

Abstract
The latency-associated nuclear antigen (LANA) is central to the maintenance of Kaposi's sarcoma-associated herpesvirus (KSHV) and to the survival of KSHV-carrying tumor cells. In an effort to identify interaction partners of LANA, we purified authentic high-molecular-weight complexes of LANA by conventional chromatography followed by immunoprecipitation from the BC-3 cell line. This is the first analysis of LANA-interacting partners that is not based on forced ectopic expression of LANA. Subsequent tandem mass spectrometry (MS/MS) analysis identified many of the known LANA-interacting proteins. We confirmed LANA's interactions with histones. Three classes of proteins survived our stringent four-step purification procedure (size, heparin, anion, and immunoaffinity chromatography): two heat shock proteins (Hsp70 and Hsp96 precursor), signal recognition particle 72 (SRP72), and 10 different ribosomal proteins. These proteins are likely involved in structural interactions within LANA high-molecular-weight complexes. Here, we show that ribosomal protein S6 (RPS6) interacts with LANA. This interaction is mediated by the N-terminal domain of LANA and does not require DNA or RNA. Depletion of RPS6 from primary effusion lymphoma (PEL) cells dramatically decreases the half-life of full-length LANA. The fact that RPS6 has a well-established nuclear function beyond its role in ribosome assembly suggests that RPS6 (and by extension other ribosomal proteins) contributes to the extraordinary stability of LANA.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is associated with primary effusion lymphoma (PEL), Kaposi's sarcoma (KS), and the plasmablastic variant of multicentric Castleman disease (MCD) (6, 8, 73). Like other herpesviruses, KSHV exhibits two distinct phases in its life cycle: latent episomal persistence and lytic replication. During latent infection, only a small subset of viral proteins is expressed (7, 12–14, 28, 56, 74, 83). One of these is the KSHV latency-associated nuclear antigen (LANA). It is encoded by open reading frame 73 (ORF73) and consistently expressed in all latently infected cells. LANA is necessary and sufficient for viral episome persistence and faithful partition of the latent KSHV genome. LANA tethers the viral plasmid to cellular chromosomes (1, 2, 24, 25). However, this is not the only function of LANA.
LANA is a multifunctional protein. Many proteins bind to LANA. These can be grouped according to their presumed functions. (i) Barbera et al. demonstrated that the N terminus of LANA docks onto cellular chromosomes by directly binding to the folded regions of histones H2A and H2B to mediate nucleosome attachment (3). Both histones H2A and H2B were necessary for LANA to bind nucleosomes. (ii) Robertson and colleagues found that LANA also binds histone H1 as well as other proteins involved in the structural remodeling of DNA (77). (iii) Cellular replication and replication licensing factors can bind to LANA (45, 75, 78), as well as the chromatin-modifying factors SAP30, mSin3A, CIR, meCP2, and DEK (35, 36). Further binding partners have been described (5, 71). These include Ku70, Ku80, and PARP-1. (iv) Cellular transcription factors can bind to LANA, such as Sp-1 (33), RBP-jκ (also known as CSL) (40), glycogen synthase kinase 3β (GSK-3β) (21), CBP/p300 and ATF4/CREB2 (43), Ring3 (48, 53, 60, 79), and KSHV Rta/orf50 (41). (iv) LANA binds to Rb (61) as well as to p53 (18, 80). The LANA-p53 complex can be destroyed by the mdm-2/p53 interaction inhibitor nutlin (9, 58, 67), which leads to p53-dependent apoptosis in PEL. Because of its ability to decorate host chromosomes, LANA can induce chromosome instability phenotypes that are akin to p53 inactivation (54, 70).
Here we find a new binding partner of LANA: the ribosomal protein S6 (RPS6). RPS6 (32 kDa) is a component of the 40S ribosomal subunit and the major phosphoprotein of the ribosome (59). The phosphorylation sites in RPS6 have been mapped to five clustered residues: Ser235, Ser236, Ser240, Ser244, and Ser247 (34, 66). RPS6 phosphorylation and function are highly regulated. RPS6 has been implicated in the regulation of translational initiation and protein synthesis in response to extracellular stimuli such as TRAIL and gamma interferon (IFN-γ), as well as upon activation of the phosphatidylinositol 3-kinase (PI3K)–Akt–mTOR pathway (4, 38, 42). Ribosome biogenesis and translation are regulated at multiple levels and are associated with cell growth and proliferation (65). Several ribosomal proteins are overexpressed in a variety of tumors. It remains to be determined whether this represents a cause or a consequence of tumor formation (66). Phosphorylated RPS6 is a biomarker for mTOR-targeted therapy in sarcoma (26), including KS (unpublished data). RPS6 is consistently phosphorylated in PEL and KS (72). Importantly, and perhaps underappreciated, the RPS6 protein is both cytoplasmic and nuclear localized (57). The RPS6 protein consists of three modules: a nuclear localization signal (NLS), a nucleolar binding sequence (Nobis), and the C-terminal serine cluster of phosphorylation sites, which is evolutionarily conserved (38, 44, 68).
We find that RPS6 exists in complex with the KSHV LANA protein in PEL. This complex was stable after multiple chromatographic purification steps and was resistant to DNase and RNase treatment. This interaction suggests a new role for LANA in protein translation and ribosome biogenesis and vice versa a role for RPS6 in LANA function and, as we report here, LANA protein stability.
MATERIALS AND METHODS
Cell culture.
BC-3 cells were cultured in RPMI 1640 medium (Cellgro, Inc.) containing 2 mM l-glutamine, 10% fetal bovine serum, penicillin G (100 U/ml), and streptomycin sulfate (100 μg/ml) and supplemented with 0.05 mM 2-mercaptoethanol (Sigma, Inc.), 0.075% sodium bicarbonate (Life Technologies, Inc.), and 1 U/ml human interleukin-6 (IL-6) (Roche, Inc.). HeLa cells (American Tissue Culture Collection [ATCC] no. CCL-2) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). All cells were grown at 37°C under 5% CO2 atmosphere.
Antibodies.
Anti-LANA monoclonal antibody (MAb) LN53 was purchased from Advanced Biotechnology, Inc., and used at a dilution of 1:2,000. Anti-LANA polyclonal rabbit antiserum was raised against the LANA repeat region (9). The additional antibodies used were as follows: anti-p53 MAb DO1 (1:2,000) (Santa Cruz, Inc.) and anti-phospho RPS6 rabbit antiserum (1:2,000 Ser 235/236 and 1:2,000 Ser 240/244), as well as total anti-RPS6 antibody (1:2,000) (Cell Signaling), anti-β-actin mouse MAb ((1:3,000) (Sigma, Inc.), and anti-p21 mouse MAb (1:1,000) (Santa Cruz). Mouse anti-RPL11 (clone 3a4a7) was from Invitrogen, Inc., and antihemagglutinin (anti-HA; clone HA-7) and anti-FLAG (clone M2) were from Sigma, Inc.
Plasmids.
The human RPS6 ORF was PCR amplified from RpS6-FLAG-VRP (a gift from R. Johnston [50]). To construct HA-tagged RPS6, we fused RPS6 with HA-tagged sequence (YPYDVPDYA) into plasmid pcDNA3 (Invitrogen, Inc.) at the N-terminal portion of RPS6 ORF (pDD1902). This procedure used the primers F1 (5′-GCATACCCATACGATGTTCCAGATTACGCTCTGAACATCTCCTTCCC; HA tag is underlined) and F2 (5′-CGACCCAAGCTTACCATGGCATACCCATACGATGTTCC), which incorporates a HindIII site (underlined), as well as the common reverse primer R (5′-GTCACGGGATCCCGTTATTTCTGACTGGATTC), which incorporates a BamHI site (underlined). The full-length plasmid of FLAG-LANA was a gift from D. Hayward (20). A series of mutants (amino acids [aa] 1 to 930, 1 to 428, 1 to 329, 330 to 1162, and 930 to 1162) of LANA were individually constructed into the pFLAG-CMV plasmid (Invitrogen, Inc.) using HindIII and EcoRI sites to yield pDD1926, pDD1927, pDD1928, pDD1929, and pDD1931, respectively.
Chromatography and MS/MS.
