

Abstract
Bcr–Abl is a chimeric oncoprotein that is strongly implicated in acute lymphoblastic (ALL) and chronic myelogenous leukemias (CML). This deregulated tyrosine kinase selectively causes hematopoietic disorders resembling human leukemias in animal models and transforms fibroblasts and hematopoietic cells in culture. Bcr–Abl also protects cells from death induced on cytokine deprivation or exposure to DNA damaging agents. In addition, the antiapoptotic function of Bcr–Abl is thought to play a necessary role in hematopoietic transformation and potentially in leukemogenesis. The transcription factor NF-κB has been identified recently as an inhibitor of apoptosis and as a potential regulator of cellular transformation. This study shows that expression of Bcr–Abl leads to activation of NF-κB-dependent transcription by causing nuclear translocation of NF-κB as well as by increasing the transactivation function of the RelA/p65 subunit of NF-κB. Importantly, this activation is dependent on the tyrosine kinase activity of Bcr–Abl and partially requires Ras. The ability of Bcr–Abl to protect cytokine-dependent 32D myeloid cells from death induced by cytokine deprivation or DNA damage does not, however, require functional NF-κB. However, using a super-repressor form of IκBα, we show that NF-κB is required for Bcr–Abl-mediated tumorigenicity in nude mice and for transformation of primary bone marrow cells. This study implicates NF-κB as an important component of Bcr–Abl signaling. NF-κB-regulated genes, therefore, likely play a role in transformation by Bcr–Abl and thus in Bcr–Abl-associated human leukemias.
Keywords: Bcr–Abl, NF-κB, IκB, tumorigenesis, apoptosis
In 1960, Nowell and Hungerford identified an abnormally small chromosome in patients with chronic myelogenous leukemia (CML) (Nowell and Hungerford 1960). This abnormal chromosome, which is known as the Philadelphia (Ph1) chromosome, was later identified as the result of a reciprocal translocation between the bcr gene on chromosome 22 and the abl gene on chromosome 9. The fusion of the bcr and abl genes produces a chimeric protein known as Bcr–Abl (Kurzrock et al. 1988; Rosenberg and Witte 1988; Ramakrishnan and Rosenberg 1989). Three different fusion sites result in the formation of different Bcr–Abl protein products named p185, p210, and p230, each differing in the amount of bcr-encoded sequences they contain (Wada et al. 1995; Melo 1996; Pane et al. 1996). p185 Bcr–Abl has been observed in 15%–25% of patients with a very aggressive, short latency leukemia known as acute lymphoblastic leukemia (ALL). The p210 form of Bcr–Abl is the causative mutation in 95% of cases of CML, a less aggressive and longer latency disease (Ramakrishnan and Rosenberg 1989). A 230-kD protein (p230) is the most recent form of Bcr–Abl discovered and is associated with a mild chronic neutrophilic leukemia (Wada et al. 1995; Pane et al. 1996).
The ability of Bcr–Abl to initiate leukemogenesis has been established through extensive studies in cell culture and animal models (Daley et al. 1990; Heisterkamp et al. 1990; Voncken et al. 1995). Bcr–Abl is a deregulated tyrosine kinase that transforms both fibroblasts and hematopoietic cells in culture and cells transformed by Bcr–Abl can form tumors in nude mice (Daley and Baltimore 1988; Pendergast et al. 1993b; Afar et al. 1994; Cortez et al. 1995). Irradiated mice transplanted with bone marrow cells expressing Bcr–Abl produce a myeloproliferative disease resembling CML (Daley et al. 1990). Additionally, animal models have been produced showing that mice expressing p185 Bcr–Abl produce a leukemic disease with a shorter onset than those expressing p210 Bcr–Abl (Kelliher et al. 1991; Voncken et al. 1995). These observations resemble the clinical differences observed in Ph1+ ALL and CML.
Bcr–Abl is an inhibitor of hematopoietic cell death normally caused by growth factor removal and DNA damaging agents in vitro (McGahon et al. 1994; Cortez et al. 1995). The inhibition of programmed cell death, or apoptosis, is likely an important component of oncogenesis because its inhibition may provide a selective growth advantage to tumors (Thompson 1995). In fact, the inhibition of apoptosis is thought to play a critical role in Bcr–Abl-mediated leukemogenesis in vivo (Clarkson and Strife 1993). Ras is a necessary component of Bcr–Abl-mediated inhibition of apoptosis in hematopoietic cells and its activation is also necessary for cellular transformation in fibroblasts (Pendergast et al. 1993b; Cortez et al. 1995; Gishizky et al. 1995; Goga et al. 1995; Cortez et al. 1996).
