR reactions were performed according to the manufacturer's instructions. For Southern blot analysis genomic DNA was digested with BglII, transferred to nylon membrane, and hybridized to a genomic probe corresponding to intron 4 (Fig. 2A).
Generation of Msh4−/− mice
Embryonic stem cells derived from three independently targeted clones were injected into C57Bl/6 blastocysts. All three cell lines gave rise to male chimeric animals that were mated to C57Bl/6 females. Chimeric males derived from all three cell lines transmitted the mutation through their germ line. F1 heterozygous animals were intercrossed to obtain Msh4 homozygous mutant animals.
Northern blot analysis
Mouse multiple tissue Northern blots (Origene) were hybridized with a full-length human cDNA probe to determine Msh4 expression. For analysis of Msh4 expression in 23-day-old male testis poly(A) RNA was separated on 1.0% agarose formaldehyde gels and transferred to nitrocellulose membranes. For hybridization a human cDNA probe spanning the entire MSH4 coding region and a human β-actin probe was used.
Histology
For analysis of the first meiotic wave in the testis, Msh4+/+ and Msh4−/− males were taken between day 17 pp and day 23pp, corresponding to the end of meiosis I and meiosis II, respectively. Adult males were used at between 10 and 14 weeks of age. For analysis of female meiosis, embryos were taken between E16 and E19 and at day 1 through to day 5 pp. Older females were also used at day 25 pp and adulthood (4 weeks to 7 months of age). For histological analysis, ovaries and testes were fixed in Bouin's fixative or 4% buffered formalin for periods of 1 hr until 12 hr, depending on the size of the specimen. Fixed tissues were processed for immunohistochemistry by routine methods and the paraffin-embedded tissues were sectioned at 3–5 μm, depending on the tissue.
Chromosome analysis
Testes were removed from mice between the ages of day 17 pp and day 25 pp, decapsulated and rinsed in α-MEM. Tubules were chopped coarsely on dental wax and then more finely with watchmaker's forceps. Large clumps were removed and the supernatant was centrifuged to pellet the germ cells, which were then resuspended in 20 μl of fresh α-MEM. Aliquots of 4 μl were then applied to a 400-μl bubble of hypotonic (0.5%) saline on parafilm to burst open the cells and to spread the nuclear contents across the concave surface of the bubble. Spread nuclei were then picked up on precleaned slides or on to formvar-coated 200-mesh nickel electron microscopy grids. The nuclei were then fixed twice in 1% paraformaldehyde (pH 8.2) for 3 min each on ice, followed by three 1-min washes in 0.4% photoflo-200 (Kodak). Fixed nuclei were air dried overnight and then used immediately or stored at −70°C for up to 3 weeks. Nuclei were subjected to either silver staining in 50% AgNO3 at 55°C for 1 hr, or used for immunofluorescent analysis of chromosome-associated proteins (see below).
For analysis of female meiotic chromosomes, embryonic ovaries were removed between E16 and E19 and minced finely in cold α-MEM on precleaned microscope slides. Cell suspensions were then applied to a small bubble of hypotonic (0.5%) saline on a clean microscope slide, stirred gently, and then rested for 3 hr to allow the germ cell nuclei to sink through the saline and adhere to the slide. Fixation procedures were the same as for male germ cells. Slides were used for immunofluorescence as described below.
Immunofluorescence and immunohistochemistry
Slides containing chromosome spreads were subjected to immunofluorescent staining as previously described (Edelmann et al. 1999). Primary antibodies used were: (1) a mouse monoclonal antibody against COR1, a component of the mouse SC (1:1000); (2) a rabbit polyclonal antibody directed against the last 12 amino acids of mouse MSH4 (1:400); and (3) a rabbit polyclonal antibody raised against mouse RAD51 (1:500).
Paraffin sections were subjected to immunohistochemistry using a rat hybridoma supernatant against germ cell nuclear antigen-1 (GCNA-1) (Enders and May 1994). Alternatively, slides were stained with hematoxylin and eosin to reveal more detailed histological architecture.
Acknowledgments
We thank Peter Moens and Barbara Spyropoulos (University of York, Toronto, Canada) for the gift of anti-COR1 antibody; George Enders (University of Kansas) for the gift of anti-GCNA1 antibody; Veronique Paquis-Flücklinger and Sabine Darmanin for the gift of anti-MSH4 antibodies and helpful discussions. The authors gratefully acknowledge the assistance of Shailesh Shenoy (Department of Anatomy and Structural Biology) with the cooled CCD microscope and various members of the Einstein Analytical Imaging Facility (Frank Macaluso, Leslie Gunther, Carolyn Marks) for their technical support and helpful advice. J.W.P. is a Betty and Sheldon Feinberg Senior Faculty Scholar in Cancer Research. This work was supported by the National Institutes of Health (CA 76329 to W.E., CA 84301 to R.K. and W.E., GM26017 to R.D.K., and Cancer Center grant CA 13330 to AECOM) and a Wyeth-Ayherst grant to J.W.P.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL edelmann@aecom.yu.edu; FAX (718) 430-8972.
