

Abstract
The FG-repeat domain of the yeast Rip1 protein (Rip1p) was identified initially as a possible target for the nuclear export signal (NES) of the HIV-1 Rev protein in a yeast two-hybrid assay. Rip1p is inessential, associated with nuclear pore complexes, and structurally related to the FG-nucleoporin family of pore proteins. It contributes to HIV-1 Rev-mediated RNA export and is also important for the export of heat shock RNAs at 42°C. We show here that Rip1p is essential for the export of heat shock RNAs, and this function is fulfilled by the unique carboxyl terminus of Rip1p with no substantial contribution from the FG-repeat region. Genetic interactions between Rip1p and the RNA export mediator Gle1p are described, which support a role of the carboxyl terminus of Rip1p in poly(A)+ RNA export. Finally, this domain of Rip1p also contributes to Rev-mediated RNA export. The data suggest that Rip1p promotes the nuclear export of different classes of substrates by contributing to optimal pore function.
Keywords: Yeast, RNA export, Rev, Rip1p, nuclear pore complex, heat shock RNA
Macromolecular exchange between the nucleus and the cytoplasm occurs through nuclear pore complexes (NPCs) (Davis 1995; Pante and Aebi 1995). Whereas molecular details of protein import into the nucleus have begun to be elucidated, less is known about the mechanisms of nuclear export of RNA as well as proteins (Melchior and Gerace 1995; Gorlich and Mattaj 1996; Pante and Aebi 1996; Corbett and Silver 1997). Early oocyte injection competition experiments indicated that the transport of each RNA class (i.e., mRNA, U snRNA, 5S RNA, or tRNA) is dependent on one or more specific factors, thus defining distinct RNA export pathways (Jarmolowski et al. 1994). It is also known that RNAs are exported as ribonucleoprotein (RNP) complexes (Visa et al. 1996a,b) in a process that is saturable and energy dependent. The RNP proteins or the RNAs themselves are therefore likely to contain different signals that direct the complexes to and through the nuclear pore in distinct ways (Corbett and Silver 1997; Nigg 1997).
There is evidence that several RNA-binding proteins play a role in RNA export. The mammalian hnRNPA1 (Michael et al. 1996; Izaurralde et al. 1997) and the yeast hnRNP-like NPL3 (Lee et al. 1996) proteins are involved in mRNA export, the cap-binding complex (CBC) is important for U snRNA export (Izaurralde et al. 1995), and both TFIIIA and the ribosomal L5 protein contribute to 5S RNA export (Guddat et al. 1990). However, no clear connection has yet been established between these RNAbinding proteins and the export machinery or the NPC.
The best characterized protein directly involved in RNA export is the HIV-1 Rev protein. The role of Rev in the viral life cycle is to promote the export of unspliced or partially spliced viral transcripts, which allows the expression of the structural proteins Gag, Pol, and Env, as well as the packaging of the RNA genome into new viral particles (Sodroski et al. 1986). Rev’s amino-terminal basic region binds the Rev response element (RRE), a highly structured RNA sequence within the target viral transcripts; Rev’s carboxyl terminus contains a short leucine-rich motif, the effector domain, directly involved in export (Cullen and Malim 1991). This leucine-rich region has been defined as a nuclear export signal (NES), based on the rapid nuclear export of heterologous proteins fused to that sequence (Fischer et al. 1995; Gerace 1995). The NES peptide also inhibits U snRNA and 5S RNA export in Xenopus oocytes but has no effect on mRNA or tRNA export, suggesting that Rev uses a specific export pathway (Fischer et al. 1995). Rev-like NESs have been identified in a number of proteins some of which have no known relevance to RNA export (Wen et al. 1995; Bogerd et al. 1996; Fridell et al. 1996; Fritz and Green 1996; Murphy and Wente 1996; Iovine and Wente 1997; Segref et al. 1997). This suggests that Rev exploits a more general cellular protein export pathway for the transport of viral RNAs. This pathway has been evolutionarily conserved, as Rev and Rev-like NES sequences are functional in the yeast Saccharomyces cerevisiae (Stutz and Rosbash 1994; Fritz and Green 1996).
The human hRIP/RAB1 (Bogerd et al. 1995; Fritz et al. 1995) and yeast Rip1p (Stutz et al. 1995) proteins interact with the Rev NES in the yeast two-hybrid assay and have been identified as potential mediators of Rev-mediated RNA export in mammals and yeast, respectively. Both proteins contain a series of phenylalanine–glycine dipeptide repeats (FG repeats), which are important for the two-hybrid interaction with Rev NES (Stutz et al. 1996). FG repeats are a characteristic feature of a family of NPC proteins, the FG nucleoporins, implicated in the process of nucleocytoplasmic transport (Rout and Wente 1994; Davis 1995; Doye and Hurt 1995; Corbett and Silver 1997). Whereas yeast Rip1p (yRip1p) is associated with the NPC (Stutz et al. 1995; see below), hRip1/RAB1 has been detected in the nucleoplasm as well as at the nuclear envelope (Bogerd et al. 1995; Fritz et al. 1995).
In addition to hRip1/RAB1 and Rip1p, Rev NES interacts with the FG repeats of multiple mammalian and yeast FG nucleoporins in the two-hybrid assay (Stutz et al. 1995, 1996; Fritz and Green 1996). Although it is unclear at present which and how many of these interactions are relevant to Rev function in vivo, it is possible that other FG nucleoporins, in addition to hRIP1/RAB1 and Rip1p, contribute to Rev-mediated export. In this context, it is interesting that injection into Xenopus oocyte nuclei of a GST–Rip1p fusion protein, like injection of a Rev NES peptide, interferes with U snRNA but not mRNA or tRNA export, consistent with a role for Rip1p or Rip1p-related FG repeats in Rev-mediated export (Stutz et al. 1996).
The ability of Rev NES or other NESs to function in yeast and the identification of Rip1p as a cellular target implied the existence of endogenous yeast export factors with comparable properties. A possible candidate is the recently identified Gle1 protein from yeast. Gle1p contains an essential Rev-like NES, and mutations therein result in the nuclear retention of poly(A)+ RNA, suggesting that Gle1p participates in mRNA export (Murphy and Wente 1996). However, a role for Gle1p in the export of other classes of RNAs cannot be excluded. Gle1p localizes to the NPC as well as the cytoplasm (Del Priore et al. 1996; Murphy and Wente 1996), consistent with the possibility that this protein shuttles between the two compartments. Rip1p and Gle1p interact in the yeast two-hybrid system as well as in an in vitro blot overlay assay (Murphy and Wente 1996). This further supports a role for Rip1p in mRNA export. We note, however, that there is no evidence to date for an interaction between Gle1p and Rip1p during this process. Moreover, because the disruption of RIP1 induces no obvious growth phenotype between 18°C and 37°C (Stutz et al. 1995), this contribution is inessential and therefore likely to be modest under normal laboratory culture conditions.
Cole and colleagues have recently uncovered an important role for Rip1p in the selective export of heat shock RNAs. In a wild-type strain at 42°C, heat shock RNAs are distributed in both the nucleus and cytoplasm, whereas bulk poly(A)+ RNA is retained within the nucleus (Saavedra et al. 1996). In a RIP1-deleted strain at 42°C, however, heat shock RNAs are also prominent within the nucleus and little heat shock RNA-specific signal is detectable in the cytoplasm (Saavedra et al. 1997). These experiments suggest that Rip1p is a major component of a pathway dedicated to heat shock RNA export.
In this paper, we pursue these observations by investigating heat shock protein synthesis. Our results confirm the in situ hybridization observations and, surprisingly, reveal no substantial requirement for the Rip1p FG repeats but rather point to the unique carboxy-terminal region of Rip1p as being essential for the heat shock RNA export process. We identified several Gle1 alleles in a screen for mutations that are synthetically lethal in combination with a RIP1 gene disruption, suggesting that Rip1p also contributes to the mRNA export pathway. Genetic analyses indicate that this function, like heat shock RNA export, relies predominantly on the unique carboxyl terminus of Rip1p. Finally, we show that this region also contributes to Rev-mediated export. In summary, the data suggest that Rip1p participates in multiple export pathways by providing optimal pore structure and/or function through its unique carboxyl terminus.
Results
The absence of Rip1p eliminates heat shock protein synthesis at 42°C
The in situ hybridization experiments of Saavedra et al. (1997) indicate that Rip1p is important for heat shock RNA export. An expected consequence of this selective block is a reduction in heat shock protein synthesis at 42°C in a RIP1 deletion strain (ΔRIP1). To verify this prediction, a wild-type and a__ΔRIP1 strain were incubated at 25°C, 37°C, or 42°C for 15 min followed by a 15-min labeling pulse with [35S]methionine at the same temperature. Total cell extracts were then fractionated on SDS-PAGE and autoradiographed.
