

Abstract
Background
Trichohepatoenteric syndrome (THES) is an autosomal recessive disorder characterised by life-threatening diarrhoea in infancy, immunodeficiency, liver disease, trichorrhexis nodosa, facial dysmorphism, hypopigmentation and cardiac defects. We attempted to characterise the phenotype and elucidate the molecular basis of THES.
Methods
Twelve patients with classical THES from 11 families had detailed phenotyping. Autozygosity mapping was undertaken in 8 patients from consanguineous families using 250k single nucleotide polymorphism (SNP) arrays and linked regions evaluated using microsatellite markers. Linkage was confirmed to one region from which candidate genes were analysed. The effect of mutations on protein production and/or localisation in hepatocytes and intestinal epithelial cells from affected patients was characterised by immunohistochemistry.
Results
Previously unrecognised platelet abnormalities (reduced platelet α-granules, unusual stimulated alpha granule content release, abnormal lipid inclusions, abnormal platelet canalicular system and reduced number of microtubules) were identified. The THES locus was mapped to 5q14.3 – 5q21.2. Sequencing of candidate genes demonstrated mutations in TTC37, which encodes the uncharacterised tetratricopeptide repeat protein, thespin. Bioinformatic analysis suggested thespin to be involved in protein-protein interactions or chaperone. Preliminary studies of enterocyte brush-border ion transporter proteins (NHE2, NHE3, Aquaporin 7, Na/I symporter and H / K ATPase) showed reduced expression or mislocalisation in all THES patients with different profiles for each. In contrast the basolateral localisation of Na/K ATPase was not altered.
Conclusion
THES is caused by mutations in TTC37. TTC37 mutations have a multisystem effect which may be due to abnormal stability and / or intracellular localisation of TTC37 target proteins.
Keywords: phenotypic diarrhoea of infancy, platelet alpha granules, ion transporter proteins, thespin
INTRODUCTION
Diarrhoea is a major cause of morbidity and mortality throughout the world: an estimated 1.87 million young children die of it each year1. Whilst infection is the most common cause of diarrhoea, investigation of rarer causes of diarrhoea can increase understanding of the normal gastrointestinal function and the molecular pathogenesis of diarrhoea. Inherited diarrhoea syndromes may be associated with phenotypic abnormalities of the enterocyte (e.g. microvillous inclusion disease2 and tufting enteropathy3) or with a normal cellular phenotype but abnormal solute transporter function (e.g. congenital chloridorrhea4 and congenital sodium diarrhoea5).
Trichohepatoenteric syndrome (THES, also known as phenotypic diarrhoea of infancy; MIM 222470) is an autosomal recessively inherited disorder with an estimated incidence of 1 in 400,000 to 1 in 500,000 live births. Stankler et al. (1982) first described THES in 2 siblings with low birth weight, dysmorphic features and abnormal “woolly” hair with abnormalities on microscopy indicating weakness of the hair shaft. Diarrhoea began in the third postnatal week and resulted in death. The livers were fibrotic with marked haemosiderosis6. A description of 8 similar children7 and subsequent case reports have extended the clinical spectrum8–13 which comprises distinctive facial features (hypertelorism; broad flat nasal bridge; prominent forehead), abnormal hair (coarse, sparse and fragile with trichorrhexis nodosa) and diarrhoea which manifests not at birth but weeks to months thereafter. On microscopy intestinal changes are non-specific; initial subtotal villous atrophy improves with time (although severity of diarrhoea does not correlate with the histological change) and a mixed inflammatory infiltrate varies. The enterocyte brush border is ultrastructurally normal. All affected children require parenteral nutrition to maintain life and growth. In some, parenteral nutrition is a life long requirement; others it can be weaned to full enteral feeding.
Immunodeficiency is a consistent feature with low serum concentrations of immunoglobulins (which often, but not always, improves with age) and a poor immunological response to childhood vaccination.
Hepatic involvement is inconsistent, even among siblings, and irrespective of parenteral nutrition can include hepatomegaly, fibrosis, siderosis and cirrhosis (even before the onset of diarrhoea).
Prenatal manifestations include polyhydramnios and placentomegaly, and other clinical findings include cardiac lesions (tetrology of Fallot, ventricular and atrial septal defects),hypopigmentation of the skin, inguinal and umbilical hernia, mental retardation and delayed puberty.
THES is life limiting. Death results from hepatic failure or secondary to the complications associated with the need for long term parenteral nutrition6–13.
Defining the molecular genetic basis of THES will facilitate diagnosis and management. It will help in counselling regarding prognosis and will enable prenatal and preimplantation diagnosis in families at risk. It might also provide critical insight into the mechanisms of diarrhoea and thus treatment.
METHODS
Patients
DNA was extracted from whole blood (Gentra Puregene DNA purification system: Qiagen, Crawley, West Sussex, UK) in 12 children with THES and their parents. In unaffected siblings DNA was extracted from either a blood sample or mouthswab. Lymphocyte RNA (PAXgene Blood RNA Kit: Qiagen, Crawley, West Sussex, UK) was available from four affected children. Jejunal specimens were taken at the time of initial diagnostic investigation for intractable diarrhoea and liver specimens in the course of evaluation of clinical liver disease. This study was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the South Birmingham Research Ethics Committee. In all cases written consent was obtained for sample collection and subsequent analysis.
Analytical methods
Molecular Genetic Studies
Genomic DNA was extracted by standard techniques. A genome-wide linkage scan was undertaken using the Affymetrix 250k SNP microarray with DNA from 8 affected individuals in 7 consanguineous families. Regions of homozygosity >3 cM that were shared by all the affected children were further investigated (n=3). Microsatellite markers were used to confirm or refute linkage within individual families and to fine-map the region of interest at 5q14.3 – 5q21.2. Direct sequencing of the 45 genes within the identified region was prioritised according to putative function (predicted ion transport, DNA repair and chaperone functions), site of expression and position. The genomic DNA sequence of candidate genes was taken from Ensembl (http://www.ensembl.org/index.html) and primer pairs for the translated exons were designed using ExonPrimer software (http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html). Individual exons and flanking sequences (primer details and conditions in supplemental data 1) were amplified using standard polymerase chain reaction (PCR). PCR products were directly sequenced by the Big Dye Terminator Cycle Sequencing System (Applied Biosystems, Foster City, CA) with the use of an ABI PRISM 3730 DNA Analyzer (Applied Biosystems). DNA sequences were analyzed using Chromas software (http://www.technelysium.com.au/chromas.html).
Reverse-transcriptase PCR was used to identify the effect of splice site mutations on gene expression.
Immunohistochemistry of liver specimens
Samples of formalin-fixed, paraffin-embedded liver, sectioned at 4–5µm and picked up on glass slides, were immunostained and counterstained as described previously14 for the canalicular transport proteins bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2).
