

Abstract
The Trp-cage, as the smallest miniprotein, remains the subject of numerous computational and experimental studies of protein folding dynamics and pathways. The original Trp-cage (NLYIQWLKDGGPSSGRPPPS, Tm = 42°C) can be significantly stabilized by mutations; melting points as high as 64°C are reported. In helical portions of the structure, each allowed replacement of Leu, Ile, Lys or Ser residues by Ala results in a 1.5 (±0.35) kJ/mol fold stabilization. No changes in structure or fluxionality of the core results upon stabilization. Contrary to the initial hypothesis, specific Pro/Trp interactions are not essential for core formation. The entropic advantage of Pro versus Ala (ΔΔSU = 11 ± 2 J/mol K) was measured at the solvent-exposed P17 site. Pro–Ala mutations at two of the three prolines (P12 and P18) that encage the indole ring result in less fold destabilization (2.3–3.4 kJ/mol). However, a P19A mutation reduces fold stability by 16 kJ/mol reflecting a favorable Y3/P19 interaction as well as Trp burial. The Y3/P19 hydrophobic staple interaction defines the folding motif as an 18-residue unit. Other stabilizing features that have been identified include a solvent-exposed Arg/Asp salt bridge (3.4–6 kJ/mol) and a buried H-bonded Ser side chain (≈10 kJ/mol).
Keywords: buried polar sidechain, fast-folding miniprotein, hydrophobic core formation, motif minimization, proline-tryptophan interactions
Introduction
We have previously reported the design of a 20-residue peptide, TC5b (NLYIQ WLKDG GPSSG RPPPS), which adopts the ‘Trp-cage fold’ (Fig. 1) (Neidigh et al., 2002). This novel structural motif includes a hydrophobic core (Tyr3, Trp6, Leu7, Gly11, Pro12, Pro18 and Pro19), a helical segment spanning residues Leu2 through Lys8, and a very dense web of long-range tertiary contacts (and NOEs) which reflect an unusually high contact order (Plaxco et al., 2000; Koga and Takada, 2001; Ivankov et al., 2003) for a relatively short peptide sequence. On the basis of CD and NMR data, TC5b displays two-state folding with a melting temperature (Tm) of 42°C in pH 7 aqueous buffer (Neidigh et al., 2002). This was subsequently confirmed (Qiu et al., 2002) by independent CD- and fluorescence-monitored melts; although some studies by other spectroscopic methods (Ahmed et al., 2005; Neuweiler et al., 2005) have suggested a more complicated folding scenario. IR- and fluorescence-detected T-jump experiments established TC5b as one of the fastest folding systems known (Andersen et al., 2001; Qiu et al., 2002; Kubelka et al., 2004; Ghosh et al., 2007): a P12W mutant was recently reported to be a sub-microsecond folding system (Bunagan et al., 2006).
Fig. 1.

The Trp-cage fold showing the secondary structure features and the buried Trp side chain as well as the residues that shield the Trp indole ring from solvent exposure.
It was posited that the key stabilizing features of the Trp-cage are: the burial of the Trp side chain as part of the hydrophobic core, the minimization of ΔSU (associated with the inclusion of four prolines in the sequence) and an Asp9/Arg16 salt-bridge interaction. As the smallest fully protein-like polypeptide, TC5b has emerged as a paradigm for studying the ability of molecular dynamics to simulate protein folding (Simmerling et al., 2002; Searle and Ciani, 2004). At least 15 groups (Simmerling et al., 2002; Snow et al., 2002; Carnevali et al., 2003; Chowdhury et al., 2003; Nikiforovich et al., 2003; Pitera and Swope, 2003; Schug et al., 2003, 2005; Zhou, 2003; Ota et al., 2004; Ding et al., 2005; Linhananta et al., 2005; Chen et al., 2006; Juraszek and Bolhuis, 2006; Ulmschneider et al., 2006; Kentsis et al., 2007) have reported folding simulations that reproduce structural features of the Trp-cage. These computational studies have yielded a diversity of hypotheses concerning which specific interactions are important for fold stabilization and to facilitate rapid folding.
Mutations at Trp6 established that the Trp side chain is required for fold stabilization: (W6H)- and (W6F)-TC5b mutants were completely and largely unfolded in water (Barua and Andersen, 2002). In the case of the W6F mutant, fold population estimates could be obtained in both water and 30% TFE and the extent of fold destabilization was the same in the two media, ΔΔGF = 12.5 ± 0.6 kJ/mol. The indole ring can be replaced by a naphthalene ring with some degree of success: both regioisomers, the (W6αNal)- and (W6βNal)-TC5b mutants, were fully folded in aqueous TFE. However, the naphthylalanine analogs were only 50%-folded in water, compared with circa 98% for TC5b (ΔΔGF = 9 kJ/mol). Trp association with DPC micelle head groups affects unfolding of Trp-cage structures (Neidigh et al., 2001; Neidigh and Andersen, 2002). These studies indicate that indole ring burial may contribute up to 12 kJ/mol of the driving force for Trp-cage formation. It was also noted that the W6H mutant and a C-terminally truncated peptide (NLYAQ WLKDG GPSK), which have the same N-terminal helical segment as TC5b but cannot form the Trp-cage, display only modest levels of helicity (Barua and Andersen, 2002). This suggested that Trp-cage formation provides stabilization to the helix and that helix stabilization contributes to Trp-cage formation. The latter has become the basis of a novel method for determining helix propensities based on the Trp-cage scaffold (Lin et al., 2004).
The Trp-cage sequence was derived starting with the sequence of exendin-4, a naturally occurring 39-residue peptide displaying a partially formed Trp-cage and a tendency toward aggregation in water (Neidigh et al., 2001; Hudson and Andersen, 2004). Synthetic exendin-4 (exenatide, Amylin Pharmaceuticals) is now an approved treatment for diabetes. Three serial truncations and mutations at five sites produced the 20-residue TC5b system (Neidigh et al., 2002). Since then, data for 27 Trp-cage mutants have been published (Barua and Andersen, 2002; Neidigh and Andersen, 2002; Hudson and Andersen, 2004; Lin et al., 2004; Liu et al., 2004; Bunagan et al., 2006; Naduthambi and Zondlo, 2006).
In the present account, we present previously unpublished details regarding the design of the Trp-cage and mutations that test the hypotheses regarding its stabilizing features, namely the relationship between intrinsic helix stability and overall fold stability, the entropic effect of the prolines on the latter, the contribution of a D9/R16 salt bridge and the effects of stabilization of local and secondary structural features on the folding scenario. Additional goals were to produce even smaller constructs that would still fold yielding the same hydrophobic core and to optimize the Trp-cage sequence to make constructs with increased fold stabilities suitable for pharmocophore display in drug discovery studies.
Results
Additional Trp-cage mutants (bringing the total number of reported sequences to 64, of which 11 have been reported from other laboratories) were synthesized for the present study. At this point, mutations at all positions in the original sequence, except the three glycines, have been examined. Of those retaining the Trp residue, all but five adopt the Trp-cage fold as the predominant conformation in water. As in previous studies, there is a remarkable consistency in both the chemical shifts (Supplementary Table available at PEDS online) and long-range NOEs observed for these species under conditions that favor folding (pH 6–7, 280 K). As a result, we provide detailed chemical shift assignments for only four analogs (Supplementary data available at PEDS online) and an NOE-based structure determination for only a single stabilized species. The large structuring shifts, due to ring-current effects in the hydrophobic core, are tracked as chemical shift deviations (CSD = δobs − δrc, where δobs is the observed chemical shift and δrc is a reference random coil chemical shift (Wishart and Sykes, 1994; Andersen et al., 1997; Fesinmeyer et al., 2004) and are used to measure the extent of folding in the Trp-cage. It was apparent that we had nearly achieved the fully folded shift values for the ring-current affected sites at the stage of TC5b. Only minor increases in structuring shifts at 280 K are observed as the fold stability is increased by mutation or by the addition of TFE (Supplementary Table available at PEDS online). There is similarly excellent agreement in the magnitude of the upfield shifts for key Hα sites within the helix (Y3, Q5, W6, K8); the sum of these four CSDs provides a measure of the extent of helix formation.
The Gly11 methylene is a particularly diagnostic site. The diastereotopic Δδ for the Gly11 methylene (G11ΔδHα = 2.3–2.55 p.p.m., for species judged to be >92% folded) appears to be a sensitive probe of both structure and fluxionality. G11ΔδHα is maximal for the more stable analogs in water, with G11Hα3 displaying upfield CSDs <1.01 p.p.m. (versus 3.20–3.46 p.p.m. for G11Hα2). In almost all cases, G11ΔδHα decreases in the TFE containing medium and upon mutational destabilization of the Trp-cage structure. These changes predominantly reflect a decrease in the ring-current shielding of the more shielded G11Hα2 and, in many cases, a further upfield ring-current shift of G11Hα3. G11Hα3 also displays unexpected melting behavior (vide infra). Both effects can be rationalized by increased backbone fluxionality with some motional averaging of the G11-CH2 ring-current shifts. As a result, we only use the CSD of 11Hα2 to monitor Trp-cage formation.
Measuring fold populations and melting curves
The stabilities of the Trp-cage constructs reported herein are measured using NMR determinations of the extent of amide NH exchange protection afforded by folding and by NMR and CD-monitored melting studies. Typical NMR melting data (chemical shift versus temperature) for a stable and less stable Trp-cage appear in Fig. 2. In the NMR melting studies, the fold populations are obtained from CSDs. Each site experiencing a structuring CSD provides a site-specific melting temperature (Tm, defined as the temperature at which the chemical shift for that site is exactly halfway between the folded and statistical coil value). In the present, as well as previous studies (Neidigh et al., 2002; Lin et al., 2004), there is excellent agreement between melts for sites throughout the structure. This is the case for the numerous upfield shifted sites as well as those displaying downfield ring-current shifts such as the P12α, P12β3 and R16α protons. The only exceptions to uniform melting throughout the sequence are the P12δ3 and G11α3 sites, vide infra.
Fig. 2.

Representative plots of CSDs versus temperature for a stable Trp-cage (TC10b, dark lines) and its destabilized S14A-mutant (grey lines and symbols). a illustrates the L7α shift melts, the melting of the downfield shifts at P12Hβ3 and R16α are illustrated in b, the melting of the larger upfield ring-current shifts due to the indole ring appear in (c) and (d). The lines through the data are polynomial fits with no theoretical significance. Similar effects are observed for acidification-induced decreases in extent of folding (see Fig. S1, Supplementary data available at PEDS online).
The largest CSDs in aqueous buffer were typically observed for TC10b (DAYAQWLKDGGPSSGRPPPS, see Table I) at 280 K at pH 7, conditions which yielded amide NH exchange protection factors (PF) greater than 100 (vide infra); as a result, these CSDs were employed as folded-state reference values, indicative of a 99.5%-folded population (Lin et al., 2004). The fraction-unfolded values (χU) for each construct were calculated as follows:
Table I.
