

Abstract
Over-replication of two clusters of chorion genes in Drosophila ovarian follicle cells is essential for rapid eggshell biosynthesis. The relationship of this amplification to the follicle cell cycles has remained unclear. To investigate the regulation of amplification, we developed a technique to detect amplifying chorion genes in individual follicle cells using BrdU incorporation and FISH. Amplification occurs in two developmental phases. One of the gene clusters begins to amplify periodically during S phases of follicle cell endocycles. Subsequently, after endocycles have ceased, both clusters amplify continuously during the remainder of oogenesis. In contrast to the early phase, late amplification commences synchronously among follicle cells. The pattern of Cyclin E expression mirrors these two phases. We present evidence that Cyclin E is required positively for amplification. We suggest that Cyclin E also acts negatively to inhibit refiring of most origins within a cycle, and that specific factors at chorion origins allow them to escape this negative rereplication control. Our findings suggest that chorion amplification is a model for understanding metazoan replicons and the controls that restrict replication to once per cell cycle.
Keywords: Drosophila, oogenesis, chorion, amplification, replication, cyclin E
To maintain euploid gene balance, DNA sequences must be replicated every cell cycle but not more than once. Recent evidence indicates that cell cycle control of DNA replication is effected by a two-step mechanism (for review, see Diffley 1996). Origins first become competent to replicate by assembling proteins comprising prereplication complexes onto chromatin in G1, and then later, during S, those origins initiate replication (Diffley et al. 1994). Replication from or through an origin dissociates functional prereplication complexes. Once destroyed, these complexes cannot reassemble until the subsequent G1, thereby precluding refiring of an origin in a single cycle. Several lines of evidence suggest that cyclin dependent kinases (CDKs), in addition to being required positively for cell cycle progression, act negatively and are responsible for blocking reassembly of replication complexes in S, G2, and M (Broek et al. 1991; Hayles et al. 1994; Moreno and Nurse 1994; Dahmann et al. 1995; Sauer 1995; Hua et al. 1997; Jallepalli et al. 1997; Tanaka et al. 1997). It is only after passage through mitosis, during a period in G1 when kinase levels are low, that complexes can reassemble onto chromatin. This two-step mechanism of assembly and firing linked to kinase levels ensures that each region of the genome is replicated only once per cycle.
In Drosophila melanogaster, as in many multicellular eukaryotes including humans, certain tissues become polyploid by entering an endocycle characterized by alternating S and G phases without intervening mitoses (for review, see Carminati and Orr-Weaver 1996). As in other cycles, Cyclin E (CycE), with its kinase partner CDK2, is required for S phase of the endocycle (Sauer et al. 1995; Lilly and Spradling 1996) but the mitotic Cyclins A and B are absent (Lehner et al. 1990; Knoblich et al. 1994; for review, see Follete and O’Farrell 1997). Although some heterochromatic sequences fail to replicate and become under-represented during endocycles (Gall et al. 1971; Hammond and Laird 1985; Lilly and Spradling 1996), euchromatic sequences replicate no more than once with each successive endocycle (Spierer and Spierer 1984; Spradling and Orr-Weaver 1987). Thus, although apparently lacking an incomplete replication checkpoint, polyploid cells appear to have a mechanism that restricts DNA replication to once per endocycle.
A striking exception to balanced replication of euchromatin is amplification of the chorion genes in follicle cells during Drosophila oogenesis (for review, see Spradling 1993). During the last hours of oogenesis, these polyploid cells rapidly synthesize and secrete high levels of chorion proteins that comprise the Drosophila eggshell (Spradling and Mahowald 1980; for review, see Orr-Weaver 1991). Two clusters of chorion genes on the X and third chromosomes (hereafter X chorion and third chorion) are amplified above the copy number of the remainder of the follicle cell genome before and during a time of high-level transcription. This increase occurs through repeated initiation of replication forks from these loci. Because successively initiated replication forks continue to move outward, by the end of oogenesis each gene cluster lies at the peak of a gradient of copy number that extends ∼40 kb in both directions. The final amplification level for the third chorion genes is 60- to 80-fold, and the X genes 15- to 20-fold, above the remainder of the genome. Several partially redundant cis sequences that mediate high-level amplification have been identified interspersed among the chorion transcription units (Orr-Weaver and Spradling 1986; Delidakis and Kafatos 1987; Spradling et al. 1987; Delidakis and Kafatos 1989; Heck and Spradling 1990), suggesting amplification is an amenable model for investigation into the little understood nature of metazoan origins of DNA replication.
It has remained unclear, however, whether chorion gene amplification is a model for the cell cycle regulation of DNA replication. It is possible that the mechanism for amplification of chorion genes differs from the cell cycle regulation that governs normal S phase. Further, it is unknown whether amplification occurs during follicle cell endocycles. The recent finding that amplification requires the Drosophila homolog of ORC2 (Landis et al. 1997), an essential component of prereplication complexes in yeast and other organisms, encourages the view that amplification resembles cell cycle-regulated DNA replication.
Here, we find that chorion gene amplification is closely tied to follicle cell cycles. We propose that chorion origins locally escape from normal rereplication controls that continue to operate on the rest of the genome resulting in selective amplification.
Results
Cessation of follicle cell endocycles
To understand the cell cycle regulation of chorion gene amplification it was first necessary to re-examine follicle cell endocycles. Follicle cells originate from stem cells, mitotically proliferate up until stage 7 of oogenesis, and subsequently undergo several rounds of endoreplication (see Fig. 1; for review, see Spradling 1993). Most amplification takes place during the final hours of oogenesis, a time when endocycles have ceased. Because of the uncertainty in time of onset for amplification (Orr et al. 1984; Spradling and Leys 1988), and the number and timing of endocycles (Mahowald et al. 1979; Hammond and Laird 1985; Mulligan and Rasch 1985), however, it could not be determined if these two processes overlap (see Fig. 1B).
Figure 1.

Three phases of follicle cell development during oogenesis. (A) ∼650 follicle cells form an epithelial layer that surrounds each maturing Drosophila egg chamber; in late stages most surround the oocyte, although a few follicle cells sheath the 15 nurse cells. Within follicle cells over the oocyte, two clusters of chorion genes on the X and third chromosomes amplify 15- and 60-fold, respectively. (B) Schematic representation of an ovariole is shown. Egg chambers originate in the germarium and migrate posteriorly down the ovariole as they develop. Follicle cells originate from two somatic stem cells in the germarium and complete approximately eight mitotic divisions over a 6-day period to achieve their final number during stage 6 (S6). Thereafter, follicle cells undergo three endocycles (see C); the exact stage at which endocycles are complete is uncertain (broken vertical line). Follicle cells then enter a postendocycle stage for the remainder of oogenesis. Amplification is known to occur during this phase (bottom solid line), but whether it initiates during the endocycles is not known (broken line). (C) FACS profile of follicle cell nuclear DNA content, showing 4C, 8C, and 16C classes produced by three endocycles. Egg chamber drawings modified from King (1970).
Flow sorting of follicle cell nuclei clearly shows that follicle cells undergo three endocycles (Fig. 1C; Lilly and Spradling 1996). To investigate the timing of these endocycles, dissected ovaries were incubated in BrdU, and labeled nuclei examined by immunofluorescent microscopy. Throughout stages 7–9, developing egg chambers contained both labeled, S-phase, and unlabeled, G-phase, follicle cells. This indicates that the timing of endocycles is not synchronized among follicle cells within an egg chamber. The fraction of BrdU-positive cells after a 1 hr labeling period averaged 30% up until mid-stage 9 (Fig. 2A–C). After mid-stage 9, however, the number of BrdU-positive follicle cells steadily decreased, indicating that the last endoreplication is completed asynchronously among cells in an egg chamber. The fraction of BrdU-positive cells averaged 15% in late stage 9, whereas by the first part of stage 10, known as stage 10A, chambers contained few or no labeled follicle cells (Fig. 2D–F). Thus, all follicle cells complete endoreplication by the end of stage 10A. Variability in the position of labeled cells throughout this period indicated that most cell cycles were not strictly linked to developmental patterning.
Figure 2.