Immunohistochemistry of jejunal specimens
4µm sections of archival material originally obtained for clinical diagnosis were deparaffinized and heat fixed. Slides were microwaved for antigen recovery in 10mM sodium citrate buffer, pH6 (Sigma Chemical Company, St Louis, MO) at power level setting 9 (Panasonic Model NN-C980B Conventional Microwave Oven, Secaucus, NJ), for 2–5minutes. After cooling for 30 minutes, sections were washed in phosphate buffered saline (PBS) and blocked with 5% normal goat serum in phosphate buffered saline – Tween (PBS-T). Sections were incubated with rabbit polyclonal antibodies against NHE2 (Ab597), NHE3 (Ab1381), Na/I symporter (NIS) and H/K ATPase and chicken polyclonal antibody against Aquaporin 7 (Aqp7) as well as with monoclonal antibodies against villin and the Na/K ATPase (The Na/K ATPase developed by Fambrough, DM was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242), as previously descibed15, 16,17. After 2 washes with PBS, sections were incubated with AlexaFluor 488 anti-rabbit and AlexaFluor 568 anti-mouse secondary antibodies (Invitrogen Ltd, Paisley, UK). Sections were washed, counterstained with Hoechst 33342 (Invitrogen Ltd, Paisley, UK) for nuclei, and mounted with glass coverslips mounting medium. Immunofluorescence images were obtained using a 63x water objective and a LSM 510 Meta confocal microscope. Similar settings for laser power, gain and resolution were used for each sample analysed. Jejunal samples from four patients and 20 aged matched controls (archival material initially taken for the clinical consideration of coeliac disease and found to be normal on histological examination) were analysed by a single investigator blinded to the clinical diagnosis.
Platelet studies
Blood was available for the investigation of the structure of platelets by electron microscopy as previous described18, in three patients (1C, 1D and 4C). 3.2% citrate anticoagulant blood was centrifuged (150g for 20 minutes) to obtain platelet rich plasma (PRP). PRP was fixed with 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS pH 7.4 at 4°C for 1 hour. Platelets were washed with 0.1M phosphate buffer (pH 7.4), followed by dH2O. Platelets were then postfixed with 2% osmium tetroxide, dehydrated in graded acetones and embedded in Epon (Electron Microscopy Sciences). Thin sections were examined with JEOL JEM-1011 electron microscope with uranyl acetate and lead citrate staining (Electron Microscopy Sciences) Digital images were captured with a side mounted Advanced Microscopy Techniques (AMT) Advantage HR CCD cameras (Advanced Microscopy Techniques Corp., Danvers, MA).
Platelet aggregation was measured in response to PAR1-specific peptide (SFLLRN, Alta Bioscience, Birmingham, UK), ADP (Sigma-Aldrich, Poole, UK) and collagen (Nycomed Austria, Linz, Austria) in two patients (4C and 5C). Secretion from dense granules was measured in a dual channel lumi-aggregometer (460VS, Chronolog) (Chrono-log Corporation, Havertown, PA) using a luciferase assay that detects released ATP (Chrono-log Corporation, Havertown, PA) 19. The level of expression of CD62P (P-selectin; α granule secretion indicator) was measured by flow cytometry using a specific antibody (Fluorescein isothiocyanate (FITC)-conjugated anti mouse P-selectin antibody, Emfret Analytics, Wuorzburg, Germany) following stimulation by a collagen related peptide (CRP) (Dr Richard Farndale Cambridge University, UK).
RESULTS
Clinical findings
The principal clinical findings are listed in table 1. All children had the typical facial features and trichorrhexis nodosa of the hair on microscopy. Cystine, serine and proline concentrations of the hair were reduced whilst leucine and aspartate concentrations were increased (the amino acid concentration of the hair is remarkably similar to that of trichothiodystropy and a graph comparing the amino acid compositions is provided in supplementary data 2). There was a high incidence of preterm delivery and intrauterine growth retardation. The age at presentation with diarrhoea was between 2 weeks and 7 months. Diarrhoea was characterised as having both secretory and osmotic components with no evidence to suggest a specific ion transport defect or protein loss. All affected children required parenteral nutrition in infancy but in 2 children the diarrhoea improved and parenteral nutrition was discontinued between 5 months and 3 years of age.
Table 1.
Clinical features of the study cohort of THES patients. Child 11C was a recent diagnosis and parenteral nutrition had not begun. Where no information was available the boxes have been left blank. In this cohort of THES patients, two thirds of children are born to consanguineous families, with Pakistan being the most common country to originate from (5 patients). 2 affected children are siblings (1C and 1D). All others are single affected children within individual kindred’s.
| Identification number | 1C | 1D | 2C | 3C | 4C | 5C | 6C | 7C | 8C | 9C | 10C | 11C |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Indian | Indian | Dutch | Pakistani | Kurdish | Pakistani | Pakistani | Pakistani | English | Italian | Flemish | Pakistani |
| Consanguinity | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | No | No | No | Yes |
| Family history | Sibling | Sibling | No | No | Cousin | No | No | No | No | No | No | No |
| Sex | Female | Female | Male | Female | Female | Male | Female | Male | Male | Female | Male | Male |
| Current age | died aged 6 months |
Died aged 6 months |
14 years | 11 years | 2 ½ years |
3 1/2 years | 14 onths |
Died aged 8 years |
3 years | 13 years | 2 years | 1 year |
| Gestation | 40/40 | 40/40 | 35/40 | 34/40 | 34/40 | 30/40 | 40/40 | 37/40 | 33/40 | 34/40 | 35/40 | |
| Birth weight | IUGR | IUGR | 1345g IUGR |
1410g IUGR |
1220g IUGR |
980g IUGR |
2400g IUGR |
3580g | 780g IUGR |
1700g IUGR |
1375g IUGR |
|
| Dysmorphology | ||||||||||||
| Wide forehead | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypertelorism | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Skin | Café-au-lait spots | Café-au-lait spots |
Diffuse hypopigmentation |
fair skin | ||||||||
| Trichorrhexis nodosa | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Nutrition | ||||||||||||
| Age when PN started | 7 months | 1 month | 2 months | 3 months | 1 month | 5 months | 4 months | 1 month | 2 weeks | |||
| Current weekly PN regime | off | 5 nights | 3 nights | 5 nights | 7 nights | off | 5 nights | 7 night | ||||
| PN stopped | 3 years | 5 | ||||||||||
| months | ||||||||||||
| Jejunal biopsies | ||||||||||||
| Villous atrophy | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Haematology | ||||||||||||
| Low immunoglobulins | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Thrombocytosis | Yes | Yes | No | No | No | Yes | No | No | No | Yes | No | |
| Large platelets | Yes | Yes | No | Yes | Yes | Yes | No | No | No | No | No | |
| Liver and spleen | ||||||||||||
| Fibrosis | Yes | Yes | No | Yes | Yes | Yes | No | No | No | No | ||
| Siderosis | Yes | Yes | No | No | No | No | No | No | ||||
| Splenomegaly | No | No | Yes | No | No | Yes | No | No | No | |||
| Other systems | ||||||||||||
| Cardiac anomalies | VSD | no | Fallots tetrology |
Mild aortic insufficency |
No | Pulmonary stenosis |
No | No | No | Aortic insufficiency | No | No |
| Skeletal anomalies | No | No | Perthes disease | No | No | No | No | No | No | No | No | No |
| Renal anomlalies | No | No | Small right kidney | Mild reflux | No | No | No | No | No | No | No | Bilateral inguinal hernia. |
| Development | Delayed | Delayed | No | Mild delay | Normal | Delayed | Mild delay |
Delayed | Mild delay |
IUGR – intrauterine growth retardation; PN- parenteral nutrition; VSD- ventricular septal defect;
Histological examination of jejunal biopsies showed mild villous atrophy and a mild mixed inflammatory infiltrate but no specific diagnostic features. Microscopy of serially obtained biopsy specimens in some children suggested that villous atrophy can improve with age and that the inflammatory infiltrate is not consistent. Two children (1C and 1D) had hepatic iron overload and died of liver failure. Child 4C presented at 3 weeks postnatal age with a nodular cirrhotic liver 4 weeks before the onset of diarrhoea. Liver disease developed in patient 7C, who had received long term parenteral nutrition, and who was considered for liver and small bowel transplantation.