Helix mutations and optimization of the ‘Trp-cage’ − Tm’s (°C) in aqueous buffer
| TC# | Sequence | pH 7
|
pH 2.5
|
||||
|---|---|---|---|---|---|---|---|
| Cage | Helix | CD | Cage | Helix | CD | ||
| 5a | NLFIQWLKD GGPSSG RPPPS | 32 | 37 | 31 | 14 | 16.5 | 16 |
| 5b | NLYIQWLKD GGPSSG RPPPS | 42 | 43 | 42 | 23 | 25.5 | n.d. |
| 8a | NLYAQWLKD GGPSSG RPPPS | 46 | 48 | 46 | 28.5 | 32 | 26 |
| 8b | DLYAQWLKD GGPSSG RPPPS | 50 | 53.5 | 53 | 32 | 37 | 31 |
| 9b | NAYAQWLKD GGPSSG RPPPS | 51 | 51 | 50 | 34.5 | 39 | 36 |
| NAYAAWLKD GGPSSG RPPPS | 41 | 42 | 43 | 27 | 30 | 28 | |
| NAYAQWLAD GGPSSG RPPPS | 54 | 57 | 53 | n.d. | |||
| NAUYUQWLKD GGPSSG RPPPSa | 53.5 | 59 | 55 | n.d. | |||
| 10b | DAYAQWLKD GGPSSG RPPPS | 56 | 61 | 56 | 39 | 44 | 41 |
| DAFAQWLKD GGPSSG RPPPS | 47 | 51 | 46 | n.d. | |||
| DALAQWLKD GGPSSG RPPPS | 39 | 41.5 | n.d. | ||||
| DAAAQWLKD GGPSSG RPPPS | 6b | 8.5 | n.d. | ||||
| DAYUQWLKD GGPSSG RPPPSa | 60 | 67 | 61 | n.d. | |||
| DAYAQWAKD GGPSSG RPPPS | 11 | 23c | n.d. | ||||
| DAYAQWLAD GGPSSG RPPPS | 62 | 66 | n.d. | 46 | 52 | n.d. | |
| 11b1 | GAYAQWLKD GGPSSG RPPPS | 39 | 41 | 41 | n.d. | ||
| GAFAQWLKD GGPSSG RPPPS | 29 | 32 | 26 | n.d. | |||
U = α-aminobutyric acid (Aib).
The fraction of Y3A-TC10b in the cage conformation at 280 K is in the 0.44–0.50 range based on the shifts observed for P18α,β3.
The CD indicates a lower helicity, at 280 K the [θ]223 value is 41% of that TC10b.
Fraction-unfolded values are plotted against temperature to give melting curves, from which Tm’s were calculated. For those few constructs that displayed CSDs larger than those for TC10b at some sites, the CSDs at 280 K and pH 7 for that construct were equated with a 99%-folded population for calculating χU at higher temperatures.
Since the agreement between individual site-specific measures of folding is excellent, we rely on sums of CSDs (Neidigh et al., 2002; Hudson and Andersen, 2004; Lin et al., 2004) for the Tm’s reported herein. For each construct, two Tm-measures, ‘cage’ and ‘helix’, were calculated. The ‘cage’ Tm’s (Tmcage) are derived from the sum of the CSDs of five protons (L7α, P18α, P18β3, P19δ3 and P19δ2) experiencing large upfield ring-current shifts associated with the indole ring of Trp6. The G11α2 proton has an even larger structuring CSDs (circa 3.4 p.p.m.) associated with cage formation, but CSDs of G11α2 are not available for all mutants since this signal undergoes extensive line broadening due to exchange as the population of the unfolded state increases. The G11α2 signal becomes too broad to be observed in 2D NMR spectra at high temperatures in some constructs. When available, the G11α2 CSD melt provides essentially the same Tm values as the other ‘cage-monitoring’ signals used in this study. The ‘helix’ Tm’s (Tmhelix) monitor helix formation and were obtained from the CSDs of Hα sites within the helix. For the vast majority of systems examined, these two measures of the melting behavior of Trp-cage constructs gave very similar results (Fig. 3). The few exceptions to this rule will be discussed in the later sections.
Fig. 3.
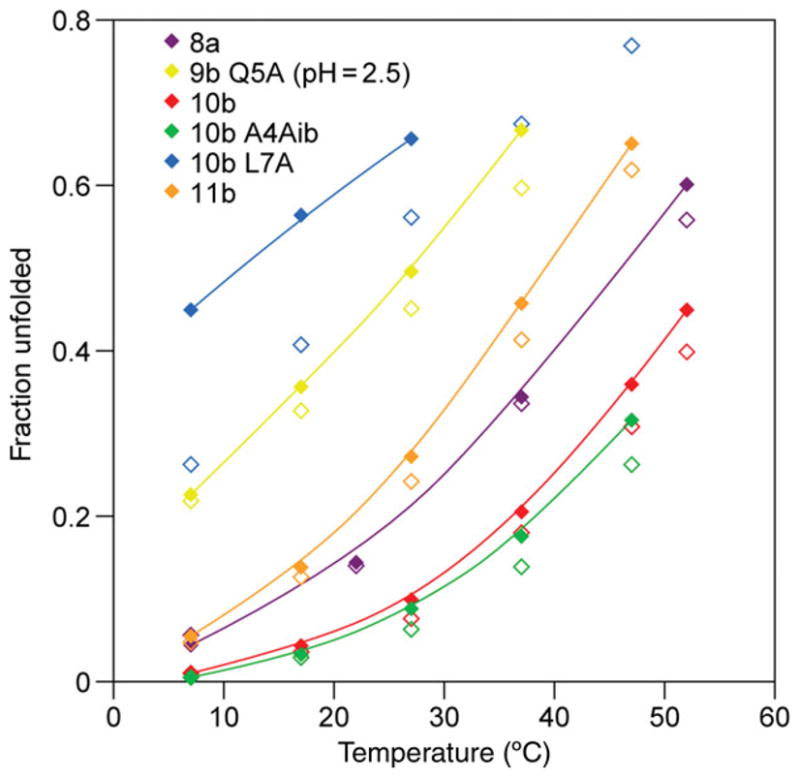
‘Cage’ (best fit lines through solid symbols) and ‘helix’ (open symbols) CSD melts, χU versus T (°C), for representative mutants from Table I, with the exception of Q5A-TC9b (at pH 2.5), all melts were measured at pH 7. The (L7A)-mutant of TC10b illustrates the large deviations between helix and cage melts that can be observed for mutants with significantly destabilized cage structures. In more typical Trp-cage sequences, the helix measure yields χU values that are only slightly smaller than the values based on the shifts that measure cage formation.
Optimization of the ‘Trp-cage’
A series of alanine substitutions were performed in the helical portion of the TC5b sequence with two different N-capping residues, Asn1 and Asp1. The effects of these mutations on fold stability, as monitored by CSD melts, are shown in Table I. Similar melting behavior is revealed by CD spectroscopy, with residue-molar ellipticities at the 222.5–223.6 nm minimum ([θ]223) used to monitor helicity (see, Methods).
The L2A, I4A and K8A mutations increased fold stability, whereas the Q5A and L7A mutations resulted in somewhat and significantly lessened fold stability, respectively. Since the double mutation, L2A and I4A, resulted in constructs with highly stable folds (TC9b and TC10b) without altering outwardly directed polar groups, all subsequent mutations were carried out with the TC9b or TC10b sequences instead of the TC5b sequence. The increase in overall fold stability due to the stabilizing Ala mutations and the Ala→Aib (α-aminobutyric acid) mutations [Aib also increases the intrinsic helix propensity (Karle and Balaram, 1990; Andersen and Tong, 1997)] indicate that the intrinsic stability of the helical portion is coupled (Lin et al., 2004) to the net fold stability.
Throughout, the constructs with an Asp N-cap were more stable than those with the Asn N-cap. The stability differences associated with the N-capping residue (Asp1 versus Asn1), ΔTmcage = 4–8°C and ΔTmhelix = 5.5–9°C, can be attributed to the greater helix N-capping propensity of Asp (particularly in its ionized state) (Doig and Baldwin, 1995; Andersen and Tong, 1997). Likewise, the decreased stability of the D1G mutants (TC11b1 and its Y3F mutant) reflects the lesser N-capping efficiency of glycine (Lin et al., 2004). The destabilization (ΔTm = −8.9 ±1.8°C) observed for the Q5A mutation may be attributable to a helix-stabilizing QxxxD hydrogen-bonding interaction between the Gln5 and Asp9 side chains (Huyghues-Despointes et al., 1995). Additional destabilization is observed upon acidification (ΔTmcage = −14°C, for the Q5A mutant, −17.4 ±1.0°C for the other systems in Table I).
Representative chemical shift melts for helix mutants appear in Fig. 3. With a few exceptions, the melting temperatures obtained from cage measures and helical measures agree within 5–6°C of each other for these constructs, implying that helix formation and formation of the tertiary structure are cooperative.
The two most destabilizing Ala substitutions in Table I were at Y3 and L7, sites that are within, or flank, the hydrophobic core of the Trp-cage. The specific implications of these large effects (ΔΔGU 280 = 13 and 11 kJ/mol, respectively) appear later.
Structural characterization of TC10b
Of the stabilized constructs in Table I, TC10b was selected for more extensive structural and folding thermodynamics evaluation. An NMR structure ensemble for TC10b (Fig. 4a) was generated from 186 NOE distances, of which 28 were long-range (i/i+n, n > 4) constraints. Over the accepted structures (28 structures from 40 random starting points) in the ensemble, the pairwise RMSD for residues 3–19 was 0.41 (±0.14) Å for the backbone atoms and 1.02 (±0.17) Å for all heavy atoms. The acceptance criteria and the method employed for generation of constraints appear in the Methods section. The structural and refinement statistics, the constraints employed and the dihedral angles observed in the ensemble appear in the Supplementary data available at PEDS online.
Fig. 4.

Trp-cage NMR structure ensembles: (a) The TC10b structure ensemble (28 structures): heavy atoms are displayed for all residue side chains and the backbone, the hydrophobic cluster residues are shown in color in the version of this figure in the Supplementary data available at PEDS online. (b) An overlay of one member each of the TC10b and TC5b structure ensembles. All atoms are displayed for Tyr3, Trp6, Leu7, Pro12, Pro18 and Pro19 with only N, CA, C′ shown for the remaining residues.
All of the TC10b structures in the ensemble displayed the full set of characteristic Trp-cage features that were reported for TC5b: an α-helix from Ala2 to Lys8, a short 310-helix (residues 12–14), an indole-NHε1 hydrogen bond to the i + 10 (Arg16) backbone carbonyl and the placement of Pro12 and Pro18,19 flanking and directly above, respectively, the indole ring, with the Tyr3 ring completing the hydrophobic cluster. In our prior reports of Trp-cage structural features, the exchange protection at Gly11-NH was attributed to a Gly11-NH→O=C-Trp6 H-bond which was present in most members of the TC5b and exendin-4 structure ensembles. In the TC10b ensemble, Gly11-NH→(O=C-Trp6 and/or O=C-Leu7) H-bonds were present in 22 of the 28 structures. A comparison of the NOE-derived structures of TC10b and TC5b (Fig. 4b) reveals ensembles that are not significantly different: the inter-ensemble pairwise residues 3–19 backbone RMSD between TC5b and TC10b was 0.68 (±0.12) Å, which is only slightly larger than the corresponding intra-ensemble RMSDs, 0.39 (±0.12) and 0.41 (±0.14) Å, respectively for TC5b and TC10b. Subsequent studies (to be reported elsewhere) have revealed that as few as 18 long-range constraints, with the addition of 36 medium- and short-range constraints, can generate Trp-cage structure ensembles within a 0.63 ±0.17 Å backbone RMSD of the published TC5b ensemble.