BrdU incorporation associated with endocycles and amplification. (A) In this stage 9 egg chamber, BrdU incorporation reveals that follicle cell endocycles are not synchronized. Some follicle cell nuclei are positive for BrdU incorporation and are in S, whereas others are blank for BrdU incorporation and, therefore, in G phase (see below). (B) Higher magnification to show the difference between S- and G-phase labeling. (C) DAPI of B. The DAPI bright dots correspond to follicle cell chromocenters that contain peri-centric heterochromatin. (D,E) Stage 10A chamber with the last few cells completing endocycles incorporating BrdU. The number of BrdU-positive cells decreases during mid-stage 9–10A as cells complete endocycles at different times. By late stage 10A, some chambers are entirely blank for BrdU incorporation. (F) DAPI of panel E. (G,H) In stage 10B, within hours after asynchronous completion of endocycles, punctate BrdU incorporation appears synchronously in every follicle cell over the oocyte but not those over the nurse cells (arrowhead). Notice the extreme difference between the BrdU incorporation in H and the absence of labeling in nuclei only a few hours earlier in E. (I) DAPI of H.
Amplification can be visualized within individual follicle cells
In contrast, a high degree of developmental regulation was revealed by BrdU labeling immediately following the end of stage 10A. Strong BrdU incorporation limited to subnuclear foci appeared in every follicle cell over the oocyte beginning in stage 10B (Fig. 2G–I, see also Fig. 3). This labeling continued through stage 13. Because chorion gene amplification is occurring at a rapid rate during stage 10 of oogenesis (Heck et al. 1990), and continues through stage 13, we predicted that the subnuclear BrdU incorporation represents amplification of chorion genes.
Figure 3.
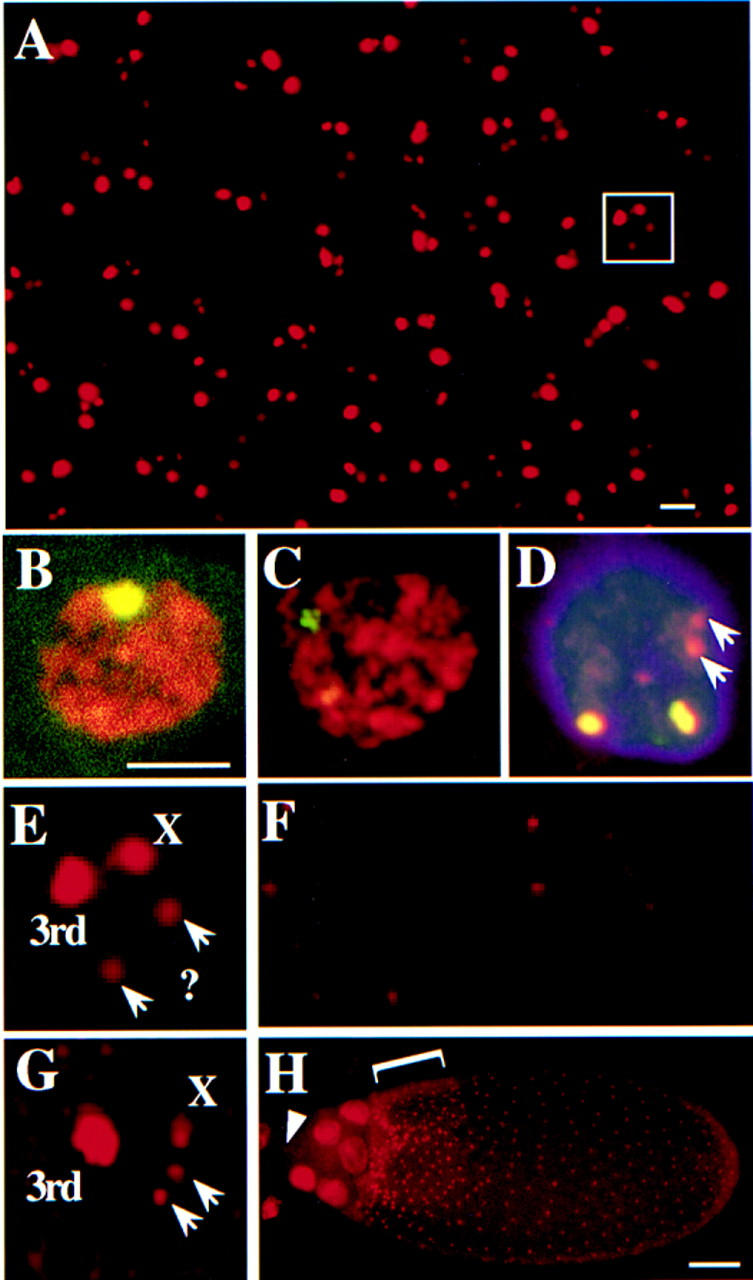
Amplification detected by BrdU and FISH. (A) Whole mount BrdU labeling in stage 10B follicle cells. Most nuclei have four spots of different intensities: one bright, one intermediate, and two faint. (B) FISH labeling. Hybridization of a chorion probe from the third chromosome to a squashed amplifying follicle cell nucleus. Total DNA is in red and hybridization signal is in yellow. (C) FISH with a chorion probe from the X chromosome to a squashed follicle cell that gives a less intense signal than the third locus commensurate with lower levels of amplification. (D) FISH/BrdU double labeling. Detection of BrdU incorporation (red) and FISH with both X and third chorion probes (green, overlap, yellow) indicating the bright BrdU focus corresponds to the highly amplified third chromosome chorion locus, the intermediate BrdU labeling to the less amplified X locus, and the two fainter spots to neither locus (arrows). The very faint extra BrdU labeling present in the center of this nucleus was not reproducibly observed. (E) Enlarged nucleus from whole mount BrdU labeling in A (white box), showing the four spot patterns and their corresponding identities. (F) Whole mount of BrdU incorporation in ovaries mutant for the Drosophila homolog of ORC2 [fs(3)293]. Compare with same magnification of wild type in A. Here, BrdU incorporation is mosaic in intensity among cells and is most easily seen at the third chorion locus. (G) BrdU incorporation in a stage 12 follicle cell. Notice that incorporation at the third locus is greater than earlier stages, X locus incorporation is reduced. (H) A dorsolateral aspect of a stage 11 egg chamber demonstrating brighter incorporation in the dorsal–anterior cells (bracket). BrdU foci are absent from follicle cells that remain over the nurse cells (for example, see arrowhead). The larger nuclei in the anterior of the chamber are those of the nurse cells. Scale bar in A for A and F, 5 μm; and in B for B–E and G, 5 μm; in H, 50 μm.
We wished to test whether the subnuclear BrdU foci, in fact, corresponded to amplifying chorion genes. Follicle cell nuclei surrounding the oocyte usually contained four subnuclear BrdU incorporation foci of different intensities: one bright, one intermediate, and two faint (Fig. 3A). We postulated that the more intense labeling represented the third chromosome chorion locus, which amplifies 60- to 80-fold, and the intermediate intensity labeling corresponds to the X chromosome locus, which amplifies 15- to 20-fold. To test this, squashed or whole mount preparations of ovaries incubated previously in BrdU were fluorescently double-labeled with anti-BrdU antibodies and chorion DNA probes. As predicted, the third chorion probes labeled the brightest BrdU spot and the X probes corresponded to the spot with intermediate intensity (Fig. 3B–D). Less than 1% of nuclei in these experiments showed two closely spaced sites of equal labeling for a given chorion probe, suggestive of homolog asynapsis as seen in other polytene cells (data not shown). Intriguingly, the faint BrdU foci did not hybridize to probes from the known chorion clusters. They apparently correspond to two previously unknown amplifying loci.
To further verify that the BrdU foci correspond to amplifying chorion genes, we examined the effects of several mutations that are known to disrupt this process. Labeling was greatly reduced in ovaries derived from fs(3)293 females, a female sterile mutation in the k43 gene that encodes the Drosophila homolog of ORC2 (Landis et al. 1997) and is known to be required for amplification (Fig 3F; Orr et al. 1984). Instead of uniform reductions, BrdU labeling was mosaic, with some follicle cells having much greater labeling than others. We examined the effect of mutations in other genes known to be required for amplification but for which the gene products are unknown. Similar to fs(3)293, BrdU incorporation was reduced in a mosaic fashion in four of these mutants [chiffon, fs(3)272, fs(1)K451, fs(1)K1214], but was reduced uniformly in a fifth [fs(2)PA77] (data not shown).