Serum immunoglobulin levels were low in all patients but 11C. Supplemental immunoglobulins were given in the neonatal period. Serum levels increased with age discontinuing the need for immunoglobulin supplementation in all but patient 2C who continues to require immunoglobulin infusions at age 14 years. The production of antibodies to Haemophilus influenzae, tetanus toxoid and Pneumococcus vaccinations was poor and revaccination failed to produce an immunological response.
Five children had cardiac developmental anomalies (aortic insufficiency n=2, peripheral pulmonary stenosis n=1, ventricular septal defect n=1, tetrology of Fallot n=1). Developmental delay was present in 7 children (3 were too young to assess their intellectual development). It is not clear if the developmental delay was secondary to long term hospitalisation and chronic illness or is a feature of the THES.
Platelet studies
Five children (1C, 1D, 3C, 4C and 5C) of the 10 THES patients studied were noted intermittently to have enlarged platelets on light microscopy of blood films. Platelets from patients 1C and 1D at the time of platelet enlargement and patients 4C when the platelets were of a normal size were examined by thin section transmission electron microscopy (TEM). Figure 1 shows the transmission electron micrographs of a representative normal platelet (A) and of platelets from 3 different THES patients (B–D). The black bar represents 500nm. Normal α-granules (white arrowheads) are observed in the control platelet (A) and occasionally in a THES patient (B) but are frequently absent in THES platelets (C, D). Whereas the membrane surface–connected canalicular system appears normal in control platelets (A, arrow) it was disrupted, with prominent tubules and small membranous vesicles, in THES platelets (B–D, arrows). Lipid inclusions were frequently observed in THES platelets (B, black arrowheads). Electron-dense lysosomal bodies fused with an α-granule were often seen (C, arrowhead). The platelets of patients 4c and 5c showed aggregation responses to high concentrations of a peptide specific to the PAR 1 thrombin receptor (100 µM), ADP (100 µM) and an intermediate collagen concentration (3 µg/ml) were similar to that of the control. No secretion defect from dense granules was detected in response to any of the three agonists as measured by release of ATP. The level of expression of CD62P during stimulation showed 2 populations of platelets, of which one expressed P-selectin and the other did not (Figure 2). The level of expression in a non-age matched control measured on the same day was within the normal range. Further, we have previously shown that activated platelets in an age matched control generate a single population of P-selectin similar to that in the control (not shown).
Figure 1.
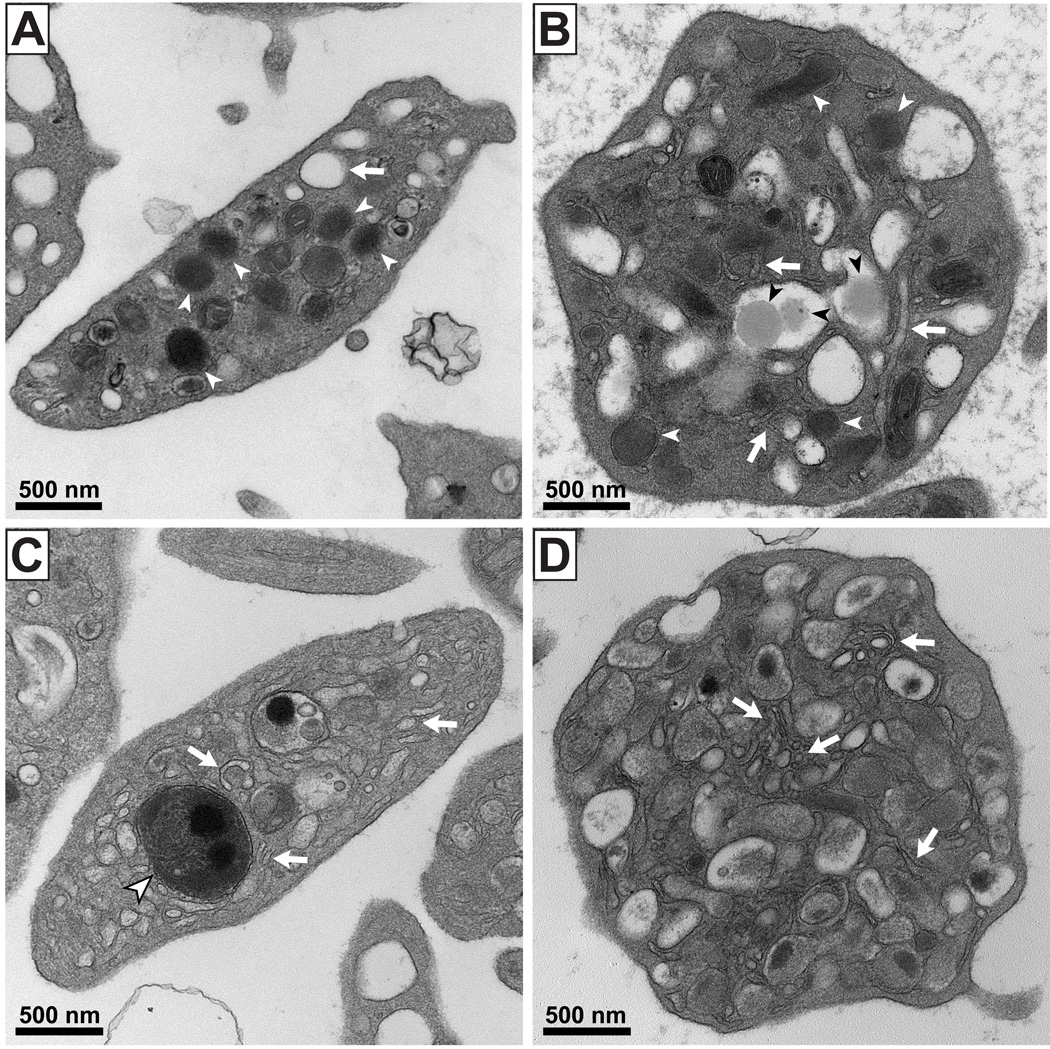
Shows the platelet abnormalities in patients with trichohepatoenteric syndrome (THES). Thin section electron microscopy of a representative normal (A) and three different THES patients (B–D). The black bar represents 500 nm. Normal α-granules are observed in control platelets (A, white arrowheads) and occasionally in some THES platelets (B) but are frequently absent in others (C, D). Whereas the membranous surface-connected canalicular system appears normal in control platelets (A, white arrow), it was disrupted with prominent tubules and smaller membranous vesicles in THES patient platelets (B–D, white arrows). Lipid inclusions were frequently observed in THES platelets (B, black arrows). Electron dense lysosomal bodies fused with an α-granule were often seen (C, large white arrowhead).