We turned to 1H/2H NH-exchange experiments to confirm the greater stability of TC10b that was apparent in melting curves and to determine the folding thermodynamic parameters at 280 K. At pD = 5.0, the PF for the backbone NHs of residues A4, L7, K8 and D9 ranged from 101.48–102.01, with slightly larger PFs (102.51 and 102.16, respectively) for the backbone NH of G11 and NHε1 of W6 corresponding to folded-fraction (χF) estimates of 0.997 and 0.993, respectively. At pD = 5.5, the PF for G11 NH and W6 NHε1 were 102.51 and 102.11 respectively; at pD = 6.6, we could only obtain an accurate rate for W6 NHε1, yielding a PF of 102.14. The thermodynamic stability (ΔGU280) for TC10b calculated from the PF is +12.2 ±1.1 kJ/mol. CD-monitored melts provide an alternative measure of the thermodynamics of unfolding; a two-state fit assuming a temperature invariant ΔCp (see Methods) affords ΔGU298 = 5.56 ±0.11 kJ/mol: ΔHU298 = 65 ±2 kJ/mol, ΔSU298 = 198 ±8 J/mol K and ΔCp = −220 ±110 J/mol K.
Probing origins of the pH effect on fold stability
A comparison of the Tm’s at pH 7 and 2.5 in Table I shows that all of the Trp-cage constructs are significantly more stable at pH 7 (ΔTm ≅ 7°C). Chemical shift melts illustrating this pH effect appear in Fig. S1 (Supplementary data available at PEDS online). CD-detected melts and pH titrations gave corroborating results (Fig. 5). The mid-point (circa pH 3.6) of the titration curve for TC5a, Fig. 5a, corresponds to the pKa of an Asp (or a C-terminal carboxylate). The complete CD melts for TC5a at pH 7 and 2.5 are shown in Fig. 5b, which also includes the common unfolded baseline observed in the presence of a chemical denaturant (see Methods) and the CD melt for TC10b and its less stable L7A mutant at pH 7.
Fig. 5.

(a) CD-monitored pH titration of fold stability for TC5a at 25°C; comparable effects were observed at both the maximum (190 nm) and the classical helix minimum (observed at 223.4 nm in this case). (b) Structure melting monitored at the 223 nm CD minimum; the unfolded baseline assumed for calculating fraction folded values (see Methods) is also given. The systems illustrated, in order of increasing stability, are: L7A-TC10b, TC5a at pH 2.5, TC5a at pH 7 and TC10b.
For constructs that have an Asp N-cap, a host of pH effects can be suggested: (a) protonation at the D9/R16 salt bridge, (b) the N-capping capability of Asp is highly pH dependent (NAsp0 = 2.8 versus NAsp− = 6.5) (Andersen and Tong, 1997), (c) the reported pH dependence (Huyghues-Despointes et al., 1995) of a helix-stabilizing QxxxD interaction, (d) some effect due to the protonation of the C-terminus and (e) a potentially stabilizing D1/K8 Coulombic interaction that would be lost upon protonation. The last of these is eliminated from consideration by the observation that the K8A mutation is equally stabilizing for both Asn- and Asp-capped Trp-cage sequences.
In order to reduce the number of potential pH effects, further mutations (Table II) were performed with Asn, rather than Asp, as the N-cap. The aim was to clarify the role of the QxxxD and D9/R16 interactions in the pH dependence of fold stability. In this series, a Q5A mutation would eliminate the helix-stabilizing QxxxD interaction but still allow salt-bridge formation, whereas an R16Nva (Arg→norvaline) mutation would allow the QxxxD interaction but eliminate any salt bridging possibility. A D9N mutant with and without an amidated C-terminus was also examined. Throughout this series, both NMR and CD (data not shown) reveal similar melting behavior. Table II lists the NMR-derived melting points for these constructs at pH 7 and 2.5. Table II also includes ΔΔGU measures of the effect of acidification for each construct and the ΔΔGU associated with each mutation.
Table II.
Mutational and pH effects on stability: the QXXXDXXXXXXR sequence of the Trp-cage
| Sequence | pH 7
|
pH 2.5
|
ΔΔ GF (pH)b (kJ/mol) | mΔΔ GF (pH)c (kJ/mol) | ||||
|---|---|---|---|---|---|---|---|---|
| %-Cagea | Tm
|
%-Cagea | Tm
|
|||||
| Cage | Helix | Cage | Helix | |||||
| NAYAQWLKDGGPSSG RPPPS at 280 K | 86 | 51 | 51 | 62 | 34.5 | 39 | 3.3 | |
| 98.9 | 85 | 6.4 (4.3)d | ||||||
| NAYAAWLKDGGPSSG RPPPS at 280 K | 72 | 41 | 42 | 50 | 27 | 30 | 2.4 | 2.2 (2.6)d |
| 90 | 78 | 2.5 (3.6)d | ||||||
| NAYAQWLKDGGPSSG Nva PPPS at 280 K | 56 | 32 | 35.5 | 59 | 33 | 36 | −0.3 | 3.9 (4.9)d |
| 81 | 80 | 0.2 | ||||||
| NAYAQWLKNGGPSSG RPPPS at 280 K | 65 | 38 | 41 | 47 | 25 | 29 | 1.8 | 3.0 (3.4)d |
| 81 | 73 | 0.9 | ||||||
| NAYAQWLKNGGPSSG RPPPS-NH2 at 280 K | 55 | 30 | 35 | 45 | 22 | 27 | 1.0 | 4.0 (5.4)d |
| 75 | 69 | 0.7 | ||||||
The %-fold estimate (from all CSDs that probe the cage region) in the top listing are at 300 K for each construct.
ΔΔGF (pH) = ΔGUpH 7− ΔGpUpH 2.5 for each construct at 300 and 280 K, based on %-cage values.
The mΔΔ GF values (= ΔGUTC9b− ΔGUmutant at pH 7 with T = 300 K) corresponds to the destabilizing effect of the mutation and is based on %-cage values.
Since %-fold estimates greater than 90% have a larger uncertainty, particularly in energy units, we also use a prior calibration of the relationship between Tm and ΔGU. On this basis, ΔΔGF (pH) at 280 K = (TmpH 7− TmpH 2.5) × 0.26 ±0.02 kJ/deg/mol and mΔΔGF at 280 K = ΔTmTC9b− Tmmutant) × 0.26 ± 0.02 kJ/deg/mol.
Destabilizing effects of truncations and other single-site mutations
The effects (typically destabilizing) of other mutations, and further truncation, of the Trp-cage are collected in Table III. These include mutations at all of the proline sites and also additional mutations in the loop region that connects the N-terminal α helix and C-terminal polyProII helix.
Table III.
Other destabilizing effects: %-folded (280 K) and Tm’s (°C), pH 7 aqueous
| TC# | Sequence | %-folded
|
Tm (°C)
|
ΔΔGFa (ΔΔGFb) | |||
|---|---|---|---|---|---|---|---|
| 280 K | 300 K | Cage | Helix | CD | |||
| Proline substitution effects | |||||||
| 10b | DAYAQWLKD GGPSSG RPPPS | 99.5 | 90 | 56 | 61 | 56 | Reference |
| DAYAQWLKD GGPSSG RAPPS | 94 | 76 | 42 | 47 | 46 | 2.6 (3.6) | |
| DAYAQWLKD GGPSSG RGPPS | 85 | 60 | 32 | 38 | n.d. | 4.5 (6.2) | |
| DAYAQWLKD GGPSSG RPLPS | 88 | 65 | 34 | 40 | 33.5 | 3.9 (5.7) | |
| DAYAQWLKD GGPSSG RPAPS | ~98 | 86 | 47 | 50 | 47 | 1.0 (2.3) | |
| DAYAQWLKD GGASSG RPPPS | 94 | 77 | 43 | 47 | 46 | 2.5 (3.4) | |
| DAYAQWLKD GGASSG RPAPS | 93.5 | 73 | 38.5 | 42 | n.d. | 3.0 (4.6) | |
| DAYAQWLKD GGPSSG RPPAS | ≤39c | ≤21 | (−9) | 15 | 15 | 11c ≥14d,e | |
| Minimization of the ‘Trp-cage’ | |||||||
| DAYAQWLKD GGPSSG RPPPNH2 | 97 | 85 | 53 | 56 | 54 | (0.8) | |
| 9b | NAYAQWLKD GGPSSG RPPPS | 98.3 | 86 | 51 | 51 | 50 | Reference |
| 12a | AYAQWLKD GGPSSG RPPPS | 27 | 13 | (−24) | 1 | 9.3 (13.6)d | |
| 12b | NYAQWLKD GGPSSG RPPPS | 84 | 63 | 35 | 42 | 37 | 3.2 (4.2) |
| tr12b | NYAQWLKD GGPSSG RPPPNH2 | 73 | 48 | 26.5 | 35 | 4.7 (6.4) | |
| Other mutations in the residue 13–16 span | |||||||
| DAYAQWLKD GGPSSG KPPPS | 91f | 76 | 45 | 48 | 2.5 (2.9) | ||
| DAYAQWLKD GGPASG RPPPS | >99 | 91 | 63 | 68 | 61 | (−1.8) | |
| DAYAQWLKD GGPSAG RPPPS | 98 | 44 | 22.5 | 34 | 20 | 6.1 (8.0)d,g | |
| NAYAQWLKD GGPSTG RPPPS | 93h | 76 | 42 | 46 | 45 | 1.7 (2.3) | |
| NAYAQWLKDG GPSalloTG RPPPS | ≥96 | 82 | 49 | 52 | 48 | 0.7 (≤0.5) | |
ΔΔ GF = ΔGUTC10b/9b− ΔGU mutant at pH 7 and 300 K, based on χF values at 300 K.
ΔΔGF at 280 K. Values in parentheses are derived as ΔΔGF280 = (TmTC10b/9b−Tmmutant) × 0.26 ±0.02 kJ/deg, where the Tm’s are taken from cage measures.
The largest %-folded measure at 280 K is that from Gly11α2 (39%, with no correction for ‘half-cage’ contributions, vide infra); the two Pro18 probes both indicate χF = 0.17. The ΔΔGF 300 value quoted is based on the Pro18 probes; χF≤0.21 provides ΔΔGF300 ≥ 8.8 kJ/mol.
ΔΔGF at 280 K from the %-folded measures at this temperature.
The ΔΔGF280.value derived from ΔTmcage is 17 kJ/mol.
This analog displays a significant spread of %-folded values depending on the probe site, with Gly11α2 and Pro18β3 reporting 85–87% folded at 280 K.
The ΔΔGF280.value derived from ΔTmcage is 8.7 kJ/mol.
The diminished %-folded values reflect a more substantial decrease in the upfield shift of Pro18β3, which appears to be altered much more than the other sites.
Mutations at proline sites
Proline residues often stabilize the folded states of proteins due to a reduction in the loss of backbone conformational entropy upon folding (Matthews et al., 1987). Pro17 appeared to be the ideal locus for testing main chain entropy effects in the Trp-cages since its side chain is solvent exposed and is clearly not part of the hydrophobic cluster (Fig. 4). The sequential substitution of P17 (in TC10b) with Ala and Gly led to increasingly less stable constructs: Tmcage decreases in the order P17 (56°C)>A17 (42°C)>G17 (32°C). Alanine mutations were also made at Pro12,18,19. These results are included in Table III. It is immediately apparent: (i) that alanine substitution is best tolerated at P18, the proline that sits immediately above, and in particularly close contact with, the indole ring; (ii) the destabilizing effects at P12 and P18 are not fully additive and (iii) that alanine substitution at the C-terminal proline is particularly destabilizing.
