

Abstract
Telomerase is a specialized reverse transcriptase that catalyzes telomeric repeat addition at the ends of existing telomeres or fragmented chromosomes. Two telomerase proteins from Tetrahymena thermophila, p80 and p95, were identified on the basis of their association with telomerase activity and telomerase RNA. Here we have produced recombinant versions of these proteins to characterize their functions in the ribonucleoprotein. Our findings indicate that the two proteins can form a complex whose association is independent of RNA. Each protein interacts directly with telomerase RNA, but the p80/p95 complex binds RNA with an affinity substantially greater than either protein alone. We have also characterized the DNA binding properties of p95 and show that it interacts with telomeric substrate DNAs with a specificity characteristic of the functionally defined Tetrahymena telomerase substrate anchor site. Together, these findings suggest a model in which protein–nucleic acid interactions separable from the active site contribute to positioning the template and primer, rather than exclusively the direct nucleic acid–active site interaction typical of other polymerases.
Keywords: Tetrahymena, telomerase, RNA-binding proteins, p95, p80
Telomeres are dynamic structures that define the ends of chromosomes and have critical functions in maintaining chromosome stability. A particular length of telomeric DNA sequence and its associated proteins are important in preventing chromosome loss, end-to-end fusions, and improper segregation (Zakian 1995). Although the complete set of factors governing telomere length is not known, DNA replication, recombination, and degradation are each thought to play a role (Greider 1996). Replication of chromosome ends in most cells and organisms requires telomerase, a specialized reverse transcriptase that uses an intrinsic RNA moiety to reiteratively template the extension of DNA 3′ ends (Blackburn 1992; Greider et al. 1996). Regulation of telomerase-dependent telomere length maintenance has been postulated to have a role in both senescence and cancer (Harley and Villeponteau 1995; Autexier and Greider 1996).
The RNA component of telomerase has been cloned from multiple organisms, facilitated by the complementarity of the telomerase RNA template region and the G-rich strand of telomeric DNA repeats (Greider 1996). Phylogenetic comparisons of telomerase RNA sequences have revealed elements of conserved secondary structure that may be important for assembly and/or function of the ribonucleoprotein (RNP) (e.g., in Tetrahymena; Romero and Blackburn 1991). In vitro and in vivo experiments have demonstrated the importance of RNA template region residues in modulating telomerase activity (Autexier and Greider 1994, 1995; Gilley et al. 1995; Gilley and Blackburn 1996; Prescott and Blackburn 1997) and the impact of altered telomeric repeat synthesis on telomere function (Yu et al. 1990; McEachern and Blackburn 1995; Gilley and Blackburn 1996; Kirk et al. 1997; Prescott and Blackburn 1997). Although these studies have investigated RNA sequences that influence template use, little is known about how RNA interacts with telomerase proteins to define the functional properties of the enzyme.
Knowledge of telomerase protein components is also limited. In Tetrahymena, the first telomerase proteins were identified on the basis of copurification with both telomerase activity and the intrinsic RNA (Collins et al. 1995). One of these two proteins, p80, has homologs in human, mouse, and rat that are also associated with telomerase activity (Harrington et al. 1997; Nakayama et al. 1997). Tetrahymena p80 can be cross-linked to Tetrahymena telomerase RNA (Collins et al. 1995), and the murine homolog of p80 interacts with murine telomerase RNA in a yeast three-hybrid system (Harrington et al. 1997). The other Tetrahymena protein, p95, was shown to crosslink specifically to telomeric DNA primers in an RNA-independent manner (Collins et al. 1995).
Biochemical purification of proteins associated with telomerase activity and RNA in Euplotes aediculatus identified two proteins, p123 and p43 (Lingner and Cech 1996), one of which, p123, also crosslinked to telomeric DNA in the context of the purified enzyme (Hammond et al. 1997). The Saccharomyces cerevisiae homolog of p123, Est2p, was independently identified in a genetic screen for defects in telomerase-dependent telomere maintenance in vivo (Lendvay et al. 1996) and has been shown to be required for telomerase activity in vitro (Counter et al. 1997; Lingner et al. 1997). Although p123 and Est2p share limited overall homology, the pattern of conserved residues highlights a set of reverse transcriptase active site motifs (Lingner et al. 1997). Schizosaccharomyces pombe and human homologs of Est2p (Trt1p and hTRT/hEST2) have also been identified (Meyerson et al. 1997; Nakamura et al. 1997). Like Est2p, S. pombe Trt1p is required for telomerase-dependent telomere maintenance in vivo (Nakamura et al. 1997). Together, these genes define a component of telomerase likely to contribute to the catalytic active site.
In addition to Est2p, three other S. cerevisiae proteins were identified in genetic screens for factors involved in telomerase-dependent telomere maintenance (Lundblad and Szostak 1989; Lendvay et al. 1996). Two of these, Est1p and Cdc13p, have been shown to possess distinct single-stranded telomeric DNA binding capabilities (Lin and Zakian 1996; Nugent et al. 1996; Virta-Pearlman et al. 1996). Est1p has also been shown to interact with the S. cerevisiae telomerase RNA in vitro and in vivo (Lin and Zakian 1995; Steiner et al. 1996; Virta-Pearlman et al. 1996). One allele of CDC13, isolated as Est4p, has the standard phenotypes of a deficiency in telomerase-dependent telomere maintenance (Lendvay et al. 1996; Nugent et al. 1996). In contrast, the original CDC13 allele demonstrates different phenotypes, suggestive of a function for this protein in the regulation of C-strand degradation rather than telomerase activity (Garvik et al. 1995). Est1p is not required for at least some of the S. cerevisiae in vitro telomerase activity assays (Cohn and Blackburn 1995). Thus, specific role(s) for both Est1p and Cdc13p in telomerase-dependent telomere maintenance remain to be determined.
Both telomerase DNA substrate specificity and elongation processivity have been characterized by use of single-stranded primer extension assays in vitro and are likely to involve protein as well as RNA interactions with DNA (Greider et al. 1996). Studies of the nucleotide addition activities of Tetrahymena thermophila, Euplotes crassus, and human telomerases suggest that the enzyme has at least two distinct binding sites on a substrate DNA, one that aligns substrate with the RNA template and one that interacts with residues 5′ to the aligned region and serves as an anchor site for high-affinity binding and processive elongation (Harrington and Greider 1991; Morin 1991; Collins and Greider 1993; Lee and Blackburn 1993; Melek et al. 1996). Binding of a primer to this anchor site is a prerequisite for telomerase-mediated nucleolytic cleavage of nontelomeric primer 3′ ends (Collins and Greider 1993; Melek et al. 1996), which cannot anneal to the RNA template nor provide the affinity for substrate interaction.
Although telomerase activity has been studied in a variety of organisms, the lack of individual, purified, recombinant proteins has hampered detailed study of the assembly and biochemical properties of the enzyme and its interactions with substrate. Although a Tetrahymena homolog of the proposed catalytic component remains to be described, we have characterized two other Tetrahymena telomerase components, p80 and p95, as recombinant proteins. Here, we have reconstructed the genes encoding p80 and p95 for expression in Escherichia coli. Using recombinant proteins and in vitro-transcribed telomerase RNA, we have analyzed the biochemical interactions of the three known components of Tetrahymena telomerase with each other and with DNA substrates. We propose that a complex of p80 and p95 interacts with telomerase RNA to help define and position the RNA template and DNA substrate at the enzyme active site.
Results
Recombinant p80 and p95 form a complex independent of RNA
The unusual codon usage of Tetrahymena includes a biased utilization of universal codons as well as a distinct stop codon usage (Martindale 1989). Only UGA specifies a stop codon in Tetrahymena; both UAG and UAA specify glutamine. Furthermore, the coding regions of the cDNA sequences of p80 and p95 reflect an atypical codon usage even for Tetrahymena (Collins et al. 1995). Together, these features make recombinant expression from the Tetrahymena cDNA sequences unfeasible. Therefore, we redesigned the complete coding sequence of both genes to reflect an optimal codon usage for E. coli expression. To create the new sequences, we adopted a strategy for synthetic gene construction based on methods used for a variety of smaller polypeptides (e.g., Dillon and Rosen 1993; Holler et al. 1993), which entails PCR amplification of overlapping oligonucleotides to construct double-stranded DNAs suitable for cloning (Fig. 1A ; see Materials and Methods).
Figure 1.