Patterns of BrdU incorporation in wild type indicated that the process of amplification is spatially and temporally regulated in a more precise manner than realized previously. The relative intensity of labeling of the X locus decreased at stage 12, when the genes in this cluster are expressed (Fig. 3, cf. E and G). Follicle cells that reside over the nurse cells, which do not synthesize chorion proteins (Parks and Spradling 1987), did not display punctate BrdU labeling (Fig. 3H). Regional differences in BrdU incorporation rate were also observed. Total fluorescence intensity at the third chorion locus in dorsal–anterior follicle cells was 1.5- to 2-fold brighter than in other cells in the egg chamber during stages 11–12 (Fig. 3H). Most of the follicle cells that contain brighter spots produce the dorsal appendages, a specialized eggshell structure. BrdU incorporation was also less affected in these cells by several of the amplification mutants (data not shown), suggesting that the regulation of amplification in these cells differs from the remaining follicle cells.
Chorion genes begin amplification during follicle cell endocycles
To further evaluate whether amplification responds to cell cycle regulation that governs normal S phase, we asked if amplification begins during endocycles. Although an examination of follicle cells earlier than stage 10B did not reveal subnuclear incorporation characteristic of amplification (Fig. 2B,E), we entertained the possibility that amplification could be occurring during endocycles, but remain undetected. If chorion genes amplify only during endocycle S phases, detection would be obscured by BrdU incorporation occurring throughout the rest of the nucleus.
Previous measurements of amplification by quantitative Southerns relied on isolation of DNA from whole egg chambers, the majority of which is derived from highly polyploid nurse cells that do not amplify chorion genes. This complicates detection of the onset of amplification when chorion copy number is only slightly increased within follicle cells. To circumvent this problem, we recovered nuclei corresponding to endocycling follicle cells by FACS (see Fig. 1C), and measured chorion gene copy number. The four major ploidy peaks represent a mixture of nuclei from mitotic (2C and 4C), endocycling (4C, 8C, and 16C) and postendocycling (16C) cells. G2 mitotic and endocycling 4C nuclei differ in DNA content because of satellite DNA under-representation, giving rise to a double 4C peak (Fig. 1C; M. Lilly and A. Spradling, unpubl.). The 8C peak consists of follicle cell nuclei that have completed two endocycles but still have one endocycle remaining. Consequently, we examined whether amplification had begun by the second endocycle by purifying DNA from 8C nuclei and measuring the copy number of chorion gene sequences by quantitative Southern blotting (Fig. 4A). This indicated that third chorion genes are 1.8- to 2.2-fold amplified in 8C follicle cell nuclei, but no amplification was found for the X chorion gene cluster (see Table 1).
Figure 4.

Amplification begins during endocycles. (A) Representative Southern blot of SalI digested genomic DNA comparing the relative copy numbers of a 3.8-kb SalI fragment from the third chromosome chorion gene locus (ch) to a 7.3-kb SalI fragment from the rosy locus as a control (c). Chorion DNA is amplified two-fold in 8C endocycling follicle cells (Table 1). (Lane 1–3) Decreasing loadings of male genomic DNA that served as a nonamplifying diploid control; (lane 4) DNA from 8C flow-sorted ovarian nuclei (see Fig. 1C); (lane 5) DNA from 16C flow sorted ovarian nuclei. (B) Similar analysis of copy number in stage 10A egg chambers indicates third chromosome chorion genes are amplified fourfold by this stage (Table 1). Notice that the measured level in B needs to be corrected for the fraction of DNA derived from follicle cells whereas that in A does not. (Lanes 1–3) Decreasing loadings male diploid control; (lane 4) stage 10A DNA. Molecular weights of marker DNAs are indicated to left in A and B (in kb).
Table 1.
Amplification during endocycles
| Control probe
|
8C nuclei
|
Stage 10A chambersa
|
||
|---|---|---|---|---|
| X chorion
|
third chorion
|
X chorion
|
third chorion
|
|
| ry | 1.1 | 1.9 | 0.7 | 4.1 |
| slbo | 1.0 | 1.8 | 0.8 | 4.7 |
| hts | 1.2 | 2.2 | N.D. | N.D. |
Fold amplification of chorion genes in follicle cells was compared to several probes. Probe specific activity was normalized using nonamplifying male DNA. Amplification was calculated as the ratio chorion/control follicle cells divided by chorion/control males. No amplification is defined as a ratio of 1.
The absolute amplification values indicated for whole egg chambers were corrected for the fraction of DNA derived from follicle cells using the formula absolute amplification = (amplification observed − 0.58)/0.42 for observed amplification values >1 (see Materials and Methods).
We could not use the 16C peak to measure amplification that occured solely in the final endocycle. This peak contains cells from late in oogenesis when chorion genes are highly amplified as well as those that have just completed the final endocycle. Therefore, to confirm that amplification does occur during endocycles, we measured chorion copy number in DNA derived from whole stage 10A egg chambers. BrdU labeling indicated that at this point in oogenesis, most follicle cells have just completed the 8C–16C endoreplication, but the subnuclear foci corresponding to chorion gene amplification have not yet appeared. After correction for the fraction of DNA represented by follicle cells, this analysis indicated that the third chorion genes are amplified 4.1- to 4.7-fold, but the chorion genes on the X are not amplified (Fig. 4B). Therefore, amplification of the third chorion genes does begin during follicle cell endocycles.
These results also suggest that amplification responds to cell cycle control. Although BrdU labeling corresponding to chorion amplification was easily detected beginning in stage 10B (estimated fivefold above background), no incorporation was observed during endocycle G phases, including those cells that were developmentally only a few hours younger (Fig 2, cf. H and E). Thus, within the sensitivity of these experiments, early amplification is confined to S phase during the endocycles. Therefore, there appears to be two discrete phases to amplification: an early phase in which third chorion genes amplify periodically during S phases occurring asynchronously among cells in an egg chamber, and a later, continuous phase that begins synchronously in all follicle cells over the oocyte.
CycE persists throughout the late phase of amplification
To investigate cell cycle mechanisms that control the two phases of amplification, we examined the expression of CycE by immunofluorescence. During endocycles, oscillations of CycE, complexed with its kinase partner CDK2, control periodic S phases (Knoblich et al. 1994; Sauer et al. 1995; Lilly and Spradling 1996). A fraction of follicle cells in stages 7–9 egg chambers exhibited nuclear CycE staining commensurate with asynchronous endoreplications (Fig. 5A,B). During stages 9–10A, as cells complete the last endocycle, a decreasing number contain high CycE levels, similar to the results for BrdU labeling (Fig. 5C,D). Follicle cells in stage 10A chambers, however, appeared to retain a low level of staining. In contrast, in stage 10B, as late amplification begins, all follicle cells over the oocyte simultaneously displayed levels of CycE comparable with earlier S phases (Fig. 5E,F). CycE persisted and failed to cycle at least until stage 14; however, the level of staining slowly diminished from stage 10B onward (Fig. 5E,F). In older chambers, the CycE staining became more punctate. By stage 12, several subnuclear foci of high-level staining were clearly observed (Fig 5H), resembling the sites of localized BrdU incorporation observed at this time in number and intensity (cf. Fig. 3E). CycE antibody staining did not work under conditions required for BrdU or FISH labeling and, thus, we were unable to colocalize CycE with chorion loci. Nonetheless, the possible accumulation of CycE at chorion loci, the restriction of amplification to endocycle S phases, and alteration of CycE behavior during late amplification suggested CycE may be required for chorion genes to amplify.
Figure 5.

Changes in CycE behavior during the two phases of amplification. CycE protein levels within follicle cells were determined by confocal immunofluorescence. (A,B) Nuclear CycE levels oscillate during asynchronous follicle cell endocycles. (C,D) Stage 10A chamber showing that after completion of the final endocycle CycE levels drop, but a low level of staining remains. The area of less intense staining in the nuclei of this and later stages corresponds to the large nucleolus in these cells. (E,F) Shortly thereafter in stage 10, CycE rises to high levels in every follicle cell over the oocyte. The large patches of staining in A, C, and E correspond to nurse cell and oocyte nuclei. (G,H) Throughout later amplification, CycE levels slowly diminish but fail to cycle. Shown here is a stage 12 chamber demonstrating the 4 spot punctate labeling reminiscent of BrdU incorporation (inset in H, single nucleus).