Although the antiapoptotic function of Bcr–Abl plays a role in its ability to transform cells, recent evidence suggests that Bcr–Abl also signals to activate mitogenic signaling pathways (Cortez et al. 1997). In CML, chronic phase cells undergo additional cycles of cell division and in ALL, Bcr–Abl-expressing cells exhibit increased cellular proliferation compared with normal cells (Ribeiro et al. 1987; Clarkson and Strife 1993). Also, Bcr–Abl-expressing hematopoietic cells are able to proliferate in a cytokine-independent manner (Carlesso et al. 1994; Laneuville et al. 1994; Cortez et al. 1995). These observations suggest that Bcr–Abl may not only function to inhibit apoptosis but may also contribute to leukemogenesis by inducing cellular proliferation. A combination of these functions may be necessary for oncogenesis (Cortez et al. 1995).
NF-κB is a transcription factor that regulates genes involved in immune and inflammatory responses, cell proliferation, and cell differentiation (Grilli et al. 1993; Baeuerle and Henkel 1994; Siebenlist et al. 1994; Baldwin 1996). Presently, there are five members of this family: p50/NF-κB1, p52/NF-κB2, RelA/p65, RelB, and c-Rel (Gilmore 1990; Kieran et al. 1990; Neri et al. 1991; Nolan et al. 1991; Ruben et al. 1991; Schmid et al. 1991; Bours et al. 1992; Ryseck et al. 1992). Each of these proteins shares homology in a amino-terminal 300 amino acid region known as the Rel homology domain, which is important for DNA binding and dimerization between family members. This homo- and heterodimerization produces a variety of transcription factors with varying affinities for different NF-κB DNA binding sites. Classic NF-κB is a heterodimer composed of a RelA (p65) and p50 subunit (Kawakami et al. 1988; Baeuerle and Baltimore 1989). The regulation of NF-κB family members is achieved through interactions with a family of inhibitory proteins known as IκB (Baeuerle and Baltimore 1988). There are multiple members of the IκB family, including IκBα and IκBβ, which bind to NF-κB and sequester it in the cytoplasm (Baldwin 1996). Stimulation of cells with inducers such as tumor necrosis factor α (TNFα) and interleukin-1 (IL-1) initiate the activation of a signal transduction cascade that culminates in the phosphorylation of IκBα by a recently identified IκB-specific kinase known as IKK/CHUK (DiDonato et al. 1997; Regnier et al. 1997). Phosphorylation of IκBα targets it for ubiquitination and subsequent degradation by the 26S proteasome (Palombella et al. 1994; Traenckner et al. 1994; Alkalay et al. 1995; Chen et al. 1995; DiDonato et al. 1995; Lin et al. 1995). Once IκBα is degraded, the nuclear localization signal (NLS) of NF-κB is revealed allowing the translocation of NF-κB from the cytoplasm into the nucleus where it regulates the transcription of target genes (Beg et al. 1992; Ganchi et al. 1992; Henkel et al. 1992; Zabel et al. 1993). Similar mechanisms are thought to occur for other members of the IκB family. In addition, recent reports have suggested that an alternative method to NF-κB activation exists. These data show that NF-κB-dependent gene expression is regulated not only by nuclear translocation but also at the level of transactivation of NF-κB in the nucleus (Yoza et al. 1996; Finco et al. 1997; Wesselborg et al. 1997).
Recently, evidence has emerged that NF-κB plays a role in apoptosis by controlling genes involved in the inhibition of cell death (Arsura et al. 1996). Work from our laboratory and others has shown that inhibition of NF-κB sensitizes cells to killing by stimuli such as TNFα and cancer therapy drugs (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996). This suggested that NF-κB may activate genes whose products function to inhibit apoptosis. Additionally, the use of antisense oligonucleotides directed against IκB to inhibit its activity leads to oncogenic transformation (Beauparlant et al. 1994), and the use of antisense oligonucleotides against NF-κB results in inhibition of tumorigenesis induced by HTLV-1 Tax (Kitajima et al. 1992). These observations and others have lead to the proposal that NF-κB may play a fundamental role in controlling oncogenesis (La Rosa et al. 1994; Lee et al. 1995; Baldwin 1996; Finco et al. 1997; Mayo et al. 1997). Together, these data suggest that NF-κB may be an important target in the signal transduction pathways utilized by the products of oncogenes.