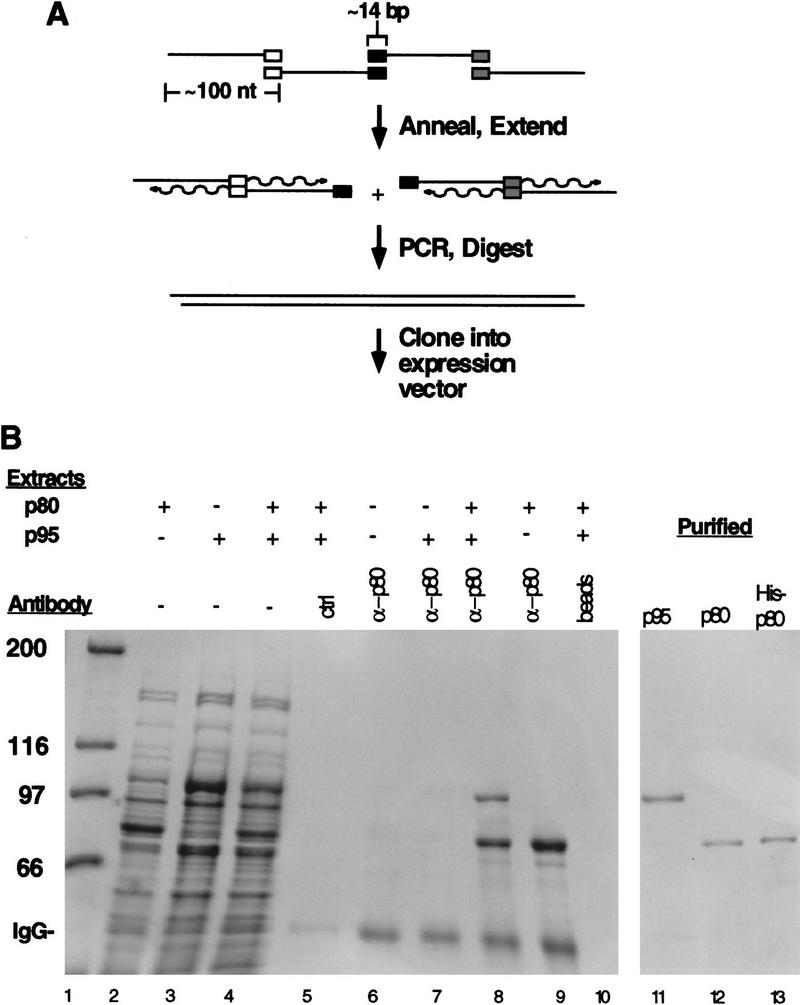
Gene construction, expression, and purification of p80 and p95. (A) Oligonucleotides of ∼100 nucleotides with an overlap of ∼14 bp were annealed and extended in pairs. Pairwise extension reactions were pooled and amplified by PCR to generate DNA for restriction digestion and cloning. (B) E. coli extracts from cells expressing p80 (lane 2), p95 (lane 3), or mixed p80 and p95 extracts (lane 4) were used in immunoprecipitation reactions. Antibodies were covalently coupled to CNBr-activated Sepharose at equivalent concentrations for these experiments; p80 antibody beads alone are shown in lane 6. Some antibody was dissociated from the resin in reducing SDS-PAGE loading buffer, migrating at the position indicated by IgG. Anti-p80 beads were used to immunoprecipitate from p95 extract (lane 7), mixed p80 and p95 extracts (lane 8), or p80 extract (lane 9). Control reactions included an antibody raised against an unrelated protein that was used to immunoprecipitate from mixed p80 and p95 extracts (lane 5) and Sepharose beads used to immunoprecipitate from the same mixed extracts in the absence of binding and conjugation of p80 antibodies (lane 10). The three purified proteins used in this study are shown in lanes 11–13. (Lane 1) Molecular mass markers indicated at left in kD.
The p80 subunit was expressed in E. coli either as an untagged protein purified in two steps or as an amino-terminally 6×-His-tagged fusion protein purified over Ni2+–agarose (see Materials and Methods). The p95 subunit was expressed as an untagged protein and purified by several chromatographic steps (see Materials and Methods). By Coomassie staining of an SDS-PAGE gel, the three recombinant proteins appeared >95% pure (Fig. 1B, lanes 11–13). We assayed for interaction between p80 and p95 using antibodies made against recombinant p80 (Fig. 1B). Antibodies against p80 could immunoprecipitate the protein from a crude E. coli extract of cells expressing p80 (lane 9), but did not precipitate any protein from an extract of cells expressing only p95 (lane 7). In mixed extracts containing both p80 and p95, p80 antibodies co-precipitated both proteins (lane 8). As expected, a control antibody against an unrelated protein precipitated neither p80 nor p95 from mixed extracts (lane 5). Thus, p80 and p95 specifically interact with one another even in the absence of telomerase RNA.
RNA binding activity of p80 and p95
Initial studies showed that p80 crosslinked to a synthetic telomerase RNA added back after nuclease treatment of the purified enzyme (Collins et al. 1995). Using recombinant proteins, we examined the RNA binding properties of p80, p95, and the two-protein complex with a direct method. Electrophoretic mobility shift assays were used to probe the interaction of purified p80 and p95 with T. thermophila telomerase RNA (Fig. 2). Synthetic telomerase RNA radiolabeled with 32P was used in a mobility shift assay with increasing amounts of p80 (lanes 2–5), p95 (lanes 6–9), or the p80/p95 complex formed from individually purified proteins (lanes 10–13). RNA binding by p80 or p95 alone generated several mobility shifts that accumulated with increasing protein concentration. This multiplicity of shift forms may derive from multiple proteins interacting with different sites on the same RNA, although aggregates based on protein-protein interactions are also possible. The p80/p95 complex bound to telomerase RNA to generate predominantly a single, discrete mobility shift. A second, more slowly migrating mobility shift appeared at the highest protein to RNA ratios when RNA was limiting in the binding reaction; this could derive from the binding of one RNA to two protein complexes at different sites. The affinity of p80 and p95 individually for RNA was similar, but the two-protein complex bound with an affinity that was substantially greater than either protein alone (∼15–40 nm). Thus, the two proteins can independently interact with telomerase RNA, but the complex binds telomerase RNA with unique, nonadditive properties.
Figure 2.
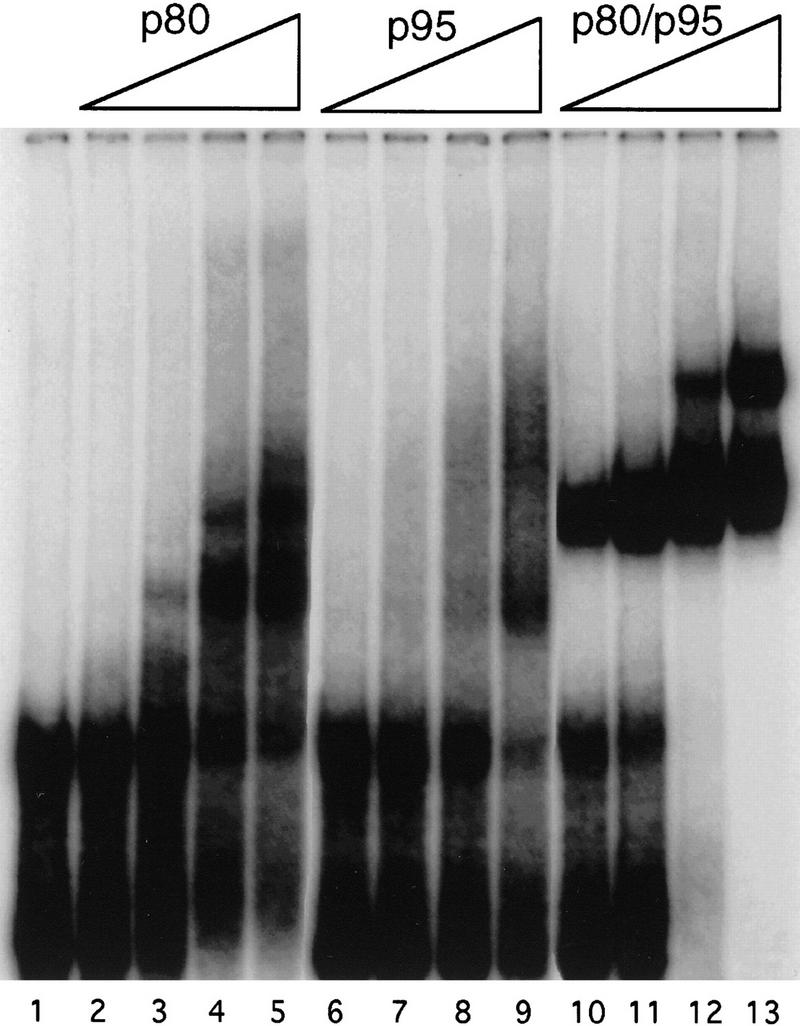
Both p80 and p95 bind to telomerase RNA. A mobility shift assay was used to test the interaction of telomerase proteins with in vitro-transcribed T. thermophila telomerase RNA. (Lane 1) Radiolabeled RNA probe (10 nm) in the absence of protein. The p80 (lanes 2–5), p95 (lanes 6–9), or p80/p95 complex (lanes 10–13) was titrated by twofold increases from 40 to 320 nm with a constant amount of RNA (10 nm). This experiment included unlabeled nonspecific competitor 5S RNA at 2 μm. Some Tetrahymena telomerase RNA transcription reactions produced two mobility forms of telomerase RNA; results were the same with RNA transcription reactions yielding RNA of only one mobility.
To test the specificity of these interactions, we first constructed several sequence variants of the telomerase RNA that eliminated evolutionarily conserved features of primary and secondary structure, such as the pseudoknot. Although the binding affinity of these mutant RNAs demonstrated some length dependence, no obvious domains singularly important for binding were found (data not shown). We also examined binding of the p80/p95 complex to telomerase RNA in the presence of various unlabeled competitor RNAs at 50- or 500-fold mass excess over the radiolabeled full-length telomerase RNA (Fig. 3A). Although unlabeled telomerase RNA competed for binding efficiently, total yeast RNA competed even better (cf. lanes 3–4 with lanes 13–14). A random transcript of similar length to the telomerase RNA also competed for binding (Random-146; lanes 9–10). Neither tRNA (lanes 5–6) nor 5S RNA (lanes 7–8), however, showed any significant competition. Although both tRNA and 5S RNA are shorter than the synthetic telomerase RNA, their lack of competition is not merely a feature of length as a 64-nucleotide region of the telomerase RNA from its 5′ end through the template region competed well (Tel RNA 1–64; lanes 11–12). These data indicate that the in vitro interaction of p80/p95 with telomerase RNA shows a limited degree of specificity. Various protocols for refolding the in vitro-transcribed telomerase RNA, such as heating and slow cooling in the presence of different ions and salt concentrations, did not alter the binding specificity (data not shown).