CycE is required but not sufficient for amplification
We used several approaches to test the functional role of CycE in amplification. Because CycE is required in mitotically dividing cells (Knoblich et al. 1994; Duronio and O’Farrell 1995), it was not feasible to generate loss-of-function clones by standard mitotic recombination. Incubation of ovaries in the kinase inhibitor 6-DMAP eliminated detectable chorion BrdU incorporation within 30 min (data not shown) suggesting continued kinase activity is important for amplification.
To test more directly if CycE is required for amplification, we induced expression of a specific inhibitor of CycE, the dacapo gene (dap) (de Nooij et al. 1996; Lane et al. 1996), using the GAL4 activation system (Brand and Perrimon 1993). The c323:GAL4 strain is an enhancer trap that induces high-level expression in all follicle cells beginning in late stage 9 before the onset of late amplification (Manseau et al. 1997). When c323:GAL4 was combined with UAS:dap, amplification was severely inhibited. c323:GAL4; UAS:dap females were sterile and laid flaccid eggs with thin eggshells and smaller dorsal appendages (data not shown), a phenotype shared by all mutations that disrupt amplification. Moreover, BrdU incorporation at chorion loci was undetectable in virtually all follicle cells in these egg chambers (Fig. 6A,B). Reminiscent of regional differences observed in wild-type and amplification mutants, however, egg chambers often had dorsal–anterior follicle cells with reduced BrdU labeling (data not shown). To determine whether CycE was required continuously, we also misexpressed Dap using a heat-shock-inducible hsp70:GAL4 transgene. BrdU staining at chorion foci was abolished within 2 hrs after heat induction (data not shown). As with c323:GAL4, the dorsal–anterior follicle cells were more often positive for BrdU incorporation at chorion loci, suggesting they contain more robust amplification activity. These experiments strongly suggest that CycE is required continuously for the late phase of amplification.
Figure 6.
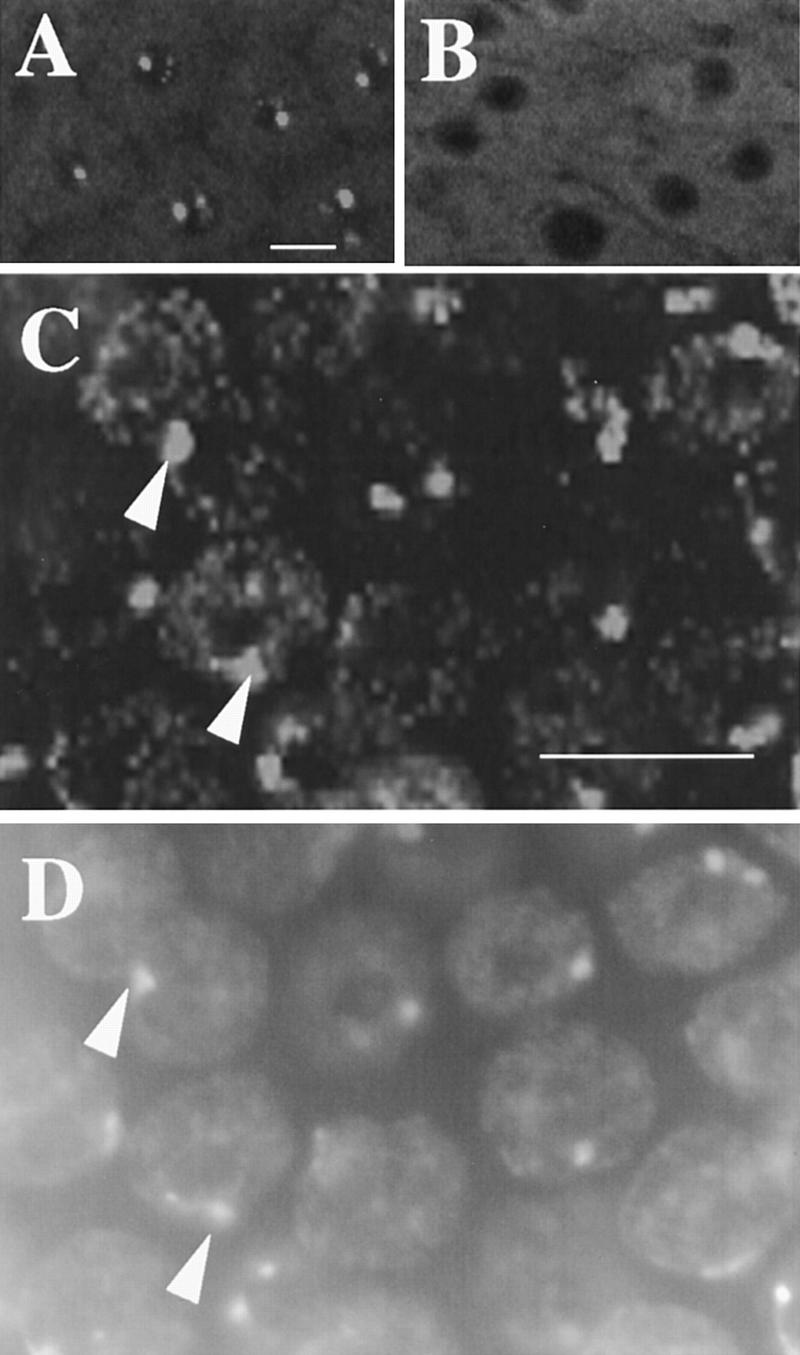
CycE is necessary, but not sufficient, for amplification. (A) BrdU foci in wild-type stage 12 nuclei. (B) Stage 12 chamber from a c323GAL4; UAS:Dap female showing that misexpression of the CycE inhibitor, Dacapo, drastically reduces BrdU labeling. (C) Overexpression of CycE in stage 10A. High magnification shows robust late replication corresponding to heterochromatic chromocenter but no BrdU incorporation characteristic of amplification. (D) DAPI image of C to show correspondence of labeling with DAPI bright chromocenter (cf. arrowheads in C and D). Scale bar, 10 μm.
To ask if CycE is sufficient to induce amplification, we misexpressed it using either c323:GAL4 or hsp70:GAL4 in combination with UAS:cycE (Lane et al. 1996) during stage 10A when CycE levels are normally low and amplification is not observed. Despite the premature expression of CycE, incorporation of BrdU into chorion foci was not observed in stage 10A egg chambers (Fig. 6C,D). With prolonged CycE expression, some stage 10A and later nuclei also had low-level speckles of BrdU incorporation distributed throughout the nucleus. These speckles did not appear similar to chorion foci and likely result from earlier disruption of endocycles (see below). Consistent with previous results (Lilly and Spradling 1996), however, CycE overexpression did induce robust late replication of heterochromatin in stage 10A follicle cells as evidenced by increased BrdU labeling corresponding to the chromocenter (Fig. 6C,D). The combined results for UAS:dap and UAS:cycE suggest that CycE is necessary, but not sufficient, for amplification.
Inhibition of replication from nonchorion origins is associated with persistent CycE
Another characteristic of the late phase of amplification is the absence of replication throughout most of the follicle cell genome. In other cell cycles, S phase kinases act positively to initiate replication, but also inhibit rereplication within a cycle (for review, see Diffley 1996; Hua et al. 1997; Tanaka et al. 1997). A period when kinase levels are low is required for assembly of prereplication complexes. It has been suggested that oscillations of CycE during endocycles control once-per-cycle replication (Su and O’Farrell 1995; Lilly and Spradling 1996). In support of this, although misexpression of CycE initially resulted in more S phase follicle cells during endocycles, prolonged misexpression inhibited entry into subsequent S phases (data not shown). In wild type, it is likely that the persistent CycE observed beginning in stage 10B inhibits assembly of new prereplication complexes at most origins during this period. As in previous endocycle S phases, chorion origins can escape this negative regulation. To explain the absence of one round of genomic replication at the onset of the late phase of amplification, we postulated that CycE/CDK2 kinase activity must remain sufficiently high to inhibit origin resetting after the final endocycle. Although we did observe low level CycE antibody staining after the final endocycle S phase in stage 10A chambers, the sensitivity of these experiments did not allow us to determine whether levels remained higher than during previous G phases when origins can reset. Thus, we sought other ways of examining CycE/CDK2 activity during this period.