Figure 2.
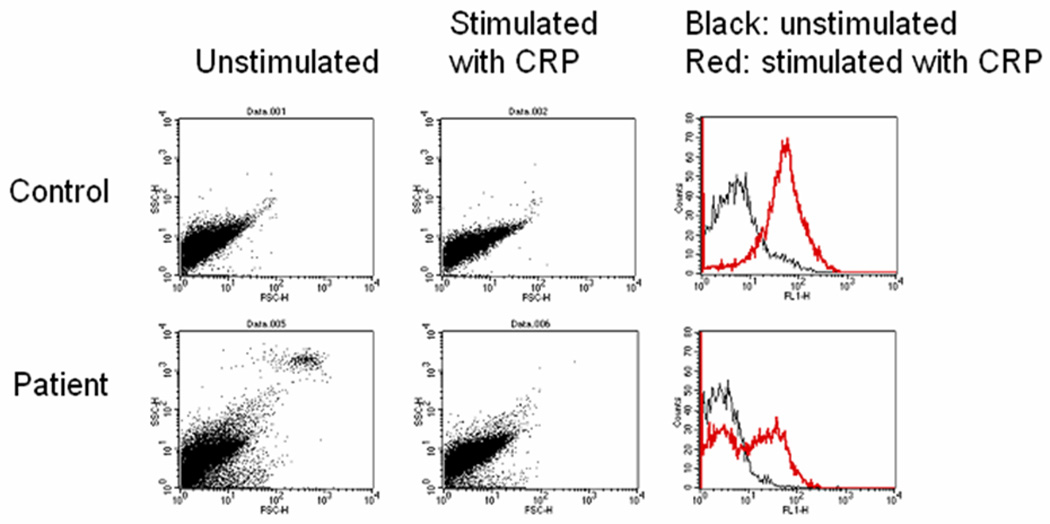
The fluorescence-activate cell sorting (FACS) analysis of the platelets shows that in a control, stimulation by collagen related peptide (CRP) for 90 seconds induces a single, sharp peak in release of α-granule content from the platelet. In THES patients (n=2) there are two peaks due to the absence of secretion in a subpopulation of platelets.
Molecular genetic studies
A genome-wide linkage screen using Affymetrix 250k SNP microarrays in 8 children with THES revealed 3 extended regions of homozygosity shared by all 8: a 15.1 Mb region on chromosome 5 (from 87858428 to 102981136bp); a 15.4 Mb region on chromosome 16 (from 31629915 to 47050362bp) and a 9.5 Mb region on chromosome 19 (from 23551431 to 33083786bp). Further genotyping in all available family members, using microsatellite markers mapping within the homozygous regions eliminated linkage to chromosomes 16 and 19 and confirmed linkage to 5q14.3 to 5q21.2 and narrowed the candidate region to a 12.9 Mb interval between markers D5S815 and D5S409. Multipoint linkage analysis gave a maximum lod score at D5S1462 of 5.8 at theta=0.0. The target region contained 57 genes. After sequencing 42 genes (details on request) in 4 affected individuals (3C, 4C, 5C and 6C) we detected mutations in the hypothetical gene KIAA0372 (subsequently, TTC37). The mutations detected are listed in table 2; their positions within TTC37 are shown in Figure 3. Mutations were detected in 12 affected individuals from 11 families. Mutations were detected in both alleles in 10/12 affected children with a total of 20 alleles having sequence changes. Nine different mutations were identified (3 nonsense due to the formation of a premature stop codon, 2 non synonymous missense changes and 4 splice site changes resulting in aberrant splicing and formation of a premature stop codon). All mutations segregated with disease status within the families. None of the mutations identified were detected in at least 350 ethnically matched South Asian and Caucasian control chromosomes. A nonsense mutation in exon 28 (c.2808G>A: p.W936X) was detected in three apparently unrelated families (1, 7 and 11) of South Asian origin. The SNP haplotypes in the affected individuals were identical over a 974 kb region (from rs255375 to rs34897) containing TTC37, consistent with a founder mutation. An intron 28 splice site mutation was detected in two apparently unrelated families from Pakistan (3 and 5) with identical SNP haplotypes over a 457 kb interval encompassing TTC37 (from rs116286 to rs7736948).
Table 2.
A summary of mutations in TTC37 found in children with THES, their ethnicity and consanguinity (NM_014639.2; NP_055454.1)
| Family Identifier |
Number of affected individuals |
Ethnicity | Consanguinity | Mutation 1 | Mutation 2 |
|---|---|---|---|---|---|
| 1 | 2 | Indian | No | c.2808G>A p.Trp936X |
c.2808G>A p.Trp936× |
| 2 | 1 | Dutch | No | c.3847G>A p.Asp1283Asn |
|
| 3 | 1 | Pakistani | Yes | c.2779-2G>A p.Glu974Glyfs*19 |
c.2779-2G>A p.Glu974Glyfs*19 |
| 4 | 1 | Kurdish | Yes | c.1632+1delG p.Glu545Phefs*40 |
c.1632+1delG p.Glu545Phefs*40 |
| 5 | 1 | Pakistani | Yes | c.2779-2G>A p.Glu974Glyfs*19 |
c.2779-2G>A p.Glu974Glyfs*19 |
| 6 | 1 | Pakistani | Yes | c.751G>A p.Phe215Glufs*14 |
c.751G>A p.Phe215Glufs*14 |
| 7 | 1 | Pakistani | Yes | c.2808G>A p.Trp936X | c.2808G>A p.Trp936× |
| 8 | 1 | English | No | c.1300_1301delAA p.Lys434Lysfs*14 |
c.4514T>C p.Leu1505Ser |
| 9 | 1 | Italian | No | c.439C>T p.Gln147X |
c.2251C>T p.Gln751× |
| 10 | 1 | Flemish | No | c.3847G>A | |
| p.Asp1283Asn | |||||
| 11 | 1 | Pakistani | Yes | c.2808G>A p.Trp936X |
c.2808G>A p.Trp936X |
Figure 3.