Figure 3.

The specificity of telomerase RNA association with recombinant p80/p95 complex or with purified, endogenous Tetrahymena telomerase proteins is similar in vitro. (A) The p80/p95 complex was used in a mobility shift assay at a concentration of 100 nm with 40 nm radiolabeled telomerase RNA. (Lane 1) Probe in the absence of proteins. The mobility shift caused by p80/p95 in the absence of competitors is shown in lane 2. Because molar equivalents could not be determined for total yeast RNA, unlabeled competitor RNAs were added into the binding reaction at 100 ng and 1 μg (5- and 50-fold the mass amount of probe): (lanes 3,4) total yeast RNA; (lanes 5,6) tRNA; (lanes 7,8) 5S RNA; (lanes 9,10) a random 146 nucleotide transcript (see Materials and Methods); (lanes 11,12) a 64 nucleotide region of the T. thermophila telomerase RNA; (lanes 13,14) 159-nucleotide full-length T. thermophila telomerase RNA. All reactions except that in lane 15 were conducted with a 5-min heating step at 37°C with 5 mm EDTA (conditions for reconstitution of telomerase activity with recombinant RNA and endogenous proteins). The lane 15 reaction has the EDTA step omitted to show that these conditions do not affect the nature of the shift. (B) Native telomerase was reconstituted with in vitro-transcribed telomerase RNA in the presence of the same RNA competitors as in A. Native telomerase activity on d(G4T2)3 is shown in lane 1. Lanes 4–15 have the indicated RNAs added during the reconstitution step. These RNAs were added at 400 ng and 1 μg (two- and fivefold the level of wild-type telomerase RNA). Lanes 14 and 15 have 400 ng and 1 μg of additional wild-type telomerase RNA to show that excess RNA per se does not inhibit the reaction. Some of the activity in lanes 12 and 13 is attributable to the ability of the competitor RNA to reconstitute an active enzyme (data not shown).
Strikingly, reconstitution of telomerase activity with in vitro-transcribed telomerase RNA and purified endogenous proteins from Tetrahymena cells demonstrated the same specificity profile of competition as in vitro telomerase RNA binding by recombinant p80/p95. Tetrahymena telomerase activity can be partially reconstituted in vitro, by use of micrococcal nuclease (MNase) to degrade the endogenous RNA followed by substitution with in vitro-transcribed telomerase RNA (Autexier and Greider 1994). Reconstitution of purified Tetrahymena telomerase proteins with the same synthetic RNA as used in the binding studies above was performed in the presence of the same nonspecific RNA competitors as in Figure 3A. Telomerase activity in the purified sample (Fig. 3B, lane 1) was completely abolished by MNase treatment (lane 2) and partially restored by the addition of in vitro-transcribed telomerase RNA (lane 3). The same nonspecific RNAs that competed for recombinant protein–synthetic RNA interactions also competed for reconstitution of telomerase activity with endogenous proteins and synthetic RNA (Fig. 3, cf. A and B). Addition of full-length telomerase RNA at the same concentrations as the competitors did not inhibit reconstitution, demonstrating that higher RNA concentrations per se were not inhibitory (Fig. 3B, lanes 14–15). In addition, we have observed that like p80/p95, MNase-treated, purified telomerase shows a competition of telomerase RNA mobility shift by nonspecific RNAs (data not shown). Thus, although it is not yet possible to recapitulate in vitro the specific telomerase RNA association evident in vivo, the RNA binding specificity of recombinant p80/p95 reflects that of RNA association with endogenous proteins required to generate an active, reconstituted enzyme.
Recombinant p95 preferentially binds telomeric sequences
The mechanisms for DNA substrate recognition by telomerase have been a point of earnest study. Because p95 can crosslink specifically to single-stranded telomeric DNA in the context of the purified enzyme, we tested the DNA binding properties of recombinant p95 using mobility shift assays with typical elongation substrates. Mixing of radiolabeled, single-stranded d(G4T2)3 with p95 in the presence of excess double-stranded DNA (poly [d(I-C)]) resulted in a discrete mobility shift (Fig. 4A, lane 2) that could be supershifted with antibodies against p95 (data not shown). Unlabeled d(G4T2)3 competed for the shift of radiolabeled d(G4T2)3 (lanes 3–4), whereas even a 500-fold excess of nontelomeric sequence of similar length could not compete (lanes 5–6). Total yeast RNA, which is likely to contain some G-rich sequences, competed to some extent (lanes 7–8; see below), as did telomerase RNA (lanes 11–12).
Figure 4.
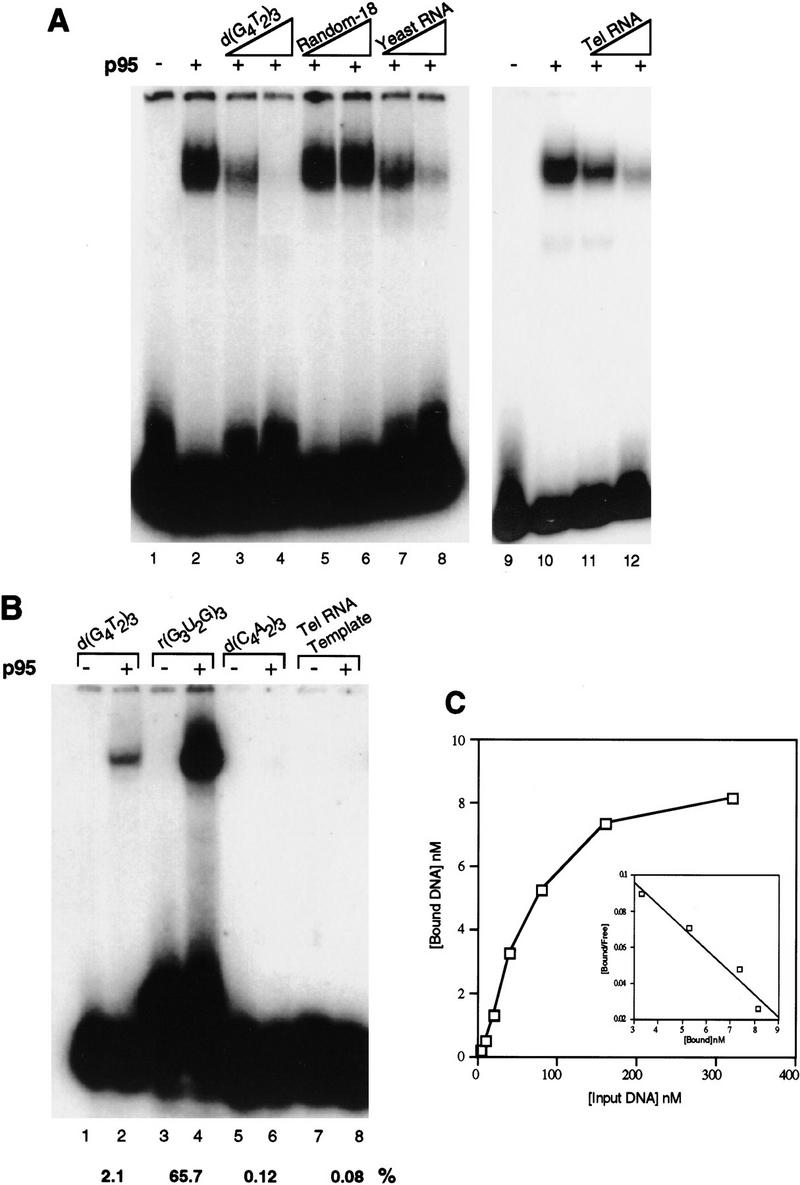
Single-stranded, G-rich telomeric sequence is preferred for interaction with p95. (A) Mobility shift assay with p95 at 100 nm and radiolabeled d(G4T2)3 at 20 nm (double-stranded DNA competitor poly[d(I–C)], only; see Materials and Methods). (Lanes 1,9) probe alone; (lanes 2,10) mobility shift induced by p95. Lanes 3–6 and 11 and 12 have the indicated unlabeled competitor DNA or RNA added at 50- or 500-fold the molar concentration of probe: (lanes 3,4) d(G4T2)3; (lanes 5,6) a random 18-nucleotide DNA sequence (Random-18); (lanes 11,12) T. thermophila telomerase RNA. Because molar equivalents could not be determined for total yeast RNA, lanes 7 and 8 have total unlabeled yeast RNA added at 100 ng and 1 μg [roughly double the mass of d(G4T2)3 in (lanes 3 and 4)]. The p95 used in lanes 7 and 8 was a side fraction; the weak, lower mobility shift band in these lanes likely represents binding to a breakdown product of p95 in this fraction. (B) Direct mobility shift assays of radiolabeled telomeric and nontelomeric DNA and RNA sequences by p95 (double-stranded DNA competitor poly[d(I–C)], only). Probes were used at 20 nm with 100 nm p95 as follows: (lanes 1,2) d(G4T2)3; (lanes 3,4) r(G3U2G)3; (lanes 5,6) d(C4A2)3; (lanes 7,8) the telomerase RNA template region, rCAACCCCAAAAAUCUAGUGC. The percentage probe bound in each case was quantitated and indicated below the appropriate lane as %. (C) The binding of p95 to single-stranded, G-rich telomeric DNA was measured using 25 nm protein and twofold increases of d(G4T2)3 from 5 to 320 nm. A Scatchard plot (inset) was derived from data at the four highest protein concentrations (40–320 nm).