We have found that MPM-2 monoclonal antibody is a useful marker for CycE activity. This antibody recognizes a variety of cell cycle-dependent phosphoepitopes in an array of organisms (Davis et al. 1983). Although originally raised against HeLa cells in mitosis, it also stains interphase cells (Ding et al. 1997 and references therein). MPM-2 labels both diploid and polyploid cells in Drosophila (Millar et al. 1987). We find that MPM-2 stains a subnuclear sphere of unknown identity in a fraction of endocycling follicle cells, suggesting that this labeling is cell cycle restricted (Fig 7A). Overproduction of CycE with either UAS:CycE or hsp70:CycE increased the intensity and number of MPM-2 sphere-positive follicle cells. Within 45 min of induction of CycE expression, virtually every follicle cell displayed MPM-2 spheres (Fig. 7C). Conversely, inhibition of CycE/CDK2 activity by misexpression of Dap resulted in abolition of MPM-2 sphere staining in follicle cells (Fig. 7D). Thus, this MPM-2 epitope responds to alterations in CycE levels. Although it remains unclear whether this putative phosphoepitope is directly produced by CycE/CDK2, MPM-2 staining is a useful marker to monitor CycE activity.
Figure 7.

MPM-2 reveals synchronization of the cell cycle and responds to CycE. (A) Endocycling follicle cells have cell cycle-dependent staining of an MPM-2 reactive nuclear sphere. (B) A field of cells from stage 10A. The number of MPM-2 sphere containing cells increases throughout stage 9 so that by stage 10 every follicle cell is positive. (C) Overexpression of CycE with a hsp70:cycE transgene results in an increase in the number of stage 8 endocycling follicle cells that are MPM-2 positive. (D) Stage 10 follicle cells showing that inhibition of CycE activity in hsp70:GAL4; UAS:Dap flies results in dramatic reductions in MPM-2 sphere staining. Scale bar, 10 μm.
In wild-type chambers, we observed an interesting transition in MPM-2 staining preceding and continuing into the late phase of amplification. During stages 9–10A, as more cells have entered and completed the last endoreplicative S phase, there was an increase in the number of cells positive for MPM-2 spheres. By stage 10A, before the onset of late amplification, every follicle cell over the oocyte was positive for MPM-2 staining (Fig.7B). The MPM-2 staining in follicle cells over the oocyte increased in intensity into stage 10B when late amplification begins and high levels of CycE protein were observed. Similar to CycE, MPM-2 staining slowly diminished after stage 11, so that by late stage 12 to 13 intensity was greatly reduced (data not shown). As with BrdU incorporation at chorion loci, MPM-2 staining was greater in dorsal–anterior cells of the egg chamber. Moreover, although Dap misexpression abolished MPM-2 staining in most cells during late amplification, the dorsal–anterior cells were often positive for staining. Thus, in many ways, MPM-2 staining reflects CycE activity. The increasing number of MPM-2 positive cells from stages 9–10A suggests CycE activity persists following the final endocycle. This could inhibit most origins from reassembling prereplication complexes explaining the absence of replication throughout most of the follicle cell genome during the late phase of amplification.
Discussion
Because amplification is most easily detected late in oogenesis when follicle cell replication has ceased, it has been thought of as a continuous process unconnected to the cell cycle. Our results reveal a very different picture. Amplification begins within the S phases of asynchronously cycling follicle cells and undergoes a developmentally regulated transition into a late continuous phase. Both phases are closely connected to the behavior of the major regulator of follicle cell endocycles, CycE, whose activity is necessary for amplification. Finally, our results suggest that origins at chorion loci escape the normal mechanisms that prevent rereplication. Thus, further study of this system as an exception to once-per-cycle replication will likely reveal aspects of the normal mechanisms governing rereplication control.
Amplification and follicle cell cycles
It had been an open question as to whether chorion amplification occurs exclusively after the completion of endocycles. Our measurements indicate third chorion genes do amplify during endocycles, but X chorion genes do not. Amplification, therefore, may begin within the first endoreplicating follicle cells, perhaps as early as stages 7–8 of oogenesis. The data suggest that chorion origins can reinitiate only during periodic S phases explaining why there are only small increases in third chorion copy number during endocycles. The absence of amplification for the X chorion genes indicates that the X and third chorion origins are distinguished by different propensities to rereplicate during the time allotted by endocycle S phase. The observation of amplification during endocycle S phases is significant because it suggests the cell cycle-regulated mechanism of amplification resembles normal replication, but that chorion origins can locally escape rereplication control that operates on the remainder of the genome at this time.
The appearance of two foci of BrdU incorporation that are distinct from the X and third chorion gene clusters indicates that there are additional loci that differentially replicate. These loci may correspond to genes required for late oogenesis and could encode other minor chorion proteins. Identification of these loci may aid identification of cis sequences that mediate amplification.
It is interesting to contrast our findings with amplification that occurs in Sciarid flies. In the salivary gland polytene chromosomes of these insects, genes encoding cocoon proteins rereplicate within visible DNA puffs (for review, see Gerbi and Urnov 1996). This occurs during a final endocycle S phase when the remainder of the genome doubles in copy number. Unlike chorion gene amplification, however, Sciarid genes are not known to amplify periodically during multiple endocycles nor do they amplify in two distinct phases.
The positive role for CycE
The inhibition of amplification by 6-DMAP and Dap suggests that CycE is required for amplification. Although it is possible that Dap overexpression inhibits amplification by an unknown CycE-independent mechanism, the observation that early and late amplification occurs only when CycE is high supports the conclusion that it is required. CycE is not sufficient for amplification, however, because premature expression did not induce precocious amplification in stage 10A follicle cells, nor in follicle cells associated with the nurse cells. We know that overexpression of CycE resulted in higher kinase activity because stage 10A did have increased replication of heterochromatin. This may be the result of initiation from late S phase origins that had assembled prereplication complexes in the previous G phase, or elongation from early replicating regions of the genome. Replication of heterochromatin is not readily observed after stage 10A in normal chambers nor those overexpressing CycE. This suggests that conditions required for late replication of heterochromatin do not persist into the continuous phase of amplification. Moreover, the absence of amplification after CycE overexpression in stage 10A predicts that levels of another factor required for amplification increase with CycE at the onset of continuous amplification in stage 10B.
In our antibody labeling experiments, we observed subnuclear concentrations of CycE reminiscent of chorion gene BrdU labeling. Because of technical impediments, we could not colocalize CycE with BrdU or FISH in double labeling experiments. Nevertheless, this reproducible similarity in appearance suggests that local concentrations of CycE contribute to the late phase of amplification. The mechanisms by which CycE/CDK2 induces S phase have not been entirely uncovered (for review, see Weinberg 1995; Duronio et al. 1996; Lukas et al. 1997). In yeast, CDK complexes that promote S phase physically interact with ORC2 (Leatherwood et al. 1996). The punctate antibody labeling we observe suggests that CycE/CDK2 may, in part, promote amplification by modifying proteins that are closely associated with chorion loci during amplification. Our finding that CycE is required for amplification, taken with the previous report that Drosophila ORC2 is also required (Landis et al. 1997), suggests that many of the same components controlling S phase during normal cycles are required for amplification.
Developmental modification of follicle cell cycles during oogenesis
The transition from early to late amplification is associated with cessation of endoreplication and synchronization of follicle cell behavior (Fig. 8A). During this transition period, most follicle cells migrate posteriorly to cover the growing oocyte. Synchronous late amplification was specific for these follicle cells; we never observed BrdU subnuclear incorporation in follicle cells that remain associated with nurse cells. Additional regional specialization was observed for the dorsal–anterior cells over the oocyte that had greater BrdU incorporation than other cells in wild-type and compromised backgrounds. These cells synthesize specialized chorion structures and participate in oocyte/follicle cell signaling required for proper patterning of both the eggshell and embryo (for review, see Ray and Schüpbach 1996). These observations suggest signals emanating from the germline coordinate amplification with egg chamber development.