Defining the minimum sequence length of the Trp-cage
Deletions at both the N-terminus and the C-terminus of the sequence were examined (Table III). The deletion of the N-terminal helix-capping residue produced a construct (TC12a) that was <40%-folded at 280 K in aqueous buffer at pH 7. However, deleting Ala2 rather than Asn1 of TC9b (and thus retaining a helix N-cap) afforded a folded 19-residue system (TC12b, 84%-folded at 280 K). Another construct was prepared by the truncation of the sequence at the C-terminus, with the Ser20 replaced by an amide function (tr10b). This construct was ~97%-folded at 280 K with a Tm of 53°C, indicating a circa 1 kJ/mol stability loss. These two truncations were then combined in the same sequence to prepare an 18-mer (tr12b) that was found to be 73%-folded at 280 K.
Probing other features of the ‘trp-cage’
We examined an R16K mutation as an alternative to the D9/R16 salt bridge which would be less likely to display alternate cation/π interactions with the indole ring. This mutation reduced fold stability (ΔΔGU280 = 2.9 kJ/mol) less than the R16Nva mutation. As it turns out (see Discussion), this effect may be related to interactions with residues in the 310-helix. The loop region between the N-terminal helix and the C-terminal RPPPS sequence consists of a short 310-helix between residues 12–14. Of these residues, Pro12 forms one edge of the Trp-cage hydrophobic cluster, whereas the potential roles of the other two residues, Ser13 and Ser14, in the short 310-helix were not readily apparent from the NMR structures. The Ser13 side chain is fully solvent exposed in both of our Trp-cage structure ensembles; in analogy to α helix sites, the S13A mutation was fold stabilizing (by 1.8 kJ/mol).
In contrast, the S14 side chain is internalized in all of our structure ensembles. Moreover, Neidigh reported (Neidigh, 1999) that the NOESY and TOCSY spectra of (S14T)-TC5b in pH 7 aqueous buffer (with 30% TFE added) display a prominent threonine hydroxyl proton resonance indicating that this proton is protected from exchange with the solvent. However, (S14T)-TC5b was too unstable for NMR characterization in the absence of TFE. To probe the interactions of the S14 side chain, we prepared the S14A of TC10b and S14T/S14allo-Thr mutants of TC9b. The allo-Thr mutation was fully tolerated with no apparent loss (≤0.7 kJ/mol) in cage stability; the S→T mutation was slightly destabilizing (ΔΔGF280 = 2 kJ/mol); but the S14A mutation, with a ΔΔGF280 of 8.4 kJ/mol, was the most destabilizing effect observed for any residue that was not an obvious apolar component of the hydrophobic core.
Discussion
The Trp-cage structure is retained in both optimized and truncated sequences
The sequence of the Trp-cage can be optimized for fold stability without causing any significant structural changes. The NMR ensembles generated for TC5b (Tm = 42°C) and TC10b, which bears three stabilizing mutations N1D/L2A/I4A (Tm = 58 ±2°C), are identical to within a 0.6 Å backbone RMSD. A few detectable differences in the residues 12–17 span are evident by examining the backbone φ/ψ values (Supplementary Table available at PEDS online); given the sparseness of NOE constraints in this loop, these minor differences may not be significant. TC10b also retains all of the notably ring-current shifted signals of TC5b and these hardly increase in magnitude due to the stabilizing mutations: accounting for the unfolded fraction of TC5b present (estimated from the NH protection data), the sum of the structuring shifts for the folded state of TC5b is >95% of the sum of maximum CSDs at each position observed in higher melting constructs. We view this as evidence that the less stable TC5b structure does not show dramatically enhanced structural fluxionality in the folded state. MD simulations starting from the Trp-cage fold (Fesinmeyer, 2005), which will be reported elsewhere, reveal a host of collapsed, near-native folds (backbone RMSD = 0.8–2 Å versus the NMR structure) that do not reproduce the chemical shifts observed. Structures with 1–2 Å Cα or backbone RMSDs from the NMR structure, which also fail to predict the observed ring-current shifts, have been equated with the folded state in a number of literature accounts of computational Trp-cage folding simulations.
The 19-mer and 18-mer constructs that were obtained by the truncation of the TC10b sequence also displayed the characteristic long-range NOEs and the CSDs associated with the Trp-cage constructs. For example, the ratios of the ring-current shifts at G11Hα2/P18Hα and P18Hβ3/P19Hδ2 (1.53 and 3.12, respectively) in the 18-mer were identical (±0.02) to those observed for TC10b with the absolute values indicating a 0.72 ±0.02-fold-population for the 18-mer at 280 K. This implies that truncation did not lead to any significant structural changes; rather ΔGF became less negative. As a result, we view the Trp-cage as an 18-residue folding motif.
Confirmation of key assumptions about the Trp-cage fold
Except for Y3A, Q5A, L7A and presumably W6A [not examined since W6F is highly destabilizing (Barua and Andersen, 2002)], mutations to alanine in the N-terminal helix of the Trp-cage results in improved fold stability. This suggests that these side chains are involved in specific fold-stabilizing interactions. The L7A construct is only 55%-folded at 280 K in aqueous buffer; it appears that Leu7 (sandwiched between the Tyr3 and Trp6 side chains) is an important contributor to the burial of the indole ring. Interactions within the helix may be responsible for the decrease in stability associated with the Q5A mutation: Gln–Asx (i, i+4) hydrogen-bonding interactions have been reported to occur in helices (Huyghues-Despointes et al., 1993; Stapley and Doig, 1997). It has been reported that this interaction is more stabilizing when the Gln is N-terminal to the Asx, particularly for an ionized Asp (Huyghues-Despointes et al., 1995). The very large Y3A effect (≥14 kJ/mol) is attributed primarily to favorable Y3/P19 hydrophobic interactions, vide infra.
The introduction of N9D and A16R mutations during the initial Trp-cage optimization procedure had a profound effect leading to the D9/R16 salt-bridge hypothesis (Neidigh et al., 2002). A D9/R16 salt bridge has also been a prominent feature in most of the MD folding simulations reported for the Trp-cage, either as an interaction that was viewed as facilitating rapid folding (Snow et al., 2002; Chowdhury et al., 2003; Nikiforovich et al., 2003) or as a kinetic trap (Zhou, 2003; Ding et al., 2005; Linhananta et al., 2005). However, the NMR structure ensembles fail to provide a well-defined H-bonded salt-bridging geometry. This reflects the apparent absence of side chain/side chain NOEs across the bridging sites. Inserting a restraint that keeps the D9/R16 side chain termini within interaction distance, established that a better defined salt bridge is fully consistent with the observed NOEs (vide infra).
Our efforts to rationalize the substantial pH effects on Trp-cage fold stability [initially reported (Neidigh et al., 2002) to be 5.8–7.5 kJ/mol] focused on mutational effects in the QxxxDxxxxxxR unit of Trp-cages, and the effects of C-terminal amidation (Table II) and how these alter the fold population dependence on pH. The most instructive mutation was the replacement of Arg16 with norvaline (the desguanidino equivalent). The fold-population of the R16Nva mutant of TC9b was pH-independent (χF = 0.56 at 300 K, versus 0.86 for TC9b at pH 7). Assuming that D9/R16 interaction is the only fold-stabilizing interaction involving the guanidino group, implies a 4–5 kJ/mol fold stabilization associated with the salt bridge. This result also suggests that the helix-stabilizing QxxxD interaction is not pH-dependent in our construct. The data for the Q5A and D9N mutants were less clear-cut. The D9N mutation produced a smaller destabilization (ΔΔGF280 = 3.4 kJ/mol) and the mutant retained some pH dependence to its fold stability (≤2 kJ/mol), even though the C-terminal carboxyl is the only ionizable site. Indeed, terminal amidation reduced fold stability by 1.5 ±0.5 kJ/mol. In sum, based on the R16Nva, Q5A, and D9N mutations, the D9/R16 salt-bridge stabilization is bracketed in the 3.4–6 kJ/mol range. There may also be a stabilizing Coulombic interaction between the C-terminal carboxylate and the N-terminal ammonium ion in the Asn-capped sequences, which is masked in species with Asp as the N-cap. Assuming no other Q5 side-chain interactions that affect fold stability, the stability decrease for (Q5A)-TC9b (2.4 kJ/mol) represents a measure of the free energy contribution of the QxxxD H-bond to helix stability. The validity of any of these estimates is also complicated by the general concern that this type of mutational analysis cannot factor out all context effects and thus attribution to particular side chain/side chain interactions is always less than fully justifiable.
In our initial report, we posited the inclusion of four prolines in the TC5b sequence as a significant factor contributing to the unexpected fold stability of this 20-residue sequence. The importance of the entropic effect of the proline residues on Trp-cage fold stability was confirmed by mutations of the P17 residue. The substitution of P17 by either Ala or Gly resulted in less folded constructs, with ΔΔGF280 estimated as 3.6 and 6.2 kJ/mol, respectively; the reduction in fold stability associated with mutations at this site is attributed solely to increases in ΔSU. From the protein stability data of Matthews et al. (1987), the expected magnitude of the ΔSU effects of a P→A and P→G mutation are 3.5–6 and 5.5–9 kJ/mol, respectively, at this temperature. The agreement between our mutational results at P17 and the expectation values for entropic effects in proteins is excellent. However, extension of this conclusion to the other proline sites in not straight-forward.
Does the Trp-cage provide evidence for uniquely stabilizing Pro/Trp or Pro/Tyr interactions?
The locations of the apolar ring portions of P12 and P18, in tight association with the central indole ring of the hydrophobic core, and some structural analogies to geometries observed in the complexes of polyPro peptides and protein domains that recognize such sequences led us to suggest (Neidigh et al., 2001, 2002) that these represented particularly favorable interactions. Thus, we were surprised to discover that mutations at these two prolines were tolerated. The first mutation examined was P18L, based on the notion that the aliphatic chain of Leu might be a reasonable replacement for the proline ring placed immediately above the indole ring (Fig. 4). The P18L mutation was only slightly more destabilizing (ΔΔGF = 5.7 kJ/mol) than the P17A effect attributed to the ΔΔSU effect. However, Pro→Ala mutations at either (or both) Pro12,18 were distinctly less stabilizing than the P17A mutation. In both (P18A)-TC10b and the double mutant, the diagnostic ring-current shifts of the Trp-cage are retained; the CSD at 18Hα (as large as −2.6 p.p.m.) was in fact greater in the P18A mutants than when this site is a proline (Hα CSD = −2.3 in TC10b). The Ala18 alpha methine appears to be even closer to, and centered within the shielding cone of, the indole ring. The P19Hδ2 site also experiences a larger upfield shift. No other changes in ring-current shifts are apparent, for example, the CSDs of L7Hα, G11Hα2, G11Hα3 and R16Hα agree within 0.02 p.p.m. for TC10b and its P18A mutant. The effect of the P18A mutation (ΔΔGF280 = 2.3 kJ/mol) must, therefore, be rationalized as a minor, enthalpically favorable change in the docking of the residues 17–19 segment onto the indole ring that compensates for the ΔΔSU effect. This result and the well-tolerated P12A mutation raise the possibility that the solvent shielding of the indole ring (and the resulting favorable enthalpic contribution of Trp-cage formation) may largely be due to interactions between the ring and the peptide backbone. [This is similar to aryl rescue of glycine at β-sheet sites (Kemmink et al., 1993; Merkel and Regan, 1998; Jackups and Liang, 2005) which also requires an unsubstituted Gly.] It should be noted, however, that the ΔΔSU effect could be muted for a PPP→PAP mutation due to a narrower unfolded state φ/ψ distribution at the central alanine in a sequence with a significant preference for a polyProII conformation.