A number of normal cellular protein bands were detected at 25°C, which are identical in both wild-type and ΔRIP1 (Fig. 1A, lanes 1,2). In the 42°C shifted samples, however, several heat shock proteins (Lindquist 1986; Lindquist and Craig 1988), including Hsp104p, Hsp82p, and SSA4 (a member of the Hsp70 family of proteins), were strongly induced in the wild-type strain but remained completely absent in the ΔRIP1 strain (lanes 3,4). This result provides a biochemical confirmation of the in situ hybridization data and indicates an apparent absolute Rip1p requirement for heat shock RNA transport at 42°C. It is interesting to note that there is no detectable Rip1p requirement for heat shock protein synthesis at 37°C (lanes 5,6).
Figure 1.
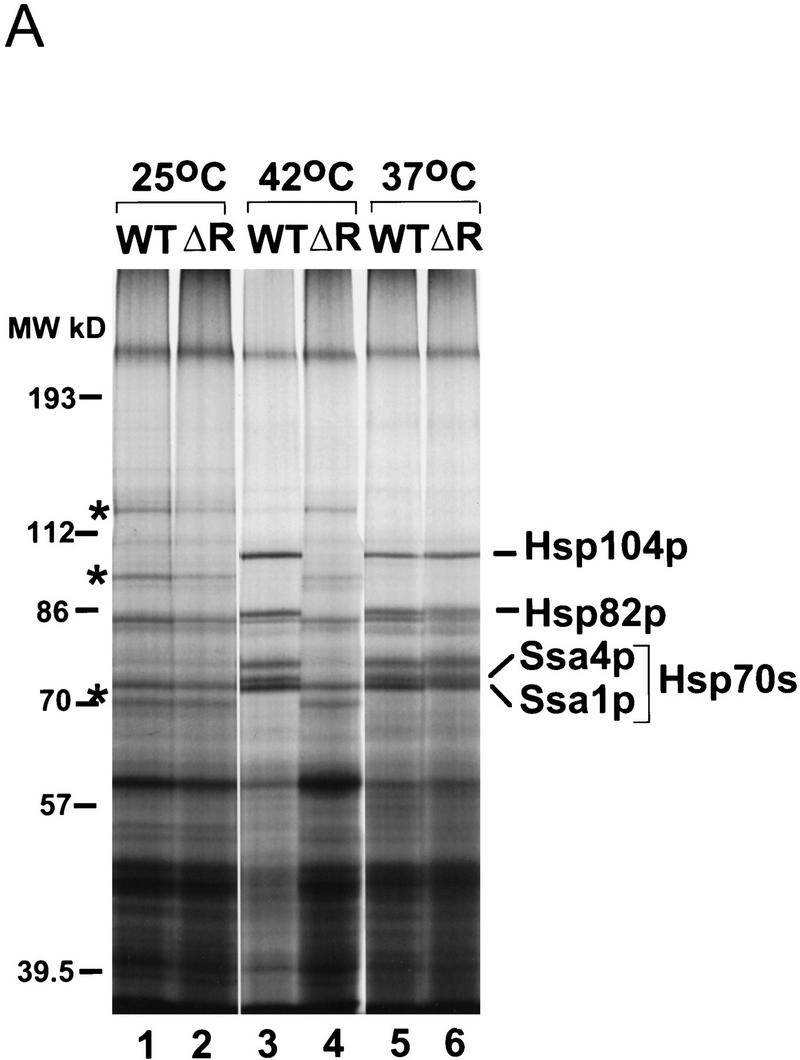

(A) Analysis of heat shock protein synthesis in wild-type and ΔRIP1 strains. Cultures of wild-type (WT) or ΔRIP1 (ΔR) strains were preheated for 15 min and subsequently labeled for 15 min with [35S]methionine at the indicated temperatures. Total protein extracts were fractionated by SDS-PAGE and autoradiographed. The positions of the high-molecular-weight heat shock proteins (Hsp104p, Hsp82p, and the Hsp70 proteins Ssa1p and Ssa4p) are indicated. Asterisks (*) correspond to non-heat shock bands that persist at 42°C in the ΔRIP1 strain. (B) Heat shock RNA levels in wild-type and ΔRIP1 strains.Total RNA from wild-type (WT, lanes 1–3) or ΔRIP1 (lanes 4–6) cultures grown at 25°C or shifted to 37°C or 42°C for 30 min was analyzed by primer extension with a primer specific for the HSP104 heat shock mRNA. A primer specific for U2 snRNA was used in the same reactions as an internal control for loading. The reactions were fractionated on a denaturing polyacrylamide gel and autoradiographed. The positions of the HSP104 and U2 RNA primer extension products are indicated.
The [35S]methionine-labeling experiment (Fig. 1) also shows that the synthesis of heat shock proteins at both 37°C and 42°C is accompanied by the disappearance of a number of proteins normally synthesized at 25°C (Fig. 1, cf. lanes 1 and 3; the most prominent bands are indicated by asterisks). Interestingly, these bands persist at 42°C in the ΔRIP1 strain. The synthesis of these proteins also persists for at least 45 min in the RNA polymerase II temperature-sensitive mutant rpb1-1 (Nonet et al. 1987) after a shift to 42°C (data not shown). This observation suggests that these proteins correspond to long-lived cytoplasmic RNAs rather than to newly synthesized and recently exported transcripts. In a wild-type strain at 42°C, the translation of these transcripts is most likely competed by the heat shock RNAs.
To determine whether the block in heat shock RNA export in ΔRIP1 was accompanied by changes in heat shock RNA levels, primer extension analysis was used to examine the RNAs encoding Hsp104p in wild-type or _ΔRIP1 cells after a 30-min temperature shift (Fig. 1B). HSP104 RNA was undetectable at 25°C, and virtually identical HSP104 RNA levels were observed in the wild-type and mutant strains at 37°C; at 42°C, the HSP104 mRNA levels were three to four times lower in the ΔRIP1 compared with the wild-type strain (cf. lanes 2 and 5, or 3 and 6). This modest decrease is likely to result from a higher turnover of these RNAs when sequestered within the nucleus of the ΔRIP1 strain.
Heat shock RNA export at 42°C is sensitive to low levels of Rip1p and is not dependent on the FG-repeat domain of Rip1p
To define more precisely the Rip1p requirement in heat shock RNA export, we compared different Rip1p-expressing plasmids for their ability to restore heat shock RNA export after transformation into a ΔRIP1 strain. Construct RIP1–Lo (low) contains the RIP1 gene on an ∼1.9-kb genomic fragment with only 380-bp 5′-flanking sequences (Fig. 2A); Western blot analysis indicates that this construct expresses five to ten times lower levels of Rip1p than a wild-type strain (Fig. 2B, cf. lanes 1 and 3; note that lane 1 is slightly overloaded compared with the other lanes). Construct RIP1–Hi (high) contains the RIP1 gene on a 3.5-kb HindIII genomic fragment with 780-bp 5′-flanking sequences (Fig. 2A); this plasmid expresses nearly wild-type levels of Rip1p (Fig. 2B, cf. lanes 1 and 4). As expected, the latter construct fully rescues heat shock RNA export when transformed into the ΔRIP1 strain, as assayed by [35S]methionine labeling of heat shock proteins after a shift to 42°C (Fig. 2C, lane 4). In contrast, no heat shock protein synthesis is observed in the presence of the low Rip1p-expressing plasmid RIP1–Lo (Fig. 2C, lane 3). The data indicate a requirement for minimum levels of Rip1p for heat shock RNA export at 42°C.
Figure 2.

(A) Schematics of RIP1 constructs. The RIP1–Lo (pFS724) and RIP1–Hi (pFS398) constructs contain the RIP1 gene with 380 bp and 780 bp of 5′-flanking sequences, respectively, on a centromeric plasmid. (B) Rip1p levels in a wild-type (WT) or in a ΔRIP1 strain transformed with Rip1p-expressing constructs. Total protein extracts were prepared from a wild-type strain transformed with vector p366 alone (WT + V) (lane 1), or from the ΔRIP1 strain transformed with vector (V), the RIP1–Lo (Lo), or the RIP1–Hi (Hi) centromeric constructs (lanes 2–4) and fractionated on a 10% SDS–polyacrylamide gel; Rip1p was detected by Western blot analysis using a rabbit anti-Rip1p polyclonal antibody directed against the FG-repeat region of Rip1p. (C) Rescue of heat shock protein synthesis in the _ΔRIP1 strain by the Rip1p-expressing plasmids. Cultures from the wild-type strain transformed with vector (WT + V), or with the ΔRIP1 strain transformed with vector (V), the RIP1–Lo (Lo), or the RIP1–Hi (Hi) centromeric constructs, were preheated for 15 min at 42°C and subsequently labeled with [35S]methionine for 15 min at 42°C. Total protein extracts were fractionated by SDS-PAGE and autoradiographed.