BC-3 cells (5 × 109) were harvested and the nuclear extracts were obtained as described previously (11). The supernatant was loaded onto a Sepharose 6B column (Sigma, Inc.) and washed with 0.1 M KCl in buffer D (20 mM HEPES-KOH [pH 7.9], 10% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 0.5 mM dithiothreitol [DTT], 0.5% cocktail protein inhibitor). Sepharose 6B was used not as an affinity resin, but as a sizing matrix for high-molecular-weight complexes. This choice was based on the initial use of Sepharose 6B prior to its discovery as an affinity resin. When using 6B resin to separate large protein complexes such as LANA, multimers (220 kDa × 4) and multimer-containing complexes elute close to the void volume, monomers, and broken complexes tend to be retained. Column fractions containing LANA were collected and loaded onto Hiprep 16/10 heparin FF column (Amersham, Inc.), followed by a wash with 20 column volumes of 0.2 M KCl in buffer D and elution with 0.5 M KCl in buffer D. The fractions were then loaded onto a Mono-Q column (Sigma, Inc.). We collected two peaks at 0.4 M buffer D and 0.7 M KCl in buffer D. Immunoprecipitation with anti-LANA antibody (LN53; ABI, Inc.) was from a the second peak as described previously (9). Proteins were resolved by 8 to 16% gradient SDS-PAGE and stained with Coomassie blue (Bio-Rad, Inc.). Visible bands were cut and further subjected to mass spectrometry at the University of North Carolina—Chapel Hill core facility. They were further digested with trypsin, and the resulting peptides were analyzed on an ABI 4800 matrix-assisted laser desorption ionization–tandem time of flight (MALDI TOF/TOF) MS analyzer. MS spectra were executed in a reflector positive-ion mode, with a mass range of 700 to 4,000 Da, and with a total number of shots/spectrum of 1,250. MS/MS spectra were executed in a positive-ion mode at 2 kV and with a total number of shots/spectrum of 1,350 or less. The peptides with signal/noise ratio above 20 at the MS mode were selected for MS/MS run; a maximum of 45 MS/MS was allowed per spot. The precursor mass window was 200 relative resolution (fwhm). All spectra were searched against taxonomy, NCBInr database, using GPS Explorer software version 3.6 (ABI) and the Mascot (MatrixScience) search engine. Variable modifications included oxidation (M). Mass tolerance was 80 ppm for precursor ions and 0.5 Da for fragment ions; two missed cleavages were allowed.
Immunoprecipitation and Western blot analysis.
For immunoprecipitation, the collected transfected cells were lysed with cold radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40, 50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, 0.5% cocktail protein inhibitor [Sigma, Inc.]) for 20 min. After centrifugation at 12,000 × g, the supernatants (or column fractions where indicated) were precleared with 30 μl protein G-agarose beads (Sigma) for 2 h and then incubated first with the appropriate antibodies overnight and then with 40 μl of protein G-agarose beads with gentle rotation for 2 h at 4°C. The beads were collected by centrifugation at 5,000 × g for 1 min, and the pellets were washed four times with ice-cold RIPA buffer.
For immunoprecipitation of FLAG-tagged proteins, the cell lysates were incubated with 30 μl EZview anti-FLAG M2 affinity resin (Sigma, Inc.) for 4 h or overnight at 4°C. FLAG-tagged proteins were eluted by incubation with 50 μl of 150 μg/ml 3× FLAG peptide (catalog no. F 4799; Sigma, Inc.) in Tris-buffered saline (TBS; 10 mM Tris-HCl, 100 mM NaCl [pH 8.0]) for 1 h at 4°C. Samples were then added to 5× SDS protein sample buffer (300 mM Tris-HCl [pH 8.0]), 10% SDS, 25% β-mercaptoethanol, 0.1% bromophenol blue, 50% glycerol) and analyzed with 8 to 16% SDS-PAGE Tris-HCl gel, and transferred to Hybond P membranes (GE Life Sciences, Inc.). After blocking with 5% nonfat milk, the membrane was incubated with the corresponding primary antibody in TBS-Tween (TBST; 10 mM Tris-HCl [pH 8.0], 100 mM NaCl, 0.1% Tween 20) plus 5% nonfat dry milk and washed three times for 10 min with TBST. After incubation with a horseradish peroxidase-linked secondary antibody (1:3,000) (VWR, Inc.), an enhanced chemiluminescence system (Thermo Scientific, Inc.) was used for detection via X-ray film (Genesee, Inc., catalog no. 30-101) for visualization.
ChIP assay.
For chromatin immunoprecipitation (ChIP), BC-3 cells (1 × 106) were cross-linked with formaldehyde at a final concentration of 1% for 10 min at 37°C. After being washed with phosphate-buffered saline (PBS) twice, cellular chromatin was sheared to approximately 500 bp using the Sonifier 450 sonicator (Branson output, 3.0; duty cycle, 30%) on ice four times for 30 s each. Immunoprecipitation, washing, and recovery of bound DNA were performed according to the manufacturer's protocol (Upstate Biotechnology, Inc.). The antibodies used were mouse monoclonal anti-LANA (LN53; ABI, Inc.) at 1 μg/reaction and anti-RPS6 (54D2; Cell Signaling, Inc.) at 1 μg/reaction. Mouse IgG at 1 μg/reaction was used as a control. Equal amounts of DNA pellets were used for PCR (Promega, Inc., catalog no. M7122). The forward primer for the LANA promoter (LANAp) was 5′-AGATCGCAGACACTGAAACGCTGA-3′, and the reverse primer was 5′-GCAAAGCAGACACGCCTTCTTCAGT-3′. β-Actin was used for a control with forward primer 5′-GGCATCGTGATGGACTCCG-3′ and reverse primer 5′-GCTGGAAGGTGGACAGCGA-3′.
shRNA knockdown of RPS6.
A set of pLKO.1 lentiviral vectors for RPS6 (NM_001010) were obtained from Open Biosystems/Thermo, Inc. The reconstructed lentiviruses were produced by the Lenti-shRNA Core Facility of University of North Carolina. BC-3 cells (5 × 105) were grown in six-well plates, infected with either of the respective lentiviruses (titer of 106 IU/ml) and Polybrene at a final concentration of 10 μg/ml, and incubated at 37°C for 6 h. After infection for 6 h, BC-3 cell medium was replaced with fresh RPMI 1640 supplemented with 10% fetal bovine serum. After 24 h, puromycin (5 μg/ml) was added to the medium, and cells were harvested 3 or 4 days after transfection. The numbers of viable cells were counted throughout the time course. Lentivirus short hairpin RNA (shRNA) against green fluorescent protein (GFP) or untreated BC-3 cells were used as a control.
Reporter assays.
SLK cells (1 × 105 to 5 × 105) were seeded in 12-well plates at 37°C. After 24 h, the cells were transfected with reporter (pDD83) (62) and effector plasmids (pDD104 expressing LANA and pDD1902 expressing RPS6), using 7.5 μl Superfect reagent (Qiagen, Inc.) according to previously published methods (29). pcDNA3 (Invitrogen, Inc.) was used to adjust total DNA concentration, and the medium was changed 24 h after transfection. The cells were harvested 48 h posttransfection, washed twice with ice-cold PBS, and lysed in 200 μl of 1× reporter lysis buffer, and then the luciferase substrate was added (Promega, Inc.). Luciferase activity was quantified via a luminometer (Fluorstar Optima; BMG, Inc.). The background (pGL3 basic vector) activity was below 100 relative light units (RLU). The transfection efficiencies were normalized by cotransfecting a lacZ expression plasmid (pDD173) and analyzing β-galactosidase activity using the Galacto-Light plus β-galactosidase assay kit (Applied Biosystems, Inc.). The data series represents one of three transfections. We used robust regression to establish a linear dose response relationship (64).
RESULTS
Identification of nuclear LANA binding proteins under physiological conditions.
During viral latency in KSHV-infected PEL cells, LANA is expected to interact with multiple cellular proteins. We hypothesized that there exist multiple distinct complexes of LANA and one or more associated cellular proteins, each for the different physiological functions of LANA. Because of their different enzymatic functions, these complexes are expected to have different biochemical properties. Since these complexes may have different biochemical properties, they are enriched by different biochemical purification procedures. Because LANA has a distinct domain structure with the two terminal globular domains separated by a large variable, and for the most part dispensable repeat region, prior studies (3, 31) used domain-based baits rather than the whole protein to identify interaction partners.