We also compared the interaction of p95 with G-rich, telomeric-sequence DNA relative to other single-stranded nucleic acids of similar length (Fig. 4B). These included a telomeric ribonucleotide primer r(G3U2G)3 (lanes 3–4), a d(C4A2)3 sequence to mimic the C-rich strand of telomeric DNA (lanes 5–6), and a 20-nucleotide RNA sequence spanning the template region of telomerase RNA (lanes 7–8). Neither d(C4A2)3 nor the template RNA sequence were bound by p95. These are not substrates for telomerase elongation, and the RNA template region is not expected to interact strongly with telomerase proteins on the basis of its accessibility to modification in the RNP (Zaug and Cech 1995). The binding of p95 to an 18-nucleotide G-strand telomeric-sequence RNA, however, exceeded that to a comparable 18-nucleotide telomeric DNA by at least 30-fold in our standard assay conditions (Fig. 4B, cf. lanes 3–4 with lanes 1–2). This preference for telomeric-sequence RNA relative to DNA reflects the specificity of substrate interaction at the Tetrahymena telomerase anchor site: Telomeric RNA primers have a Km for elongation well below that of the corresponding DNA primers and can serve as potent competitive inhibitors of DNA primer elongation (Collins and Greider 1995). These experiments also underscore the point that p95 shows preferential binding to single-stranded, G-rich telomeric sequence, whether DNA or RNA.
The affinity of p95 for d(G4T2)3 was measured by quantitation of mobility shifts with constant protein and a titration of DNA concentration (Fig. 4C). A Scatchard plot of the relevant concentration range in this experiment yielded a Kd value of 85 nm for p95–DNA interaction. The Km of telomerase for elongation of a single-repeat primer, which binds to the enzyme predominantly through RNA template interactions, is in the micromolar range (Collins and Greider 1993). The Km for elongation of longer primers that can interact with both the anchor and template sites is 10–50 nm (Collins and Greider 1993). Thus, the observed Kd of DNA interaction with p95 reasonably reflects the expected contribution of protein components to substrate binding.
The interaction of p95 with single-stranded, G-rich telomeric DNA also appears to occur in the context of the p80/p95 complex. In either E. coli lysate (Fig. 5A, lane 3) or as a purified protein (Fig. 5B, lane 2), p80 itself showed a weak mobility shift of the single-stranded telomeric DNA d(G4T2)3 in the presence of excess competitor nucleic acids. This shift was distinct from those of other proteins in a mock E. coli extract (Fig. 5A, lane 2) and was distinct from the stronger p95 mobility shift from E. coli extract (Fig. 5A, lane 4) or purified p95 (Fig. 5B, lane 3). We were unable to estimate a dissociation constant for p80 DNA binding, however, because of the low affinity of the interaction. Although p80 showed a preference for single-stranded telomeric sequence similar to that of p95 (data not shown), the significance of such a low-affinity interaction remains to be established. The complex formed by the two proteins and DNA (Fig. 5A, lane 6 and B, lane 4) had only a slight mobility difference from p95–DNA alone (Fig. 5A, lane 4 and 5B, lane 3). The affinity of the p80/p95 complex for DNA, however, appeared less than the affinity of p95 alone. As a control, no change in p95–DNA interaction was induced by mixing of p95 extract with mock extract lacking p80 (Fig. 5A, lane 5). Also, the p80/p95 DNA mobility shift was specifically supershifted into the well of the gel by antibodies against p80 (Fig. 5A, cf. lanes 5 and 7), whereas the mixed p95/vector extract mobility shift was not (data not shown). Therefore, p80 must be a part of the weaker mobility shift complex formed when the two proteins are mixed together. Furthermore, the DNA mobility shift of purified, endogenous MNase-treated or native telomerase (Fig. 5B, lanes 5 and 6) appeared remarkably similar to that of the mixed recombinant proteins and could also be supershifted by p80 antibodies (data not shown). Because p95 DNA binding demonstrated the highest affinity, the p80/p95 complex was not altered in specificity relative to p95, and p95 cross-links to substrates in the endogenous RNP (Collins et al. 1995), our characterization focused on more detailed studies of the p95–DNA interaction.
Figure 5.

The p80/p95 complex binds single-stranded, G-rich telomeric sequence DNA. (A) Approximately 2 μg of total protein from E. coli lysates of the empty vector, pRSET (lane 2), p80 (lane 3), or p95 (lane 4), were mixed with radiolabeled d(G4T2)3 at 20 nm final concentration in a mobility shift assay. For combined extract shifts (as indicated in lanes 5 and 6), 2 μg of each extract was used. Extracts were comparable to those shown in Fig. 1C, with similar amounts of p80 or p95 per microgram of extract protein. Assays were conducted in the presence of a mix of single-stranded 18-nucleotide competitor DNAs and double-stranded poly[d(I–C)] (see Materials and Methods). The p80/p95 complex mobility shift (lane 6) was supershifted to the well by the presence of p80 antibodies (lane 7). (B) Purified proteins at a concentration of 100 nm were mixed as indicated with radiolabeled d(G4T2)3 at a concentration of 20 nm. All reactions contained the same mixture of competitor DNAs as in A. MNase-treated telomerase with degraded endogenous RNA (lane 5) and native telomerase (lane 6) are purified fractions containing substantially less protein.
Recombinant p95 DNA binding specificity correlates with telomerase substrate specificity
To compare p95 binding specificity to that of telomerase substrate elongation, the standard 18-nucleotide elongation primer, d(G4T2)3, was used as probe in a mobility shift assay and competed with similar single-stranded telomeric sequences of varying length (Fig. 6A). Competition with three repeats (lanes 3–5) was better than competition with two repeats (lanes 6–8), and a single repeat did not significantly compete for the three-repeat probe (lanes 9–11). This length preference of p95 binding is comparable to that of purified telomerase activity on the same primers (Fig. 6B, lanes 1–4). The ability of a single repeat, dG4T2, to compete for p95 binding or to be elongated was dramatically enhanced by the addition of 18 nucleotides of nontelomeric 5′ sequence (Fig. 6A, cf. lanes 9–11 and 21–23; Fig. 6B, cf. lanes 4 and 8). Relatively efficient elongation of primers with limited telomeric 3′ end sequence and longer nontelomeric 5′ ends has been observed previously (e.g., Harrington and Greider 1991) and is likely attributable to a reduced overall sequence specificity for anchor site binding relative to primer 3′ end annealing at the template.
Figure 6.

DNA binding by p95 and substrate elongation by purified telomerase show a similar length dependence. (A) Radiolabeled d(G4T2)3 was used at a concentration of 20 nm with 100 nm p95. Assays contained single-stranded competitor DNA (Random-18) as well as double-stranded poly[d(I–C)]. In lanes 3–23, the indicated single-stranded competitors were added at 5-, 50-, and 500-fold that of the probe concentration. (B) Telomerase activity assays were performed with purified telomerase with the indicated single-stranded DNA primers at a concentration of 600 nm.
Although DNA length is an important factor in p95 binding specificity, sequence is also critical. While a single dG4T2 repeat did not compete for p95 interaction with a three-repeat telomeric substrate, addition of a dG residue to the 3′ end of the repeat allowed some degree of competition (Fig. 6A, lanes 18–20). Addition of two dT residues to the 5′ end of a single repeat, however, did not significantly improve competition (Fig. 6A, lanes 15–17), even though this primer is slightly longer than dG4T2G. This same sequence preference is also characteristic of telomerase activity. Although neither primer is processively elongated, addition of the first nucleotides onto dG4T2G is more efficient than addition to dT2G4T2, the longer primer (Fig. 6B, lanes 6,7; Collins and Greider 1993). Additional experiments will be required to investigate more precisely the sequence specificity of p95–DNA interaction to determine whether the number or spacing of G residues or the specific permutation of telomeric sequence is most important for binding.
We next investigated whether the position of telomeric repeats within a DNA sequence influenced p95 interaction. We compared the relative binding of p95 to four 36-nucleotide oligonucleotides that contained 12 nucleotides of G-strand telomeric sequence at the 3′ end, the 5′ end, internally, or not at all. To make this comparison, we radiolabeled the 3′ telomeric oligonucleotide and used unlabeled DNA of each sequence as competitor (Fig. 7A). Interestingly, the strongest competitor was the 5′ telomeric primer (lanes 3–5), whereas 3′ (lanes 6–8) and internally telomeric sequence (lanes 9–11) competed better than fully nontelomeric sequence (lanes 12–14). At the anchor site, this pattern of binding would allow stronger interaction with the 5′ region of the substrate, to provide a greater flexibility in positioning of the primer 3′ end.
Figure 7.