The rescue of heat shock protein synthesis with plasmid RIP1–Hi was used as an assay to define the regions of Rip1p that are essential for function. To this end, deletions were created within the Rip1p coding region of construct RIP1–Hi (Fig. 2A). In construct RIP1–1FG, codons 38 through 364 were deleted and replaced by a SalI linker; this construct encodes a 104-amino-acid protein that lacks all the FG repeats except for one FG dipeptide at positions 4 and 5 of Rip1p (Fig. 3A). In construct RIP1–0FG, codons 2 through 364 were deleted and replaced by a XhoI linker; this plasmid encodes a 68-amino-acid protein from which all the FG repeats have been eliminated (Fig. 3A). The RIP1–1FG and RIP1–0FG plasmids were introduced into a ΔRIP1 strain, and heat shock protein synthesis was assayed at 42°C. Surprisingly, the RIP1–1FG construct efficiently rescued heat shock RNA export, as indicated by the potent induction of heat shock proteins (Fig. 3B, lane 3). The RIP1–0FG protein rescued export very poorly when expressed from a centromeric plasmid (lane 4) but was fully functional when expressed from a high-copy 2-μm plasmid (lane 5). This suggests that the 68-amino-acid protein is active but probably less stable than the 104-amino-acid protein encoded by RIP1–1FG. We have been unable to detect the truncated Rip1p proteins by Western blot analysis with a polyclonal antibody directed against the carboxyl terminus of Rip1p or by HA-tagging the mutant proteins (data not shown). One possibility is that the truncated proteins are degraded during the extraction procedure; alternatively or in addition, these antibody reagents may be less sensitive than the rabbit polyclonal antibody specific for the FG repeats of Rip1p (Fig. 2B). However, we have been able to detect a biologically active fusion protein between RIP1-0FG and GFP (green fluorescent protein; not shown). A high-copy number plasmid expressing only the FG-repeat region of Rip1p (Stutz et al. 1995) was unable to rescue heat shock protein synthesis in a RIP1 deletion strain (data not shown). All the data indicate that overproduction of the short unique Rip1p carboxy-terminal domain is sufficient to support robust heat shock RNA export at 42°C.
Figure 3.
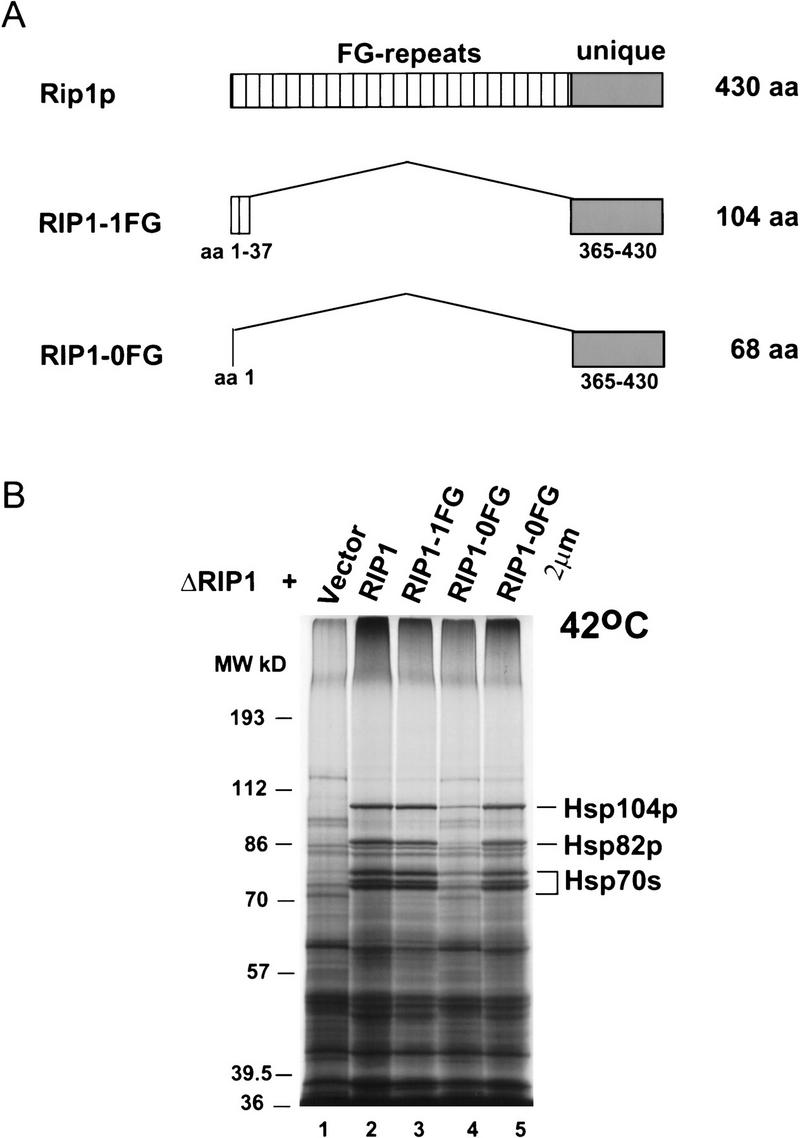
(A) Schematic drawing of full-length Rip1p and the deleted Rip1 proteins encoded by the RIP1–1FG (pFS662) and RIP1–0FG (pFS730 and pFS733) constructs. The lengths of the proteins are indicated at right. The FG-repeat region and the unique region of Rip1p are represented by striped and shaded boxes, respectively. The amino acids contained in the deleted proteins are indicated below. The RIP1–1FG protein contains a single FG dipeptide at amino acid positions 4 and 5. (B) The unique carboxyl terminus of Rip1p rescues heat shock protein synthesis at 42°C in the ΔRIP1 strain. The ΔRIP1 strain was transformed with vector alone (V, lane 1) or with the Rip1p-expressing constructs RIP1–Hi (pFS398, lane 2), RIP1–1FG (FS662, lane 3), RIP1–0FG (pFS730, lane 4), or RIP1–0FG 2μ (pFS733, lane 5). Heat shock protein synthesis at 42°C was analyzed as described in Fig. 2.
The carboxyl terminus of Rip1p is important for Rev-mediated RNA export
We have shown previously that Rev promotes the export of RRE-containing transcripts in yeast. In this assay, Rev’s activity is monitored by changes in the copper resistance of an engineered yeast strain (Stutz and Rosbash 1994). This copper-sensitive strain contains a Revexpressing plasmid and a CUP1 reporter plasmid (PC–CUP–RRE). The transcripts generated by the PC–CUP–RRE plasmid contain the CUP1 coding sequence interrupted at its 5′ end by a small in-frame intron (Legrain and Rosbash 1989), followed by the RRE after the CUP1 stop codon. The CUP1 protein is encoded only from the pre-mRNA (the spliced mRNA is out-of-frame) so that the copper resistance of this reporter strain directly reflects the levels of cytoplasmic pre-mRNA. In the presence of wild-type Rev, the reporter strain grows in up to 1.2 mm copper, whereas it grows up to 0.8 mm copper with no Rev or with the Rev M10 mutant. (Rev M10 contains a mutation in the NES that inhibits Rev’s export activity; Table 1). These Rev effects on yeast pre-mRNA export parallel the Rev effects on viral gene expression in mammalian cells (Stutz et al. 1995).
Table 1.
The carboxyl terminus of Rip1p is important for Rev-mediated RNA export
| URA3/2μ | ←————————————————————————— PC–CUP–RRE —————–——–——————————→ | ||||||
|---|---|---|---|---|---|---|---|
| TRP1/CEN | V | Rev | M10 | ←————————————————— Rev —––——–—————————————→ | |||
| LEU2/CEN | V | V | V | RIP1 | RIP1–1FG | RIP1–0FG | RIP1–0FG 2μ |
| WT | 0.8 | 1.2 | 0.8 | 1.2 | 1.2 | 1.2 | 1.2 |
| ΔRIP1 | 0.8 | 1.0 | 0.8 | 1.2 | 1.1 | 1.0 | 1.1 |
Rev-mediated RNA export is reflected by an increase in the copper resistance of the yeast reporter strain. WT (Y59ΔCUP) or ΔRIP1 (Y59ΔCUPΔRIP1) strains were transformed with the PC–CUP–RRE reporter plasmid (URA3/2 μ) and with Rev-expressing plasmids (TRP1/CEN) as described earlier (Stutz et al. 1995). In the wild-type strain (WT), the export of the PC–CUP–RRE transcript is stimulated by Rev and results in an increase of copper resistance. This effect is weakened in the ΔRIP1 strain.
The rescue of Rev-mediated export was examined by transforming both strains with the Rip1p-expressing plasmids pFS398 (RIP1), pFS662 (RIP1–1FG), pFS730 (RIP1–0FG CEN), or pFS733 (RIP1–0FG 2 μ) or with empty vector controls (V). The transformed strains were grown to saturation and spotted onto Ura− Leu− Trp− plates containing increasing concentrations of CuSO4. The numbers correspond to the copper resistance [Cu2+ (mm)] of each transformed strain.
As reported previously, the Rev-mediated copper resistance is reduced to 1 mm copper in the ΔRIP1 strain, indicating a contribution of Rip1p to Rev-mediated export (Stutz et al. 1995). To determine which region of Rip1p is involved in Rev function, constructs expressing total Rip1p or truncated versions of the Rip1p protein were introduced into the ΔRIP1 strain together with the PC–CUP–RRE reporter construct and a plasmid expressing wild-type Rev. The full-length RIP1 construct (RIP1–Hi) increased copper resistance to 1.2 mm; RIP1–1FG and the 2-μm version of RIP1–0FG increased copper resistance to 1.1 mm, indicating substantial rescue, whereas the centromeric RIP1–0FG had no effect (Table 1). This indicates that the carboxyl terminus of Rip1p contributes to Rev-mediated export as well as to heat shock mRNA export. The lack of a major role for the FG-repeat region of Rip1p is surprising (see Discussion).