Our data indicates that Bcr–Abl activates NF-κB-dependent gene expression in a tyrosine-kinase dependent manner. Bcr–Abl signaling leads to an increase in NF-κB-dependent gene expression by enhancing the nuclear translocation of NF-κB in 32D myeloid cells as well as by increasing the transactivation function of RelA/p65 in Baf3 pro-B and 32D cells. In addition, Ras is at least partially required for Bcr–Abl-mediated NF-κB activation. Although NF-κB has been shown to be antiapoptotic, NF-κB activity is not a necessary component of the Bcr–Abl mediated antiapoptotic pathway following growth factor deprivation or exposure to DNA damaging agents in 32D cells. These data suggest that Bcr–Abl utilizes an alternative pathway or multiple signaling pathways to protect cells from death. Importantly, tumor challenge studies in nude mice and primary bone marrow transformation assays reveal a requirement for NF-κB in tumorigenesis and transformation by Bcr–Abl. Therefore, the involvement of NF-κB in oncogenesis could have important implications for the development of therapies to treat Ph1+-leukemias and potentially other cancers.
Results
Bcr–Abl activates NF-κB-dependent gene expression
Activation of NF-κB is associated with its dissociation from IκB molecules in the cytoplasm and subsequent translocation into the nucleus (Finco and Baldwin 1995; Verma et al. 1995). Translocation of NF-κB into the nucleus results in DNA binding at NF-κB-responsive elements, an event that can be monitored through electrophoretic mobility shift assays (EMSAs). To address whether Bcr–Abl activates NF-κB, EMSAs were performed on nuclear extracts obtained from 32D myeloid cells stably expressing p185 or p210 Bcr–Abl (32D/p185 or 32D/p210). Parental 32D cells contain little NF-κB DNA-binding activity whereas 32D/p185 or 32D/p210 cells contain increased DNA-binding activity (Fig. 1A). However, Bcr–Abl does not increase NF-κB DNA-binding activity in the pro-B cell line, Baf3 (Fig. 1B). Supershift analyses were performed to identify the components of the induced complexes in 32D cells. Antibodies specific for the p50 and RelA/p65 subunits of NF-κB supershifted the DNA-binding complex activated by Bcr–Abl in 32D cells (Fig. 1A). Therefore, the presence of Bcr–Abl in 32D myeloid cells leads to an increase in authentic NF-κB (p50/p65) DNA-binding activity.
Figure 1.


Bcr–Abl expression leads to an increase in NF-κB DNA binding in 32D myeloid cells but not in Baf3 pro-B cells. (A) EMSA with nuclear extracts from 32D, 32D/p185, and 32D/p210 cells and an oligonucleotide probe containing a NF-κB binding site. Supershift analysis was performed by incubating nuclear extracts from 32D/p185 cells with antibodies specific for p50 or p65. (B) EMSA with nuclear extracts from Baf3 and Baf3/p185 cells and an oligonucleotide probe containing a NF-κB binding site. NF-κB complexes are indicated with arrows.
Translocation of NF-κB into the nucleus and subsequent binding to NF-κB-responsive elements leads to an increase in the transcription of NF-κB-regulated genes (Baldwin 1996). Luciferase reporter assays were performed to determine whether the increase in nuclear NF-κB DNA binding found in 32D cells correlates with an increase in NF-κB-dependent gene expression. These assays were performed by use of a luciferase reporter construct fused to a promoter containing three NF-κB binding sites (WT κB luc). Parental 32D cells were transiently cotransfected with WT κB luc and expression constructs for p185 or p210 Bcr–Abl. Expression of either form of Bcr–Abl activates gene expression (Fig. 2A). In addition, 32D/p185 and 32D/p210 stable cell lines also exhibit increased NF-κB-dependent gene expression relative to the parental cell controls (Fig. 2B). A reporter construct with three mutated NF-κB binding sites (mutant κB luc) is not activated by Bcr–Abl expression, indicating that activation of gene expression is NF-κB-dependent (Fig. 2B). Bcr–Abl transforming activity is dependent on the constitutively active tyrosine kinase activity of the Abl portion of the protein (Pendergast et al. 1993b; Afar et al. 1994). Importantly, expression of a mutated form of p210 (p210K-R), which lacks Bcr–Abl tyrosine kinase activity, is unable to activate NF-κB-dependent gene expression (Fig. 2A).