In distinct contrast, the P19A mutation reduces fold stability by ≥14 kJ/mol. Pro19 displays interactions with both Trp6 and Tyr3. The Y3/P19 interaction, evidenced by numerous side chain/side chain NOEs and a strong Y3α/P19γ connectivity, defines the limits of the 18-residue Trp-cage motif and completes the encaging of the indole ring. Although we had not anticipated it, the observation that the favorable enthalpic interaction with the greatest contact order is also the interaction with the largest effect on thermodynamic stability seems entirely reasonable: it can be viewed as a hydrophobic staple holding the Trp-cage structure together. We also probed the Y3/P19 interaction by examining Y3L and Y3A mutations. The Y3L mutation was only modestly destabilizing (ΔΔGF280 = 4.4 kJ/mol), and a L3/P19 interaction was evidenced by stereospecific side chain/side chain NOEs. The Y3A mutation was nearly as destabilizing (ΔΔGF280≥ 13 kJ/mol) as the P19A mutation. Taken together, these observations suggest that the Y3/P19 interaction is indeed hydrophobic in nature and the major contributor to the cage stability differences observed for both the Y3A and the P19A mutations. Y3 and P19 also make contacts with the W6 side chain, but the data do not allow the parsing of the mutational ΔΔGF data into components reflecting three possible interactions (Y3/W6, Y3/P19 and W6/P19).
The contribution of buried H-bonds
The NH exchange protection due to two buried long-range H-bonds, Trp6 indole-NHε1 to O=C-Arg16 and Gly11-NH→(O=C-Trp6 and/or O=C-Leu7), is particularly diagnostic of Trp-cage formation; to these we can now add H-bonding interactions of the buried Ser14 side chain hydroxyl. Although the contribution of H-bonding to protein fold stability is generally viewed as rather small, on the order of 5 kJ/H-bond or less (Baldwin, 2003), reflecting the compensating effect of H-bonding interactions with water in the unfolded state, it is clear that virtually all polar backbone group of proteins are H-bonded; one estimate of the cost of breaking a solvating H-bond without replacing it with an intermolecular one is ≥20 kJ (Fleming and Rose, 2005). The mutations of S14 and R16 reported in Table III were, in part, based on observations made by computational chemists examining the Trp-cage motif. Pitera and Swope noted that the S14 hydroxyl was positioned for H-bonding (to the backbone carbonyls of G10 or G11) in their simulated folded state geometries. In the NMR structure ensemble of TC10b, based strictly on NOE connectivities, S14Hγ is within H-bonding distances of either the Asp9 or Gly11 backbone carbonyls in most of the structures. Simmerling and Roitberg (personal communication) brought another feature, an Arg-NHε to Ser-Oγ H-bond (Fig. S2, Supplementary data available at PEDS online) to our attention; such an interaction would be expected to increase the strength of any H-bond involving S14Hγ. We examined an R16K mutation to probe this region; the Lys side chain could still have a stabilizing Coulombic interaction with D9, but no side-chain/side-chain hydrogen bonding interaction with Ser would be expected. Consistent with an Arg16-NHε H-bonding interaction, the R16K mutation was destabilizing (3 kJ/mol), but other rationales are available.
The S14 mutational studies support an H-bonded status: both Thr and allo-Thr were well tolerated at this position, whereas the S14A mutation (ΔΔGF280 = 8.4 ±0.4 kJ/mol) was the most destabilizing effect observed for any residue that was not an obvious, apolar contributor to the hydrophobic core. Furthermore, in aqueous pH 7 buffer, the diastereomeric S14T mutants display a side-chain hydroxyl proton in the 2D-NMR experiments [A search of the BMRB database reveals that Ser-OH groups are observed in only 2.1% of the proteins examined and that these observations are restricted to systems with large hydrophobic cores. The statistics for observing Thr-OH are only slightly more favorable (3.8%). Hevein, a tetra-disulfide 43-mer (Cao, 1993; Asensio et al., 1995), is, to our knowledge, the smallest protein to display side-chain OH signals in its NMR spectra under the conditions routinely used for protein resonance assignment. Alternative experiments that allow even random coil Ser and Thr OHs to be observed are available (e.g. Liepinsh et al., 1992).], indicating an equivalent and significant degree of exchange protection for this proton. The internalized position of the Thr side chain in the S14T mutant is supported by close contact with the Asp9-CH2 evident as a very intense NOE with Thr methyl; the stereochemistry change at Cβ reduces the NOE intensity to the Asp9-CH2 and produces indole-CH to CγH3 NOEs that are not observed for the S14T mutant. Apparently, the Thr hydroxyl forms the same H-bond in both diastereomers, with the Cβ diastereotopic change only altering the location of the methyl group. Our estimate of the stabilizing effect of the Ser14 hydroxyl based on the S14A mutation includes the fold improvement expected for a Ser→Ala mutation within the 310-helix (1.8 kJ/mol for S13A); thus the ‘measured’ buried H-bond effect is 10.2 ±0.7 kJ/mol for Ser14 of the Trp-cage: ~50% of the folding impairment expected for an internalized backbone amide lacking an H-bonding interaction (Fleming and Rose, 2005).
The Thr and allo-Thr side-chain CSDs further corroborate the assignment and provided insights into the position of the Ser14 side chain in the Trp-cage. The Ser14 Hβ’s have disparate CSD values (ΔδHβ3 = −0.393, ΔδHβ2 = −0.012 in TC9b); CSDs as large as −0.45 have been observed for S14Hβ3 in more stable Trp-cage analogs. Within the IUPAC naming convention, Hβ of Thr corresponds to Ser Hβ2; Hβ3 in the case of allo-Thr: the Ser14 CSD values reflect the similar magnetic environments experienced by corresponding nuclei in allo-Thr14 Hβ (−0.490) and Thr14 Hβ (−0.005 p.p.m.). The methyl signals (Hγ2) show, as expected, the opposite trend - allo-Thr14 Hγ2 slightly down-field (+0.033) and that of Thr14 significantly upfield (−0.160 p.p.m.). These observations were used to test alternative computational models of the Trp-cage conformation.
The NMR ensemble of TC10b was calculated without H-bond restraints. Since neither the D9/R16 salt bridge nor a consistent pattern of H-bonds involving the Ser14 side chain appeared in this or the prior Trp-cage structure ensemble, we examined the extent to which specific H-bonding patterns are consistent with the NOE constraints and ring-current shift at the internalized S14 side chain (data not shown). With the exception of the S14Hγ→O=C-Gly10 suggestion of Pitera and Swope, all α and 310-helical H-bonding networks as well as S14Hγ and R16Hε H-bonding interactions could be accommodated. Added H-bond restraints (and distance restraints maintaining the Asp and Arg side chains within salt-bridging distance) did not introduce any significant increases in the violations of NOE-derived distance constraints; in fact, a number of modified CNS runs with a full complement of H-bonds specified resulted in decreased ENOE terms. The resulting models for TC10b are viewed as ones ‘incorporating other NMR data besides the NOEs’. The quality of ensembles calculated with alternative H-bond restraints was judged based on: ETOT and ENOE, the extent to which H-bonds displayed normal geometries, agreement with the 49 medium- and long-range NOEs, and compatibility with the S14 mutation data, see Supplementary data available at PEDS online. The lowest energy model resulted when salt bridging and 14Hγ-9O′/16Hε-14Oγ/9HN-5O′/11HN-7O′ H-bond restraints were applied. The resulting ensemble (intra-ensemble 3–19 backbone RMSD = 0.31 ±0.12 Å) was still very similar to the ensemble generated with only NOE constraints (inter-ensemble RMSD = 0.47 ±0.16 Å) but displayed improved energies versus the NOE-constraints-only ensemble and rationalizes all other NMR observations. However, distinguishing between a 14Hγ-9O′ and 14Hγ-11O′ H-bond is not really possible; as this corresponds to nothing more than a change in χ2 of S14. Views of a representative member of this ensemble, as well as atomic coordinates of this model of the Trp-cage structure, appear in the Supplementary data available at PEDS online.
Cooperativity between helix formation and Trp-cage formation
The ‘helix’ and ‘cage’ melting temperatures of most of the Trp-cage constructs reported in this study are within 2–5°C of each other, implying cooperativity between helix formation and formation of the tertiary structure involving the hydrophobic cluster about the Trp side chain. The requirement of an efficient helix N-cap for cage formation also supports this hypothesis. Further evidence for this cooperativity came from the observation that stabilization of the helical part of the sequence resulted in improving overall fold stability (the L2/I4/K8→Ala and the A4→Aib mutations). Introduction of an alanine in the 310-helix (S13→Ala) also led to an increase in fold stability. The average stabilization effect, based on the changes in melting temperatures, was 1.5 ±0.35 kJ/mol per alanine substitution.
All Trp-cage species bearing the D9/R16 unit with (Tmhelix − Tmcage) <7°C displayed two other consistent correlations: the Tm from the CD melt was closer to the Tmcage (rather than Tmhelix) value, and the (Tmhelix − Tmcage) difference increased by 1.5–2.5°C upon protonation of the Asp side chain. The better correlation between TmCD and Tmcage, rather than Tmhelix, can be rationalized as follows. NMR measures of helicity are sensitive only to local φ/ψ values whereas the CD measure of helicity requires a longer contiguous string of helical φ/ψ values. Apparently, when Asp protonation decreases the extent of cage formation; unfolded or partially folded states, which retain some local helicity, make a larger contribution to the NMR melts. A well-defined complete helix is present when the Trp-cage is folded; however, in the absence of Trp-cage formation (e.g. C-terminally truncated species and the W6F mutant of TC5b), only partial local helicity has been reported (Barua and Andersen, 2002). To provide a measure of the intrinsic helicity of the Cap-AYAQWLADG unit in our better Trp-cage species in the absence cage formation, we compared the 13C shift melts of truncated species NAYA′QWLA′DGKK, A′=(13C=O)-Ala, in pH 7 buffer with and without 6 M urea added to that of fully folded DAYA′QWLA′DGGPS SGRPPPS. This indicated the maximal fH at the labeled ala-nines was 0.35 in the truncated species under the most favorable folding conditions: cage formation increases helix formation by at least a 12 kJ/mol ΔΔG increment.
Turning to those Trp-cage analogs that display (Tmhelix − Tmcage) ≥ 7°C, these fall into two categories: (i) mutational or solvent effects that increase the intrinsic stability of the N-terminal α-helix with an opposite (or lesser) effect on the tendency for Trp-cage formation and (ii) mutation in the residues 14–19 span that destabilize the Trp-cage. Representatives of the first category are the introduction of Aib residues, N-terminal helix extensions (Lin et al., 2004), the L7A mutation and the addition of 30% TFE. The TC10b mutants with the largest difference, (Tmhelix − Tmcage), were S14A-TC10b (ΔTm = 11.5), L7A-TC10b (ΔTm = 12) and P19A-TC10b (ΔTm = 24°C). The helix and cage melts for L7A-TC10b are shown in Fig. 3. In contrast to the L2A substitution (ΔΔGF = −1.3 kJ/mol), mutation of the hydrophobic L7 side chain reduces the Trp-cage population to 0.56 at 280 K (ΔΔGF ≅ +12 kJ/mol), whereas the helix Hα sites report a Tm of 296 K. The latter represents partial formation of the N-terminal helix in states lacking the Trp-cage. Melting data comparisons for TC10b and its S14A and P19A mutants appear in Fig. 6. In the case of the P19A mutant at 280 K, the helix measures indicate a 60 ±4%-fold population, even though the sum of the Pro18α/β3 CSDs indicate(s) χF = 0.17.