Relationship of Rip1p to the nuclear pore and to Gle1p
Previous immunolocalization experiments suggested that Rip1p is associated with the NPC, which is also consistent with its sequence features. To confirm this notion, we used a biochemical approach to address Rip1p’s subcellular localization. In this fractionation procedure (Fig. 4), Rip1p is undetectable in the low-speed supernatant (S) of a total cell lysate (in which >50% of the nuclei are lysed) and is released from the pellet by extraction with 1 m NaCl (P). The behavior of Rip1p in this assay is identical to that described for other nuclear pore proteins (Belanger et al. 1994). The observations are consistent with the association of the majority of Rip1p protein with the NPC.
Figure 4.

Rip1p is part of an insoluble and salt labile high-molecular-weight complex. Wild-type yeast cells were fractionated as described in Materials and Methods. The soluble fraction (S) represents proteins released into a low-speed supernatant after cell lysis. The particulate fraction (P) represents material released from the low-speed pellet by extraction with buffer containing 1 m NaCl. All the nucleoporins are solubilized under these conditions (Belanger et al. 1994). Aliquots of total (T) cell lysate (0.15 OD600 cells; lane 1), fraction S (2.4 OD600 cells; lane 2), or fraction P (3 OD600 cells; lane 3) were subjected to Western blot analysis with a mouse polyclonal antibody directed against the unique carboxyl terminus of Rip1p. The Rip1p protein was identified by parallel analysis of control (Co) total cell extracts prepared from a wild-type (WT; lane 5) or the ΔRIP1 (lane 6) strain by TCA precipitation.
To identify components genetically related to Rip1p, we isolated mutations that are lethal in the absence of Rip1p (Bender and Pringle 1991; see Materials and Methods). One of the identified genes encodes Nup85p (data not shown), a nuclear pore protein with a role in poly(A)+ RNA export (Goldstein et al. 1996; Siniossoglou et al. 1996). Although incomplete at present, this synthetic lethal screen also gave rise to a high proportion (75%) of mutants that mapped to the previously defined mRNA export mediator Gle1p (Del Priore et al. 1996; Murphy and Wente 1996). This result provides genetic evidence that Rip1p, like Gle1p, contributes to mRNA export.
Three of the Gle1 alleles identified in this screen (gle1-1, gle1-2, and gle1-3) were chosen at random and characterized in more detail. These three alleles affect Gle1p function to different extents, because they exhibit different growth phenotypes in the presence of wild-type levels of Rip1p (Table 2). With different RIP1-rescuing plasmids, there was an effect on nuclear retention of poly(A)+ RNA as well as on growth rate; both of these phenotypes were exacerbated in the Gle1 mutants by limiting levels of Rip1p (data not shown; Table 2). Some poly(A)+ RNA nuclear staining was also observed in the presence of low levels of Rip1p at 25°C. The mutations in gle1-1, gle1-2, and gle1-3 were all determined to be amino acid substitutions in the carboxy-terminal part of Gle1p, beyond the proposed NES (Table 2; Fig. 5A). This identifies a novel and unanticipated domain of Gle1p, as previous results from others had focused on the NES-like region of Gle1p that is important for its interaction with nuclear pore FG repeats (Murphy and Wente 1996). The NES mutation L356A in Gle1p (Murphy and Wente 1996) is also lethal in the absence of Rip1p (data not shown).
Table 2.
Rescue of the RIP1 synthetic lethal alleles of GLE1 with different RIP1 constructs
|
gle1 mutant
|
Mutation
|
Growth
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| no. RIP1 | RIP1 high | RIP1 low | RIP1–1FG | RIP1–0FG | RIP1–0FG 2μ | ||||||||
| (25°C) | (37°C) | (25°C) | (37°C) | (25°C) | (37°) | (25°C) | (37°C) | (25°C) | (37°C) | (25°C) | (37°C) | ||
| gle1-1 | F381S | − | − | + | + | + | slow | + | very slow | + | − | + | very slow |
| gle1-2 | T468I | − | − | + | + | + | + | + | + | + | − | + | + |
| gle1-3 | M398I | − | − | + | − | + | − | + | − | + | − | + | − |
The mutation in each of the three Gle1 mutants is shown; the growth of the Gle1 mutant strains in the presence of different Rip1p-expressing plasmids was examined by replacing the screening plasmids pFS652 or pFS478 (URA3/ADE3/CEN) with the following LEU2/CEN plasmids: p366 vector (no RIP1), pFS398 (RIP1 high), pFS724 (RIP1 low), pFS662 (RIP1–1FG), pFS730 (RIP1–0FG) or with the LEU/2μ construct pFS733 (RIP1–0FG). The phenotype of these strains was examined by growth on Leu− plates at 25°C or 37°C.
Figure 5.
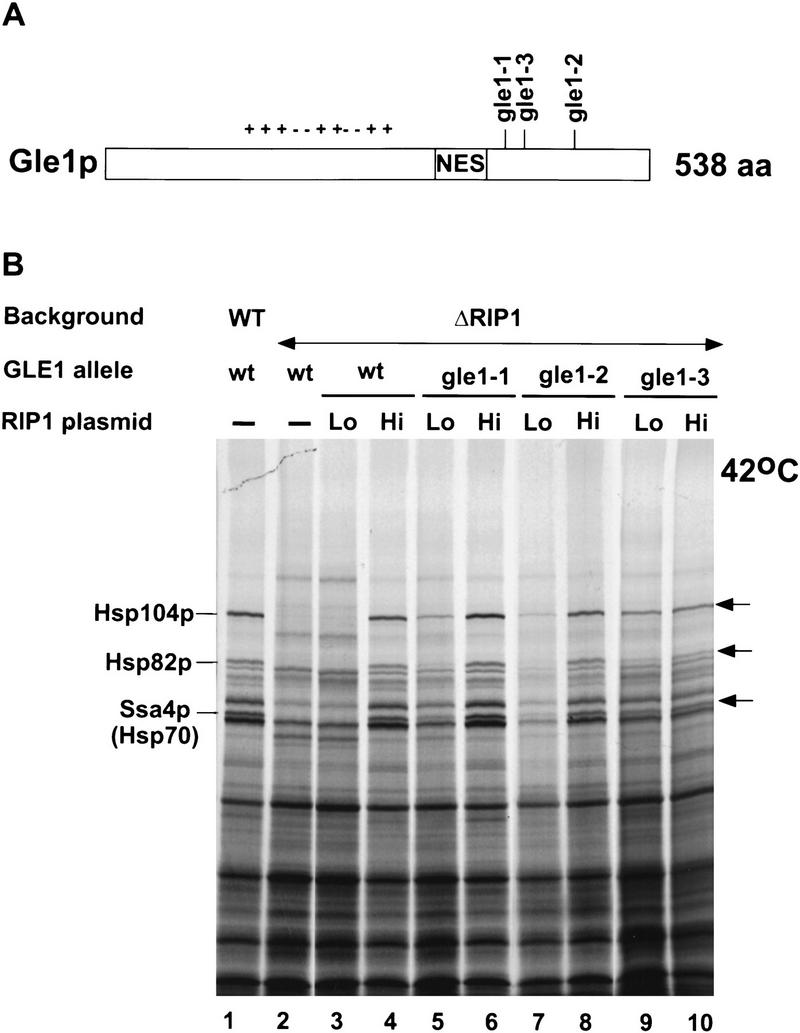
(A) Schematic drawing of the Gle1 protein. The positions of the three Gle1 mutations (gle1-1, gle1-2, and gle1-3) that are synthetically lethal with a RIP1 deletion are indicated. The NES (amino acid positions 351–356) is boxed. (+/−) A central domain rich in charged residues. (B) The Gle1 mutations synthetically lethal with a RIP1 deletion restore heat shock protein synthesis at 42°C in the presence of low amounts of Rip1p. Heat shock protein synthesis at 42°C was assayed as in Fig. 2C. Lanes 1 and 2 correspond to wild-type and ΔRIP1 strains transformed with the p366 vector alone; lanes 3, 5, 7, and 9 correspond, respectively, to the ΔRIP1, gle1-1, gle1-2, and gle1-3 strains carrying construct RIP1–Lo (Lo) and expressing low levels of Rip1p (Fig. 2A); lanes 4, 6, 8, and 10 correspond, respectively, to the ΔRIP1, gle1-1, gle1-2, and gle1-3 strains carrying construct RIP1–Hi (Hi) and expressing high levels of Rip1p (Fig. 2).