Figure 2.




Bcr–Abl activates NF-κB-dependent gene expression. (A) Luciferase reporter assay of 32D cells coelectroporated with a NF-κB luciferase reporter (WT κB luc) and empty expression vector or expression vectors for p185, p210, or p210K-R Bcr–Abl. (B) Luciferase reporter assay of 32D, 32D/p185, and 32D/p210 stable cell lines coelectroporated with WT κB luc reporter or a mutant NF-κB luciferase reporter (mutant κB luc) and a LacZ expression vector. Luciferase results were normalized to β-galactosidase activity. (C) Luciferase reporter assay of Baf3 cells coelectroporated with WT κB luc and empty expression vector or expression vectors for p185 or p185K-R Bcr–Abl. (D) Luciferase reporter assay of Baf3 and Baf3/p185 stable cell lines coelectroporated with WT κB luc or mutant κB luc and a LacZ expression vector. Luciferase results were normalized to β-galactosidase activity. Data shown are from representative experiments where electroporations were performed in triplicate. Fold activation is indicated ±s.d..
In addition, we tested whether Bcr–Abl could activate NF-κB-dependent gene expression in Baf3 cells. Transient or stable expression of Bcr–Abl in Baf3 cells results in a significant increase in NF-κB-dependent gene expression by use of the luciferase reporter assay (Fig. 2C,D). This result is intriguing because stable expression of Bcr–Abl in Baf3 cells does not lead to an increase in nuclear NF-κB (Fig. 1B). In addition, a kinase inactive form of p185 (p185K-R) is unable to activate NF-κB-dependent gene expression (Fig. 2C). This data suggests that Bcr–Abl may activate NF-κB in Baf3 cells by a mechanism other than nuclear translocation.
v-Abl is the viral transforming counterpart of the cellular gene, c-Abl, and unlike c-Abl it exhibits constitutive tyrosine kinase activity (Rosenberg and Witte 1988). Because Bcr–Abl and v-Abl both exhibit deregulated tyrosine kinase activity and this activity is necessary for NF-κB activation by Bcr–Abl, we were interested in determining whether v-Abl could activate NF-κB activity in 32D myeloid and Baf3 pro-B cells. Transient coexpression of v-Abl and WT κB luc in 32D cells results in an increase in NF-κB-dependent gene expression in both cells types whereas overexpression of c-Abl has no effect on gene expression (data not shown). In addition, coexpression of v-Abl with the mutant κB luc reporter reveals that activation is NF-κB-dependent (data not shown). These experiments indicate that constitutive Abl tyrosine kinase activity results in activation of NF-κB.
Bcr–Abl activates the transactivation domain of RelA/p65 in a Ras-dependent manner
Despite an apparent lack of NF-κB nuclear accumulation in Baf3 cells expressing Bcr–Abl, a significant increase in NF-κB-dependent gene expression is observed. Luciferase reporter assays were performed to determine whether Bcr–Abl can stimulate NF-κB-dependent gene expression by activating the transactivation domain of pre-existing nuclear RelA/p65. Parental Baf3 cells were transiently cotransfected with a luciferase reporter construct fused to a promoter containing five Gal4 DNA-binding sites (Gal–luc), along with expression constructs encoding the Gal4 DNA-binding domain that lacks a transactivation domain (Gal4–DB) or the Gal4 DNA-binding domain fused to transactivation domain 1 (TA1 of RelA/p65 (Gal4–p65) (Schmitz et al. 1995). These transfections were performed in the presence or absence of expression constructs encoding p185 Bcr–Abl. Transient expression of p185 Bcr–Abl activates the transcription function of Gal4–p65 but has no effect on gene expression in the absence of a transactivation domain (Gal4–DB) (Fig. 3A). In addition, p185K-R is unable to activate Gal4–p65 (Fig. 3A). Also, transient expression of p185 Bcr–Abl does not significantly activate (1.3-fold) Gal4 fused to the transactivation domain of SP1 (Gal4–SP1-B) (data not shown). Whereas 32D cells show an increase in NF-κB nuclear translocation in the presence of Bcr–Abl (Fig. 1A), luciferase reporter assays were performed to determine whether activation of RelA/p65 also plays a role in NF-κB-dependent gene expression by Bcr–Abl in these cells. Expression of p185 Bcr–Abl is able to activate Gal4–p65 in 32D cells without affecting transcription of Gal4–DB (data not shown).