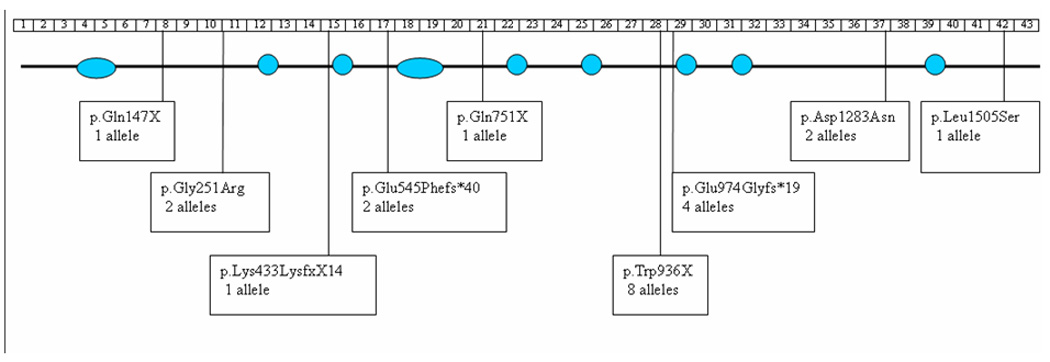
TTC37 is comprised of 43 exons. The position of each of the mutations is shown on this schematic drawing. The TPR domains in relation to the exons and mutations are shown in blue.
RNA studies were undertaken to investigate putative splice site mutations in 4 affected individuals (Figure 4b). In families 3 and 5 an intronic c.2779-2G>A sequence change resulted in skipping of exon 29 and a predicted truncated protein (p.Glu974Glyfs*19) (Figure 4c). In family 4, a c.1632+1delG variation at the first nucleotide of intron 17 resulted in skipping of exon 18 producing a frameshift and a premature stop codon, p.Glu545Phefs*40. In family 6 the c.751G>A substitution, predicted to cause a missense substitution (p.Gly251Arg) involved the final nucleotide of exon 10 and resulted in skipping of exon 10 (Figure 4c) resulting in a frameshift and a predicted a truncated protein (p.Phe215Glufs*14).
Figure 4.
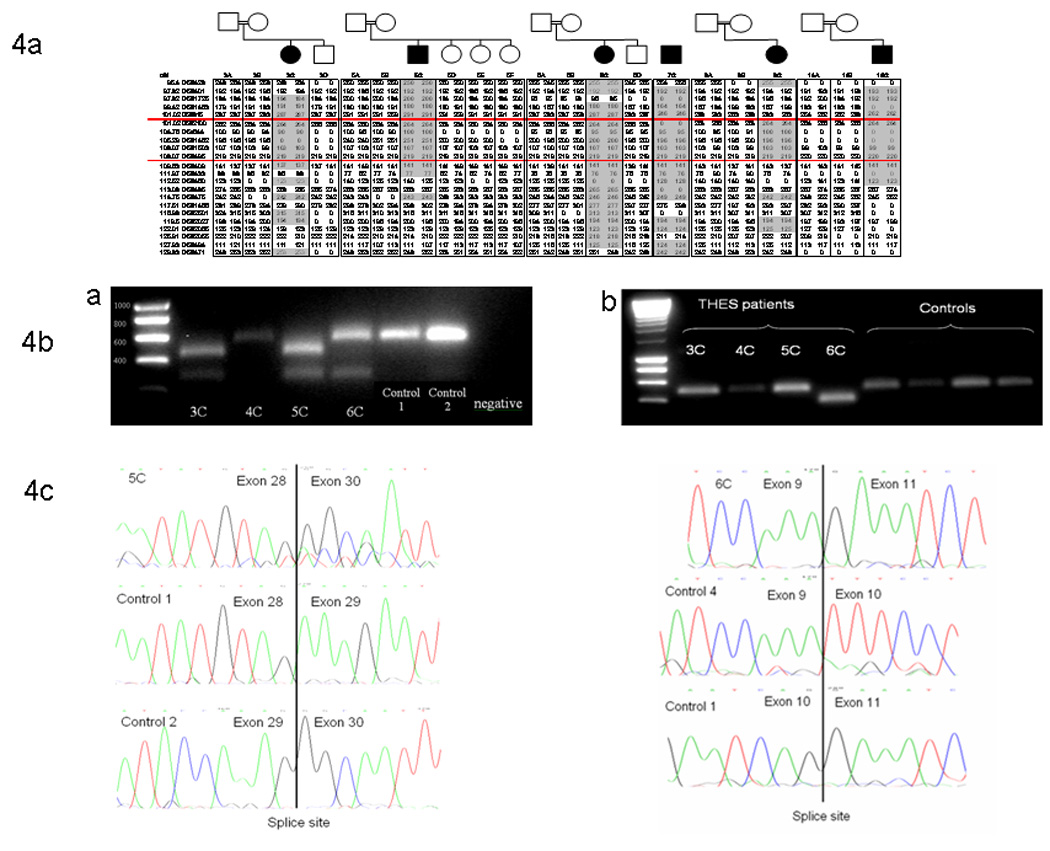
Microsatellite markers haplotype from the probands of Pakistani origin are shown in 4a. The regions of homozygosity are highlighted in grey. At D5s2100 all the probands have identical haplotype. 5b shows a gel electrophoresis of the reverse transcriptase PCR products. 4b.a. shows small PCR products in patients 3C and 5C as compared to the other affected children 4C, 6C and the control samples. 3C and 5C have c.2779-2G>A sequence change in intron 27. 5b.b shows that 6C who has a c.751G>A mutations has a smaller PCR product as compared to other affected children and controls. Figure 4c shows the sequence of the reverse transcriptase PCR’s. Exon 29 is skipped in patients 3C and 5C which corroborates the gel electrophoresis. Exon 10 is skipped in patient 6C and corresponds to the smaller PCR product seen on gel electrophoresis.
Bioinformatic analysis and immunohistochemical studies
In view of the association of THES with mutations in TTC37, we named the TTC37 gene product thespin (Tricho-Hepatic-Enteric Syndrome ProteIN). Thespin is predicted to contain 20 tetratrico peptide repeat (TPR) domains. TPR domain containing proteins have been implicated in a variety of functions including protein-protein interactions and chaperone activity. BLAST analysis (Uniprot identifier Q6PGP7, 1564 amino acids) against the UniProtKB/SwissProt database identified 83 homologous of known function (E-values < 0.01), of which all contained TPR domains. These homologous performed diverse functions, including mediation of chaperone association.
Preliminary immunohistochemical studies
We hypothesised that the multisystem THES phenotype might result from secondary effects on the stability or localisation of TTC37 target proteins and proceeded to undertake immunohistochemical studies on intestinal and liver transport proteins in patients with germline TTC37 mutations.