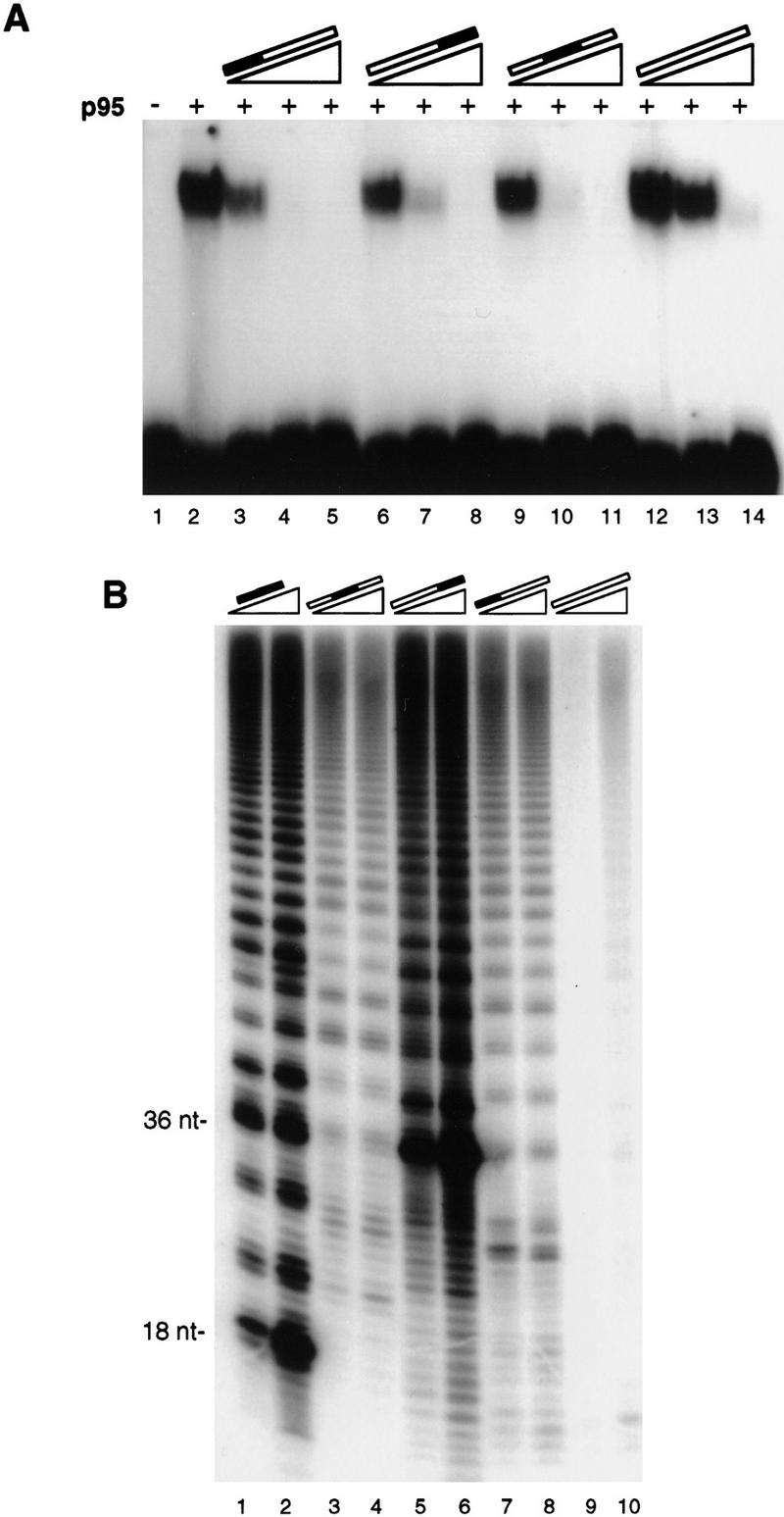
Substrates containing telomeric repeats at any position are bound by p95 and elongated by telomerase. (A) A 36-nucleotide oligonucleotide with two repeats of telomeric sequence at the 3′ end and two repeats of a 12-nucleotide nontelomeric cassette at the 5′ end was radiolabeled and assayed at 40 nm with 100 nm p95. The p95 shift of this probe (lane 2) was competed (lanes 3–14) with sequences altered in the position of the telomeric repeats. Competitor concentrations were 5-, 50-, and 500-fold that of the probe concentration. Open bars indicate nontelomeric sequence; solid bars indicate telomeric sequence. (B) The same 36-nucleotide primers as in A were used as substrates for elongation in activity assays with purified telomerase. Each primer was used at a concentration of 200 nm or 2 μm (equal to the lowest two competitor concentrations above). (Lanes 1,2) Elongation of the standard 18-nucleotide d(G4T2)3 for comparison.
For comparison with p95 binding, we examined the relative elongation of these 36-nucleotide primers using purified telomerase. Because nontelomeric sequence at a primer 3′ end disallows base-pairing as a mechanism for recruitment to and alignment with the template, oligonucleotides with nontelomeric 3′ ends must interact with the anchor site to become substrates for elongation. Their elongation, however, will be less efficient than that of an oligonucleotide with telomeric 3′ end, even if bound to the anchor site equally well. As anticipated, the 3′ telomeric primer was extended most efficiently of the set when compared to the standard 18-nucleotide primer (Fig. 7B, lanes 5,6 and 1,2). Yet, both the internally telomeric and 5′ telomeric primers were extended by telomerase (lanes 3,4 and 7,8). At least part of the extension of these primers occurred subsequent to telomerase-mediated cleavage of the substrate to generate an appropriate 3′ end. The internally telomeric primer yielded products beginning 12 nucleotides below the 36-nucleotide primer length, indicative of cleavage of the entire nontelomeric 3′ end to the position of the internal telomeric repeats. The 5′ telomeric primer also yielded products shorter than primer length, but not wholly corresponding to cleavage through all 24 nucleotides preceding the telomeric sequence. Instead, these short products appear to represent cleavage through 8 nucleotides 5′ of the 36-nucleotide primer 3′ end to a fortuitous dG, which served as a starting point for extension. The fully nontelomeric substrate could be weakly elongated only at a relatively high concentration of 2 μm. Many nontelomeric sequences can be elongated by telomerase, but with the requirement for greater length and/or higher primer concentration (e.g., Harrington and Greider 1991; Wang and Blackburn 1997). Reflecting this requirement, the 36-nucleotide nontelomeric sequence competed better than the 18-nucleotide nontelomeric sequence for p95 binding (cf. Fig. 7A, lanes 12–14, with Fig. 4A, lanes 5,6) and served as a better elongation substrate (cf. Fig. 7B, lanes 9,10 with Fig. 6B, lane 10).
We also examined p95 binding to double-stranded DNA substrates and double-stranded DNA with single-stranded, G-rich telomeric repeat overhangs. Both of these are potential substrates for telomerase in vivo, yet in vitro, telomerase cannot elongate double-stranded DNAs at all and elongates overhangs of less than 19 nucleotides poorly (Wang and Blackburn 1997; data not shown). To address the relative affinity of p95 for these different types of DNAs, we used a single-stranded nontelomeric cassette of 16 or 15 nucleotides to anneal with complementary oligonucleotides that would leave single-stranded, G-rich telomeric 3′ overhangs of 5, 11, 17, or 29 nucleotides or 5′ overhangs of 11 or 17 nucleotides. Also, fully double-stranded DNAs were constructed with telomeric sequence at the 5′ or 3′ end of the G-rich strand, or lacking any telomeric sequence. In a direct binding assay, both 5′ and 3′ telomeric overhang substrates showed a length-dependent increase in affinity for p95, although the 3′ overhangs were bound more strongly than the 5′ ones (Fig. 8A). This differs from the preference for a 5′ telomeric block in single-stranded sequences (Fig. 7A). DNA binding by p95 demonstrated no distinct preference for 3′ overhangs relative to the telomeric single strand of these partial duplexes alone (Fig. 8B). Therefore, inefficient use of 3′ overhang substrates by the native enzyme could be attributable either to some factor other than p95 binding or to altered p95 affinity for overhangs in the context of the holoenzyme. Double-stranded DNAs with 5′ or 3′ telomeric sequence and nontelomeric double-stranded DNAs were bound poorly by p95 (Fig. 8A), and none of these DNAs was elongated by purified telomerase (data not shown).
Figure 8.

Partial duplex DNAs but not double-stranded DNAs are bound by p95. Assays contained single-stranded competitor DNA (Random-18) as well as double-stranded poly[d(I–C)]. The percentage of probe bound was quantitated and indicated below the appropriate lane as %. (A) Reactions were performed in sets of three, with the first lane showing probe alone (20 nm); the second with added p95 at 50 nm; and the third with added p95 at 150 nm. The fully telomeric sequence used in lanes 1–3 is d(G4T2)3. Other substrates contain G-rich, telomeric overhangs of 5 nucleotides (lanes 4–6); 11 nucleotides (lanes 7–9); 17 nucleotides (lanes 10–12); or 29 nucleotides (lanes 13–15) at the 3′ end or at the 5′ end of 11 nucleotides (lanes 16–18); or 17 nucleotides (lanes 19–21). Oligonucleotides were also annealed to generate fully double-stranded DNAs with 17 nucleotides of 3′ telomeric sequence (lanes 22–24); 11 nucleotides of 5′ telomeric sequence (lanes 25–27); or fully nontelomeric sequence (lanes 28–30). Open bars indicate nontelomeric sequence; solid bars indicate telomeric sequence. (B) The G-rich strands of the 3′ overhang substrates in A were radiolabeled and compared to their partial duplex counterparts. Reactions are in sets of two, with the first lane showing the probe alone (20 nm), and the second with added p95 at 100 nm.