Figure 8.
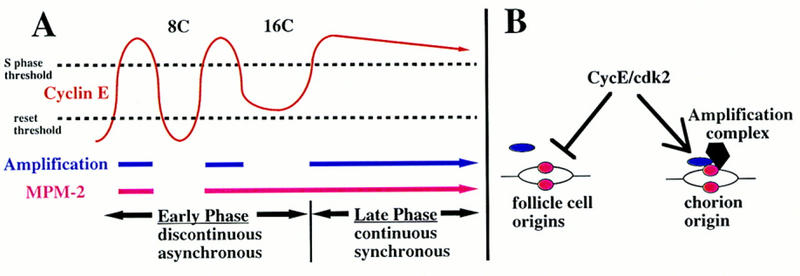
A model for the two phases of amplification and escape from rereplication control. (A) In the early phase, third chorion genes amplify periodically during follicle cell endocycle S phases occurring asynchronously among cells within an egg chamber. In the late phase, chorion amplification commences synchronously among cells and replicates continuously (blue line). During the end of the early phase, follicle cells acquire synchrony as they complete the final endoreplication, but do not enter a subsequent normal endocycle. These changes in periodicity are associated with alterations in behavior of CycE oscillations (red waves above) and MPM-2 reactivity (red lines below). CycE levels must be greater than the S-phase threshold for replication (top broken line). CycE levels also must decline below the reset threshold for origins to reset (bottom broken line). If CycE levels remain above this threshold after the final endoreplication, it would explain the absence of widespread origin firing when late phase amplification begins. (B) For replication from most follicle cell origins CycE likely plays dual roles—S-phase promotion and rereplication inhibition. Here, after initiation of replication, some proteins (pink ovals) remain associated with follicle cell origins, whereas association of others (blue ovals) into prereplication complexes is inhibited by CycE. Amplification complexes resident at chorion origins escape inhibition by CycE but require its positive activity to initiate replication. It may be that in addition to proteins common to other origins, proteins specific to amplification complexes (black polygon) impart resistance to CycE inhibition.
Developmental signals must act in part to modify CycE levels during the transition to late amplification. Ubiquitin-mediated degradation is an important system for restricting cyclins to precise periods during the cell cycle (for review, see Hoyt 1997). If degradation of CycE is modified during the final endocycle, this would explain its continued presence in cells during the transition between early and late amplification (Fig. 8A). In the absence of ubiquitin-mediated degradation, induction of a single burst of cycE transcription during stage 10 would be sufficient to account for the high, but slowly declining, protein levels during late amplification. Differential degradation within the nucleus may also account for the punctate CycE labeling. Although the period of late amplification has been viewed previously as the end of endocycles, it may be more accurate to describe this as entry into a synchronized and prolonged final endocycle S phase during which only a few origins are prepared to replicate.
A model for amplification as an escape from rereplication control
Considerable evidence supports the idea that CDKs that are required for S phase also inhibit rereplication. (Dahmann et al. 1995; Piatti et al. 1996; Hua et al. 1997, Tanaka et al. 1997; for review, see Diffley 1996). Given the absence of Cyclins A and B, it has been suggested that CycE is responsible for once-per-cycle replication in endocycles (Su and O’Farrell 1995; Lilly and Spradling 1996). Consistent with this, we found continued misexpression of CycE blocked endocycling follicle cells from entering subsequent S phases. We suggest chorion amplification is essentially an escape from rereplication control mediated by CycE/CDK2 complexes in follicle cells (Fig. 8B). Thus, during a single endocycle S phase, and later when CycE levels fail to oscillate, chorion origins initiate DNA replication more than once. The persistence of CycE in late follicle cells may, therefore, inhibit most origins, but not those at chorion loci, from assembling prereplication complexes. This predicts that if CycE activity were inhibited transiently during late amplification, most origins would reset, and an additional endocycle S phase would ensue. Although we were able to inhibit CycE activity with currently existing fly strains, we could not restore activity to sufficient levels to induce a subsequent S phase to test this hypothesis.
How might chorion loci escape rereplication control? One critical aspect of rereplication control models is dissociation of replication complexes from chromatin when origins fire. It may be that special amplification complexes at chorion origins are not destroyed with a single firing, or are reassembled within S phase, thus engendering local resistance to rereplication inhibition (Fig. 8B). We propose that amplification complexes resident at chorion origins are associated with unique amplification factor(s) that impart protection from inhibition exerted by the CycE/CDK2 pathway (Fig. 8B). These may be special members of the ORC, CDC6, and MCM families that associate with origins during acquisition of replication competence (for review, see Rowles and Blow 1997), or molecules that counteract inhibitory phosphorylation known to regulate these proteins (Jallepalli et al. 1997). Signals that induce expression of an amplification factor in follicle cells during endocycles would be sufficient to explain the developmental specificity for the onset of amplification. A continued investigation of amplification will likely reveal general cell cycle mechanisms controlling genome replication.
Materials and methods
Fly strains and genetic manipulations
Flies were raised at 25°C on standard cornmeal media. Abbreviations and information regarding standard strains can be found in Flybase (Gelbart et al. 1997). In most cases, y;ry was used as a standard for BrdU labeling. The sources and full genotype of the amplification mutants examined are as follows: v K1214/FM7c and v K451/FM7c (Komitopoulou et al. 1983), PA77 cn bw sp/CyO (Schüpbach and Wieschaus 1991), h th fs272 e/TM3 Sb and cu fs293 red/TM3 Sb (Snyder et al. 1986). The chiffon allele, fs(2)00233 cn/CyO; ry506, was recovered in a P-element female sterile screen, but the amplification defect does not map to the P-element insertion site at 37F. c323:GAL4 is on the X chromosome (Manseau et al. 1997) and hsp70:GAL4 is on the third chromosome. The UAS:dap (II-1) and UAS:cycE strains (III-1), on the second and third chromosome, respectively, were kindly provided by C. Lehner (Lane et al. 1996). For misexpression experiments, GAL4 females were crossed to UAS line males to create GAL4/+; UAS/+ females from which ovaries were examined. The hsp70:cycE P element is on the third chromosome (Knoblich et al. 1994). Heat induction of hsp70 transgenes was at 37°C in a water bath for 30 min.
Antibody labeling and image capture
Ovaries were isolated, fixed, and immunolabeled as described in Lin and Spradling (1993). For CycE, the monoclonal antibody 8B10 (Richardson et al. 1995) was used as described (Lilly and Spradling 1996). MPM-2 antibody (Davis et al. 1983) was purchased from DAKO Corporation, and used at a dilution of 1:100. Secondary antibodies FITC-antimouse 1:200 or CY3-antimouse 1:400 were purchased from Jackson Immunoresearch Laboratories.
BrdU labeling and measurements
BrdU labeling was for 1 hr in Grace’s medium as described previously (Margolis and Spradling 1995; Lilly and Spradling 1996). For a given BrdU focus, incorporation was defined as the sum of all pixel intensities by use of IP lab spectrum software (Scanalytics). For measurement of incorporation in dorsal–anterior versus other follicle cells, 20 foci corresponding to the third chorion locus were analyzed. For 6-DMAP inhibition, ovaries were first incubated in Grace’s medium containing 2 mm 6-DMAP for ½ hr, followed by BrdU labeling in the absence of 6-DMAP for 1 hr.
FISH
Fluorescent hybridization to squashed preparations of ovaries or staged egg chambers was according to the protocol of K. Dej and A. Spradling (unpubl., available upon request) using reagents from ONCOR. For squashed BrdU/FISH double-labeling, ovaries incubated previously in BrdU were processed as for FISH and subsequently detected with either avidin or antidigoxigenin antibodies and anti-BrdU (Becton-Dickinson).
Whole mount FISH was essentially as described (Dernburg et al. 1996) except that fixation was in 37% formaldehyde/buffer B/dH2O (1:1:4) (Lin and Spradling 1993). For BrdU/FISH whole mounts, ovaries incubated in BrdU and processed as for FISH. The third chorion probe was the 3.8-kb plasmid subclone pt2 from the Ace3 region. The X chorion probe was the plasmid 104.63, which is a 6.3-kb subclone from λ phage 104 (Spradling 1981).