In THES patients (3C, 4C, 5C, 6C and 11C) as well as age-matched controls (n=20), the apical cytoskeleton marker, villin, was expressed in the brush border of enterocytes suggesting that the morphology of the brush border was normal in all patient samples (Figure 5). NHE2, NHE3, NIS, Aqp7 and H/K ATPase were all normally expressed in the brush border of age-matched intestinal control samples. The localisation of the Na/H exchangers, NHE2 and NHE3, was significantly altered in the brush-border and remained in a subapical region in patients 3C and 6C. In addition, the Na/I symporter, NIS, which is normally expressed in the brush-border just below villin, no longer localized to the brush-border in THES patients but rather to a peri-nuclear compartment within the enterocytes (Figure 5). Similarly, apical expression of H/K ATPase was also reduced in THES patients (4C, 5C and 6C) while some intracellular staining remained present (figure 5). Alternatively, apical expression of Aqp7 was absent in patients 4C, 5C, 6C and 11C suggesting TTC37 may regulate expression of Aqp7. Based on these data, mutations in TTC37 therefore resulted in missorting and/or decreased expression of multiple apical transport proteins in jejunal enterocytes. In order to determine whether basolateral transport proteins are affected by mutations in TTC37, we examined the localization of the Na/K ATPase. As shown in Figure 5, the Na/K ATPase was normally expressed in the basolateral membrane of enterocytes from both THES patients and their age-matched controls.
Figure 5.

Expression and localization of apical and basolateral transport proteins in THES patients and age-matched controls. Paraffin-embedded jejunal samples were fixed and stained with antibodies against brush-border transport proteins, NHE2, NHE3, Aquaporin 7, Na/I Symporter, and the H/K ATPase as well as the basolateral Na/K ATPase. Villin was used as a marker of the enterocyte brush-border. Localization of NHE2, NHE3, NIS and the H/K ATPase was altered in THES patient samples compared to age-matched controls. Alternatively, expression of Aquaporin 7 appeared to be significantly reduced in THES patients versus controls. In contrast, expression and localization of the Na/K ATPase (basolateral) and villin (brush-border) were not altered in any control or THES patient samples analyzed. Inserts demonstrate 6X zoom magnification of original images obtained with 63X water immersion objective. Arrowheads indicate localization of the BB. Scale bar = 20µm.
There was no abnormality detected in the expression and localisation of the bile salt transporters BSEP and MRP2 in liver biopsies (results not shown).
DISCUSSION
We have identified mutations in TTC37 as a cause of THES. The identification of this gene will now enable an accurate molecular diagnosis of THES and facilitate patient and family management. In accompanying clinical work, involving the largest set of THES patients yet described, we have extended the clinical phenotype by identifying cardiac abnormalities to be common (45% of the study cohort). We also identified a rare abnormality of platelet α-granules as a novel aspect of the THES multisystem phenotype.
Nine different mutations in TTC37 were found in 12 children from 11 families with classical features of THES. We detected mutations in both alleles in 10 children. In two children mutations were detected in only one allele suggesting the other sequence variant in this autosomal inherited disease to may be either in an intronic or regulatory region or a mutation not detectable by sequencing. No genotype-phenotype correlations were apparent.
The identification of genes for inherited intractable diarrhoea syndromes may provide insights into mechanisms of diarrhoea. In microvillus inclusion disease (MIM 251850; caused by mutations in MYO5B which encodes type Vb myosin motor protein2) and tufting enteropathy (caused by mutations in EpCAM which encodes an epithelial cell adhesion molecule3), there is an enterocyte brush border abnormality and the diagnosis can be made by histological and electronmicroscopy examination. In contrast the intestinal histology is normal but the stool ion concentration is abnormal with mutations of the solute transporter SLC26A3 causing congenital chlordiarrhoea4 (MIM 214700) which may be regulated by the intracellular pH controlled by NHE3, and congenital sodium diarrhoea caused by mutations in SPINT2, a serine-protease inhibitor5.
The TTC37 gene product, thespin, comprises 1564 amino acid residues and 20 TPR domains. The TPR motif is present in >130 different proteins and corresponding coding sequences are known in many hypothetical genes. TPR domains were first described in the yeast cell division control protein 23 (CDC23)20. They form an α-helix structure that incorporates binding grooves; these may be sites of protein-protein interactions. TPR domains are involved in many cellular processes including the assembly of multiprotein complexes21. Disorders resulting from mutations in genes that encode TPR-containing proteins include Fanconi anaemia22, Leber’s congenital amaurosis23, Bardet-Biedl syndrome24, peroxisome biogenesis disorders25, chronic granulomatous disease26, and Charcot-Marie-Tooth disease type C27. Nevertheless relatively little is known about the normal function of proteins that contain TPR domains and about the pathogenesis of conditions in which these proteins are lacking or abnormal.
The identification of enlarged platelets in some children was further investigated in this study. The ultrastructural analysis suggests an abnormality in platelet α-granule formation and secretion of α-granule contents. An abnormal α-granule secretion pattern was also observed with agonist-stimulated THES platelets whereby only a subpopulation of platelets expressed P-selectin on their surface. It is likely that only those platelets containing some α-granules released their contents whereas platelets without α-granules could not. Clinically children with THES do not have a bleeding diathesis which can be explained by the identification of two platelet populations, one of which functions normally. Platelet α-granule deficiencies are very rare and have only previously been described in grey platelet syndrome (GPS)28 in which the molecular basis has not been elucidated, Quebec platelet disorder29 when there is abnormal break down of α-granule proteins and as a feature of Arthrogryposis-Renal dysfunction-Cholestasis (ARC) syndrome (MIM #208085). ARC syndrome is caused by mutations in VPS33B which is associated with abnormal vesicular trafficking and mislocalisation of polarised membrane proteins30. As well as α-granule abnormalities, children with ARC syndrome also have diarrhoea and liver disease which overlaps with clinical features of THES. The hypothesised role of TPR proteins in protein-protein interactions and the shared characteristics with ARC syndrome raises the possibility of an abnormal protein localisation in THES.