To identify the components of LANA complexes that bound to the whole LANA protein in latently infected cells, we first prepared nuclear extracts from BC-3 cells and then used multiple steps of chromatographic enrichment (Sepharose, heparin FF, and Mono-Q column) prior to immunoprecipitation with a LANA-specific monoclonal antibody. Figure 1 A diagrams our purification strategy. This approach requires LANA-interacting proteins to be stably bound to LANA and the interaction to withstand high-salt- and low-protein-concentration conditions. Many transcription factors were initially identified by this approach (11), and we previously used it to discern different LANA-p53 complexes (9). Coimmunoprecipitation was then used to identify the proteins bound to LANA either after chromatography on just a size exclusion column and heparin column (Table 1 ) or after chromatography that included an additional ion-exchange column (Table 2). The immunoprecipitates were ultimately analyzed by 8 to 16% gradient SDS-PAGE (Fig. 1B and C), and individual bands were subjected to tandem MS (MS/MS) analysis.
Fig. 1.

Purification and identification of LANA binding proteins. (A) A schematic of the procedures used to purify LANA complexes. (B and C) SDS-PAGE of the 8 to 16% gradient gel stained with Coomassie blue. Samples were eluted following immunoprecipitation with rat anti-LANA (α-LANA) antibody from heparin fractions (B) or after additional ion-exchange (MonoQ) purification (C). The isolated protein bands of complexes are denoted with black arrows (LANA, S6, and IgG). Co-IP, coimmunoprecipitation; M, molecular mass markers.
Table 1.
LANA binding proteins with anti-LANA immunoprecipitation after purification by heparin column chromatographya
| Lane | Database identification no. | Protein name | Size (no. of aa) | Mol mass (Da) | No. of peptides (hits) | MS/MS score | Source or referenceb |
|---|---|---|---|---|---|---|---|
| 1 | AAP69525 | Protein kinase, DNA activated | 4,128 | 468,787.9 | 49 | 195 | 34 |
| 2 | BAD52438 | Nonerythrocytic spectrin α | 2,452 | 282,108 | 44 | 155 | |
| 3 | Q53R99 | Hypothetical protein SPTBN1 | 2,314 | 268,549 | 48 | 494 | |
| 4 | MYH9_HUMAN | Myosin-9 (myosin heavy chain, IIa) | 1,960 | 226,260.5 | 58 | 732 | 34 |
| 5 | Q9HBD4_HUMAN | SMARCA4 isoform 2, BRG1 | 1,679 | 188,030.8 | 28 | 242 | 74 |
| 6 | A54854 | Ras GTPase-activating protein-related protein | NAc | 189,133.8 | 31 | 183 | |
| 7 | Q5T5X0 | Eukaryotic translation initiation factor 3 | 1,382 | 166,468.3 | 42 | 120 | |
| 8 | AAB41498 | Alpha II spectrin β | 2,477 | 284,890.5 | 55 | 556 | 34 |
| 9 | AAB48855 | RNA helicase A | 1,279 | 141,979.7 | 20 | 158 | 74 |
| 10 | Q75M86 | Hypothetical protein GTF2I | 978 | 110,211 | 35 | 527 | |
| 11 | Q59FF0 | EBNA-2 coactivator variant | 964 | 107,366.3 | 32 | 564 | |
| 12 | ILF3_HUMAN | Interleukin enhancer binding | 894 | 95,279.1 | 20 | 370 | 27 |
| 13 | A46302 | PTB-associated splicing factor | NA | 76,101.6 | 26 | 441 | |
| 14 | AAH27713 | Heterogeneous nuclear ribonucleoprotein | 804 | 90,235.8 | 23 | 159 | |
| Q86VG2 | Splicing factor | 707 | 76,140.7 | 15 | 116 | 27 | |
| Q2VPJ6 | Hsp90AA1 protein | 585 | 68,328.8 | 13 | 75 | ||
| 15 | AAB48826 | BTK-associated protein 135 | 957 | 107,812 | 25 | 204 | |
| 16 | CAH10627 | HSM803020 NID—Homo sapiens | 652 | 72,496.1 | 17 | 151 | |
| 17 | Q59GX6 | DEAD/H (Asp-Glu-Ala-Asp/His) box | 674 | 74,532.7 | 20 | 180 | 74 |
| 18 | JC1087 | RNA helicase, ATP-dependent | NA | 69,104.7 | 19 | 249 | 34, 74 |
| 19 | A25707 | U1 snRNP 70K protein | NA | 70,039.5 | 17 | 121 | |
| 20 | Q8TBR3 | Fusion [involved in t(12;16) in malignant liposarcoma] | 526 | 53,367.8 | 12 | 175 | 74 |
| 21 | Q59H57 | Fusion (involved in t(12;16) in malignant liposarcoma] | 300 | 31,974.3 | 14 | 294 | 74 |
| 22 | Q8TBR3 | Fusion [involved in t(12;16) in malignant liposarcoma] | 526 | 53,367.8 | 11 | 155 | 74 |
| 23 | AAQ05673 | Ig heavy chain variable region, VH3 | 120 | 13,308.5 | 4 | 77 | IgG |
| 24 | AAA35750 | DNA-binding protein B | 364 | 39,953.8 | 16 | 223 | |
| Q53HR5 | Eukaryotic translation EF1α | 426 | 50,093.1 | 12 | 90 | 27 | |
| 25 | AAH12854 | ACTB protein | 360 | 40,194.1 | 24 | 899 | |
| 26 | Q53FG3 | Interleukin enhancer binding | 390 | 43,021.2 | 14 | 255 | |
| 27 | CAF06488 | AX961958 NID—Homo sapiens | 285 | 30,569.1 | 12 | 214 | |
| AAH04383 | Polypyrimidine tract binding protein | 531 | 57,185.6 | 5 | 118 | ||
| 28 | Q53RW7 | Hypothetical protein HNRPA3 | 378 | 39,570.6 | 14 | 244 | 74 |
| 29 | B34504 | Heteronuclear ribonucleoprotein B1 | NA | 37,406.7 | 22 | 603 | 81 |
| 30 | Q2HJ60 | Heteronuclear ribonucleoproteins A2 | 341 | 35,983.9 | 21 | 766 | 74 |
| 31 | B34504 | Heteronuclear ribonucleoprotein A2/B1 | NA | 37,406.7 | 18 | 595 | 74 |
| 32 | Q3MIB7 | HNRPA1 protein | 267 | 29,368.2 | 22 | 781 | 74 |
| 34 | AAH12197 | Ribosomal protein L8 | 257 | 27,993.3 | 14 | 69 | ribo |
| 35 | RL7A_HUMAN | 60S ribosomal protein L7a | 266 | 29,846 | 17 | 200 | ribo |
| 36 | Q3KQU0 | RPL7 protein | 318 | 36,998.8 | 24 | 213 | ribo |
| Q53HV1 | Ribosomal protein S4 | 263 | 29,560 | 12 | 134 | ribo | |
| 37 | I38049 | MUC18 precursor | NA | 71,748.7 | 14 | 84 | |
| Q5JR95 | Ribosomal protein S8 | 188 | 21,866 | 9 | 80 | ribo | |
| 38 | CAE93910 | AX887985 NID—Homo sapiens | 209 | 25,112.9 | 16 | 193 | |
| 39 | RL10_HUMAN | 60S ribosomal protein L10 | 214 | 24,429.8 | 19 | 261 | ribo |
| RL13A_HUMAN | 60S ribosomal protein L13a | 203 | 23,431.3 | 15 | 124 | ribo | |
| 40 | RL18_HUMAN | 60S ribosomal protein L18 | 188 | 21,490 | 12 | 115 | ribo |
| 41 | RS9_HUMAN | 40S ribosomal protein S9b | 194 | 22,446.5 | 13 | 177 | 34 |
| S55916 | Ribosomal protein S5 | NA | 22,762.9 | 8 | 125 | ribo | |
| 42 | MLRM_HUMAN | Myosin regulatory light chain 2 | 171 | 19,650.4 | 7 | 214 | |
| AAB94895 | 60S ribosomal protein L12 | 129 | 14,332.8 | 5 | 100 | ribo | |
| 43 | MLRM_HUMAN | Myosin regulatory light chain 2 | 171 | 19,650.4 | 7 | 272 | |
| 44 | RS13_BOVIN | 40S ribosomal protein S13 | 151 | 17,080.6 | 8 | 150 | ribo |
| Q9BQQ5 | Ribosomal protein L27 | 106 | 12,007.4 | 5 | 109 | ribo | |
| R3RT18 | Ribosomal protein S18 | NA | 17,707.9 | 13 | 108 | 34 | |
| 45 | MOHU6 M | Myosin alkali light chain 6 | NA | 16,919.1 | 8 | 193 | |
| 46 | H2A1A_HUMAN | Histone H2A type 1-A (H2A/r) | 131 | 14,093.9 | 5 | 99 | 4, 34 |
| 47 | R3HU12 | Ribosomal protein S12, cytosolic | NA | 14,516.5 | 2 | 86 | ribo |
| 48 | S55054 | Sm protein G—human | NA | 8,490.4 | 5 | 75 |
In each case, the highest-scoring hit is shown.