Discussion
Telomerase protein–telomerase RNA interactions
We have begun the complete reconstitution of telomerase from recombinant components. Here, we describe a three-component assembly of Tetrahymena telomerase: p80/p95/RNA. This complex was isolated as a stable, stoichiometric RNP (Collins et al. 1995). The basis for the stability of this complex is evident from our characterization of the interactions between recombinant components. In yeast and likely human cells, telomerase RNA-containing RNPs assemble and accumulate at comparable levels in cells that express or lack the catalytic subunit (Lingner et al. 1997; Nakamura et al. 1997). Conventional purification protocols for mammalian telomerases frequently result in a loss of specific activity, suggesting that the active RNP may not be a highly stable RNP in vitro (Greider et al. 1996). The exception to this theme may be the ciliate E. aediculatus, for which the endogenous complex isolated by purification contains a stoichiometric quantity of p123 (Lingner and Cech 1996). It will be interesting to determine in Tetrahymena whether the apparently substoichiometric quantity of the catalytic subunit relative to p80/p95/RNA in the purified RNP (Collins et al. 1995) reflects a substoichiometric association in vivo or a consequence of the extract preparation procedure.
The similar pattern of competition for in vitro telomerase RNA binding by recombinant p80/p95 and endogenous Tetrahymena telomerase proteins, assayed by reactivation of the MNase-treated enzyme, suggests that telomerase RNA binding to the p80/p95 complex is a critical determinant of RNA association during reconstitution of telomerase activity in vitro. The reason(s) for the low specificity of RNA binding in vitro, however, are not clear. Although recombinant telomerase RNA is capable of reactivating MNase-treated Tetrahymena telomerase (Autexier and Greider 1994), this reconstitution requires only a limited region of the full-length telomerase RNA (L. Gandhi and K. Collins, unpubl.). Therefore, only minimal requirements for RNA folding and RNP assembly may be assayed in this manner. The conformation of telomerase RNA and its interaction with proteins in regions outside the template could be substantially different in endogenous and reconstituted RNPs. To create an authentic RNP, post-transcriptional RNA modifications, post-translational protein modifications, or protein–protein and protein–RNA contacts that are facilitated by a particular in vivo location or assembly pathway could each be required.
Substrate binding by p95
DNA substrate recognition is a key feature of telomerase that is thought to involve both RNA template interactions in the active site as well as protein interactions in the anchor site. Detailed investigation of the p95–DNA interaction indicated several interesting features that are likely to be relevant to the mechanisms of both substrate elongation and nucleolytic cleavage. The affinity and specificity of p95 for telomeric sequences closely correlates with that of the native enzyme. In addition, the affinity of the p80/p95 DNA interaction was reduced relative to that of p95 alone. Although additional experiments with p80, p95, and the other holoenzyme components will be required before models for primer binding and positioning can be made in detail, our results suggest that substrate interactions may be modulated in the context of the RNP to regulate positioning during elongation.
The interaction of p95 with single-stranded, G-rich telomeric DNA differs in several respects from that observed for proposed single-stranded telomeric DNA binding proteins. First, because telomerase from most species rapidly releases product DNA after elongation, telomerase would logically have a lower affinity for telomeric DNA than a stably associated telomere-binding protein. The interaction of p95 with telomeric DNA has a Kd of ∼100 nm. In contrast, the proposed single-stranded telomeric DNA binding protein S. cerevisiae Cdc13p has a binding affinity for similar DNAs that is 100 times greater than p95 (Nugent et al. 1996). The α/β telomere protein complex from the ciliate Oxytricha also binds to single-stranded telomeric DNA with nanomolar affinity (Fang et al. 1993). Second, p95 demonstrates a reduced sequence specificity of binding relative to single-stranded telomeric DNA binding proteins (Cardenas et al. 1993; Lin and Zakian 1996; Nugent et al. 1996), consistent with the relaxed sequence specificity of substrate elongation by telomerase in vitro compared with telomere function in vivo (e.g., see Hanish et al. 1994).
Single-stranded, G-rich sequence telomeric DNA binding has also been demonstrated for S. cerevisiae Est1p (Virta-Pearlman et al. 1996). DNA binding by Est1p has some similarities to p95, including a moderate affinity and an imprecise sequence specificity. Est1p has been implicated as a protein associated with telomerase activity and telomerase RNA (Lin and Zakian 1995; Steiner et al. 1996), although like p95, in vitro telomerase RNA binding by Est1p is not specific with respect to other RNAs (Virta-Pearlman et al. 1996). Although in vitro telomerase activity assays in S. cerevisiae extract require Est2p and not Est1p (Cohn and Blackburn 1995; Lingner et al. 1997), these assays may not depend as significantly on anchor site interactions. Future biochemical characterization of both primer elongation and DNA binding by S. cerevisiae telomerase will be required to determine whether Est1p contains a substrate interaction site functionally comparable to that of p95.
Potential roles of p80/p95 in the holoenzyme
Telomerase proteins are likely to carry out numerous functions in the RNP, including telomerase RNA interactions, RNA template positioning, DNA substrate interactions, nucleotide addition, and nucleolytic cleavage. Also, at least in ciliates, telomerase proteins or other associated factors must regulate the efficiency of activity on telomeric versus nontelomeric substrates. The task of assigning these functions at a molecular level is complicated by the lack of extensive evolutionary conservation of telomerase component primary sequences and the lack of obvious nucleic acid binding motifs in telomerase proteins.
The biochemical features of recombinant p80 and p95 described here suggest a model in which the p80/p95/RNA complex serves to help present the template and substrate to the active site. The RNA template itself does not appear to be tightly bound to the proteins of the RNP. In vivo, the template region is the only substantial portion of the RNA accessible to modification (Zaug and Cech 1995). Thus, the template is likely to be broadly defined by interactions of protein(s) with RNA sequences flanking the template itself. We have shown that a 20-nucleotide RNA containing the portion of the Tetrahymena telomerase RNA template region accessible to modification in the RNP is not bound by p80 or p95 (Fig. 4; data not shown). In contrast, the catalytic component is predicted to interact directly with template sequence. A 64-nucleotide region of the telomerase RNA surrounding the template, however, can compete with the full-length RNA for p80/p95 binding or for reconstitution of MNase-treated, purified telomerase (Fig. 3) and can itself serve to reactivate MNase-treated enzyme (L. Gandhi and K. Collins, unpubl.). Thus, p80/p95 can associate with RNA sequences flanking the template. Furthermore, numerous substrate elongation and nucleolytic cleavage assays have revealed that the interaction of protein with a primer 5′ region is critical for substrate affinity and 3′ end placement at the active site. We find that p95 possesses a DNA binding activity with the properties anticipated for the Tetrahymena telomerase protein site interacting with a primer 5′ end.
Polymerases described to date recognize nucleic acid templates and nucleic acid substrates by direct interaction of protein side chains in the active site cleft with the template and primer (Joyce and Steitz 1994). Whether the catalytic component of Tetrahymena telomerase alone is sufficient for defining the template region and interacting stably with substrate or whether these features require additional components within the RNP remains to be determined. Some hints of complexity in active site interactions have been suggested by the consequences of simple RNA sequence changes in the template region on the processivity and fidelity of catalytic activity (Autexier and Greider 1995; Gilley et al. 1995; Gilley and Blackburn 1996; Prescott and Blackburn 1997). A functional characterization of the biochemical features of all recombinant telomerase components should add insight in the quest to decipher how polymerases may have bridged the transition in evolution from a primordial RNA world to the current world dominated by DNA genomes.
Materials and methods
Gene construction
The complete gene sequence of both proteins (2163 bp for p80 and 2622 bp for p95) was redesigned to optimize for gene expression (sequences available on request from kcollins@socrates.berkeley.edu). Overlapping oligonucleotides of 85–100 nucleotides were synthesized by Operon Technologies and gel purified. Many strategies were attempted for synthesis; the methods used for final products all contained the steps described below. Oligonucleotide pairs were annealed and extended with Pfu or Vent polymerase. These pairs were pooled and PCR amplified. PCR products were gel purified, restriction digested, and cloned into a pRSET-based vector (Invitrogen). Mutations were corrected either by resynthesis or by a site-directed mutagenesis protocol based on the QuikChange Site-Directed Mutagenesis method (Stratagene).
Expression and purification of recombinant proteins
For bacterial expression of p80, BL21-DE3 cells were transformed with 6× His-tagged or untagged p80 in pRSET. Colonies were inoculated into LB with 0.4% glucose and 100 μg/μl ampicillin, grown at 21°C, and induced with 400 μm IPTG. Cells were harvested in 30 ml of 50 mm phosphate (pH 8.0), 300 mm NaCl with 0.1 mm PMSF and 10 μg/ml leupeptin. For tagged constructs, the sonicated, cleared lysate was loaded directly over Ni–NTA resin (Qiagen), washed with the same buffer containing 0.1% NP-40 at pH 6.3, and eluted with a step titration of 100–500 mm imidazole in the wash buffer. Lysates containing untagged protein were diluted with one-third volume water and loaded onto SP Sepharose. The column was washed in the same buffer with 0.1% NP-40 and eluted in a single step with 50 mm HEPES (pH 8.0), 500 mm NaCl, 1 mm MgCl2. This eluate was diluted to 250 mm NaCl, adjusted to 5 mm DTT, 5 mm EDTA, and 10% glycerol, and loaded on phosphocellulose. After washing, the sample was eluted in a gradient from 300–1500 mm NaCl. Fractions containing p80 were pooled and dialyzed against 20 mm Tris (pH 8.0), 1 mm MgCl2, 10% glycerol (T2MG) with 150 mm NaCl.