Interestingly, all three Gle1 mutants were rescued by the RIP1–0FG 2-μm construct nearly as well as by full-length Rip1p (RIP1 high), suggesting that the carboxyl terminus of Rip1p is also relevant to its genetic interaction with Gle1p (Table 2). The three mutants were also viable in the presence of limiting amounts of full-length Rip1p (RIP1 low), although growth at 25°C was marginally affected in the case of the more severe gle1-1 and gle1-3 alleles (not shown). Taken together with the previously defined role of Gle1p in poly(A)+ RNA export (Del Priore et al. 1996; Murphy and Wente 1996), the genetic interactions suggest that the carboxy-terminal domain of Rip1p is also the primary region of Rip1p contributing to this export pathway. The genetic interactions may reflect a physical interaction as indicated by yeast two-hybrid interaction between wild-type Gle1p and the carboxy-terminal domain of Rip1p as well as by in vitro interactions of various Rip1p–GST fusions with reticulocyte lysate-translated Gle1p (data not shown).
The Gle1 mutants allow heat shock RNA export in the presence of limiting amounts of Rip1p
Given the genetic relationship between Gle1p and Rip1p, the role of Gle1p in poly(A)+ RNA export, and the important role of Rip1p in heat shock RNA export, it was also of interest to determine whether Gle1p manifests any detectable role in heat shock RNA export. We therefore assayed heat shock protein synthesis at 42°C in the three Gle1 mutant strains, in the presence of normal or low levels of Rip1p. As described above, low (Lo) levels of Rip1p prevent heat shock RNA export in an otherwise wild-type background (Fig. 5, lane 3; Fig. 2). Interestingly, this heat shock RNA export defect was suppressed in the presence of all three Gle1 mutant alleles: Synthesis of Hsp104p and to a lesser extent Hsp82p and Ssa4p was substantially rescued after a shift to 42°C (Fig. 5B, cf. lane 3 with lanes 5, 7, and 9). With normal (Hi) Rip1p levels, heat shock protein synthesis at 42°C in the three Gle1 mutant strains was comparable with that observed in the wild-type GLE1 background (Fig. 5B, cf. lane 4 with lanes 6, 8, and 10). Rather than defining a strong role for Gle1p in heat shock RNA export, the data suggest that there is competition between the apparently Gle1p-dependent poly(A)+ RNA export pathway and the apparently Rip1p-dependent heat shock RNA export pathway: The Gle1 mutants weaken the poly(A)+ RNA export pathway thus allowing heat shock RNA export to occur even in the presence of limiting amounts of Rip1p (Fig. 6).
Figure 6.

Model for heat shock RNA and poly(A)+ RNA export under stress conditions. (A) In a wild-type strain, stress allows efficient export of heat shock RNAs but inhibits the export of poly(A)+ RNA (Saavedra et al. 1996). Both Gle1p and Rip1p are involved in heat shock RNA export (under stress) and poly(A)+ RNA export (under normal conditions). Gle1p is essential for poly(A)+ RNA export but also participates in heat shock RNA export. Rip1p is essential for heat shock RNA export but also contributes to poly(A)+ RNA export. It is proposed that heat shock and poly(A)+ RNAs compete for access to Gle1p. (B) No heat shock RNA export is observed with low or no Rip1p. (C) Mutant Gle1p affects poly(A)+ RNA export more strongly than heat shock RNA export, perhaps because mutant alleles are more deleterious to the interaction with poly(A)+ RNPs than that with heat shock RNPs. The increased availability of Gle1p then allows heat shock RNA export in the presence of limiting amounts of Rip1p.
Discussion
As discovered by Saavedra et al. (1997), the selective export of heat shock RNAs at 42°C is affected by deletion of the RIP1 gene. Our biochemical results fully confirm these in situ hybridization observations and indicate that the effect is robust. As RIP1 is an inessential gene for growth at normal temperatures, the different observations make a strong case that Rip1p is only an essential component of the heat shock mRNA transport pathway. This implies that the heat shock and cellular (i.e., non-heat shock) mRNA transport pathways are distinct. Consistent with this notion, factors such as Gsp1p (a small GTPase) and its effectors Rna1p and Prp20p (the GAP and nucleotide exchange factors for Gsp1p, respectively) are required for general RNA transport (Koepp and Silver 1996; Corbett and Silver 1997) but dispensable for heat shock mRNA export (Saavedra et al. 1996).
Interestingly, heat shock mRNA transport is not dependent on Rip1p at 37°C. This might reflect a more transient and less severe heat shock response at this lower and more physiological temperature. Experiments from the Cole laboratory suggest that the Rip1p dependence is not restricted to temperature but is a more general response to stress conditions (Saavedra et al. 1997). These must alter heat shock mRNP and/or the nuclear pore so that a role for Rip1p is revealed. Stress does not seem to affect Rip1p, as we have been unable to detect any obvious temperature-induced change in protein levels or mobility by Western blot analysis (data not shown).
A contribution of Rip1p to selective RNA transport recalls the discovery of this protein as a mediator of Rev-dependent RNA transport in yeast: The FG-repeat domain of Rip1p shows a yeast two-hybrid interaction with the NES in Rev and a deletion or over-expression of Rip1p-affected Rev-mediated RNA export (Stutz et al. 1995). As coexpression of Rev manifests a partial inhibition of heat shock mRNA transport as assayed by in situ hybridization (Saavedra et al. 1997), a Rev-like NES sequence probably mediates the nuclear export of heat shock mRNAs. Although this might indicate an involvement of Rip1p FG repeats, Rip1p carboxy-terminal region fully rescues heat shock RNA transport and at least partially rescues Rev-mediated transport. This is surprising and, at a minimum, suggests a redundant contribution of other FG repeat-containing nucleoporins. Consistent with this possibility, other FG nucleoporins bind credibly to Rev and Rev-like NES sequences in the two-hybrid assay (Stutz et al. 1995, 1996; Fritz and Green 1996). Moreover, there is evidence that the interaction between Rev and Rip FG repeats may be indirect (Neville et al. 1997). Therefore, we suspect that this unique region of the protein contributes to pore function rather than binding directly to RNA or to an RNA-relevant NES.
Yet Rip1p appears to contribute preferentially to an RNA export function. This view is based in part on the synthetic lethal relationship between a RIP1 deletion and mutants in other cellular components implicated in general poly(A)+ RNA transport. Gle1 temperature-sensitive alleles, in particular, have strong and rapid effects on poly(A)+ RNA retention within nuclei, consistent with the proposed role of this protein as a mediator of mRNA transport (Murphy and Wente 1996). Also, mutations in Nup82p (L. Davis, unpubl.) and in Nup85p (our screen; data not shown) are synthetic lethal with a RIP1 deletion; both of these nuclear pore proteins are involved in poly(A)+ RNA export (Grandi et al. 1995; Hurwitz and Blobel 1995; Goldstein et al. 1996; Siniossoglou et al. 1996). We suggest, therefore, that the export of poly(A)+ RNA is suboptimal through Rip1p-deficient pores, but this is normally not limiting for growth; in combination with any of the three Gle1 missense mutations, however, poly(A)+ RNA export becomes too low to support viability (Table 2). This is also consistent with the poly(A)+ retention phenotype of all three Gle1 alleles, which is enhanced in combination with low Rip1p levels. We cannot, however, exclude the possibility that the genetic relationship between RIP1 and GLE1 reflects the transport of a small, specific subset of the poly(A)+ RNA population or of other classes of RNAs (i.e., U snRNAs or 5S RNAs), which have features in common with heat shock mRNP substrates.
If Rip1p is a component of the poly(A)+ as well as the heat shock RNA export pathway, both sets of substrates must compete for access to shared components. The experiment in Figure 5 suggests that the Gle1 mutants depress non-heat shock mRNA transport and thereby restore detectable levels of heat shock RNA transport and heat shock protein synthesis in the presence of limiting amounts of Rip1p (Fig. 6). The results are hard to understand unless the role of Gle1p in normal poly(A)+ RNA transport is more important than any role it may play in heat shock mRNA transport. However, we cannot exclude the possibility that the observations reflect allele-specific effects of the three Gle1 mutants, and we note that Saavedra et al. (1997) show that a temperature-sensitive Gle1 mutant (rss1-37) interferes with heat shock as well as poly(A)+ mRNA export.
Rip1p, in contrast, is clearly more important for heat shock mRNA export than for poly(A)+ mRNA export. In addition to the arguments described above, the rescue of the Gle1 synthetic lethal mutants with low levels of Rip1p (Table 2) suggests that the requirement for cellular RNA transport is fulfilled with minimal Rip1p levels. These same low levels of Rip1p do not suffice for detectable heat shock mRNA transport (Fig. 2).
Rip1p contributes to different pathways, indicating that different substrates must compete for access to the shared components of a universal nuclear pore. Yet the substrates are affected differentially by the physiological and genetic manipulations. It follows that stress must reduce the transport efficiency of non–heat shock mRNAs more than that of heat shock mRNAs, which indicates a difference in the two classes of RNP structures under these conditions (Saavedra et al. 1996). Incubation at 42°C could decrease transport efficiency by affecting the mRNP substrates or the pore. Adequate RNA transport may then require an otherwise fully functional pore for the expression of normal or near-normal levels of heat shock proteins. This transport efficiency view would explain the apparently selective role of Rip1p in heat shock mRNA transport: An essential role is revealed only under stress conditions, when transport is more generally compromised. Similarly, the generic contribution of Rip1p to non–heat shock RNA export at 25°C becomes apparent only in the presence of additional mutant alleles (i.e., Gle1 alleles) that reduce the efficiency of RNA transport.