Fig. 6.
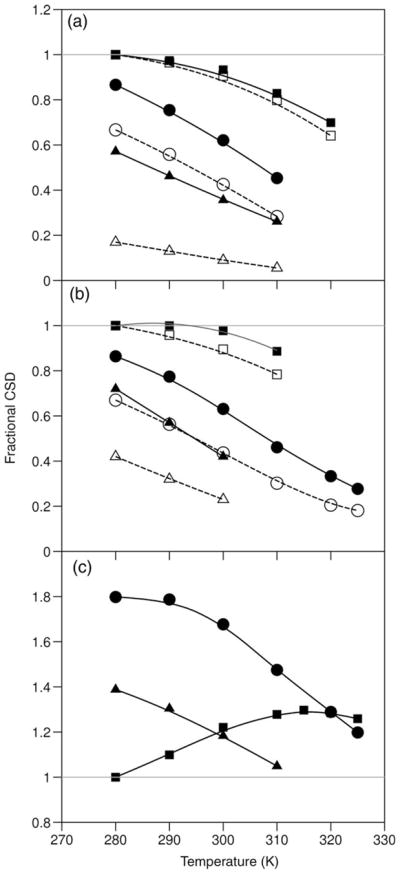
Differential melting effects upon destabilization of the Trp-cage: fractional CSDs versus T (°C) for TC10b (squares) and its S14A-(circles) and P19A-mutants (triangles)–(a) the ‘cage’ (dashed lines) and ‘helix’ (solid line, filled symbols) measures of folding, (b) the G11α2 (dashed lines) and G11α3 (solid line, filled symbols) CSDs, (c) the P12δ3 CSDs, TC10b at 7°C is the calibration standard for CSD=1.0 in all panels. The P12δ3 CSDs (upfield) for the less stable systems are larger than those observed for TC10b. In the case of the P19A mutant, the only cage measure available for (a) are the P18α and β3 CSDs.
Partial formation of the N-terminal helix in unfolded or partially folded states that lack the complete Trp-cage (evident in Fig. 6a) also rationalizes the general observation that Tmhelix values increase more than Tmcage values on TFE addition. The extreme example of this is exendin-4 in 30% TFE: the PF of the backbone NHs of the residues in the helix C-terminus were as much as 500-fold larger than the PF obtained from the indole ring Hε1 of Trp6; the latter, due to the W6Hε1→O=C-R16 H-bond, reflects full Trp-cage formation. Figure 7a shows a melting (unfolding) scenario that rationalizes the data. With the long helix of exendin-4, there is essentially no (χU < 10−5) random coil state in 30% TFE at temperatures below 30°C and cage formation (Kcage = 260) can be viewed as the docking of the triPro unit onto the Trp and Tyr rings held in place on one side of a preformed helix. In systems with shorter helices, particularly those lacking helix stabilizing Aib or Ala substitutions, cage disruption (by melting or mutation) does not produce partially helical ‘intermediates’. In fact, TC5b in aqueous buffer has no additional protection for the backbone HN near the helix C-terminus and shows nearly two-state chemical shift and CD melting behavior. In optimized Trp-cage species with more stable helices, intermediate behavior is observed.
Fig. 7.

Unfolding (melting) scenarios for Trp-cages with different degrees of intrinsic helix stability: (a) Trp-cage formation as Pro17,18,19 docking onto a stable helix, (b) Trp-cage melting producing an unfolded ensemble retaining a measurable population of a residual hydrophobic cluster, a ‘half-cage’ structure.
MD folding simulations for TC5b have indicated different sequences of events regarding the appearance of hydrophobic core features. Docking of the P17–19 unit onto the Trp side-chain figures in early hydrophobic collapse in some simulations (Snow et al., 2002; Chowdhury et al., 2003; Juraszek and Bolhuis, 2006) with, in at least one case, the formation of a non-native core which cannot reorganize to the native form without breaking a strong D9/R16 salt bridge (Zhou, 2003). Collapse of P12 (and Y3) onto the indole ring precedes interactions between W6 and the C-terminal triPro in other folding scenarios (Nikiforovich et al., 2003; Ding et al., 2005; Linhananta et al., 2005; from Prof. C.Simmerling, personal communication). In the analysis of a recent explicit solvent simulation of unfolding (Juraszek and Bolhuis, 2006), the authors suggested that dissociation of the P12/W6 interaction is the early melting event. In contrast to the latter, experimental data (prior NMR melts) led to the half-cage ‘unfolding pathway’ hypothesis (Neidigh et al., 2002). This pathway (Fig. 7b) served as a rationale for the deviant melting behavior of the G11Hα3 and P12Hδ3 protons, which is illustrated in the present cases by Fig. 6. This scenario can explain the persistence of helical populations as the C-terminal triPro unit undocks and provides a rationale for the effects of mutations in the residues 14–19 span that destabilize the Trp-cage but retain significant helicity.
Turning to the specific proton melting behavior that does not track the melting behavior of the proton shifts that are used as measures of Trp-cage folding, the upfield CSD of P12Hδ3 (for TC10b and other stable Trp-cage species) actually increases in magnitude as partial melting occurs (Fig. 6c) and G11Hα3 displays little loss of its structuring CSD on partial melting. The P19A mutant provides a compelling example of similar behavior in response to a mutation that disfavors docking of the C-terminal segment; even though the Gly11α2 measure of cage formation and the sum of the Pro18α/β3 CSDs indicate χF = 0.39 and 0.17, respectively, the CSD at Gly11α3 is fully 72% of that observed for TC10b. These chemical shift melts and mutational effects can be rationalized as the consequence of the population, in the ‘unfolded state’, of a residual hydrophobic cluster consisting of the W6 and the G11/P12 residues interacting in a non-native orientation, the ‘half-cage motif’, that reduces the ring-current shift of G11Hα2 (but not G11Hα3) and places the P12-δCH2 further into the shielding region than in the full Trp-cage structure. The P12-δCH2 protons (particularly P12Hδ3) in many destabilized mutants display negative CSDs of greater magnitude than those observed for TC10b itself, suggesting that the population of the ‘half-cage’ increases with the destabilization of the full ‘Trp-cage’.
Formation of the ‘half-cage’ requires at least partial formation of the C-terminal portion of the helix with the W6/P12 interaction acting as a helix C-cap: both the (S14A)-and (P19A)-mutants display upfield shifts at G11Hα3 that are larger than expected based on the CSDs observed at G11α2 and P18α. Further, when large P12Hδ3 CSDs are observed in mutants, they display a temperature dependence that correlates with CSDs that monitor helicity rather than cage formation. Evidence of the population of a discreet ‘half-cage’ state in C-terminally truncated species and by the P19A mutant of TC10b (as well as other mutants designed to reduce the tendency toward complete Trp-cage formation) will be presented elsewhere.
An alternative rationale for the unusual ‘melting behavior’ of the structuring shifts at P12Hδ3 and the two G11-CH2 resonances—increased G10–P12 loop fluxionality within the folded state at higher temperatures—deserves mention. Segmental motion of this type would, if the region with increased fluxionality in the high temperature folded ensemble extends to P18, rationalize much of the melting data. Mutational effects on cage stability imply that the P19/Y3 (andW6) interaction is particularly stabilizing; this may also be the more persistent hydrophobic interactions at higher temperature as we consistently find that P19δδ’ CSD melts suggest a higher χF than the P18α/β3 and G11α2 CSDs at higher temperatures. More detailed studies of additional mutants will be required to support such a rationale. In their absence, the nearly constant CSD ratios for sites in L7, G10, G11, P12, R16 and P18 in the numerous analogs examined to date support a common non-fluxional fold and a nearly two-state folding process. Recent studies (Mok et al., 2007) reveal retention of the W6/P12 (but not the W6/P18) interaction in the urea-denatured state and suggest partial hydrophobic collapse even in this ‘unfolded’ state.
Concluding remarks
It is now established that the Trp-cage is an 18-residue motif. The NMR ensemble generated for the more stable TC10b analog displays dihedral angles (and H-bonding networks) that correspond more closely to secondary structure norms; in particular, the ψ angle values for residues 13–14 are closer to the standard 310-helix value (−26°), and the residues in the 16–19 span display φ/ψ angles corresponding to a polyProII helix (−75°/+145°). In the absence of salt-bridging distance restraints, the R16 side chain displays two χ1 values and side chain arrangements only one of which is consistent with an R16/D9 ion-pairing interaction (Fig. 4a). Additional restrained MD trajectories (Supplementary data available at PEDS online) indicate that enforcing an R16/D9 ion-pairing interaction as well as a defined set of H-bonding interactions including the Ser14 hydroxyl and regularizing the H-bonding pattern within the α-helical segment yields a structural model that is fully consistent with all of the NMR observations. Our best model of TC10b appears in the Supplementary data available at PEDS online, with coordinates included so that these can serve as starting structures for initiating computational unfolding trajectories. We urge the use of the TC10b sequence for any further folding simulations and suggest that a backbone RMSDs <1 Å should be used to define the folded state; we have been unable to generate any structures that are consistent with the ring-current shifts observed for the folded state which differ from the NOE-derived structure by >0.9 Å.
Although the burial of the Trp side chain is clearly the dominant design feature of the Trp-cage motif, the present mutational study suggests that indole/backbone interactions rather than specific indole/proline ring interactions may be the key component. The high proline content of the Trp-cage clearly improves fold stability. The entropic advantage of Pro versus Ala (ΔΔSU = 11 ±2 J/mol K) was measured at the solvent-exposed P17 site. The study has also revealed two unexpected contributors to ΔGF: a buried H-bonding Ser side chain (ΔΔGF ≅ 10 kJ/mol) and a Y3/P19 hydrophobic staple interaction. Concerning the latter, both P19A and Y3A mutations are destabilizing by >13 kJ/mol, but folding can be rescued with a Y3L mutation (ΔΔGF = 4.4 kJ/mol).
The data presented herein support a cooperative melting scenario for Trp-cage species; the chemical shift melts serving as the best experimental measure: uniform structure loss over probes throughout the structure (Ferguson et al., 2005; Fesinmeyer et al., 2005; Olsen et al., 2005) requires folding cooperativity. In the present case, the excellent agreement between the observed structuring chemical shifts and those calculated for structure ensembles that are tightly converged about a single conformer lends additional validation to the structure.
The two-state character of Trp-cage folding transition gains support from a recent correlation of CD melts and differential scanning calorimetry (Streicher and Makhatadze, 2007). Apparently contrasting observations or conclusions, however, deserve some comment. A number of the folding simulations (vide supra) show initial hydrophobic collapse that could be equated with a molten globule folding intermediate. A UV-resonance Raman study (Ahmed et al., 2005) of the thermal unfolding of TC5b has been interpreted as evidence for non-two-state behavior and posits that the folded state becomes more compact on warming from 4°C to 20 °C followed by rather gradual partial melting from 25°C to 70°C with the latter corresponding to the loss of only four helical amide units. To be consistent with the observed temperature dependence of chemical shifts that include large ring-current components, the ‘more compact state’ formed at 20°C would have to have a more fluxional structure in which most of the ring-current shifts are significantly, and similarly, diminished in magnitude: a rather unlikely set of coincidences.