Bioinformatic analysis led us to hypothesise that TTC37 might be implicated in regulating the stability and/or intracellular localisation of, as yet unidentified, target proteins. To initiate investigations of this hypothesis we investigated the expression and localisation of several transporter proteins in enteral and liver tissue samples from THES patients and age-matched controls. Our preliminary studies revealed that although hepatocyte apical bile salt proteins were normally expressed and localised (including BSEP which is mislocalised in ARC syndrome), all five THES patients demonstrated variable abnormalities in expression or localisation of enterocyte brush-border transporter proteins. Reduced apical localisation of Aqp7, NIS and the H/K ATPase was the most common finding. Mislocalisation of NHE2 and NHE3 was detected in 2/4 probands tested (5C was not tested due to lack of patient material). However, localisation of the basolateral transporter, Na/K ATPase, was not affected. The NHE proteins play an integral role in neutral sodium absorption in the gastrointestinal tract. NHE2 is normally located in the apical plasma membrane and NHE3 recycles between the endosomal compartment and the apical plasma membrane. Our preliminary findings are consistent with the hypothesis that TTC37 inactivation disturbs normal intracellular enteral transporter metabolism and/or localisation pathways. NHE3 is often inhibited in diarrhoeal diseases and the NHE3 knockout mouse exhibits modest diarrhoea with increased fluid in the small intestine and colon31. However whilst all patients demonstrated an abnormality of expression or localisation of at least one enterocyte transporter protein, none of the transporters analysed was abnormal in all cases. Hence further investigations are required to confirm and extend these findings and elucidate the relationship between abnormal expression/localisation of specific target proteins and the clinical phenotype of THES patients.
The identification of TTC37 will be of immediate clinical benefit to patients and families affected by THES. Thespin will be of interest in many different research fields due to the multisystem nature of THES and the likely common molecular pathway. Studies of thespin function will provide insight into the mechanisms of diarrhoea and other features of THES.
Supplementary Material
Acknowledgements
This work was supported by a MRC Clinical Research Fellowship to JH and NIH/NIDDK K01-080930 to NZ. We also thank Birmingham Children’s Hospital Research Foundation, Children’s Liver Fund, New Life, WellChild, British Heart Foundation (PG/06/038 and RG/09/007) and the Wellcome Trust for financial support. Many thanks for technical assistance from Dr E Meyer and Dr MA Brundler; for the clinicians who initially identified patients with THE syndrome; Dr Skidmore, Dr C Spray, Dr J Puntis, Dr J Kogelmier, Dr A Brady, Dr G de Leo, Dr M Immacolata, Dr E Goh; and to S Pasha for Genetic Counselling for the families.
Abbreviations
- AMT
advance microscopy techniques
- Apq7
Aquaporin 7
- ARC
arthrogryposis-renal dysfunction-cholestasis syndrome
- BSEP
bile salt export pump
- CDC23
cell division repeat protein 23
- CRP
collagen related peptide
- FACS
fluorescence activated cell sorting
- FITC
flourescein isothiocyanate
- GPS
Grey Platelet Syndrome
- MRP2
multidrug resistance associated protein 2
- H/K ATPase
hydrogen potassium ATPase
- NHE2
sodium hydrogen exchanger 2
- NHE3
sodium hydrogen exchanger 3
- NIS
sodium iodide symporter
- PBS-T
phosphate buffered saline-tween
- PCR
polymerase chain reaction
- PRP
platelet rich plasma
- SNP
single nucleotide polymorphism
- TEM
transmission electron microscopy
- THES
tricohepatoenteric syndrome
- TPR
tetratricopeptide repeat domains
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
No author has any financial, professional or personal conflicts of interest.
Contributor Information
Jane Louise Hartley, Department of Medical and Molecular Genetics, University of Birmingham School of Medicine, Birmingham, B15 2TT, UK, and Liver Unit, Birmingham Children’s Hospital, Steelhouse Lane, Birmingham, B4 6NH, UK.
Nicholas C. Zachos, Department of Medicine, Division of Gastroenterology and Hepatology, Hopkins Centre for Epithelial Disorders, Johns Hopkins University School of Medicine, Baltimore, MD 21205.
Ban Dawood, Centre for Cardiovascular Sciences, Institute for Biomedical Research, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT.
Mark Donowitz, Department of Medicine, Division of Gastroenterology and Hepatology, Hopkins Center for Epithelial Disorders, Johns Hopkins University School of Medicine, Baltimore, MD 21205.
Julia Forman, Institut Pasteur, Unite de Bioinformatique Strucurale, Paris 75015, France.
Rodney J Pollitt, Clinical Chemistry Department, The Children’s Hospital, Sheffield, S10 2TH, UK.
Neil V Morgan, Wellchild Paediatric Research Centre and Department of Medical and Molecular Genetics, University of Birmingham School of Medicine, Birmingham, B15 2TT UK.
Louise Tee, Department of Medical and Molecular Genetics, University of Birmingham School of Medicine, Birmingham, B15 2TT, UK.
Paul Gissen, School of clinical and experimental medicine, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK and Inherited Metabolic Diseases Unit, Birmingham Children’s Hospital, Steelhouse Lane, Birmingham, B4 6NH.
Walter H.A. Kahr, Department of Paediatrics, Division of Haematology/Oncology, Hospital for Sick Children, and University of Toronto, Program in Cell Biology, Research Institute of the Hospital for Sick Children, Toronto, ON, Canada.
A.S. Knisely, Institute of Liver Studies / Histopathology, King's College Hospital, Denmark Hill, London, SE5 9RS, UK.
Steve Watson, Centre for Cardiovascular Sciences, Institute for Biomedical Research, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT.
David Chitayat, Hospital for Sick Children, Department of Pediatrics, Division of Clinical and Metabolic Genetics University of Toronto, Toronto, Ontario, Canada and Mount Sinai Hospital, Department of Obstetrics and Gynecology, The Prenatal Diagnosis and Medical Genetic Program, University of Toronto, Toronto, Ontario, Canada.
IW Booth, The Medical School, University of Birmingham, Edgbaston, Birmingham, B15 2TT.
Sue Protheroe, Department of Gastroenterology and Nutrition, Birmingham Children’s Hospital, Steelhouse Lane, Birmingham, B4 6NH.
Stephen Murphy, Division of Reproductive and Child Health, The Medical School, University of Birmingham Birmingham B15 2TT.
Esther de Vries, Department of Paediatrics, Jeroen Bosch Hospital, PO Box 90153, 5200ME, s-Hertogenbosch, The Netherlands.
Deirdre A Kelly, Liver Unit, Birmingham Children’s Hospital, Steelhouse Lane, Birmingham, B4 6NH, UK.
Eamonn R Maher, Medical and Molecular Genetics, School of clinical and experimental medicine, University of Birmingham, Edgbaston, Birmingham, B15 2TT.