Previously published in the reference(s) indicated. IgG refers to human IgG, and ribo refers to ribosomal components.
NA, not available.
Table 2.
LANA binding proteins with anti-LANA precipitation after purification by heparin and anion-exchange column chromatography
| Lane | Database identification no. | Protein name | Size (no. of aa) | Mol mass (Da) | No. of peptides (hits) | MS/MS score | pI |
|---|---|---|---|---|---|---|---|
| 1 | Q60FE2 | Myosin | 1,960 | 226,391.6 | 40 | 488 | 5.5 |
| 2 | Q9QR71_HHV8 | LANA | 1,129 | 131,267.9 | 12 | 264 | 3.9 |
| 3 | AAA59888 | Myosin | 1,337 | 154,265.7 | 30 | 492 | 5.7 |
| 4 | AAK74072 | Hsp96 precursor | 782 | 90,138.1 | 18 | 208 | 4.73 |
| 5 | AAC97490 | SRP72 | 671 | 74,560 | 12 | 203 | 9.31 |
| 6 | A27077 | Hsp70 | 646 | 70,854.2 | 24 | 662 | 5.62 |
| 7 | AAH04949 | Tubulin | 449 | 49,863.5 | 13 | 464 | 4.96 |
| 8 | AAH20946 | Tubulin | 444 | 49,639.9 | 17 | 715 | 4.75 |
| 9 | AAH20946 | Tubulin | 444 | 49,639.9 | 14 | 208 | 4.75 |
| 10 | RL4_HUMAN | Ribosomal L4 (L1) | 427 | 47,536.4 | 15 | 91 | 11.07 |
| 11 | Q96HG5 | Actin | 368 | 40,978.4 | 22 | 1,100 | 5.56 |
| 12 | AAH01127 | Ribosomal P0 | 317 | 34,252.7 | 17 | 378 | 5.42 |
| 13 | Q6NXR8 | Ribosomal S3A | 264 | 29,955.8 | 13 | 183 | 9.75 |
| 14 | AAH09427 | Ribosomal S6 | 247 | 28,403.8 | 8 | 128 | 10.84 |
| 15 | RL7A_HUMAN | Ribosomal L7a | 266 | 29,846 | 12 | 129 | 10.61 |
| 16 | Q53HV1 | Ribosomal S4 | 263 | 29,560 | 13 | 159 | 10.11 |
| 17 | S23753 | Ribosomal L13 | 211 | 24,276.5 | 8 | 129 | 11.69 |
| 18 | CAC94454 | IgG(κ) | 78 | 84,96.1 | 2 | 168 | 5.55 |
| 19 | RL10_HUMAN | Ribosomal L10 | 214 | 24,429.8 | 13 | 381 | 10.11 |
| 20 | RS13_BOVIN | Ribosomal S13 | 151 | 17,080.6 | 10 | 358 | 10.53 |
| 21 | R3HU16 | Ribosomal S16 | 146 | 16,435 | 8 | 192 | 10.42 |
We identified 48 proteins that coimmunoprecipitated with LANA after heparin fractionation (Table 1). Twenty-two (46%) were previously found in MS/MS screens for mutant LANA-associated proteins. These included the previously identified histone H2A. Ten (21%) of the bands corresponded to ribosomal proteins, and the remainder corresponded to novel potential LANA interaction partners and IgG. We did not subject bands of the expected molecular weight of LANA to MS/MS for economic reasons. Twenty-one proteins remained bound to LANA after an additional MonoQ purification step (Table 2). We recovered LANA itself (131,268 kDa), as expected, as well as IgG(κ) light chain (8,496 kDa). We recovered multiple forms of myosin, actin, and tubulin. Although it seems unlikely that these are bound to LANA, another KSHV protein, LANA-2, has been shown to interact with tubulin and to thereby affect paclitaxel resistance of PEL (51).
We recovered heat shock protein 96 (Hsp96) precursor (90,138 kDa) and Hsp70 (70,854 kDa) from LANA coimmunoprecipitates, as well as the signal recognition particle SRP72 (74,560 kDa). The significance of these results is under investigation, although it seems plausible that LANA binds to Hsps analogous to the Epstein-Barr virus (EBV) EBNA5/EBNA-LP (16, 23, 32, 46) and EBV EBNA3A (81) proteins binding Hsps.
We recovered multiple ribosomal proteins that coimmunoprecipitated with LANA. This mirrors earlier results by Kaul et al. (31). However, we did not recover exactly the same ribosomal proteins. Our collection of copurified ribosomal proteins is characterized by proteins with predominantly basic isoelectric points (pIs). With the exception of P0, the other 10 ribosomal proteins have predicted pIs of ≥9.7. In contrast, full-length LANA has a predicted pI of 3.9. Hence, it seems unlikely that the ribosomal proteins would copurify over charge-based separation columns (heparin and MonoQ) if they were not tightly bound to the LANA protein.
It may at first seem counterintuitive that ribosomal subunit proteins would bind to a nuclear transcription factor, such as LANA. However, ribosomal proteins are not exclusively cytoplasmic: some such as RPS6 shuttle in and out of the nucleus and accumulate in nuclear and nucleolar bodies (76). We focused our initial efforts on validating the interaction between LANA and RPS6 because of its known nuclear localization, because of the availability of highly specific reagents, and because the distribution of MS/MS peptide coverage made it unlikely that RPS6 was incorrectly identified (Fig. 2).
Fig. 2.

Analysis of the interaction between LANA and RPS6 in BC-3 cells. (A) Lanes 1 and 2, nuclear extracts (NE) of BC-3 cells; lanes 3 to 6, samples purified by heparin FF column. Treatments for each lane are as follows: lane 1, 37°C for 30 min and treatment with 500 U/ml DNase I and100 μg/ml RNase A; lane 2, 37°C for 30 min without nuclease treatment; lane 3, 25°C for 20 min without nuclease treatment; lane 4, 35°C for 20 min with DNase I and RNase; lane 5, 37°C for 20 min with 5 U/μl DNase I and 10 U/μl RNase; lane 6, 4°C for 30 min without nuclease treatment. All of the above buffers contain no EDTA. For the immunoprecipitation (IP) assay, lanes 1, 3, 4, and 5 were supplemented with anti-S6 antibodies and lanes 2 and 6 were used as negative controls (IgG only). (B) Immunoprecipitation and Western blot of an irrelevant ribosomal protein L11 from nuclear extract of BC-3 cells. Shown is input and immunoprecipitation with a control antibody (IgG), with anti-LANA mouse monoclonal antibody (IP LANA), or anti-RPL11 antibody (IP RPL11) followed by Western blotting with anti-LANA antibody (left) or anti-RPL11 antibody (right). (C) RPS6 protein and the sequences of its corresponding peptides identified by mass spectrometry analysis, shown in a table of peptides. (D) Alignment of peptides (underlined) with the S6 sequence (GenBank accession no. NP_001001; gene identification [ID] no. 6194 RPS6).
LANA interacts with RPS6.