The few deviations from uniform loss of structuring CSDs upon melting or the introduction of destabilizing single-site mutations are fully rationalized by the folding scenarios in Fig. 7, which allow for partial local structure in the ‘unfolded’ ensemble. The appropriate description of denatured and unfolded states of Trp-cage species remains an area of active exploration (Mok et al., 2007), as does a clarification of the details of the folding pathway(s) for this ultra-fast folding system. Two Trp-cage constructs were included in the development of a new model of ultra-fast folding (Ghosh et al., 2007) which attributes folding acceleration to the existence of a multiplicity of parallel pathways to the folded state geometry. We continue to view the Trp-cage motif as an ideal model for exploring the correlation between protein folding simulations and experimental data. For its size, the system displays a sharp melting transition and the Trp-cage has local structuring propensities in the unfolded state that are similar to those observed in a number of proteins of interest to the protein folding research community. Reproducing, in silico, the mutational effects elucidated in this study should provide a stringent test of folding computation methods.
Methods
Peptide synthesis and purification
Peptides were synthesized on an Applied Biosystem 433A peptide synthesizer using standard Fmoc solid-phase peptide synthesis methods. The resins used for the synthesis were Wang resins preloaded with the C-terminal amino acid. C-terminal amides were prepared similarly but using Rink resins. Peptides were cleaved from the resin using a 95:2.5:2.5 trifluoroacetic acid (TFA):triisopropylsilane:water mixture. The cleaved peptides were purified by reverse phase HPLC on a Varian C18 prep-scale column using gradients of water/acetonitrile (having 0.1% and 0.085% TFA, respectively). Collected fractions were lyophilized and their identity and molecular weight confirmed using a Bruker Esquire Ion Trap mass spectrometer.
CD spectroscopy and melting analysis
Stock peptide solutions for CD experiments were prepared in 10 mM phosphate buffer at the appropriate pH. The concentration of the stock solution was determined by the UV absorption of tyrosine and tryptophan (ε = 1190 M−1 cm−1 and 5580 M−1 cm−1, respectively at 278 nm). CD samples with concentrations of 20–30 μM were prepared by dilution of the stock solution. Spectra were recorded on a Jasco J715 instrument in cells with a path-length of 0.1 cm. Typical spectral accumulation parameters were a scan rate of 100 nm/min with a 2 nm band-width, and a 0.1 nm step resolution over the wavelength range 185–270 nm with eight scans averaged for each spectrum at temperatures ranging from 0°C to 90°C. The spectrophotometer was equilibrated at each temperature for 5 min before acquisition. The raw ellipticity data were converted into mean residue molar ellipticity units, (deg cm2)/(residue dmol) using standard Jasco software.
Typical changes in the CD signature of a Trp-cage that undergoes nearly complete melting over the 0–90°C range are shown in Fig. 8a.
Fig. 8.
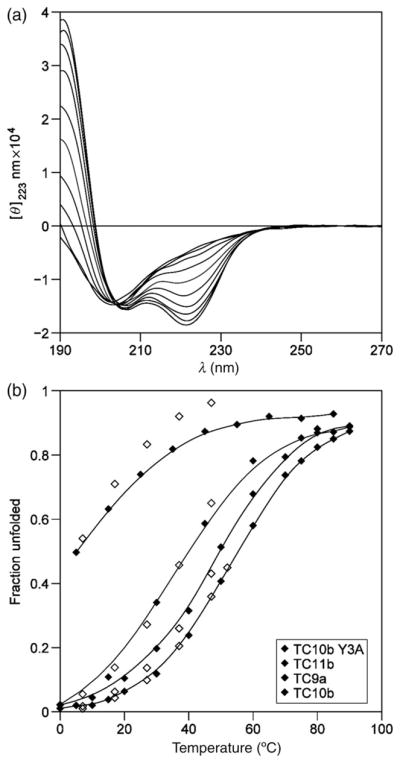
(a) The temperature dependence of the CD spectrum of TC10b at pH 7, traces are shown for every 10° increment from 0°C to 90°C. (b) Melting curves (χU versus T) for TC10b and some of its single site-mutants, in order of increasing stability: Y3A-TC10b, TC11b, TC9a and unaltered TC10b. NMR χU values from ‘cage CSDs’ as open symbols are superimposed on the CD melts. When the cage is substantially destabilized, illustrated here by Y3A-TC10b, some residual helicity remains after the cage structure melts.
CD melts are presented as plots of mean residue molar ellipticity at 222.5–223.6 nm ([θ]223) versus temperature. The melting curves for TC10b (and additional analogs) is shown in Fig. 5, which also includes an unfolded reference line. The 100%-folded baseline was obtained by assuming a temperature gradient (δ[θ]223/δT) that was 0.32% of the 100%-folded ellipticity. The latter was obtained from the lowest temperature [θ]223 observation adjusting it to 100%-folded based on the NMR measures (CSDs) of χF. The temperature dependence of [θ]223 in 7 M GdmCl was determined for three Trp-cage species, TC5b, TC10b and its less stable S14A mutant. For the less stable species, TC5b and (S14A)-TC10b, [θ]223 versus T (°C) was linear to within experimental error: −661 − 29.6·T and −997 − 26.3·T, respectively. The corresponding plot for TC10b displayed slight curvature which could be rationalized as the melting of a residual 2% helicity; the linear fit over the low temperature region was −1430 − 18·T. For the calculation of CD Tm values and melting curves (expressed as χU versus T, Fig. 8b), a common equation was used for unfolded state reference, [θ]223, U=−900 − 29.T. The interpolation for determining the Tm was a four-order polynomial fit of melting curve. For constructs with Y3 replaced by a non-aromatic residue, alternative baselines, [θ]223, U = −2050 − 21·T and [θ]223, H = −22 400 + 72·T, reflecting the known positive [θ]223-contribution of Tyr in both the coil and helical state (chakrabatty et al., 1993) were also examined.
Unfolding thermodynamics were derived by the method previously applied to two-state hairpin unfolding transitions (Andersen et al., 2006). We first determined the range of ΔCp values that produced a linear relationship for Eq. (1), as measured by R2. We report the thermodynamic parameters for T* = 298 K.
| (1) |
For TC10b, ΔCp values between −330 and −110 J/mol K displayed R2>0.998, consistent with apparent two-state behavior, over the 10–90°C range. This, in turn, provided the range of ΔS and ΔH values. A recent calorimetric study of TC5b (Streicher and Makhatadze, 2007) also found a small ΔCpU value, but it was positive (0.3 ± 0.1 kJ/mol K).
NMR spectroscopy
All NMR experiments were performed on a Bruker DRX 500 MHz spectrometer on peptide samples of 1–1.5 mM concentration in 50 mM phosphate buffer at pH 7 and 2.5, with 10% D2O. DSS was used as the internal proton reference standard and set to 0 p.p.m. for all conditions. A combination of TOCSY and NOESY spectra 2D NMR spectra recorded at temperatures ranging from 280 to 325 K were used to assign all resonances. An MLEV-17 (Bax and Davis, 1985) spinlock (12 or 60 ms) was employed for the TOCSY and mixing times of 60, 150 or 200 ms were used for the NOESY spectra.
CSD calculations and melts
The CSDs (CSD = δobs − δrc) for all the peptides were calculated using the reference random coil chemical shifts (δrc) for the specific residue with nearest neighbor corrections. With the exception of Gly11, all the Hα random coil values were obtained from our automated CSD calculation program (Fesinmeyer et al., 2004), which is available at http://andersenlab.chem.washington.edu/CSDb. The reference random coil values used for Gly11-Hα2, Pro18-Hβ3, Pro19-Hδ2 and -Hδ3 were 4.02, 2.29, 3.59 and 3.74 p.p.m. respectively; the proline side chain values are used for prolines at other positions as well.
NMR melting temperatures reported herein rely on sums of CSDs. For each construct, two Tm-measures, ‘cage’ and ‘helix’, were calculated. The ‘cage’ Tm’s are derived from the sum of the CSDs of five protons (L7α, P18α, P18β3, P19δ3 and P19δ2; when available G11α2 replaces or supplements L7α in the calculation) experiencing large upfield ring-current shifts associated with the indole ring of Trp6. The helix melt measure employs Hα sites within the helix–Y3, Q5, W6, K8. (For species with N-terminal modifications or truncations, the measure of helix formation includes only the Q5/W6/K8 Hα sites.)
NH exchange studies and PF calculations
The NH exchange experiments were performed on a Bruker DRX 500 MHz spectrometer at 280 K by adding pre-cooled D2O phosphate buffer (pD = 5.0, 5.5 or 6.6) to the lyophilized peptide sample in a pre-cooled NMR tube and then recording 1D proton spectra at various time intervals. The slowest exchanging NH’s were used for calculating the exchange rates and PF (PF = krc/kobs). Exchange rates (kobs) were obtained from 1D spectra as the slopes of plots of ln (NH signal intensity) versus time. The reference random coil rate constant for exchange (krc) was calculated using the appropriate Molday factors (Bai et al., 1993), as previously described for exendin-4 and TC5b (Neidigh et al., 2001, Neidigh et al., 2002). The relationship, χF = 1 − (1/PF) (Fezoui and Braswell, 1999), was employed to convert PF to an extent of folding measure. The mean ΔGU was calculated using the χF values obtained from the PFs of the three or four slowest exchanging protons measured at the three different pDs (5.0, 5.5 and 6.6), as described previously (Lin et al., 2004).
NMR structure ensemble calculation
NOE intensities from NOESY spectra from all available experimental conditions were converted into distance constraints. The NOE intensities were obtained directly from the NOESY spectra and these were converted to distances by an automated program, DIS (Fesinmeyer, 2005), which also calculates the allowed ranges (see Supplementary data available at PEDS online). The resulting constraints were then used in a previously reported (Neidigh et al., 2001) simulated annealing protocol using the program CNS (Brünger et al., 1998). Stereospecific assignments for the glycine Hα’s, W6-Hβ, L7-Hβ, K8-Hβ, D9-Hβ and S14-Hβ prochiral CH2 groups, the proline ring hydrogens and the L7 methyl signals were accomplished by pairwise trials accepting the assignment that gave a significantly lower ENOE. Resolution of the D9-CβH2 and S14-CβH2 assignments required trials for all four possibilities. The resulting assignments and the constraint list appear in the Supplementary data available at PEDS online.
In the present study, 40 structures were generated, out of which 28 structures were accepted. The acceptance criteria for these structures were no NOE violation >0.2 Å and agreement with idealized covalent geometry (the mean of the bond, angle and improper torsion violations could not exceed 0.01 Å, 0.55° and 0.2° in any structure with no individual values exceeding 0.02 Å, 3° and 2°, respectively). A structure ensemble was generated using the 28 accepted structures. The backbone and heavy atom RMSD measures of convergence are the mean ±s.e. for pairwise comparisons over the entire accepted ensemble. The RMSD calculations and figures depicting structure ensembles were generated using MolMol (Koradi et al., 1996). Since some bond lengths and angles are parameterized differently by CNS and the structure analysis algorithm used by the Research Collaboratory for Structural Bioinformatics (RCSB-PDB), the structures submitted to the PDB (accession #2JOF) are the CNS-generated structures modified by 500 steps of steepest-descent minimization using the SANDER application of AMBER6. This minimization has no significant effect on the NOE–constraint deviations. The largest heavy atom position change observed during such minimizations was 0.32 Å.