This interpretation is similar to a contemporary view of pre-mRNA splicing, in which a fundamentally generic set of spliceosome components processes substrates with a range of efficiencies; less efficient substrates more easily reveal contributions from a larger number of factors (Liao et al. 1993; Stutz et al. 1997). Another lesson from the more completely understood splicing pathway as well as the protein import pathway is that in vitro biochemistry will be required to clarify the contribution of individual components to the RNA export process.
Materials and methods
The DNA manipulations were performed according to standard methods (Maniatis et al. 1982; Ausubel et al. 1994); yeast media and yeast transformations were performed with standard procedures (Guthrie and Fink 1991). The strains and plasmids used in this study are described in Table 3.
Table 3.
Strains and plasmids used in this study
| Strain
|
Genotype
|
Reference
|
|---|---|---|
| W303 | ade2 his3 leu2 trp1 ura3 MATa | |
| W303ΔRIP1 | ade2 his3 leu2 trp1 ura3 MATa rip1::KANR | this study |
| Y59ΔCUP | ade2 arg4 leu2 trp1 ura3 Mata ΔCUP1 | Stutz et al. (1995) |
| Y59ΔCUPΔRIP1 (FSY2) | ade2 arg4 leu2 trp1 ura3 Mata ΔCUP1 ΔRIP1 | Stutz et al. (1995) |
| CH1305 | ade2 ade3 leu2 ura3 lys2 can1 MATa | Holm (1993) |
| CH1305ΔRIP1 (FSY5) | ade2 ade3 leu2 ura3 lys2 can1 MATa ΔRIP1 | this study |
| FSY56 | ade2 ade3 leu2 ura3 lys2 can1 MATa ΔRIP1 carrying pFS652 | this study |
| CH1462 | ade2 ade3 leu2 ura3 his3 MATα | Holm (1993) |
| CH1462ΔRIP1 (FSY6) | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 | this study |
| FSY13 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 carrying pFS478 | this study |
| FSY14 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 carrying pFS652 | this study |
| FSY15 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-1 carrying pFS478 | this study |
| FSY16 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-1 carrying pFS652 | this study |
| FSY20 | ade2 ade3 leu2 ura3 lys2 can1 MATa ΔRIP1 gle1-2 carrying pFS652 | this study |
| FSY23 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-3 carrying pFS652 | this study |
| FSY57 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-1 carrying pFS724 | this study |
| FSY58 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-1 carrying pFS398 | this study |
| FSY59 | ade2 ade3 leu2 ura3 lys2 can1 MATa ΔRIP1 gle1-2 carrying pFS724 | this study |
| FSY60 | ade2 ade3 leu2 ura3 lys2 can1 MATa ΔRIP1 gle1-2 carrying pFS398 | this study |
| FSY61 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-3 carrying pFS724 | this study |
| FSY62 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 gle1-3 carrying pFS398 | this study |
| FSY65 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 carrying pFS724 | this study |
| FSY66 | ade2 ade3 leu2 ura3 his3 MATα ΔRIP1 carrying pFS398 | this study |
| Plasmid | ||
| p366 (LEU2/CEN) | YCp50-based vector in which URA3 was replaced with LEU2. | Liao et al. (1993) |
| pFS398 (RIP1–Hi) | HindIII RIP1 genomic 3.5-kb fragment inserted into the HindIII site of p366 (LEU2/CEN); contains 0.78-kb RIP1 5‘-flanking sequences. | this study |
| pFS724 (RIP1–Lo) | HindIII RIP1 1.9-kb fragment generated by PCR inserted into the HindIII site of p366; contains 0.38-kb RIP1 5‘-flanking sequences. | this study |
| pFS662 (RIP1–1FG) | deletion of codons 38–364 of RIP1 in pFS398. | this study |
| pFS730 (RIP1–0FG) | deletion of codons 2–364 of RIP1 in pFS398. | this study |
| pFS733 (RIP1–0FG) | HindIII 2.3-kb insert of pFS730 inserted into the SalI site of JH21 vector (LEU2/2 μm) using SalI linkers. | this study |
| pCH1122 | URA3 ADE3 CEN vector. | Holm (1993) |
| pFS652 | HindIII RIP1 genomic 3.5-kb fragment inserted into the SalI site of pCH1122 using SalI linkers; contains 0.78-kb RIP1 5‘-flanking sequences. | this study |
| pFS478 | HindIII RIP1 1.9-kb fragment generated by PCR inserted into the SalI site of pCH1122 using SalI linkers; contains 0.38-kb RIP1 5‘-flanking sequences. | this study |
Plasmid constructions
Plasmid pFS398 (RIP1–Hi) contains a genomic 3.5-kb RIP1 HindIII fragment with 0.78-kb 5′-flanking sequences (Stutz et al. 1995) inserted into the HindIII site of vector p366 (LEU2/CEN). pFS724 (RIP1–Lo) contains the RIP1 gene on a 1.9-kb HindIII PCR fragment with 0.38-kb 5′-flanking sequences inserted into the HindIII site of p366. The RIP1 deletion mutant constructs pFS662 and pFS730, encoding RIP1–1FG and RIP1–0FG, respectively, were generated by PCR using the 3.5-kb insert of pFS398 as a template. pFS662 was obtained by ligation of a 5′ HindIII–SalI PCR fragment (from position −780 to codon 37 of RIP1) to a 3′ SalI–HindIII PCR fragment (containing codons 365 to the stop of RIP1 and 1.5-kb 3′ flanking sequences) and cloning into p366 cut with HindIII. pFS730 was obtained by ligation of a 5′ HindIII–XhoI fragment (from position −780 to codon 1 of RIP1) to a 3′ XhoI–HindIII fragment (containing codons 365 to the stop and 1.5-kb 3′-noncoding sequences) and cloned into p366 cut with HindIII. pFS733 was obtained by cloning the 2.3-kb HindIII RIP1–0FG insert of pFS730 into the SalI site of vector pJH21 (LEU2/2 μ) using SalI linkers.
Yeast strains
The W303ΔRIP1 strain was made using a PCR-based disruption procedure with the kanMX2 template (Wach et al. 1994) and two oligonucleotides: 5′-CAAGCTATCA TTAAATTTCG ACCCGAGCAA CGACTCAACT GTAGCCAGCT GAAGCTTCGT ACGCTG-3′ and 5′-CTATGCAACC AATGCAGGTG GTGGAGGTAT ATCAGGGACA AGGCATAGGC CACTAGTGGA TCTG-3′. This generated a PCR fragment of ∼1.4 kb containing the Escherichia coli KANR (kanamycin resistance) gene flanked at the 5′ end by 44 bp of sequences from bp −1 to −43 of RIP1 and at the 3′ end by 42 bp of sequences from bp +1251 to the stop of RIP1. One to two micrograms of this PCR fragment was transformed into W303, and transformants were selected on YPD plates containing 200 mg/l G418 (Geneticin, GIBCO BRL). Replacement of the RIP1 gene by the KANR gene was confirmed by PCR analysis and Southern blotting.
The Y59ΔCUP and Y59ΔCUPΔRIP1 (FSY2) strains have been described (Stutz et al. 1995).
In vivo protein labeling
The yeast strains were grown as 15-ml cultures in synthetic complete medium lacking methionine or in the appropriate dropout medium lacking methionine overnight at 23°C to an OD600 of 0.05–0.1 (5.105–1.106 cells/ml). Cells were collected by centrifugation, resuspended in 0.5-1 ml of medium lacking methionine and distributed into Eppendorf tubes as 0.25-ml aliquots. These cells were rapidly mixed with one volume of medium lacking methionine preheated to 49°C or 59°C and incubated at 37°C or 42°C, respectively. The 25°C control samples were mixed with one volume of medium at 25°C. After 15 min, the samples were mixed with 50 μCi of Trans 35S-label (1191 Ci/mmole, 11.02 mCi/ml, ICN Pharmaceuticals, Inc.) and incubated for another 15 min at the same temperature. Protein labeling was stopped by centrifugation at 4°C; the cell pellets were washed twice with 1 ml of culture medium in the cold, resuspended in 30–50 μl of 1× SDS sample buffer, and immediately frozen on dry ice. The samples were boiled for 10 min and fractionated on a 8% SDS–polyacrylamide gel. Gels were dried and autoradiographed.