References
- Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. hMSH2 forms specific mispair–binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci. 1996;93:13629–13634. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A, et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Barlow C, Liyange M, Moens PB, Tarsounas M, Nagashima K, Brown K, Rottinghaus S, Jackson SP, Tagle D, Ried T, Wynshaw-Boris A. Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development. 1998;125:4007–4017. doi: 10.1242/dev.125.20.4007. [DOI] [PubMed] [Google Scholar]
- Bedell MA, Brannan CI, Evans EP, Copeland NG, Jenkins NA, Donovan PJ. DNA rearrangements located over 100 kb 5′ of the Steel (Sl)-coding region in Steel-panda and Steel-contrasted mice deregulate Sl expression and cause female sterility by disrupting ovarian follicle development. Genes & Dev. 1995;9:455–470. doi: 10.1101/gad.9.4.455. [DOI] [PubMed] [Google Scholar]
- Bocker T, Barusevicius A, Snowden T, Rasio D, Guerrette S, Robbins D, Schmidt C, Burczak J, Croce CM, Copeland T, et al. hMSH5: A human MutS homologue that forms a novel heterodimer with hMSH4 and is expressed during spermatogenesis. Cancer Res. 1999;59:816–822. [PubMed] [Google Scholar]
- Buermeyer AB, Deschênes SM, Baker SM, Liskay RM. Mammalian mismatch repair. Annu Rev Genet. 1999;33:533–564. doi: 10.1146/annurev.genet.33.1.533. [DOI] [PubMed] [Google Scholar]
- Crouse GF. Mismatch repair systems in Saccharomyces cerevisiae. In: Nickoloff JA, editor. DNA repair in prokaryotes and lower eukaryotes. Totowa, NJ: Humana Press; 1998. pp. 411–448. [Google Scholar]
- de Vries SS, Baart EB, Dekker M, Siezen A, de Rooij DG, de Boer P, te Riele H. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes & Dev. 1999;13:523–531. doi: 10.1101/gad.13.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Claij N, Jansen L, van Klink Y, Radman M, Riggins G, van der Valk M, van't Wout K, te Riele H. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet. 1999;23:359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen PE, Kane MF, Lipford JR, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Cohen PE, Kneitz B, Winand N, Lia M, Heyer J, Kolodner R, Pollard JW, Kucherlapati R. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat Genet. 1999;21:123–127. doi: 10.1038/5075. [DOI] [PubMed] [Google Scholar]
- Edelmann W, Umar A, Yang K, Heyer J, Kucherlapati M, Lia M, Kneitz B, Avdievich A, Fan K, Wong E, et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000;60:803–807. [PubMed] [Google Scholar]
- Enders GC, May JJ. Developmentally regulated expression of a mouse germ cell nuclear antigen examined from embryonic day 11 to adult in male and female mice. Dev Biol. 1994;163:331–340. doi: 10.1006/dbio.1994.1152. [DOI] [PubMed] [Google Scholar]
- Flores-Rozas H, Kolodner RD. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc Natl Acad Sci. 1998;95:12404–12409. doi: 10.1073/pnas.95.21.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- Guerrette S, Wilson T, Gradia S, Fishel R. Interactions of human hMSH2 with hMSH3 and hMSH2 with hMSH6: Examination of mutations found in hereditary nonpolyposis colorectal cancer. Mol Cell Biol. 1998;18:6616–6623. doi: 10.1128/mcb.18.11.6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habraken Y, Sung P, Prakash L, Prakash S. Enhancement of MSH2-MSH3-mediated mismatch recognition by the yeast MLH1-PMS1 complex. Curr Biol. 1997;7:790–793. doi: 10.1016/s0960-9822(06)00337-x. [DOI] [PubMed] [Google Scholar]
- Her C, Doggett NA. Cloning, structural characterization, and chromosomal localization of the human orthologue of Saccharomyces cerevisiae MSH5 gene. Genomics. 1998;52:50–61. doi: 10.1006/geno.1998.5374. [DOI] [PubMed] [Google Scholar]
- Hollingsworth NM, Ponte L, Halsey C. MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes & Dev. 1995;9:1728–1739. doi: 10.1101/gad.9.14.1728. [DOI] [PubMed] [Google Scholar]