To validate the LANA-RPS6 interaction, we used nuclear extract of BC-3 cells and conducted the reciprocal immunoprecipitation using an anti-RPS6 antibody followed by Western blotting with anti-LANA antibody. LANA was associated with RPS6 in the nuclear extract prior to any purification steps (Fig. 2A, lanes 1 and 2).
Since ribosomal proteins like RPS6 are associated with rRNA (19), and since LANA may have nonspecific, as well as sequence-specific nucleic acid-binding capabilities, we examined the hypothesis that the LANA-RPS6 interaction was dependent on copurifying nucleic acids. We used RNase A and DNase I to digest nucleic acids in the partially purified LANA-positive heparin fractions prior to immunoprecipitation with anti-RPS6 antibody. Despite nuclease digestion, LANA still interacted with RPS6 in this assay (Fig. 2A, lanes 3 to 6).
To further investigate the specificity of the LANA-RPS6 interaction, we conducted immunoprecipitation with anti-RPL11 antibody (i.e., a random ribosomal protein that was not found in our MS/MS profiling) (Fig. 2B). We did not observe any interaction between LANA and RPL11, which supports the notion that the LANA-RPS6 interaction was specific and not the result of whole ribosomes binding to LANA multimers or the nonspecific anti-LANA antibody.
Next, we used the FLAG-tagged fusion protein of LANA to detect the interaction with a HA-tagged protein of RPS6 in cotransfected cells. This experiment confirms specificity and also demonstrates that the LANA-RPS6 interaction is independent of other KSHV proteins. First we generated a full-length HA-tagged version of RPS6 and cotransfected it with a FLAG-tagged version of full-length LANA. Using a FLAG-tagged LANA, rather than anti-LANA antibody, avoids the potential for nonspecific activity of the anti-LANA monoclonal antibody LN53. Using tag-specific antibodies for immunoprecipitation and Western blotting, we found that LANA coimmunoprecipitated with RPS6 and that RPS6 coimmunoprecipitated with LANA after both proteins were transiently transfected into HeLa cells (Fig. 3 A). This demonstrates that LANA and RPS6 interact in the absence of other KSHV viral proteins.
Fig. 3.
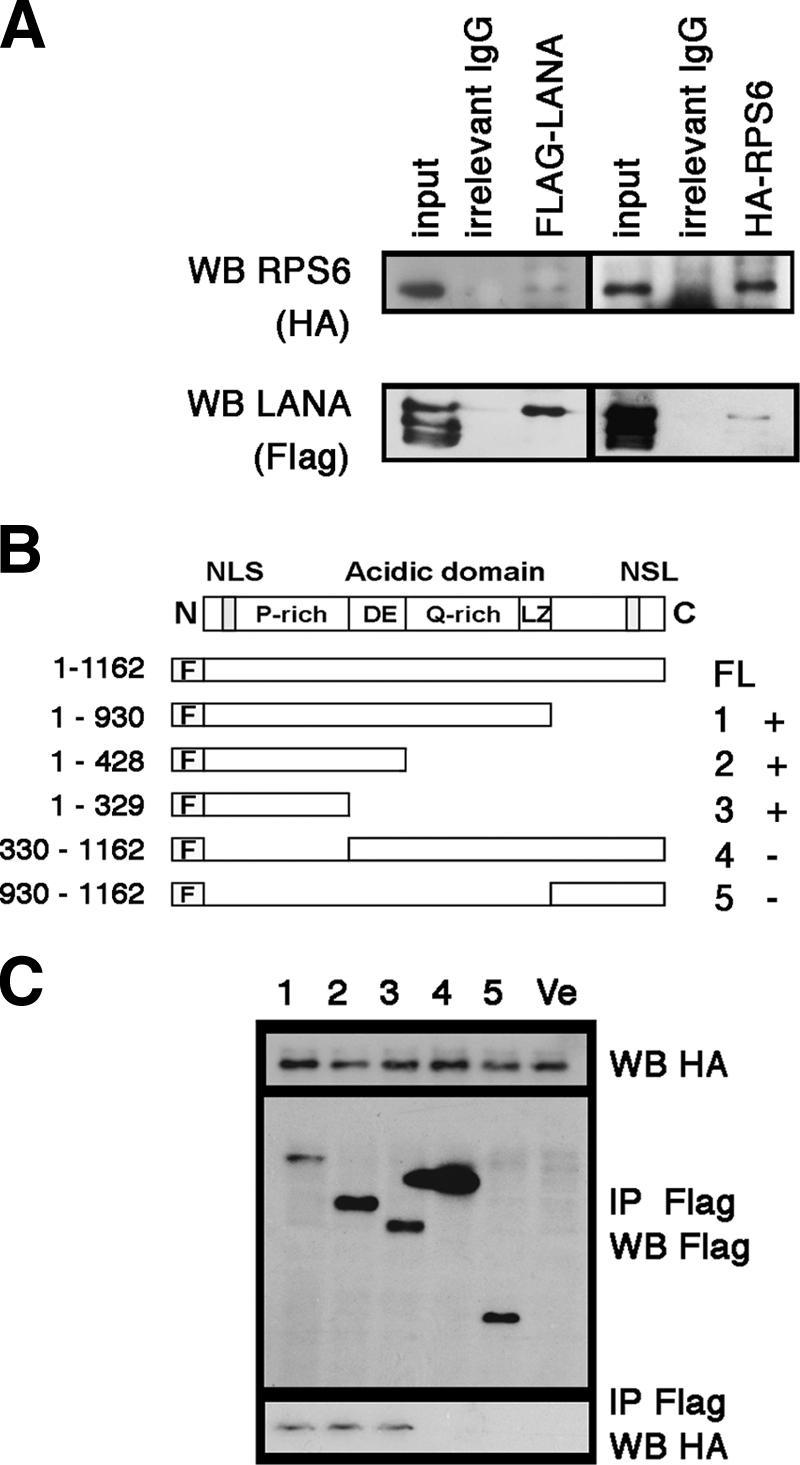
Analysis of the interaction of LANA and RPS6 in HeLa cells after transfection. (A) Coimmunoprecipitation of full-length FLAG-tagged LANA and HA-tagged S6 after cotransfection into HeLa cells. After cotransfection, protein extract was immunoprecipitated with the indicated reagents. The lanes represent input, IgG only, anti-FLAG, and anti-HA (HA). Anti-FLAG antibodies and anti-HA antibodies were used for Western blot (WB) analysis, as indicated on the left. (B) Schematic diagram of mutant LANA proteins used. All segments were fused with FLAG tag at the N terminus of LANA. The numbers on the right indicate the lane numbers in panel C and the “+” or “−” symbols summarize whether or not an interaction was detected. (C) Coimmunoprecipitation assay of HA-RPS6 and different mutants of FLAG-tagged LANA. Anti-FLAG and anti-HA antibodies were used for immunoprecipitation (IP) or Western blotting (WB) as indicated. Ve, vector.
To determine which domain of LANA interacts with RPS6, a series of LANA mutants were constructed with a FLAG tag fused to the N terminus of LANA. We also constructed RPS6 with an HA tag fused to the N terminus of RPS6 (Fig. 3B). Cotransfection of HA-tagged RPS6 and different FLAG-tagged LANA mutants into HeLa cells indicated that the N-terminal portion of LANA (aa 1 to 329), excluding the DE repeats, represents the shortest fragment that could still interact with RPS6 (Fig. 3B). Neither the C terminus nor the repeat-rich central domain of LANA interacted with RPS6.
In sum, we have validated the LANA-RPS6 interaction by multiple independent means: (i) immunoprecipitation with anti-LANA monoclonal antibody coprecipitates RPS6 at physiological levels in BC-3 cells, (ii) immunoprecipitation with anti-RPS6 antibody coprecipitates LANA at physiological levels in BC-3 cells, (iii) cotransfection and coimmunoprecipitation with different artificial epitopes (tags) show a complex of both proteins in the absence of other viral proteins, and (iv) the LANA N terminus (but not the central region or C terminus) is sufficient to bind to RPS6.
RPS6 associated with LANA on LANAp.