RNA extractions and primer extensions
The wild-type (W303) and ΔRIP1 (W303ΔRIP1) strains were grown overnight in 100 ml of YPD at 25°C to an OD600 of 0.1 (∼1.106 cells/ml). Twenty-five-milliliter aliquots of each culture were rapidly mixed with an equivalent volume of YPD at 25°C, 49°C, or 59°C and incubated at 25°C, 37°C, or 42°C, respectively, for 30 min. RNA extractions and primer extensions were performed according to published procedures (Pikielny and Rosbash 1985) using two oligonucleotide primers. Oligonucleotide primer OFS 191 is complementary to positions +63 to +85 from the ATG of the HSP104 gene. Oligonucleotide primer DT58, which is complementary to U2 snRNA and gives an ∼120-base primer extended product, was used as an internal control for loading. Extension products were analyzed on 6% polyacrylamide denaturing gels (Fig. 1B).
Rev growth assay
Strains Y59ΔCUP and Y59ΔCUPΔRIP1 were transformed with a URA3/2μ version of the PC–CUP–RRE reporter construct and different Rev-expressing plasmids (TRP1/CEN) as described earlier (Stutz and Rosbash 1994; Stutz et al. 1995). These strains were then transformed with the p366 vector (LEU2/CEN) or with the Rip1p-expressing plasmids pFS398, pFS662, pFS730 (LEU2/CEN), or pFS733 (LEU2/2μ). The triple transformants were grown to saturation in selective medium and spotted onto selective plates containing increasing concentrations of CuSO4 (from 0.1 to 1.3 mm with 0.1 mm increments). The copper resistance of each strain was scored after incubation at 30°C for 5 days.
Total cell extracts and cell fractionation
For total cell extracts, 1 ml of overnight-saturated cultures were spun in an eppendorf tube and the cells were broken with glass beads in 100 μl of cold 20% trichloroacetic acid (TCA). A TCA precipitate was obtained by addition of 1 ml of 5% TCA and centrifugation for 20 min at 4°C. The pellets were resuspended in 70 μl of 1× sample buffer and neutralized with 30 μl of 2 m Tris-base. Ten to fifteen microliters were used for fractionation on SDS-PAGE and Western blot analysis.
Yeast cells were fractionated essentially as described (Belanger et al. 1994). Briefly, yeast cells (80–100 OD600 units, 1.109 cells) were spheroplasted and lysed in cold 20 mm HEPES–HCl (pH 7.5), 5 mm MgCl2, and protease inhibitors, followed by vortexing and incubation on ice for 5 min. Lysates were spun at 9000 rpm at 4°C for 45 min. The supernatant (S) was decanted, and the pellet was resuspended in 5 ml of ice-cold 20 mm HEPES–HCl (pH 7.4), 5 mm MgCl2, 1 m NaCl, and protease inhibitors, incubated on ice for 10 min, and centrifuged at 9000 rpm for 45 min. The supernatant (P) was collected. Total lysate (T), S, and P fractions were precipitated with TCA before fractionation on SDS-PAGE and Western blot analysis.
Western blot analysis
TCA precipitates in 1× sample buffer were boiled and fractionated on 10% SDS–polyacrylamide gels. The gels were transferred to nitrocellulose filters by electroblotting according to standard methods (Ausubel et al. 1994). The filters were incubated overnight with a rabbit polyclonal antibody directed against the Rip1p FG repeats (1:2000 dilution) or with a mouse polyclonal antibody directed against the Rip1p unique carboxyl terminus (1:2000). Immunoreactive bands were detected by ECL (Amersham Life Science, Inc.).
RIP1 synthetic lethal screen and identification of Gle1 alleles
Strains
The original strains for the colony sectoring assay CH1305 and CH1462 (Table 3) were obtained from C. Holm (Kranz and Holm 1990; Holm 1993). The CH1305ΔRIP1 (FSY5) and CH1462ΔRIP1 (FSY6) were generated using the same strategy as described previously to produce the Y59ΔCUPΔRIP1 strain (FSY2) (Stutz et al. 1995).
Plasmids
Two screens were performed successively with two different screening plasmids. The screening plasmid pFS478 was constructed by inserting the 1.9-kb HindIII RIP1 fragment of pFS724 into the SalI site of pCH1122 (Kranz and Holm 1990; Holm 1993) using SalI linkers. The screening plasmid pFS652 was constructed by inserting the 3.5-kb RIP1 HindIII genomic fragment of pFS398 (see above) into the SalI site of pCH1122 using SalI linkers. pFS398 was used as the testing plasmid in both screens.
Screens
In the first screen, FSY5 and FSY6 strains transformed with pFS478 were grown in Ura− medium, plated on YPD + 4% glucose and mutagenized by UV (UV Stratalinker 1800, Stratagene) to produce 40%–50% cell death. A total of 200,000 colonies were screened for a nonsectoring (Sect−) phenotype. Nonsectoring colonies were subscreened as described (Holm 1993). A single nonsectoring mutant strain (FSY15) was selected. Backcross of this mutant with the starting wild-type strain (FSY5) indicated the presence of a single recessive mutation. To clone the mutant gene, the FSY15 strain was transformed with a p366-based (LEU2/CEN) yeast genomic library (Liao et al. 1993); transformants were grown on Leu− plates and subsequently replica-plated to 5-fluoro-orotic acid (5-FOA) plates. Plasmid DNA was recovered from strains that became 5-FOA resistant and retransformed into FSY15. The genomic sequences that rescued the Sect− phenotype in FSY15 were partially sequenced and subcloned. A 5.5-kb SphI fragment containing the GLE1 gene rescued the Sect− phenotype of FSY15.
In the second screen, FSY5 and FSY6 strains transformed with pFS652 were grown in Ura− medium to 5.107 cells/ml, mutagenized with ethylmethylsulfonate (EMS) as described (Lawrence 1991) to produce 40%–50% cell death, and plated on YPD + 4% glucose plates. Nonsectoring colonies were identified and subscreened as described above. Mating with FSY15 (gle1-1) and complementation analysis identified the mutant strains FSY20 and FSY23 as containing the gle1-2 and gle1-3 alleles, respectively. The mutations in the Gle1 alleles were identified by PCR amplification of genomic DNA prepared from FSY15, FSY20, and FSY23 and by PCR sequencing of both strands; this procedure was repeated twice.
Plasmid shuffling
The growth of the Gle1 mutant strains in the presence of different Rip1p-expressing plasmids was examined by transformation of the synthetic lethal strains with vector p366, or various RIP1 constructs. Transformants were selected on Leu− plates and subsequently streaked on 5-FOA plates to select for strains that had lost the URA3 ADE3 CEN screening plasmids (pFS478 or pFS652). The growth of the 5-FOA resistant strains was analyzed on plates at 25°C and 37°C.