Cultures were similarly grown to express p95, except that NZCYM (Sigma) was used as growth media and transformations were grown immediately in culture without plating first. Cultures were induced with IPTG at 250 μm for 2 hr. Cells were harvested in T2MG with 75 mm NaCl, 0.1% NP-40, 0.1 mm PMSF, and 10 μg/ml of leupeptin. Sonicated, cleared lysates were first diluted to 50 mm NaCl and loaded on DEAE Sepharose. The column was eluted with a gradient from 100–600 mm NaCl. Fractions containing p95 were pooled and brought to 400 mm NaCl to load on phenyl–Sepharose, from which protein was eluted with T2MG plus 1% Triton X-100. This eluate was adjusted to 100 mm NaCl and loaded on MonoQ. A gradient from 100–400 mm NaCl was used to elute from the column. Fractions containing p95 were pooled and diluted into HEPES buffer at pH 6.6. This material was loaded on MonoS and eluted with a gradient from 50–400 mm NaCl. Fractions containing p95 were pooled and dialyzed against T2MG plus 150 mm NaCl.
Immunoprecipitations of p80 and p95
Affinity-purified polyclonal rabbit antibodies (R. Mitchell, L. Gandhi, and K. Collins, in prep.) were coupled to CNBr-activated Sepharose beads according to the manufacturer’s instructions (Pharmacia). For both control and p80 antibodies, 160 μg of antibodies were coupled to 160 μl of beads. Seven microliters of antibody beads or beads alone was mixed with 100 μl of each extract made in T2MG plus 75 mm NaCl, 0.1% NP-40. Reactions were brought to a total volume of 250 μl in this buffer plus 75 mm additional NaCl. After rotation at 4°C for 3 hr, reactions were washed three times in the same buffer, twice in T2MG plus 400 mm NaCl, 0.4% NP-40, and once in T2MG plus 150 mm NaCl. Beads were resuspended in 10 μl of T2MG plus 150 mm NaCl and mixed with 5× sample buffer before loading on a 10% SDS–polyacrylamide gel.
Electrophoretic gel mobility shift assays
Binding reactions were conducted in a 10 μl total volume of T2MG plus 150 mm NaCl, 0.1% NP-40 with 5 μg of acetylated BSA. Labeled RNA or DNA was always added last (after competitors). Some RNA binding reactions also contained 1 unit of RNasin (Promega) and 5 mm DTT. Complexes of p80 and p95 used in binding reactions contained either 6× His-tagged p80 or untagged p80; results were the same with either. DNA-binding reactions were similar to RNA-binding reactions except that they contained 100 ng of poly[d(I–C)] (Pharmacia) and single-stranded DNA competitors (where indicated) at 200-fold the probe concentration. Single-stranded DNA competitors consisted of either Random-18 or a mixture of Random-18 with T7 and T3 sequencing primers. Reactions were mixed and incubated at room temperature for 20–30 min before loading on 5% acrylamide gels (37.5:1 acrylamide/bis-acrylamide) made with 4% glycerol in 0.5× TBE. Gels were run at 150–250 V at 4°C.
Most competitor RNAs and radiolabeled telomerase RNA probes were transcribed with T7 RNA polymerase as described (Autexier and Greider 1994). Tel RNA 1-64 was made by engineering of a PstI site into the telomerase RNA sequence, such that linearization with PstI would yield a T7 transcript that contained only the first 64 nucleotides of the full-length RNA. This transcript also contained an additional 2 nucleotides (CG) at the 3′ end from the PstI restriction site. Both the full-length and 1–64 transcripts also contained an additional 3 G residues at the 5′ end (Autexier and Greider 1994). Random-146 nucleotides was made by T3 RNA polymerase transcription of pBS KS+ containing the human telomerase RNA to yield an RNA with 81 nucleotides of vector sequence and 65 nucleotides of sequence antisense to the region 3′ of the processed human telomerase RNA 3′ end. All transcription reactions were extracted twice with phenol:chloroform:isoamyl alcohol and precipitated with 2.5 m ammonium acetate in ethanol. E. coli 5S rRNA was purchased from Boehringer Mannheim. The tRNA (Sigma) and total yeast RNA (BDH Chemicals) were additionally purified by repeated phenol:chloroform extractions and reprecipitation. Telomerase RNA was radiolabeled with [α-32P]CTP (800 Ci/mmole) during the transcription reaction.
DNA sequences used as probes were prepared by labeling of synthetic oligonucleotides with T4 polynucleotide kinase and [γ-32P]ATP (6000 Ci/mmole). Radiolabeling reactions were phenol:chloroform:isoamyl alcohol-extracted and ethanol precipitated or heat killed and desalted through a NAP-5 column (Pharmacia) to remove unincorporated nucleotide. The DNA competitor sequences used in DNA binding reactions (other than those given in individual figures) were as follows:
(1) pBRT2G4, 5′-AGCCACTATCGACTACGCTTGGGG-3′; (2) Random-18, 5′-TTATGCTAGTTATTGCTC-3′; (3) 3′ telomeric 36-nucleotide oligomer, 5′-TGTGATCGAGCTTGTGATCGAGCTGGGGTTGGGGTT-3′; (4) 5′ telomeric 36-nucleotide oligomer, 5′-GGGGTTGGGGTTTGTGATCGAGCTTGTGATCGAGCT-3′; (5) internally telomeric 36-nucleotide oligomer, 5′-TGTGATCGAGCTGGGGTTGGGGTTTGTGATCGAGCT-3′; (6) nontelomeric 36-nucleotide oligomer, 5′-TGTGATCGAGCTTGTGATCGAGCTTGTGATCGAGCT-3′; (7) nontelomeric cassette for 3′ overhangs, 5′-CCCCTGTGATCGAGCT-3′; (8) strand to generate 5-nucleotide 3′ overhang, 5′-AGCTCGATCACAGGGGTTGGG-3′; (9) strand to generate 11-nucleotide 3′ overhang, 5′-AGCTCGATCACAGGGGTTGGGGTTGGG-3′; (10) strand to generate 17-nucleotide 3′ overhang, 5′-AGCTCGATCACAGGGGTTGGGGTTGGGGTTGGG-3′; (11) strand to generate 29-nucleotide 3′ overhang, 5′-AGCTCGATCACAGGGGTTGGGGTTGGGGTTGGGGTTGGGGTTGGG-3′; (12) nontelomeric cassette for 5′ overhangs, 5′-TGTGATCGAGCTCCC-3′; (13) strand to generate 11-nucleotide 5′ overhang, 5′-GGGGTTGGGGTTGGGAGCTCGATCACA-3′; (14) strand to generate 17-nucleotide 5′ overhang, 5′-GGGGTTGGGGTTGGGGTTGGGAGCTCGATCACA-3′; (15) full-length complement of 10, 5′-CCCAACCCCAACCCCAACCCCTGTGATCGAGCT-3′; (16) full-length complement of 14, 5′-TGTGATCGAGCTCCCAACCCCAACCC-3′; (17) nontelomeric primer 1, 5′-AAAAAGTCGACTAGAAAAAATAAATA-3′; (18) complement of 17, 5′-TATTTATTTTTTCTAGTCGACTTTTT-3′. Double-stranded and partial duplex DNAs were made by mixing of the appropriate oligonucleotides, heating to 90°C, and slow cooling to room temperature. These annealed mixtures were then twice gel purified on an 8% native acrylamide gel and quantitated by use of a spectrophotometer.
Affinity measurements for DNA binding
Radiolabeled and unlabeled DNAs were titrated in twofold steps from 5 to 320 nm into a binding reaction with 25 nm p95 and 400 nm single-stranded competitor DNA (competitor DNA was a mixture of T7, T3, and Random-18 primers). As for all DNA binding reactions, 5 μg of acetylated BSA and 100 ng of poly[d(I–C)] were included in each reaction. Scatchard analysis was conducted with a range of DNA concentration from 40 to 320 nm.
Telomerase activity assays and reconstitution
Telomerase assays were performed as described with highly purified fractions after glycerol gradient centrifugation (Collins et al. 1995). Reactions utilized 600 nm primer unless indicated otherwise. Reconstitution assays were also performed as described (Autexier and Greider 1994). Briefly, MNase and CaCl2 were added to purified telomerase for 10 min at 30°C. EGTA was then added to inactivate the MNase. Next, in vitro transcribed telomerase RNA and 5 mm EDTA were added and the mixture was heated at 37°C for 5 min. Competitor RNAs were added to the reaction after MNase inactivation but before heating of the fraction with 5 mm EDTA and wild-type telomerase RNA at 37°C. The RNA preparations were the same as described for binding reactions. Product DNAs were analyzed on denaturing 10% acrylamide gels.
Acknowledgments
We are grateful to Rachel Mitchell for help with DNA sequencing and for affinity purified antibodies used in this study, and to Michelle Pflumm for the 1–64 telomerase RNA expression construct. We thank Donald Rio, Ed Rebar, and Shane Donovan for helpful discussions during the course of the work. We also thank Donald Rio, Robert Tjian, Daniel Pak, Michael Botchan, and members of the Collins lab for critical comments on the manuscript. L.G. is a Howard Hughes predoctoral fellow. This work was funded by a grant from the National Institutes of Health to K.C. (GM 54198).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kcollins@socrates.berkeley.edu; FAX (510) 642-7000.