- Hunter N, Borts RH. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes & Dev. 1997;11:1573–1582. doi: 10.1101/gad.11.12.1573. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes & Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, Wahlberg S, Fox EA, Peel D, Ziogas A, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- Li GM, Modrich P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci. 1995;92:1950–1954. doi: 10.1073/pnas.92.6.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin SM, Wang V, Jacoby R, Banerjee-Basu S, Baxevanis AD, Lynch HT, Elliott RM, Collins FS. MLH3: A DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. 2000;24:27–35. doi: 10.1038/71643. [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes & Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Datta A, Kolodner RD. Multiple functions of MutS- and MutL-related heterocomplexes. Proc Natl Acad Sci. 1999;96:14186–14188. doi: 10.1073/pnas.96.25.14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Q, Prolla TA, Liskay RM. Functional domains of the Saccharomyces cerevisiae Mlh1p and Pms1p DNA mismatch repair proteins and their relevance to human hereditary nonpolyposis colorectal cancer-associated mutations. Mol Cell Biol. 1997;17:4465–4473. doi: 10.1128/mcb.17.8.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquis-Flucklinger V, Santucci-Darmanin S, Paul R, Saunieres A, Turc-Carel C, Desnuelle C. Cloning and expression analysis of a meiosis-specific MutS homolog: The human MSH4 gene. Genomics. 1997;44:188–194. doi: 10.1006/geno.1997.4857. [DOI] [PubMed] [Google Scholar]
- Peltomaki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: Database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997;113:1146–1158. doi: 10.1053/gast.1997.v113.pm9322509. [DOI] [PubMed] [Google Scholar]
- Pittman DL, Cobb J, Schimenti KJ, Wilson LA, Cooper DM, Brignull E, Handel MA, Schimenti JC. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell. 1998;1:697–705. doi: 10.1016/s1097-2765(00)80069-6. [DOI] [PubMed] [Google Scholar]
- Pochart P, Woltering D, Hollingsworth NM. Conserved properties between functionally distinct MutS homologs in yeast. J Biol Chem. 1997;272:30345–30349. doi: 10.1074/jbc.272.48.30345. [DOI] [PubMed] [Google Scholar]
- Polani PE, Jagiello GM. Chiasmata, meiotic univalents, and age in relation to aneuploid imbalance in mice. Cytogenetics Cell Genet. 1976;16:505–529. doi: 10.1159/000130668. [DOI] [PubMed] [Google Scholar]
- Prolla TA, Pang Q, Alani E, Kolodner RD, Liskay RM. MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science. 1994;265:1091–1093. doi: 10.1126/science.8066446. [DOI] [PubMed] [Google Scholar]
- Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998;18:276–279. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A, Waterhouse P, Mittrucker HW, Wakeham A, Liu B, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- Rockmill B, Sym M, Scherthan H, Roeder GS. Roles for two RecA homologs in promoting meiotic chromosome synapsis. Genes & Dev. 1995;9:2684–2695. doi: 10.1101/gad.9.21.2684. [DOI] [PubMed] [Google Scholar]
- Roeder GS. Meiotic chromosomes: It takes two to tango. Genes & Dev. 1997;11:2600–2621. doi: 10.1101/gad.11.20.2600. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Roeder GS. Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell. 1994;79:1069–1080. doi: 10.1016/0092-8674(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148:1637–1646. doi: 10.1093/genetics/148.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TF, Kleckner N, Hunter N. Functional specificity of MutL homologs in yeast: Evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci. 1999;96:13914–13919. doi: 10.1073/pnas.96.24.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winand NJ, Panzer JA, Kolodner RD. Cloning and characterization of the human and Caenorhabditis elegans homologs of the Saccharomyces cerevisiae MSH5 gene. Genomics. 1998;53:69–80. doi: 10.1006/geno.1998.5447. [DOI] [PubMed] [Google Scholar]
- Woods LM, Hidges CA, Baart E, Baker SM, Liskay M, Hunt PA. Chromosomal influence on meiotic spindle assembly: Abnormal meiosis I in female Mlh1 mutants. J Cell Biol. 1999;145:1395–1406. doi: 10.1083/jcb.145.7.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Berends MJ, Mensink RG, Kempinga C, Sijmons RH, van Der Zee AG, Hollema H, Kleibeuker JH, Buys CH, Hofstra RM. Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. Am J Hum Genet. 1999;65:1291–1298. doi: 10.1086/302612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Kondoh G, Matsuda Y, Habu T, Nishimune Y, Morita T. The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol Cell. 1998;1:707–718. doi: 10.1016/s1097-2765(00)80070-2. [DOI] [PubMed] [Google Scholar]
- Zalevsky J, MacQueen AJ, Duffy JB, Kemphues KJ, Villeneuve AM. Crossing over during Caenorhabditis elegans meiosis requires a conserved MutS-based pathway that is partially dispensable in budding yeast. Genetics. 1999;153:1271–1283. doi: 10.1093/genetics/153.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]