Quantitative Southern blotting
For quantification of amplification in 8C cells, nuclei were isolated from 240 y;ry506 females and sorted as described (Lilly and Spradling 1996). For quantification in stage 10A, DNA was prepared from 110 early 10A chambers. DNA was prepared as described (Swimmer et al. 1989) and digested with SalI before standard agarose electrophoresis and Southern blotting. The sum of signal intensity above background was quantified by the Storm System (Molecular Dynamics) after exposure to PhosphorImager screens. Amplification value was calculated as chorion/control follicle cell divided by chorion/control nonamplifying male DNA. For stage 10A, ∼42% of DNA is derived from follicle cells (16C × 650 cells) whereas 58% is derived from nurse cells (average of 955C × 15 cells) (Hammond and Laird 1985). Measured values >1 were, therefore, corrected with the formula: absolute amplification = (observed amplification − 0.58)/0.42. The third chorion probe was plasmid pt2 and the X probe was plasmid 104.63 described above for FISH. Control probes were as follows: p5′rySB, a 3.2-kb SalI–BamHI subclone from the 5′ end of the rosy gene (Gray et al. 1991); p1208, a BamHI–XbaI subclone from the coding region of the slbo gene (Rorth 1994); and cDNA clone c13 from the hu-li tai shao gene (Yue and Spradling 1992).
Acknowledgments
We thank Jon Margolis for early BrdU observations and Kim Dej for squashed FISH development and instruction. Thanks to Abby Dernburg for advice on whole-mount FISH experiments. We acknowledge the generosity of the following individuals and laboratories in providing reagents and fly strains: Len Dobens, Fotis Kafatos, Christian Lehner, Iswar Hariharan, Rob Ray, Trudi Schüpbach, and Helena Richardson. We thank James Flook of the Johns Hopkins Medical Institute Cell Sorting facility for his expert assistance. Thanks to Maggie de Cuevas, Horacio Frydman, Orna Cohen-Fix, Nicole Grieder, Doug Koshland, and Phil Newmark for critical reading of the manuscript. B.R.C. thanks Anja-Katrin Bielinsky for critical opinions. B.R.C. and M.A.L. were postdoctoral fellows of the American Cancer Society. Research expenses were defrayed in part by National Institutes of Health grant GM27875 to A.C.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Spradling@mail1.ciwemb.edu; FAX (410) 243-6311.
References
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Broek D, Bartlett R, Crawford K, Nurse P. Involvement of p34cdc2 in establishing the dependency of S phase on mitosis. Nature. 1991;349:388–393. doi: 10.1038/349388a0. [DOI] [PubMed] [Google Scholar]
- Carminati JL, Orr-Weaver TL. Changes in DNA replication during animal development. In: DePamphilis ML, editor. DNA replication in eukaryotic cells. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 409–434. [Google Scholar]
- Dahmann C, Diffley JFX, Nasmyth KA. S-phase-promoting cyclin-dependent kinases prevent re-replication by inhibiting the transistion of replication origins to a pre-replicative state. Curr Biol. 1995;5:1257–1269. doi: 10.1016/s0960-9822(95)00252-1. [DOI] [PubMed] [Google Scholar]
- Davis FM, Tsao TY, Fowler SK, Rao PN. Monoclonal antibodies to mitotic cells. Proc Natl Acad Sci. 1983;80:2926–2930. doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nooij JC, Letendre MA, Hariharan IK. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87:1237–1247. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- Delidakis C, Kafatos FC. Amplification of a chorion gene cluster in Drosophila is subject to multiple cis-regulatory elements and to long-range position effects. J Mol Biol. 1987;197:11–26. doi: 10.1016/0022-2836(87)90605-x. [DOI] [PubMed] [Google Scholar]
- ————— Amplification enhancers and replication origins in the autosomal chorion gene cluster of Drosophila. EMBO J. 1989;8:891–901. doi: 10.1002/j.1460-2075.1989.tb03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dernburg AF, Broman KW, Fung JC, Marshall WF, Philips J, Agard DA, Sedat JW. Perturbation of nuclear architecture by long-distance chromosome interactions. Cell. 1996;85:745–759. doi: 10.1016/s0092-8674(00)81240-4. [DOI] [PubMed] [Google Scholar]
- Diffley JF. Once and only once upon a time: Specifying and regulating origins of DNA replication in eukaryotic cells. Genes & Dev. 1996;10:2819–2830. doi: 10.1101/gad.10.22.2819. [DOI] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH, Dowell SJ, Rowley A. Two steps in the assembly of complexes at yeast replication origins in vivo. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- Ding M, Feng Y, Vandre DD. Partial characterization of the MPM-2 phosphoepitope. Exp Cell Res. 1997;231:3–13. doi: 10.1006/excr.1996.3439. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH. Developmental control of the G1 to S transition in Drosophila: CyclinE is a limiting downstream target of E2F. Genes & Dev. 1995;9:1456–1468. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, Brook A, Dyson N, O’Farrell PH. E2F-induced S phase requires Cyclin E. Genes & Dev. 1996;10:2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- Follete PJ, O’Farrell PH. Cdks and the Drosophila cell cycle. Curr Opin Genet and Dev. 1997;7:17–22. doi: 10.1016/s0959-437x(97)80104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall JG, Cohen EH, Polan ML. Repetitive DNA sequences in Drosophila. Chromosoma. 1971;33:319–344. doi: 10.1007/BF00284948. [DOI] [PubMed] [Google Scholar]
- Gelbart WM, Crosby M, Matthews B, Rindone WP, Chillemi J, Russo TS, Emmert D, Ashburner M, Drysdale RA, Whitfield E, Millburn GH, de Gray A, Kaufman T, Matthews K, Gilbert D, Strelets V, Tolstoshev C. FlyBase: A Drosophila database. The FlyBase consortium. Nucleic Acids Res. 1997;25:63–66. doi: 10.1093/nar/25.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA, Urnov FD. Differential DNA replication in insects. In: DePamphilis ML, editor. DNA replication in eukaryotic cells. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 947–970. [Google Scholar]
- Gray M, Charpentier A, Walsh K, Wu P, Bender W. Mapping point mutations in the Drosophila rosy locus using denaturing gradient gel blots. Genetics. 1991;127:139–149. doi: 10.1093/genetics/127.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond MP, Laird CD. Chromosome structure and DNA replication in nurse and follicle cells of Drosophila melanogaster. Chromosoma. 1985;91:267–278. doi: 10.1007/BF00328222. [DOI] [PubMed] [Google Scholar]
- Hayles J, Fisher D, Woollard A, Nurse P. Temporal order of S phase and mitosis in fission yeast is determined by the state of the p34cdc2-mitotic B cyclin complex. Cell. 1994;78:813–822. doi: 10.1016/s0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- Heck MM, Spradling AC. Multiple replication origins are used during Drosophila chorion gene amplification. J Cell Biol. 1990;110:903–914. doi: 10.1083/jcb.110.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA. Eliminating all obstacles: Regulated proteolysis in the eukaryotic cell cycle. Cell. 1997;91:149–151. doi: 10.1016/s0092-8674(00)80396-7. [DOI] [PubMed] [Google Scholar]
- Hua XH, Yan H, Newport J. A role for cdk2 kinase in negatively regulating DNA replication during S phase of the cell cycle. J Cell Biol. 1997;137:183–192. doi: 10.1083/jcb.137.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli PV, Brown GW, Muzi-Falconi M, Tien D, Kelly TJ. Regulation of the replication initiator protein p65cdc18 by CDK phosphorylation. Genes & Dev. 1997;11:2767–2779. doi: 10.1101/gad.11.21.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RC. Ovarian development in Drosophila melanogaster. New York, NY: Academic Press; 1970. [Google Scholar]
- Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cyclin E controls S phase progression and its down regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- Komitopoulou K, Gans M, Margaritis LH, Kafatos FC, Masson M. Isolation and characterization of sex-linked female-sterile mutants in Drosophila melanogaster with special attention to eggshell mutants. Genetics. 1983;105:897–920. doi: 10.1093/genetics/105.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis G, Kelley R, Spradling AC, Tower J. The k43 gene, required for chorion gene amplification and diploid cell chromosome replication, encodes the Drosophila homolog of yeast origin recogntion complex subunit 2. Proc Natl Acad Sci. 1997;94:3888–3892. doi: 10.1073/pnas.94.8.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87:1225–1235. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- Leatherwood J, Lopez-Girona A, Russell P. Interaction of Cdc2 and Cdc18 with a fission yeast ORC2-like protein. Nature. 1996;379:360–363. doi: 10.1038/379360a0. [DOI] [PubMed] [Google Scholar]
- Lehner CF, O’Farrell PH. The roles of Drosophila Cyclins A and B in mitotic Control. Cell. 1990;61:535–547. doi: 10.1016/0092-8674(90)90535-m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly MA, Spradling AC. The Drosophila endocycle is controlled by Cyclin E and lacks a checkpoint ensuring S-phase completion. Genes & Dev. 1996;10:2514–2526. doi: 10.1101/gad.10.19.2514. [DOI] [PubMed] [Google Scholar]
- Lin H, Spradling AC. Germline stem cell division and egg chamber development in transplanted Drosophila germaria. Dev Biol. 1993;159:140–152. doi: 10.1006/dbio.1993.1228. [DOI] [PubMed] [Google Scholar]
- Lukas J, Herzinger T, Hansen K, Moroni MC, Resnitzky D, Helin K, Reed SI, Bartek J. Cyclin E-induced S phase without activation of the pRB/E2F pathway. Genes & Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- Mahowald AP, Caulton JH, Edwards MK, Floyd AD. Loss of centrioles and polyploidization in follicle cells of Drosophila melanogaster. Exp Cell Res. 1979;118:404–410. doi: 10.1016/0014-4827(79)90167-8. [DOI] [PubMed] [Google Scholar]
- Manseau L, Baradaran A, Brower D, Budhu A, Elefant F, Phan H, Philp AV, Yang M, Glover D, Kaiser K, Palter K, Selleck S. GAL4 enhancer traps expressed in the embryo, larval brain, imaginal discs, and ovary of Drosophila. Dev Dynamics. 1997;209:310–322. doi: 10.1002/(SICI)1097-0177(199707)209:3<310::AID-AJA6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Margolis J, Spradling AC. Identification and behavior of epithelial stem cells in the Drosophila ovary. Development. 1995;121:3797–3807. doi: 10.1242/dev.121.11.3797. [DOI] [PubMed] [Google Scholar]
- Millar SE, Freeman M, Glover DM. The distribution of a “mitosis-specific” antigen during Drosophila development. J Cell Sci. 1987;87:95–104. doi: 10.1242/jcs.87.1.95. [DOI] [PubMed] [Google Scholar]
- Moreno S, Nurse P. Regulation of progression through the G1 phase of the cell cycle by the rum1+ gene. Nature. 1994;367:236–242. doi: 10.1038/367236a0. [DOI] [PubMed] [Google Scholar]
- Mulligan PK, Rasch EM. Determination of DNA content in the nurse and follicle cells from wild type and mutant Drosophila melanogaster by DNA-Feulgen cytophotometry. Histochemistry. 1985;82:233–247. doi: 10.1007/BF00501400. [DOI] [PubMed] [Google Scholar]
- Orr W, Komitopoulou K, Kafatos FC. Mutants suppressing in trans chorion gene amplification in Drosophila. Proc Natl Acad Sci. 1984;81:3773–3777. doi: 10.1073/pnas.81.12.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL. Drosophila chorion genes: Cracking the eggshell’s secrets. BioEssays. 1991;13:97–105. doi: 10.1002/bies.950130302. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL, Spradling AC. Drosophila chorion gene amplification requires an upstream region regulating s18 transcription. Mol Cell Biol. 1986;6:4624–4633. doi: 10.1128/mcb.6.12.4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks S, Spradling AC. Spatially regulated expression of chorion genes during Drosophila oogenesis. Genes & Dev. 1987;1:497–509. [Google Scholar]
- Piatti S, Bohm T, Cocker JH, Diffley JFX, Nasmyth K. Activation of S-phase-promoting CDKs in late G1 defines a “point of no return” after which Cdc6 synthesis cannot promote DNA replication in yeast. Genes & Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- Ray RP, Schüpbach T. Intercellular signaling and the polarization of body axes during Drosophila oogenesis. Genes & Dev. 1996;10:1711–1723. doi: 10.1101/gad.10.14.1711. [DOI] [PubMed] [Google Scholar]
- Richardson H, O’Keefe LV, Reed S, Saint R. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development. 1995;121:3371–3379. doi: 10.1242/dev.121.10.3371. [DOI] [PubMed] [Google Scholar]
- Rorth P. Specification of C/EBP function during Drosophila development by the bZIP basic region. Science. 1994;266:1878–1881. doi: 10.1126/science.7997882. [DOI] [PubMed] [Google Scholar]
- Rowles A, Blow JJ. Chromatin proteins involved in the initiation of DNA replication. Curr Opin Genet Dev. 1997;7:152–157. doi: 10.1016/s0959-437x(97)80123-2. [DOI] [PubMed] [Google Scholar]
- Sauer K, Knoblich JA, Richardson H, Lehner CF. Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes & Dev. 1995;9:1327–1339. doi: 10.1101/gad.9.11.1327. [DOI] [PubMed] [Google Scholar]
- Schüpbach T, Wieschaus E. Female sterile mutations on the second chromosome of Drosophila melanogaster. II. Mutations blocking oogenesis or altering egg morphology. Genetics. 1991;129:1119–1136. doi: 10.1093/genetics/129.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder PB, Galanopoulos VK, Kafatos FC. Trans-acting amplification mutants and other eggshell mutants of the third chromosome in Drosophila melanogaster. Proc Natl Acad Sci. 1986;83:3341–3345. doi: 10.1073/pnas.83.10.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spierer A, Spierer P. Similar level of polyteny in bands and interbands of Drosophila giant chromosomes. Nature. 1984;307:176–178. doi: 10.1038/307176a0. [DOI] [PubMed] [Google Scholar]
- Spradling AC. The organization and amplification of two chromosomal domains containing Drosophila chorion genes. Cell. 1981;27:193–201. doi: 10.1016/0092-8674(81)90373-1. [DOI] [PubMed] [Google Scholar]
- Spradling AC. Developmental genetics of oogenesis. In: Bate M, Arias AM, editors. Development of Drosophila melanogaster. Vol. 1. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 1–70. [Google Scholar]
- Spradling AC, Mahowald AP. Amplification of genes for chorion proteins during oogenesis in Drosophila melanogaster. Proc Natl Acad Sci. 1980;77:1096–1100. doi: 10.1073/pnas.77.2.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Orr-Weaver TL. Regulation of DNA replication during Drosophila development. Annu Rev Genet. 1987;21:373–403. doi: 10.1146/annurev.ge.21.120187.002105. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Leys E. Slow replication fork movement during Drosophila chorion gene amplification. In: Kelly T, Stillman B, editors. Cancer cells. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. pp. 305–309. [Google Scholar]
- Spradling A, deCicco DV, Wakimoto BT, Levine JF, Kalfayan LJ, Cooley L. Amplification of the X-linked Drosophila chorion gene cluster requires a region upstream from the s38 chorion gene. EMBO J. 1987;6:1045–1053. doi: 10.1002/j.1460-2075.1987.tb04857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT, O’Farrell PH. Qualifying for the license to replicate. Cell. 1995;81:825–828. doi: 10.1016/0092-8674(95)90000-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swimmer C, Delidakis C, Kafatos FC. Amplification-control element ACE-3 is important but not essential for autosomal chorion gene amplification. Proc Natl Acad Sci. 1989;86:8823–8827. doi: 10.1073/pnas.86.22.8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Knapp D, Nasmyth K. Loading of an Mcm Protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell. 1997;90:649–660. doi: 10.1016/s0092-8674(00)80526-7. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Yue L, Spradling AC. hu-li tai shao, a gene required for ring canal formation during Drosophila oogenesis, encodes a homolog of adducin. Genes & Dev. 1992;6:2443–2454. doi: 10.1101/gad.6.12b.2443. [DOI] [PubMed] [Google Scholar]