Figure 3.


Bcr–Abl partially signals through Ras to activate the transactivation domain of RelA/p65. (A) Luciferase reporter assay of Baf3 cells coelectroporated with a Gal4 luciferase reporter (Gal–luc) and empty expression vector (open bar) or expression vectors for p185 (solid bar) or p185K-R (hatched bar) Bcr–Abl along with expression vectors for the Gal4 DNA-binding domain (Gal4) or the Gal4 DNA-binding domain fused to transactivation domain 1 of p65 (Gal4–p65). (B) Luciferase reporter assay of Baf3 cells coelectroporated with Gal–luc, Gal4–p65, and an empty expression vector (open bar) or expression vector for p185 (solid bar) Bcr–Abl in the presence or absence of 10 or 20 ng of Ras A17.
Because Ras activation is a necessary component of Bcr–Abl signaling (Pendergast et al. 1993b; Cortez et al. 1995; Gishizky et al. 1995; Goga et al. 1995; Cortez et al. 1996), we were interested in determining whether the activation of NF-κB by Bcr–Abl also relies on Ras. Parental Baf3 cells were transiently cotransfected with Gal–luc, Gal4–p65, and an expression construct for p185 Bcr–Abl. This experiment was performed in the presence or absence of a dominant negative form of Ras that contains an alanine mutation at amino acid 17 (RasA17). Expression of RasA17 blocked the ability of Bcr–Abl to fully activate NF-κB (Fig. 3C). In addition, expression of RasA17 has no effect on NF-κB-dependent gene expression in response to TNFα (data not shown), indicating that RasA17 does not decrease gene expression nonspecifically or inhibit NF-κB in Baf3 cells. Also, β-galactosidase staining revealed that no significant cell death was observed in the presence of RasA17 in Baf3 cells (data not shown). These results support a model in which Bcr–Abl leads to the activation of RelA/p65 by requiring a signaling pathway that, at least in part, utilizes a Ras or Ras-like molecule.
Bcr–Abl does not require NF-κB to protect cells from death following IL-3 withdrawal
To address the role of NF-κB activation in Bcr–Abl signaling, it was necessary to inhibit NF-κB activity in Bcr–Abl expressing cells. To block NF-κB activation, we utilized a super-repressor form of IκBα. This IκBα contains serine to alanine mutations at the sites of inducible phosphorylation (serines 32 and 36). Phosphorylation of these serines is required for IκBα degradation and subsequent NF-κB activation (Brockman et al. 1995; Brown et al. 1995; Traenckner et al. 1995; Whiteside et al. 1995; DiDonato et al. 1996). Therefore, this mutated form of IκBα (IκBα-super-repressor; IκBα-SR) is unable to be inducibly phosphorylated and subsequently degraded and, therefore, continuously sequesters NF-κB in the cytoplasm. Like IκBα, the IκBα-SR not only inhibits nuclear translocation of NF-κB but also enters the nucleus and removes NF-κB bound to DNA (Zabel and Baeuerle 1990).
To test the role that NF-κB plays in Bcr–Abl signaling we introduced the IκBα-SR into Bcr–Abl-expressing 32D cells. 32D/p185 or 32D/p210 cells were infected in the presence of IL-3 with a retrovirus expressing the IκBα-SR. Mass populations and clonal cell lines were obtained by G418 selection that express both Bcr–Abl and the IκBα-SR (32D/p185/SR; 32D/p210/SR) (Fig. 4A). Western blot analysis reveals that these cells show a dramatic loss of endogenous IκBα (Fig. 4A). Loss of endogenous IκBα in cells expressing the highly stable IκBα-SR most likely reflects a decrease in free IκBα (caused by the short half-life of uncomplexed IκBα) and a decrease in IκBα gene expression, which is regulated by NF-κB (Brown et al. 1993; Rice and Ernst 1993; Scott et al. 1993; Sun et al. 1993; Chiao et al. 1994). Therefore, the loss of endogenous IκBα is a good indication of the presence of a functional IκBα-SR. EMSAs were performed to determine whether the IκBα-SR is, in fact, functional in inhibiting NF-κB activity in Bcr–Abl-expressing cells. These analyses revealed that the increased NF-κB DNA binding observed in 32D/p185 and 32D/p210 cells is eliminated in 32D/p185/SR and 32D/p210/SR cells (Fig. 4B).