We had shown earlier that LANA could bind to a LANA-responsive DNA element within its promoter and thereby autoactivate the LANA promoter (LANAp) (30). We had found that the binding interaction between the LANA protein and this single DNA element is considerably weaker than the interaction between LANA and the multimerized, canonical LANA binding sites in the viral terminal repeats (TRs). Thus, chromatin immunoprecipitation analysis (ChIP) analysis of the LANA promoter represents a more stringent assay for the purpose of demonstrating a trimeric LANA-RBS6-DNA complex than the TR. The LANA promoter also provides for a better PCR target for ChIP, whereas we were unable to PCR across the LANA binding sites within the GC-rich terminal repeat regions (data not shown). Using ChiP, we asked whether RPS6 was present in the LANA-LANA promoter complex in vivo. After cross-linking with 1% formaldehyde, the sonicated samples were immunoprecipitated with anti-LANA or anti-RPS6 monoclonal antibodies, respectively, and we PCR amplified the coprecipitated DNA (Fig. 4 A, upper panel). We could amplify a band of the expected molecular weight for the specific fragment of the LANA promoter from immune precipitates using anti-LANA as well as anti-RPS6 antibodies, but not the IgG-negative control. We could not PCR amplify any product using primers specific for β-actin, which served as a negative control for nonspecific DNA binding (Fig. 5 A, lower panel). We conclude, by another independent assay, that RPS6 is in a complex with KSHV LANA and that at least some of the RPS6-LANA complexes are present at the LANA promoter.
Fig. 4.

Complex of LANA and S6 binds to LANA promoter. (A) BC-3 cells were cross-linked and sonicated, and chromatin immunoprecipitation (IP) was performed with mouse anti-LANA (LN53) and mouse anti-RPS6 (54D2) monoclonal antibodies. IgG was used as a control. Inputs were 5% of lysates of each sample. The DNA fragments immunoprecipitated were amplified with the respective primers for the LANA promoter (LANAp) and β-actin. (B) Cotransfection of LANA and RPS6 into SLK cells increases the activity of a LANA promoter-luciferase reporter. Relative light units (RLU) are shown on the y axis, and the amount of S6 plasmid per transfection mixture is shown on the horizontal axis. All measurements were conducted in triplicate, and each is shown. The background (pGL3 vector) activity was below 100 RLU. Also shown are the regression lines (black), which demonstrate a linear dose dependence of LANA promoter activity on RPS6. (C) Western blot analysis of SLK cells cotransfected with increasing amounts of HA-tagged RPS6 expression vector in the absence or presence of LANA. A Western blot for actin is shown as a loading control.
Fig. 5.

Analysis after knockout of RPS6. (A) BC-3 cells were transduced with five recombinant lentiviruses (S6 Sh-1 to Sh-5) encoding different shRNAs directed against RPS6 in six-well plates. shRNAs against GFP-encoding lentivirus and untreated BC-3 cells were used as controls. Western blot analysis was performed 4 days postransduction using anti-RPS6 (S6) and anti-β-actin (Actin) antibodies. (B) Western blots were performed 3 days after knockdown of RPS6 with shRNA no. 4, probing with anti-LANA, anti-p53, anti-p21, and anti-β-actin antibodies, respectively, at 3 days postransduction. (C) Proliferation of BC-3 cells was measured at different time points following shRNA-mediated RPS6, as described above. The number of cells is shown on the y axis, and the time after transduction is shown on the x axis. The cells were not refed in between time points. (D) Protein stability analysis of LANA. BC-3 cells were transduced with anti-RPS6 (S6 Sh-4) shRNA vector or control shRNA vector (GFP Sh) and exposed to cycloheximide (CHX) starting at day 3 postransduction, and LANA and actin protein levels were determined by Western blot analysis at the indicated times (in hours). (E) Protein stability analysis of p53. BC-3 cells were transduced with anti-RPS6 (S6 Sh-4) shRNA vector or control shRNA vector (GFP Sh) and exposed to cycloheximide and LANA, and actin protein levels were determined by Western blot analysis at the indicated times (in minutes). (F) Protein stability determination for LANA and p53 (G). The protein bands in panels D and E were scanned, and band intensities were calculated. The log10 band intensity of the fraction at time zero is shown on the y axis, and time is shown on the x axis. Lines represent the robust fit of the data.
By no means do we want to imply that RPS6 is a transcription factor. Rather, we use the phenotype of LANA to bind its cognate DNA as a biochemical assay to verify the RPS6-LANA interaction. As an alternative readout for the LANAp-LANA-RPS6 interaction, we modified our established transient transfection assay, using a LANA promoter-luciferase construct as the reporter. This represents a variant of a mammalian two-hybrid assay using the LANA DNA binding domain and cognate cis element. Instead of fusing RPS6 to a strong artificial transactivation domain, we used a statistical measure (dose dependence under limiting conditions) to validate the LANA-RPS6 interaction. The LANA promoter is constitutively active. Cells were cotransfected with the reporter and increasing amounts of RPS6. We used empty vector to adjust for the total DNA concentration. The background (pGL3 vector) activity was below 100 RLU. In the absence of LANA, RPS6 did not affect promoter activity (Fig. 4B, left panel). This would be expected since RPS6 is not a transcription factor. Transient transfection of LANA itself transactivates this LANA promoter report construct, albeit weakly, and we used a suboptimal amount of LANA in order to measure the effect of RPS6. In the presence of cotransfected LANA, RPS6 increased promoter activity (Fig. 4B, right panel). We observed a linear dose-response curve. Albeit not dramatic, the slope was significant at P ≤ 0.027 using robust linear regression (64). We interpret the linear dose dependence rather than the overall fold change as evidence for the interaction between LANA and RPS6. Clearly RPS6 is not a transcription factor. If we increase the amount of transfected LANA, the RPS6 becomes negligible since LANA protein is no longer rate limiting within the cell.
One possible outcome was that RPS6 would alter overall LANA protein levels. To test this hypothesis, the experiment was repeated and LANA and RPS6 levels were determined by Western blot (Fig. 4C). We observed the expected dose-dependent increase of RPS6 protein levels with increased amounts of RPS6 expression vector. In contrast, the level of LANA did not change appreciably. Using LANA's DNA binding ability as a novel, independent assay, Fig. 5 verifies that LANA binds to RPS6.
Reduction of RPS6 protein reduces LANA protein expression.
Since RPS6 was in a complex with LANA, we hypothesized that RPS6 positively supports the translation of LANA (and perhaps contributes to the extraordinary stability of LANA). To test this hypothesis under physiological conditions, BC-3 cells were infected with a series of recombinant shRNA lentiviruses that target the RPS6 mRNA. One vector (Sh-4) resulted in marked reduction of RPS6 levels at day 4 after infection (Fig. 5A) and was selected for further study. Transduction of BC-3 cells with this validated shRNA lentiviral vector against RPS6, but not an irrelevant (GFP) shRNA lentiviral vector led to a reduction of LANA protein steady-state levels as early as day 3 post-shRNA lentiviral vector transduction (Fig. 5B). Levels of an unrelated, equally stable protein (β-actin) were not affected (Fig. 5B). Transduction of BC-3 cells with this validated shRNA lentiviral vector against RPS6, but not an irrelevant (GFP) shRNA lentiviral vector, induced levels of p21 and p53 (Fig. 5B). We used p53 as a positive control to investigate the hypothesis that ablation of RPS6 does not abrogate protein translation in general but results in a specific cellular signaling event. Disturbing ribosome assembly by ablating RPS6 expression is known to activate p53 (22), which in turn induces transcription and translation of p21. This piece of data is consistent with p53 activation, not nonspecific translation inhibition.
Inhibition of RPS6 expression by shRNA inhibited cell proliferation compared to control shRNA lentiviral transduction, as shown in Fig. 5C. BC-3 cells were transduced on day 0 and then observed over time without refeeding. These experiments were conducted at high cell density so as to maximize the viability of the cells after shRNA transduction. In this experiment, we did add fresh medium after transduction, which would activate the mTOR pathway and may have changed the steady-state levels and function of RPS6. At day 1, there was no statistical difference between RPS6-specific shRNA-treated and control shRNA-treated cells. On subsequent days, RPS6-specific shRNA-infected cells exhibited cell death, whereas control shRNA-treated cells proliferated and remained viable. In sum, we have shown that RPS6 contributes to LANA protein levels, but not to actin protein levels, and that this results in the induction of p53 protein levels. Hence, it is unlikely that our knockdown of RPS6 resulted in widespread, indiscriminate translational arrest of protein synthesis. Rather, the impact of reduced RPS6 levels is most significant upon proteins that are associated with or deeply dependent upon the presence of RPS6.