REFERENCES
- 1.Boschi-Pinto C, Velebit L, Shibuya K. Estimating child mortality due to diarrhoea in developing countries. Bull of the World Health Org. 2008;86:710–717. doi: 10.2471/BLT.07.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller T, Hess MW, Schiefermeier N, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40:1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 3.Sivagnanam M, Mueller JL, Lee H, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135:429–437. doi: 10.1053/j.gastro.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoglund P, Haila S, Socha J, et al. Mutations of the Down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat Genet. 1996;14:316–319. doi: 10.1038/ng1196-316. [DOI] [PubMed] [Google Scholar]
- 5.Heinz-Erian P, Muller T, Krabichler B, et al. Mutations in SPINT2 causes a syndromic form of congenital sodium diarrhea. Am J Hum Genet. 2009;84:188–196. doi: 10.1016/j.ajhg.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stankler L, Lloyd D, Pollitt R, Gray E, Thom H, Russell G, et al. Unexplained diarrhoea and failure to thrive in 2 siblings with unusual facies and abnormal scalp hair shafts: a new syndrome. Arch Dis Child. 1982;57:212–216. doi: 10.1136/adc.57.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girault D, Goulet O, Le Deist F et al. Intractable infant diarrhoea associated with phenotypic abnormalities and immunodeficiency. J Pediatr. 1994;125:36–42. doi: 10.1016/s0022-3476(94)70118-0. [DOI] [PubMed] [Google Scholar]
- 8.Verloes A, Lombet J, Lambert Y, et al. Tricho-hepato-enteric syndrome: further delineation of a distinct syndrome with neonatal hemochromatosis phenotype, intractable diarrhea, and hair anomalies. Am J Med Genet. 1997;68:391–395. doi: 10.1002/(sici)1096-8628(19970211)68:4<391::aid-ajmg3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.Goulet OJ, Brousse N, Canioni D, Walker-Smith JA, Schmitz J, Phillips AD. Syndrome of intractable diarrhoea with persistent villous atrophy in early childhood: a clinicopathological survey of 47 cases. J Pediatr Gastroenterol Nutr. 1998;26:151–161. doi: 10.1097/00005176-199802000-00006. [DOI] [PubMed] [Google Scholar]
- 10.de Vries E, Visser DM, van Dongen JJ, Jacobs CJ, Hoekstra JH, van Tol MJ. Oligoclonal gammopathy in phenotypic diarrhea. J Pediatr Gastroenterol Nutr. 2000;30:349–350. doi: 10.1097/00005176-200003000-00030. [DOI] [PubMed] [Google Scholar]
- 11.Dweikat I, Sultan M, Maraqa N, Hindi T, Abu-Rmeileh S, Abu-Libdeh B. Tricho-hepato-enteric syndrome: a case of hemochromatosis with intractable diarrhea, dysmorphic features, and hair abnormality. Am J Med Genet A. 2007;143:581–583. doi: 10.1002/ajmg.a.31583. [DOI] [PubMed] [Google Scholar]
- 12.Fabre A, Andre N, Breton A, Broue P, Badens C, Roquelaure B. Intractable diarrhea with 'phenotypic anomalies' and tricho-hepato-enteric syndrome: two names for the same disorder. Am J Med Genet A. 2007;143:584–588. doi: 10.1002/ajmg.a.31634. [DOI] [PubMed] [Google Scholar]
- 13.Goulet O, Vinson C, Roquelaure B, Brousse N, Bodemer C, Cezard JP. Syndromic (phenotypic) diarrhea in early infancy. Orphanet J Rare Dis. 2008;3:6. doi: 10.1186/1750-1172-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerova D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–1214. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 15.Hoogerwerf WA, Tsao SC, Devuyst O, et al. NHE2 and NHE3 are human and rabbit intestinal brush border proteins. Am J Physiol. 1996;270:29–41. doi: 10.1152/ajpgi.1996.270.1.G29. [DOI] [PubMed] [Google Scholar]
- 16.Donowitz M, Singh S, Salahuddin FF, Hogema BM, Chen Y, Gucek M, et al. Proteome of murine jejunal brush border membrane vesicles. J Proteome Res. 2007;6:4068–4079. doi: 10.1021/pr0701761. [DOI] [PubMed] [Google Scholar]
- 17.Nicola JP, Basquin C, Portulano C, Reyna-Neyra A, Paroder M, Carrasco N. The Na/I Symporter mediates active iodide uptake in the intestine. Am J Physiol Cell Physiol. 2009;296:C654–C662. doi: 10.1152/ajpcell.00509.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lo B, Li L, Gissen P, et al. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet granule biogenesis. Blood. 2005;106:4159–4166. doi: 10.1182/blood-2005-04-1356. [DOI] [PubMed] [Google Scholar]
- 19.Dawood BB, Wilde J, Watson SP. Reference curves for aggregation and ATP secretion to aid diagnosis of platelet-based bleeding disorders: effect of inhibition of ADP and thromboxane A(2) pathways. Platelets. 2007;18:329–345. doi: 10.1080/09537100601024111. [DOI] [PubMed] [Google Scholar]
- 20.Sikorski RS, Bogaski MS, Goebl M, Hieter PA. A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell. 1990;60:307–317. doi: 10.1016/0092-8674(90)90745-z. [DOI] [PubMed] [Google Scholar]
- 21.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 22.Blom E, van de Vrugt HJ, de Vries Y, de Winter JP, Arwert F, Joenje H. Multiple TPR motifs characterize the Fanconi anemia FANCG protein. DNA Repair (Amst) 2004;3:77–84. doi: 10.1016/j.dnarep.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Sohocki MM, Bowne SJ, Sullivan LS, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. 2000;24:79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bin J, Madhavan J, Ferrini W, Mok CA, Billingsley G, Heon E. BBS7 and TTC8 (BBS8) mutations play a minor role in the mutational load of Bardet-Biedl syndrome in a multiethnic population. Hum Mutat. 2009 doi: 10.1002/humu.21040. epublished. [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Roach C, Hirsh IS, et al. An unexpected extended conformation for the third TPR motif of the peroxin PEX5 from Trypanosoma brucei. J Mol Biol. 2001;307:271–282. doi: 10.1006/jmbi.2000.4465. [DOI] [PubMed] [Google Scholar]
- 26.Grizot S, Fieschi F, Dagher MC, Pebay-Peyroula E. The active N-terminal region of p67phox. Structure at 1.8 A resolution and biochemical characterizations of the A128V mutant implicated in chronic granulomatous disease. J Biol Chem. 2001;276:21627–21631. doi: 10.1074/jbc.M100893200. [DOI] [PubMed] [Google Scholar]
- 27.Senderek J, Bergmann C, Stendel C, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003;73:1106–1119. doi: 10.1086/379525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurden P, Nurden AT. Congenital disorders associated with platelet dysfunctions. Thromb Haemost. 2008;99:253–263. doi: 10.1160/TH07-09-0568. [DOI] [PubMed] [Google Scholar]
- 29.Diamandis M, Veljkovic DK, Maurer-Spurej E, Rivard GE, Hayward CP. Quebec platelet disorder: features, pathogenesis and treatment. Blood Coagul Fibrinolysis. 2008;19:109–119. doi: 10.1097/MBC.0b013e3282f41e3e. [DOI] [PubMed] [Google Scholar]
- 30.Gissen P, Johnson CA, Morgan NV, Stapelbroek JM, Forshew T, Cooper WN, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, causes arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Gen. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 31.Kiela PR, Laubitz D, Larmonier CB, et al. Changes in mucosal homeostasis predisposes NHE3 knockout mice to increased susceptibility to DSS-induced epithelial injury. Gastroenterology. 2009;137:965–975. doi: 10.1053/j.gastro.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.