References
- Autexier C, Greider CW. Functional reconstitution of wild-type and mutant Tetrahymena telomerase. Genes & Dev. 1994;8:563–575. doi: 10.1101/gad.8.5.563. [DOI] [PubMed] [Google Scholar]
- ————— Boundary elements of the Tetrahymena telomerase RNA template and alignment domains. Genes & Dev. 1995;9:2227–2239. doi: 10.1101/gad.9.18.2227. [DOI] [PubMed] [Google Scholar]
- ————— Telomerase and cancer: Revisiting the telomere hypothesis. Trends Biol Sci. 1996;21:387–391. [PubMed] [Google Scholar]
- Blackburn EH. Telomerases. Annu Rev Biochem. 1992;61:113–129. doi: 10.1146/annurev.bi.61.070192.000553. [DOI] [PubMed] [Google Scholar]
- Cardenas ME, Bianchi A, de Lange T. A Xenopus egg factor with DNA-binding properties characteristic of terminus-specific telomeric proteins. Genes & Dev. 1993;7:883–894. doi: 10.1101/gad.7.5.883. [DOI] [PubMed] [Google Scholar]
- Cohn M, Blackburn EH. Telomerase in yeast. Science. 1995;269:396–400. doi: 10.1126/science.7618104. [DOI] [PubMed] [Google Scholar]
- Collins K, Greider CW. Nucleolytic cleavage and non-processive elongation catalyzed by Tetrahymena telomerase. Genes & Dev. 1993;7:1364–1376. doi: 10.1101/gad.7.7b.1364. [DOI] [PubMed] [Google Scholar]
- ————— Utilization of ribonucleotides and RNA primers by Tetrahymena telomerase. EMBO J. 1995;14:5422–5432. doi: 10.1002/j.1460-2075.1995.tb00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K, Kobayashi R, Greider CW. Purification of Tetrahymena telomerase and cloning of genes encoding the two protein components of the enzyme. Cell. 1995;81:677–686. doi: 10.1016/0092-8674(95)90529-4. [DOI] [PubMed] [Google Scholar]
- Counter CM, Meyerson M, Eaton EN, Weinberg RA. The catalytic subunit of yeast telomerase. Proc Natl Acad Sci. 1997;94:9202–9207. doi: 10.1073/pnas.94.17.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon PJ, Rosen CA. Use of polymerase chain reaction for the rapid construction of synthetic genes. In: White BA, editor. Methods in molecular biology (PCR protocols: Current methods and applications) Totowa, NJ: Humana Press, Inc.; 1993. pp. 263–268. [DOI] [PubMed] [Google Scholar]
- Fang G, Gray JT, Cech TR. Oxytricha telomere-binding protein: Separable DNA-binding and dimerization domains of the alpha-subunit. Genes & Dev. 1993;7:870–882. doi: 10.1101/gad.7.5.870. [DOI] [PubMed] [Google Scholar]
- Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley D, Blackburn EH. Specific RNA residue interactions required for enzymatic functions of Tetrahymena telomerase. Mol Cell Biol. 1996;16:66–75. doi: 10.1128/mcb.16.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley D, Lee MS, Blackburn EH. Altering specific telomerase RNA template residues affects active site function. Genes & Dev. 1995;9:2214–2226. doi: 10.1101/gad.9.18.2214. [DOI] [PubMed] [Google Scholar]
- Greider CW. Telomere length regulation. Annu Rev Biochem. 1996;66:337–365. doi: 10.1146/annurev.bi.65.070196.002005. [DOI] [PubMed] [Google Scholar]
- Greider CW, Collins K, Autexier C. DNA telomerases. In: DePamphlis M, editor. DNA Replication. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 619–638. [Google Scholar]
- Hammond PW, Lively TN, Cech TR. The anchor site of telomerase from Euplotes aediculatus revealed by photo-cross-linking to single- and double-stranded DNA primers. Mol Cell Biol. 1997;17:296–308. doi: 10.1128/mcb.17.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanish JP, Yanowitz JL, de Lange T. Stringent sequence requirements for the formation of human telomeres. Proc Natl Acad Sci. 1994;91:8861–8865. doi: 10.1073/pnas.91.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Villeponteau B. Telomeres and telomerase in aging and cancer. Curr Opin Genet Dev. 1995;5:249–255. doi: 10.1016/0959-437x(95)80016-6. [DOI] [PubMed] [Google Scholar]
- Harrington L, McPhail T, Mar V, Zhou W, Oulton R, Program E, Bass MB, Arruda I, Robinson MO. A mammalian telomerase-associated protein. Science. 1997;275:973–977. doi: 10.1126/science.275.5302.973. [DOI] [PubMed] [Google Scholar]
- Harrington LA, Greider CW. Telomerase primer specificity and chromosome healing. Nature. 1991;353:451–454. doi: 10.1038/353451a0. [DOI] [PubMed] [Google Scholar]
- Holler TP, Foltin SK, Ye QZ, Hupe DJ. HIV1 integrase expressed in Escherichia coli from a synthetic gene. Gene. 1993;136:323–328. doi: 10.1016/0378-1119(93)90488-o. [DOI] [PubMed] [Google Scholar]
- Joyce CM, Steitz TA. Function and structure relationships in DNA polymerases. Annu Rev Biochem. 1994;63:777–822. doi: 10.1146/annurev.bi.63.070194.004021. [DOI] [PubMed] [Google Scholar]
- Kirk KE, Harmon BP, Reichardt IK, Sedat JW, Blackburn EH. Block in anaphase chromosome separation caused by a telomerase template mutation. Science. 1997;275:1478–1481. doi: 10.1126/science.275.5305.1478. [DOI] [PubMed] [Google Scholar]
- Lee MS, Blackburn EH. Sequence-specific DNA primer effects on telomerase polymerization activity. Mol Cell Biol. 1993;13:6586–6599. doi: 10.1128/mcb.13.10.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvay TS, Morris DK, Sah J, Balasubramamian B, Lundblad V. Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics. 1996;144:1399–1412. doi: 10.1093/genetics/144.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zakian VA. An in vitro assay for Saccharomyces telomerase requires EST1. Cell. 1995;81:1127–1135. doi: 10.1016/s0092-8674(05)80017-0. [DOI] [PubMed] [Google Scholar]
- ————— The Saccharomyces CDC13 protein is a single-strand TG1–3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc Natl Acad Sci. 1996;93:13760–13765. doi: 10.1073/pnas.93.24.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Cech TR. Purification of telomerase from Euplotes aediculatus: Requirement of a primer 3′ overhang. Proc Natl Acad Sci. 1996;93:10712–10717. doi: 10.1073/pnas.93.20.10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- Lundblad V, Szostak JW. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell. 1989;57:633–643. doi: 10.1016/0092-8674(89)90132-3. [DOI] [PubMed] [Google Scholar]
- Martindale DW. Codon usage in Tetrahymena and other ciliates. J Protozool. 1989;36:29–34. doi: 10.1111/j.1550-7408.1989.tb02679.x. [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH. Runaway telomere elongation caused by telomerase RNA gene mutations. Nature. 1995;376:403–409. doi: 10.1038/376403a0. [DOI] [PubMed] [Google Scholar]
- Melek M, Greene EC, Shippen DE. Processing of non-telomeric 3′ ends by telomerase: Default template alignment and endonucleolytic cleavage. Mol Cell Biol. 1996;16:3437–3445. doi: 10.1128/mcb.16.7.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Morin GB. Recognition of a chromosome truncation site associated with α-thalassaemia by human telomerase. Nature. 1991;353:454–456. doi: 10.1038/353454a0. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Saito M, Nakamura H, Matsuura A, Ishikawa F. TLP1: A gene encoding a protein component of mammalian telomerase is a novel member of WD repeats family. Cell. 1997;88:875–884. doi: 10.1016/s0092-8674(00)81933-9. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Hughes TR, Lue NF, Lundblad V. Cdc13p: A single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 1996;274:249–252. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- Prescott J, Blackburn EH. Telomerase RNA mutations in Saccharomyces cerevisiae alter telomerase action and reveal nonprocessivity in vivo and in vitro. Genes & Dev. 1997;11:528–540. doi: 10.1101/gad.11.4.528. [DOI] [PubMed] [Google Scholar]
- Romero DP, Blackburn EH. A conserved secondary structure for telomerase RNA. Cell. 1991;67:343–353. doi: 10.1016/0092-8674(91)90186-3. [DOI] [PubMed] [Google Scholar]
- Steiner BR, Hidaka K, Futcher B. Association of the Est1 protein with telomerase activity in yeast. Proc Natl Acad Sci. 1996;93:2817–2821. doi: 10.1073/pnas.93.7.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virta-Pearlman V, Morris DK, Lundblad V. Est1 has the properties of a single-stranded telomere end-binding protein. Genes & Dev. 1996;10:3094–3104. doi: 10.1101/gad.10.24.3094. [DOI] [PubMed] [Google Scholar]
- Wang H, Blackburn EH. De novo addition by Tetrahymena telomerase in vitro. EMBO J. 1997;16:866–879. doi: 10.1093/emboj/16.4.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Bradley JD, Attardi LD, Blackburn EH. In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature. 1990;344:126–132. doi: 10.1038/344126a0. [DOI] [PubMed] [Google Scholar]
- Zakian VA. Telomeres: Beginning to understand the end. Science. 1995;270:1601–1607. doi: 10.1126/science.270.5242.1601. [DOI] [PubMed] [Google Scholar]
- Zaug AJ, Cech TR. Analysis of the structure of Tetrahymena nuclear RNAs in vivo: Telomerase RNA, the self-splicing rRNA intron, and U2 snRNA. RNA. 1995;1:363–374. [PMC free article] [PubMed] [Google Scholar]