Next, we tested the hypothesis that RPS6 affects the half-life of LANA. BC-3 cells were transduced with shRNA to RPS6 (S6 Sh-4) or an irrelevant shRNA lentiviral vector (shGFP) and, starting at day 3 postransduction, treated with cycloheximide to block novel protein synthesis. After the indicated times, we probed for LANA levels using Western blotting; actin was used as a control. In BC-3 cells transduced with the RPS6 shRNA vector, LANA showed a substantially decreased half-life (0.60 h) compared to BC-3 cells treated with control shRNA (28 h) (Fig. 5D and G). This complex requires the N terminus, since the LANA C terminus alone has a short half-life—on the order of hours (data not shown). This result is consistent with a model in which RPS6 is part of a complex that is responsible for the well-established, unusually long half-life (on the order of days) of the LANA protein. At the same time p53 was stabilized (Fig. 5E and G). In BC-3 cells transduced with the RPS6 shRNA vector, p53 exhibited a slighly increased half-life (8.8 h) compared to BC-3 cells treated with control shRNA (2.3 h). This is consistent with the activation of p53 and increased steady-state levels and the induction of p21 (Fig. 5B). In sum, these experiments demonstrate that RPS6 contributes to the stability and overall level of LANA in PEL cells.
DISCUSSION
LANA is a multifunctional protein, as is RPS6. Here, we find that both proteins exist in a complex together in KSHV latently infected PEL cells. Both LANA and RPS6 bind to each other in cotransfection experiments in the absence of other viral proteins. Our data reveal that the LANA N-terminal domain mediates this interaction and that this interaction is resistant to nuclease digestion and therefore independent of rRNA. Furthermore, we found that RPS6 is present in RPS6-LANA-DNA complexes on the LANA promoter and that shRNA-mediated knockdown of RPS6 is associated with a reduction in LANA protein levels and in LANA protein half-life. This implicates RPS6 in the translation and/or stability of KSHV LANA.
LANA is an extraordinarily stable protein with an estimated half-life of several days (39). The biochemical mechanism for its stability is unknown. Therefore, it is conceivable that RPS6 can contribute to LANA stability. RPS6 is a very dynamic protein. It is synthesized in the cytoplasm and then imported back into the nucleolus, where it assembles into the pre-40S ribosomal subunit. These complexes are then extruded into the nucleus proper and then into the cytoplasm. The nuclear import of RPS6 is mediated by three NLSs, and the nuclear export of RPS6 is dependent on a nuclear export signal (NES) and CRM1 (38, 44, 49, 68). CRM1-dependent nuclear export can be specifically inhibited by the pharmacological compound leptomycin B (LMB). LMB interacts with CRM1 and blocks leucine-rich, NES-mediated protein export (15, 37, 52). Similarly to normal cells, export of human RPS6 was controlled by the CRM1-mediated export pathway in PEL cells and in the KSHV-infected TIVE L1 endothelial cells (data not shown). This verifies that RPS6 is localized to the nucleus as part of its normal maturation pathway. RPS6 is phosphorylated by p70S6 kinase, which in turn is mTOR dependent. However, RPS6 can also be phosphorylated by other kinases such as p90S6 kinase.
Surprisingly, RPS6 phosphorylation does not affect overall translation efficiency or the rate of RPS6 incorporation into polyribosomes (66). Chemical inhibition of RPS6 phosphorylation (e.g., by mTOR inhibitors) does not result in widespread, indiscriminate translational arrest or cell death. Whether RPS6 phosphorylation plays a role in ribosome selectivity for specific mRNAs (27) is subject to further study.
We predict that the significance of the LANA-RPS6 and LANA-ribosomal protein interactions will reveal itself in due time, analogous to the Hdm2-ribosome interactions. The first demonstration that Hdm2 bound a ribosomal protein was published in 1994 (47). Subsequently, it was shown that Hdm2 interacts with L11 and/or L23 (10, 82), establishing a tight linkage between Hdm2, p53, and ribosome biogenesis. Recently, it has become well established that Hdm2 and p53 are sensors for ribosomal stress and ribosomal function (22). As a fraction of LANA is associated with both p53 and Hdm2 (9, 67), this provides a mechanism for KSHV to manipulate cellular ribosomal biogenesis and the stress signals that may originate from it for the purpose of latent viral persistence.
What could be the function of the LANA-RPS6 interaction, and what is its significance? We noticed that phosphorylated RPS6 in the nucleus of KSHV-infected cells was shielded from the effect of rapamycin in L1 TIVE cells (unpublished observation). Rapamycin treatment effectively inhibits phosphorylation of RPS6 by p70S6K1 as well as p70S6K1 (42, 55, 69); however, phosphorylation by p90S6K is not inhibited. Furthermore, p70S6K2 is cytoplasmic as well as nuclear and can even be centrosome associated in mammalian cells (63). It is thus conceivable that LANA may promote the phosphorylation of RPS6 by centromeric p70S6K2, which in turn increases the nuclear functionality of RPS6, perhaps during mitosis. It is during mitosis, when the nuclear envelope breaks down, that LANA decorates host chromosomes and that its function is most crucial for viral episome segregation. The discovery of the mTOR/S6K/S6 signaling arm taught us that the ribosomal proteins are not mere building blocks of ribosomes, but may have multiple functions in themselves and may be independent of steady-state protein translation.
ACKNOWLEDGMENTS
This work is supported by funding from the NIH (CA109232).
We thank members of the D. P. Dittmer and B. A. Damania labs for helpful discussions and Pauline Chugh and Debasmita Roy for critical reading of the manuscript, the UNC MassSpec core for expert analysis, and the Lenti-shRNA Core Facility for production of shRNA vectors.
Footnotes
Published ahead of print on 6 July 2011.