Acknowledgments
We thank C. Cole and colleagues for communicating data before publication. We are grateful to P. Silver, L. Davis, and I. Mattaj for critical reading of the manuscript. Our thanks also go to L. Liu for antibody preparation, E. Dougherty for help with figures, and L.A. Monaghan for secretarial assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Rosbash@binah.cc.brandeis.edu; FAX (617) 736-3164.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short protocols in molecular biology. 2nd ed. New York, NY: Greene Publishing Associates; 1994. [Google Scholar]
- Belanger KD, Kenna MA, Wei S, Davis LJ. Genetic and physical interactions between Srp1p and nuclear pore complex proteins Nup1p and Nup2p. J Cell Biol. 1994;126:619–630. doi: 10.1083/jcb.126.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Fridell RA, Madore S, Cullen BR. Identification of a novel cellular cofactor for the Rev/Rex class of retroviral regulatory proteins. Cell. 1995;82:485–494. doi: 10.1016/0092-8674(95)90437-9. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Fridell RA, Benson RE, Hua J, Cullen BR. Protein sequence requirements for function of the human T-cell leukemia virus type 1 Rex nuclear export signal delineated by a novel in vivo randomization-selection assay. Mol Cell Biol. 1996;16:4207–4214. doi: 10.1128/mcb.16.8.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett AH, Silver PA. Nucleocytoplasmic transport of macromolecules. Microbiol Mol Biol Rev. 1997;61:193–211. doi: 10.1128/mmbr.61.2.193-211.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR, Malim MH. The HIV-1 rev protein: Prototype of a novel class of eukaryotic post-transcriptional regulators. Trends Biol Sci. 1991;16:346–350. doi: 10.1016/0968-0004(91)90141-h. [DOI] [PubMed] [Google Scholar]
- Davis LI. The nuclear pore complex. Annu Rev Biochem. 1995;64:865–896. doi: 10.1146/annurev.bi.64.070195.004245. [DOI] [PubMed] [Google Scholar]
- Del Priore V, Snay CA, Bahr A, Cole CN. The product of the Saccharomyces cerevisiae RSS1 gene, identified as a high-copy suppressor of the rat7-1 temperature-sensitive allele of the RAT7/NUP159 nucleoporin, is required for efficient mRNA export. Mol Biol Cell. 1996;7:1601–1621. doi: 10.1091/mbc.7.10.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye V, Hurt EC. Genetic approaches to nuclear pore structure and function. Trends Genet. 1995;11:235–241. doi: 10.1016/s0168-9525(00)89057-5. [DOI] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Luhrmann R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Fridell RA, Fischer U, Luhrmann R, Meyer BE, Meinkoth JL, Malim MH, Cullen BR. Amphibian transcription factor IIIA proteins contain a sequence element functionally equivalent to the nuclear export signal of human immunodeficiency virus type 1 Rev. Proc Natl Acad Sci. 1996;93:2936–2940. doi: 10.1073/pnas.93.7.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz CC, Green MR. HIV Rev uses a conserved cellular protein export pathway for the nucleocytoplasmic transport of RNAs. Curr Biol. 1996;6:848–854. doi: 10.1016/s0960-9822(02)00608-5. [DOI] [PubMed] [Google Scholar]
- Fritz CC, Zapp ML, Green MR. A human nucleoporin-like protein that specifically interacts with HIV Rev. Nature. 1995;376:530–533. doi: 10.1038/376530a0. [DOI] [PubMed] [Google Scholar]
- Gerace L. Nuclear export signals and the fast track to the cytoplasm. Cell. 1995;82:341–344. doi: 10.1016/0092-8674(95)90420-4. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, Snay CA, Heath CV, Cole CN. Pleiotropic nuclear defects associated with a conditional allele of the novel nucleoporin Rat9p/Nup85p. Mol Biol Cell. 1996;7:917–934. doi: 10.1091/mbc.7.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Grandi P, Emig S, Weise C, Hucho F, Pohl T, Hurt EC. A novel nuclear pore protein Nup82p which specifically binds to a fraction of Nsp1p. J Cell Biol. 1995;130:1263–1273. doi: 10.1083/jcb.130.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guddat U, Bakken AH, Pieler T. Protein-mediated nuclear export of RNA: 5S rRNA containing small RNPs in Xenopus oocytes. Cell. 1990;60:619–628. doi: 10.1016/0092-8674(90)90665-2. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194:389–398. [PubMed] [Google Scholar]
- Holm C. A functional approach to identifying yeast homologs of genes from other species. Methods. 1993;5:102–109. [Google Scholar]
- Hurwitz ME, Blobel G. NUP82 is an essential yeast nucleoporin required for poly(A)+ RNA export. J Cell Biol. 1995;130:1275–1281. doi: 10.1083/jcb.130.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovine MK, Wente SR. A nuclear export signal in Kap95p is required for both recycling the import factor and interaction with the nucleoporin GLFG repeat regions of Nup116p and Nup100p. J Cell Biol. 1997;137:797–811. doi: 10.1083/jcb.137.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, Gamberi C, Jarmolowski A, McGuigan C, Mattaj IW. A cap-binding protein complex mediating U snRNA export. Nature. 1995;376:709–712. doi: 10.1038/376709a0. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Jarmolowski A, Beisel C, Mattaj IW, Dreyfuss G, Fischer U. A role for the M9 transport signal of hnRNP A1 in mRNA nuclear export. J Cell Biol. 1997;137:27–35. doi: 10.1083/jcb.137.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmolowski, A., W.C. Boelens, E. Izaurralde, and I.W. Mattaj. 1994. Nuclear export of different classes of RNA is mediated by specific factors. J. Cell Biol. 124:(5) 627–635. [DOI] [PMC free article] [PubMed]
- Koepp DM, Silver PA. A GTPase controlling nuclear trafficking: Running the right way or walking randomly. Cell. 1996;87:1–4. doi: 10.1016/s0092-8674(00)81315-x. [DOI] [PubMed] [Google Scholar]
- Kranz JE, Holm C. Cloning by function: An alternative approach for identifying yeast homologs of genes from other organisms. Proc Natl Acad Sci. 1990;87:6629–6633. doi: 10.1073/pnas.87.17.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CW. Classical mutagenesis techniques. In: Guthrie C, Fink GR, editors. Methods in enzymology. San Diego, CA: Academic Press; 1991. pp. 273–281. [DOI] [PubMed] [Google Scholar]
- Lee MS, Henry M, Silver PA. A protein that shuttles between the nucleus and the cytoplasm is an important mediator of RNA export. Genes & Dev. 1996;10:1233–1246. doi: 10.1101/gad.10.10.1233. [DOI] [PubMed] [Google Scholar]
- Legrain P, Rosbash M. Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell. 1989;57:573–583. doi: 10.1016/0092-8674(89)90127-x. [DOI] [PubMed] [Google Scholar]
- Liao XC, Tang J, Rosbash M. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: Implications for snRNP protein function in pre-mRNA splicing. Genes & Dev. 1993;7:419–428. doi: 10.1101/gad.7.3.419. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- Melchior F, Gerace L. Mechanisms of nuclear protein import. Curr Opin Cell Biol. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- Michael MW, Choi M, Dreyfuss G. A nuclear export signal in hnRNP A1: A signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1996;83:415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383:357–360. doi: 10.1038/383357a0. [DOI] [PubMed] [Google Scholar]
- Neville M, Stutz F, Lee L, Davis LI, Rosbash M. Evidence that the importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export in S. cerevisiae. Curr Biol. 1997;7:767–775. doi: 10.1016/s0960-9822(06)00335-6. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nucleocytoplasmic transport: Signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- Nonet M, Scafe C, Sexton J, Young R. Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol Cell Biol. 1987;7:1602–1611. doi: 10.1128/mcb.7.5.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pante, N. and U. Aebi. 1995. Exploring nuclear pore complex structure and function in molecular detail. J. Cell Sci (Suppl.) 19: 1–11. [DOI] [PubMed]
- ————— Toward the molecular dissection of protein import into nuclei. Curr Opin Cell Biol. 1996;8:397–406. doi: 10.1016/s0955-0674(96)80016-0. [DOI] [PubMed] [Google Scholar]
- Pikielny CW, Rosbash M. mRNA splicing efficiency in yeast and the contribution of nonconserved sequences. Cell. 1985;41:119–126. doi: 10.1016/0092-8674(85)90066-2. [DOI] [PubMed] [Google Scholar]
- Rout MP, Wente SR. Pores for thought: Nuclear pore complex proteins. Trends Cell Biol. 1994;4:357–365. doi: 10.1016/0962-8924(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Saavedra C, Tung K-S, Amberg DC, Hopper AK, Cole CN. Regulation of mRNA export in response to stress in Saccharomyces cerevisiae. Genes & Dev. 1996;10:1608–1620. doi: 10.1101/gad.10.13.1608. [DOI] [PubMed] [Google Scholar]
- Saavedra, C.A., C.M. Hammell, C.V. Heath, and C.N. Cole. 1997. Export of heat shock mRNAs following stress in Saccharomyces cerevisiae employs a distinct pathway defined by Rip1p and also requires a subset of factors essential for export of poly(A)+ mRNA. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Segref A, Sharma K, Doye V, Hellwig A, Huber J, Luhrmann R, Hurt E. Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J. 1997;16:3256–3271. doi: 10.1093/emboj/16.11.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniossoglou S, Wimmer C, Rieger M, Doye V, Tekotte H, Weise C, Emig S, Segref A, Hurt EC. A novel complex of nucleoporins, which includes Sec13p and a Sec13p homolog, is essential for normal nuclear pores. Cell. 1996;84:265–275. doi: 10.1016/s0092-8674(00)80981-2. [DOI] [PubMed] [Google Scholar]
- Sodroski J, Goh WC, Rosen C, Dayton A, Terwilliger E, Haseltine W. A second post-transcriptional trans-activator gene required for HTLV-III replication. Nature. 1986;321:412–417. doi: 10.1038/321412a0. [DOI] [PubMed] [Google Scholar]
- Stutz F, Rosbash M. A functional interaction between Rev and yeast pre-mRNA is related to splicing complex formation. EMBO J. 1994;13:4096–4104. doi: 10.1002/j.1460-2075.1994.tb06727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz F, Neville M, Rosbash M. Identification of a novel nuclear pore-associated protein as a functional target of the HIV-1 Rev protein in yeast. Cell. 1995;82:495–506. doi: 10.1016/0092-8674(95)90438-7. [DOI] [PubMed] [Google Scholar]
- Stutz F, Izaurralde E, Mattaj IW, Rosbash M. A role for nucleoporin FG repeat domains in export of human immunodeficiency virus type 1 Rev protein and RNA from the nucleus. Mol Cell Biol. 1996;16:7144–7150. doi: 10.1128/mcb.16.12.7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz F, Tang J, Rosbash M. Synthetic lethal/enhancer screening to identify snRNA-protein and protein-protein interactions in yeast pre-mRNA splicing. In: Smith C, editor. The practical approach series. Oxford, UK: Oxford University Press; 1997. . (in press). [Google Scholar]
- Visa N, Alzhanova-Ericsson AT, Sun X, Kiseleva E, Bjorkroth B, Wurtz T, Daneholt B. A pre-mRNA-binding protein accompanies the RNA from the gene through the nuclear pores and into polysomes. Cell. 1996a;84:253–264. doi: 10.1016/s0092-8674(00)80980-0. [DOI] [PubMed] [Google Scholar]
- Visa N, Izaurralde E, Ferreira J, Daneholt B, Mattaj IW. A nuclear cap-binding complex binds balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. J Cell Biol. 1996b;133:5–14. doi: 10.1083/jcb.133.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]