Supplementary Material
Acknowledgments
We thank Prof. Carlos Simmerling (Dept of Chemistry and Center for Structural Biology, Stony Brook University) for continuing insights into Trp-cage folding from molecular dynamics that have been useful in designing and analyzing mutational studies. The US National Science Foundation provided funds for the upgrade of NMR instrumentation in the UW Chemistry Department facility.
Funding
US National Institutes of Health [grant GM050658 to NHA].
Footnotes
For Permissions, please journals.permissions@oxfordjournals.org
Supplementary data are available at PEDS online.
References
- Ahmed Z, Ilir AB, Mikhonin AV, Asher SA. J Am Chem Soc. 2005;127:10943–10950. doi: 10.1021/ja050664e. [DOI] [PubMed] [Google Scholar]
- Andersen NH, Tong H. Protein Sci. 1997;6:1920–1936. doi: 10.1002/pro.5560060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen NH, Neidigh JW, Harris SM, Lee GM, Liu Z, Tong H. J Am Chem Soc. 1997;119:8547–8561. [Google Scholar]
- Andersen NH, Fesinmeyer RM, Neidigh JW, Barua B. In: The Trp-Cage: A Notably Stable Mini-Protein Fold in Peptides 2000. Martinez J, Fehrentz J-A, editors. EDK; Paris, France: 2001. [Google Scholar]
- Andersen NH, Olsen KA, Fesinmeyer RM, Tan X, Hudson FM, Eidenschink LA, Farazi SR. J Am Chem Soc. 2006;128:6101–6110. doi: 10.1021/ja054971w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio JL, Cañada FJ, Bruix M, Rodriguez-Romero A, Jimenez-Barbero J. Eur J Biochem. 1995;230:621–633. doi: 10.1111/j.1432-1033.1995.tb20604.x. [DOI] [PubMed] [Google Scholar]
- Bai Y, Milne JS, Mayne L, Englander SW. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin RL. J Biol Chem. 2003;278:17581–17588. doi: 10.1074/jbc.X200009200. [DOI] [PubMed] [Google Scholar]
- Barua B, Andersen NH. Lett Pept Sci. 2002;8:221–226. [Google Scholar]
- Bax A, Davis DG. J Magn Reson. 1985;65:355–360. [Google Scholar]
- Brünger AT, et al. Acta Crystallogr D Biol Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Bunagan MR, Yang X, Saven JG, Gai F. J Phys Chem B. 2006;110:3759–3763. doi: 10.1021/jp055288z. [DOI] [PubMed] [Google Scholar]
- Cao B. PhD. 1993. NMR structural studies of bound peptides and hevein, a small protein. [Google Scholar]
- Carnevali P, Toth G, Toubassi G, Siavash N. J Am Chem Soc. 2003;125:14244–14245. doi: 10.1021/ja036647b. [DOI] [PubMed] [Google Scholar]
- Chen J, Im W, Brooks CL., III J Am Chem Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Lee MC, Xiong G, Duan Y. J Mol Biol. 2003;327:711–717. doi: 10.1016/s0022-2836(03)00177-3. [DOI] [PubMed] [Google Scholar]
- Ding F, Buldyrev SV, Dokholyan NV. Biophys J. 2005;88:147–155. doi: 10.1529/biophysj.104.046375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig AJ, Baldwin RL. Protein Sci. 1995;4:1325–1336. doi: 10.1002/pro.5560040708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson N, Sharpe TD, Schartau PJ, Sato S, Allen MD, Johnson CM, Rutherford TJ, Fersht AR. J Mol Biol. 2005;353:427–446. doi: 10.1016/j.jmb.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Fesinmeyer RM. PhD. 2005. Chemical shifts define the structure and folding thermodynamics of polypeptides. [Google Scholar]
- Fesinmeyer RM, Hudson FM, Andersen NH. J Am Chem Soc. 2004;126:7238–7243. doi: 10.1021/ja0379520. [DOI] [PubMed] [Google Scholar]
- Fesinmeyer RM, Hudson FM, White GWN, Olsen KA, Euser A, Andersen NH. J Biomol NMR. 2005;33:213–231. doi: 10.1007/s10858-005-3731-7. [DOI] [PubMed] [Google Scholar]
- Fezoui Y, Braswell EH. Biochemistry. 1999;38:2796–2804. doi: 10.1021/bi9823838. [DOI] [PubMed] [Google Scholar]
- Fleming PJ, Rose GD. Protein Sci. 2005;14:1911–1917. doi: 10.1110/ps.051454805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh K, Ozkan SB, Dill KA. J Am Chem Soc. 2007;129:11920–11927. doi: 10.1021/ja066785b. [DOI] [PubMed] [Google Scholar]
- Hudson FM, Andersen NH. Biopolymers (Pept Sci) 2004;76:298–308. doi: 10.1002/bip.20126. [DOI] [PubMed] [Google Scholar]
- Huyghues-Despointes BM, Scholtz JM, Baldwin RL. Protein Sci. 1993;2:1604–1611. doi: 10.1002/pro.5560021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyghues-Despointes BM, Klinger TM, Baldwin RL. Biochemistry. 1995;34:13267–13271. doi: 10.1021/bi00041a001. [DOI] [PubMed] [Google Scholar]
- Ivankov DM, Garbuzynskiy SO, Alm E, Plaxco KW, Baker D, Finkelstein AV. Protein Sci. 2003;12:2057–2062. doi: 10.1110/ps.0302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackups RJ, Liang J. J Mol Biol. 2005;354:979–993. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- Juraszek J, Bolhuis PG. Proc Natl Acad Sci USA. 2006;103:15859–15864. doi: 10.1073/pnas.0606692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karle IL, Balaram P. Biochemistry. 1990;29:6747–6756. doi: 10.1021/bi00481a001. [DOI] [PubMed] [Google Scholar]
- Kemmink J, van Mierlo CPM, Scheek RM, Creighton TE. J Mol Biol. 1993;230:312–322. doi: 10.1006/jmbi.1993.1144. [DOI] [PubMed] [Google Scholar]
- Kentsis A, Gindin T, Mezei M, Osman R. PLoS ONE. 2007;2:e446. doi: 10.1371/journal.pone.0000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga N, Takada S. J Mol Biol. 2001;313:171–180. doi: 10.1006/jmbi.2001.5037. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Kubelka J, Hofrichter J, Eaton WA. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Otting G, Wüthrich K. J Biomol NMR. 1992;2:447–465. doi: 10.1007/BF02192808. [DOI] [PubMed] [Google Scholar]
- Lin JC, Barua B, Andersen NH. J Am Chem Soc. 2004;126:13679–13684. doi: 10.1021/ja047265o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhananta A, Boer J, MacKay I. J Chem Phys. 2005;122:114901/114901–114901/114915. doi: 10.1063/1.1874812. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu Z, Androphy E, Chen J, Baleja JD. Biochemistry. 2004;43:7421–7433. doi: 10.1021/bi049552a. [DOI] [PubMed] [Google Scholar]
- Matthews BW, Nicholson H, Becktel WJ. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel JS, Regan L. Fold Des. 1998;3:449–455. doi: 10.1016/s1359-0278(98)00062-5. [DOI] [PubMed] [Google Scholar]
- Mok KH, Kuhn LT, Goez M, Day IJ, Lin JC, Andersen NH, Hore PJ. Nature. 2007;447:106–109. doi: 10.1038/nature05728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naduthambi D, Zondlo NJ. J Am Chem Soc. 2006;128:12430–12431. doi: 10.1021/ja0648458. [DOI] [PubMed] [Google Scholar]
- Neidigh JW. PhD. 1999. Chemical shift tools in peptide folding and miniature protein design. [Google Scholar]
- Neidigh JW, Andersen NH. Biopolymers. 2002;65:354–361. doi: 10.1002/bip.10272. [DOI] [PubMed] [Google Scholar]
- Neidigh JW, Fesinmeyer RM, Prickett KS, Andersen NH. Biochemistry. 2001;40:13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
- Neidigh JW, Fesinmeyer RM, Andersen NH. Nat Struct Biol. 2002;9:425–430. doi: 10.1038/nsb798. [DOI] [PubMed] [Google Scholar]
- Neuweiler H, Doose S, Sauer M. Proc Natl Acad Sci USA. 2005;102:16650–16655. doi: 10.1073/pnas.0507351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforovich GV, Andersen NH, Fesinmeyer RM, Frieden C. Proteins. 2003;52:292–302. doi: 10.1002/prot.10409. [DOI] [PubMed] [Google Scholar]
- Olsen KA, Fesinmeyer RM, Stewart JM, Andersen NH. Proc Natl Acad Sci USA. 2005;102:15483–15487. doi: 10.1073/pnas.0504392102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota M, Ikeguchi M, Kidera A. Proc Natl Acad Sci USA. 2004;101:17658–17663. doi: 10.1073/pnas.0407015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Baldwin RL. J Mol Biol. 1994;241:706–713. doi: 10.1006/jmbi.1994.1545. [DOI] [PubMed] [Google Scholar]
- Pitera JW, Swope W. Proc Natl Acad Sci USA. 2003;100:7587–7592. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxco KW, Simons KT, Ruczinski I, Baker D. Biochemistry. 2000;39:11177–11183. doi: 10.1021/bi000200n. [DOI] [PubMed] [Google Scholar]
- Qiu L, Pabit SA, Roitberg AE, Hagen SJ. J Am Chem Soc. 2002;124:12952–12953. doi: 10.1021/ja0279141. [DOI] [PubMed] [Google Scholar]
- Schug A, Herges T, Wenzel W. Phys Rev Lett. 2003;91:158102/158101–158104. doi: 10.1103/PhysRevLett.91.158102. [DOI] [PubMed] [Google Scholar]
- Schug A, Wenzel W, Hansmann UHE. J Chem Phys. 2005;122:194711/194711–194717. doi: 10.1063/1.1899149. [DOI] [PubMed] [Google Scholar]
- Searle MS, Ciani B. Curr Opin Struct Biol. 2004;14:1–7. doi: 10.1016/j.sbi.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Simmerling CL, Strockbine B, Roitberg AE. J Am Chem Soc. 2002;124:11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- Snow CD, Zagrovic B, Pande VS. J Am Chem Soc. 2002;124:14548–14549. doi: 10.1021/ja028604l. [DOI] [PubMed] [Google Scholar]
- Stapley BJ, Doig AJ. J Mol Biol. 1997;272:465–473. doi: 10.1006/jmbi.1997.1262. [DOI] [PubMed] [Google Scholar]
- Streicher WW, Makhatadze GI. Biochemistry. 2007;46:2876–2880. doi: 10.1021/bi602424x. [DOI] [PubMed] [Google Scholar]
- Ulmschneider JP, Ulmschneider MB, Di Nola A. J Phys Chem B. 2006;110:16733–16742. doi: 10.1021/jp061619b. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- Zhou R. Proc Natl Acad Sci USA. 2003;100:13280–13285. doi: 10.1073/pnas.2233312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.