REFERENCES
- 1. Ballestas M. E., Chatis P. A., Kaye K. M. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644 [DOI] [PubMed] [Google Scholar]
- 2. Ballestas M. E., Kaye K. M. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barbera A. J., et al. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861 [DOI] [PubMed] [Google Scholar]
- 4. Blenis J., Spivack J. G., Erikson R. L. 1984. Phorbol ester, serum, and Rous sarcoma virus transforming gene product induce similar phosphorylations of ribosomal protein S6. Proc. Natl. Acad. Sci. U. S. A. 81:6408–6412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai Q. L., Knight J. S., Verma S. C., Zald P., Robertson E. S. 2006. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cesarman E., Chang Y., Moore P. S., Said J. W., Knowles D. M. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 7. Chandriani S., Ganem D. 2010. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 84:5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang Y., et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 9. Chen W., Hilton I. B., Staudt M. R., Burd C. E., Dittmer D. P. 2010. Distinct p53, p53:LANA, and LANA complexes in Kaposi's sarcoma-associated herpesvirus lymphomas. J. Virol. 84:3898–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai M. S., et al. 2004. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 24:7654–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dignam J. D., Lebovitz R. M., Roeder R. G. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dittmer D. P. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 [PubMed] [Google Scholar]
- 13. Dupin N., et al. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fakhari F. D., Dittmer D. P. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fornerod M., Ohno M., Yoshida M., Mattaj I. W. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060 [DOI] [PubMed] [Google Scholar]
- 16. Forsman A., Ruetschi U., Ekholm J., Rymo L. 2008. Identification of intracellular proteins associated with the EBV-encoded nuclear antigen 5 using an efficient TAP procedure and FT-ICR mass spectrometry. J. Proteome Res. 7:2309–2319 [DOI] [PubMed] [Google Scholar]
- 17. Franco R., Rosenfeld M. G. 1990. Hormonally inducible phosphorylation of a nuclear pool of ribosomal protein S6. J. Biol. Chem. 265:4321–4325 [PubMed] [Google Scholar]
- 18. Friborg J., Jr., Kong W., Hottiger M. O., Nabel G. J. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894 [DOI] [PubMed] [Google Scholar]
- 19. Fromont-Racine M., Senger B., Saveanu C., Fasiolo F. 2003. Ribosome assembly in eukaryotes. Gene 313:17–42 [DOI] [PubMed] [Google Scholar]
- 20. Fujimuro M., Hayward S. D. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 77:8019–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujimuro M., et al. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300–306 [DOI] [PubMed] [Google Scholar]
- 22. Fumagalli S., et al. 2009. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 11:501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han I., et al. 2001. EBNA-LP associates with cellular proteins including DNA-PK and HA95. J. Virol. 75:2475–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu J., Garber A. C., Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu J., Renne R. 2005. Characterization of the minimal replicator of Kaposi's sarcoma-associated herpesvirus latent origin. J. Virol. 79:2637–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwenofu O. H., et al. 2008. Phospho-S6 ribosomal protein: a potential new predictive sarcoma marker for targeted mTOR therapy. Mod. Pathol. 21:231–237 [DOI] [PubMed] [Google Scholar]
- 27. Jefferies H. B., Reinhard C., Kozma S. C., Thomas G. 1994. Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc. Natl. Acad. Sci. U. S. A. 91:4441–4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jenner R. G., Alba M. M., Boshoff C., Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeong J., Papin J., Dittmer D. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 75:1798–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeong J. H., et al. 2004. Regulation and autoregulation of the promoter for the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 279:16822–16831 [DOI] [PubMed] [Google Scholar]
- 31. Kaul R., Verma S. C., Robertson E. S. 2007. Protein complexes associated with the Kaposi's sarcoma-associated herpesvirus-encoded LANA. Virology 364:317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kitay M. K., Rowe D. T. 1996. Protein-protein interactions between Epstein-Barr virus nuclear antigen-LP and cellular gene products: binding of 70-kilodalton heat shock proteins. Virology 220:91–99 [DOI] [PubMed] [Google Scholar]
- 33. Knight J. S., Cotter M. A., II, Robertson E. S. 2001. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus transactivates the telomerase reverse transcriptase promoter. J. Biol. Chem. 276:22971–22978 [DOI] [PubMed] [Google Scholar]
- 34. Krieg J., Hofsteenge J., Thomas G. 1988. Identification of the 40 S ribosomal protein S6 phosphorylation sites induced by cycloheximide. J. Biol. Chem. 263:11473–11477 [PubMed] [Google Scholar]
- 35. Krithivas A., Fujimuro M., Weidner M., Young D. B., Hayward S. D. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krithivas A., Young D. B., Liao G., Greene D., Hayward S. D. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kudo N., et al. 1999. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. U. S. A. 96:9112–9117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kundu-Michalik S., et al. 2008. Nucleolar binding sequences of the ribosomal protein S6e family reside in evolutionary highly conserved peptide clusters. Mol. Biol. Evol. 25:580–590 [DOI] [PubMed] [Google Scholar]
- 39. Kwun H. J., et al. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81:8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lan K., Kuppers D. A., Robertson E. S. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lan K., et al. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee-Fruman K. K., Kuo C. J., Lippincott J., Terada N., Blenis J. 1999. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 18:5108–5114 [DOI] [PubMed] [Google Scholar]
- 43. Lim C., Gwack Y., Hwang S., Kim S., Choe J. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 276:31016–31022 [DOI] [PubMed] [Google Scholar]
- 44. Lipsius E., et al. 2005. Evolutionary conservation of nuclear and nucleolar targeting sequences in yeast ribosomal protein S6A. Biochem. Biophys. Res. Commun. 333:1353–1360 [DOI] [PubMed] [Google Scholar]
- 45. Lu F., Day L., Gao S. J., Lieberman P. M. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mannick J. B., Tong X., Hemnes A., Kieff E. 1995. The Epstein-Barr virus nuclear antigen leader protein associates with hsp72/hsc73. J. Virol. 69:8169–8172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marechal V., Elenbaas B., Piette J., Nicolas J. C., Levine A. J. 1994. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell. Biol. 14:7414–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mattsson K., et al. 2002. Latent nuclear antigen of Kaposi's sarcoma herpesvirus/human herpesvirus-8 induces and relocates RING3 to nuclear heterochromatin regions. J. Gen. Virol. 83:179–188 [DOI] [PubMed] [Google Scholar]
- 49. Meyuhas O. 2008. Physiological roles of ribosomal protein S6: one of its kind. Int. Rev. Cell Mol. Biol. 268:1–37 [DOI] [PubMed] [Google Scholar]
- 50. Montgomery S. A., Berglund P., Beard C. W., Johnston R. E. 2006. Ribosomal protein S6 associates with alphavirus nonstructural protein 2 and mediates expression from alphavirus messages. J. Virol. 80:7729–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Munoz-Fontela C., et al. 2008. Induction of paclitaxel resistance by the Kaposi's sarcoma-associated herpesvirus latent protein LANA2. J. Virol. 82:1518–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ossareh-Nazari B., Bachelerie F., Dargemont C. 1997. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 278:141–144 [DOI] [PubMed] [Google Scholar]
- 53. Ottinger M., et al. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80:10772–10786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pan H., Zhou F., Gao S. J. 2004. Kaposi's sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 64:4064–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park I. H., Bachmann R., Shirazi H., Chen J. 2002. Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J. Biol. Chem. 277:31423–31429 [DOI] [PubMed] [Google Scholar]
- 56. Paulose-Murphy M., et al. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843–4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pende M., et al. 2004. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 24:3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Petre C. E., Sin S. H., Dittmer D. P. 2007. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J. Virol. 81:1912–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pian J. P., et al. 2004. A 32 kDa protein—whose phosphorylation correlates with oncogenic Ras-induced cell cycle arrest in activated Xenopus egg extracts—is identified as ribosomal protein S6. J. Cell. Physiol. 201:305–319 [DOI] [PubMed] [Google Scholar]
- 60. Platt G. M., Simpson G. R., Mittnacht S., Schulz T. F. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Radkov S. A., Kellam P., Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121–1127 [DOI] [PubMed] [Google Scholar]
- 62. Renne R., et al. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:458–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rossi R., et al. 2007. Identification of S6K2 as a centrosome-located kinase. FEBS Lett. 581:4058–4064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rousseeuw P., Lerou A. 2003. Robust regression and outlier detection. Wiley & Sons, Hoboken, NJ [Google Scholar]
- 65. Ruggero D., Pandolfi P. P. 2003. Does the ribosome translate cancer? Nat. Rev. Cancer 3:179–192 [DOI] [PubMed] [Google Scholar]
- 66. Ruvinsky I., et al. 2005. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 19:2199–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sarek G., et al. 2007. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J. Clin. Invest. 117:1019–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schmidt C., Lipsius E., Kruppa J. 1995. Nuclear and nucleolar targeting of human ribosomal protein S6. Mol. Biol. Cell 6:1875–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shima H., et al. 1998. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 17:6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Si H., Robertson E. S. 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J. Virol. 80:697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Si H., Verma S. C., Robertson E. S. 2006. Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J. Virol. 80:9017–9030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sin S. H., et al. 2007. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 109:2165–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Soulier J., et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 74. Staskus K. A., et al. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J. Virol. 73:4181–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stedman W., Deng Z., Lu F., Lieberman P. M. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566–12575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thomas F., Kutay U. 2003. Biogenesis and nuclear export of ribosomal subunits in higher eukaryotes depend on the CRM1 export pathway. J. Cell Sci. 116:2409–2419 [DOI] [PubMed] [Google Scholar]
- 77. Verma S. C., et al. 2006. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus recruits uracil DNA glycosylase 2 at the terminal repeats and is important for latent persistence of the virus. J. Virol. 80:11178–11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Verma S. C., Choudhuri T., Kaul R., Robertson E. S. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80:2243–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Viejo-Borbolla A., et al. 2005. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 79:13618–13629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wong L. Y., Matchett G. A., Wilson A. C. 2004. Transcriptional activation by the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J. Virol. 78:10074–10085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Young P., Anderton E., Paschos K., White R., Allday M. J. 2008. Epstein-Barr virus nuclear antigen (EBNA) 3A induces the expression of and interacts with a subset of chaperones and co-chaperones. J. Gen. Virol. 89:866–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Y., et al. 2003. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 23:8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhong W., Wang H., Herndier B., Ganem D. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. U. S. A. 93:6641–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]