Figure 4.




32D/Bcr–Abl cells do not require NF-κB for survival in the absence of IL-3. (A) Production of 32D/Bcr–Abl/IκBα-SR cells. 32D/p185 and 32D/p210 cells were infected with a retrovirus expressing IκBα-SR. Mass population and clonal stable cell lines were obtained by G418 selection. Cell extracts were prepared from 32D, 32D/p185, 32D/p210, 32D/p185/IκBα-SR, and 32D/p210/IκBα-SR cells by lysing equal cell numbers in protein SDS sample buffer. Extracts were analyzed by Western blot analysis with an Abl monoclonal antibody and a carboxy-terminal IκBα polyclonal antibody. Mobility of p210, p185, endogenous c-Abl, IκBα-SR, and endogenous IκBα are indicated with arrows; (n.s.) indicates a nonspecific band. (B) 32D/Bcr–Abl cells expressing the IκBα-SR no longer contain NF-κB DNA binding. EMSAs were performed with nuclear extracts from 32D, 32D/p185, 32D/p210, 32D/p185/IκBα-SR, and 32D/p210/IκBα-SR stable cell lines and an oligonucleotide probe containing an NF–κB binding site. NF-κB complexes are indicated with arrows. (C) 32D/Bcr–Abl/IκBα-SR cells undergo apoptosis in response to TNFα. 32D (open bars), 32D/p210 (hatched bars), 32D/p210/IκBα–SR–mp (vertical bars), and 32D/p210/IκBα-SR clonal cell line H3 (solid bars) were treated with TNFα for 24 hr. Cells were stained with propidium iodide at 0 hr and 24 hr post-TNFα treatment and DNA content was analyzed by flow cytometry. Cells within the sub-G1 peak were scored as apoptotic. (D) 32D/p185/IκBα-SR cells survive in the absence of IL-3. 32D and 32D/p185/IκBα-SR cells were incubated in the presence or absence of IL-3 and cell viability was monitored by trypan blue exclusion 24 hr and 48 hr post-IL-3 withdrawal.
It has been shown that TNFα treatment of cells expressing the IκBα-SR or of cells that lack expression of RelA/p65 (p65−/−) results in apoptosis (Beg and Baltimore 1996; Van Antwerp et al. 1996; Wang et al. 1996). Therefore, as an additional test to determine whether the IκBα-SR is functional in 32D/Bcr–Abl cells, 32D, 32D/p210, a 32D/p210/SR mass population (mp), and a 32D/p210/SR clonal cell line (H3) were treated with TNFα. Percent viability and apoptosis were monitored at various times following TNFα treatment. 32D and 32D/p210 cells remained viable and showed little apoptosis whereas the 32D/p210/SR-mp and the 32D/p210/SR clonal cell line (H3) significantly lost cell viability and readily underwent apoptosis following 24 hr TNFα treatment (Fig. 4C). Similar results were obtained in 32D/p185/SR-mp cells and in all clones analyzed (data not shown). Interestingly, the 32D/p210/SR-mp cells did not undergo as extensive apoptosis as the clonal cell lines did. We found, through Western blot analysis, that cells exist in the mass population that express very little or no IκBα-SR (data not shown). These cells would not be expected to die in response to TNFα and the population, as a whole, would exhibit decreased apoptosis as observed in the data obtained above. The difference between the IκBα-SR mass populations and the clonal cell lines will become an important point in a later section (see below). Therefore, the IκBα-SR is functional because it inhibits NF-κB activation by Bcr–Abl and inhibits NF-κB-dependent gene expression that is required to inhibit apoptosis in response to TNFα treatment (Beg and Baltimore 1996; Liu et al. 1996; Van Antwerp et al. 1996; Wang et al. 1996).
32D cells rapidly undergo apoptosis when grown in the absence of IL-3 (Valtieri et al. 1987). However, the requirement for IL-3 is abolished when the cells express Bcr–Abl because apoptosis is blocked by Bcr–Abl in these cells (Daley and Baltimore 1988; Cortez et al. 1995). Coexpression of the IκBα-SR with Bcr–Abl in 32D cells blocked Bcr–Abl-mediated activation of NF-κB (Fig. 4A–C). To determine the effect of NF-κB inhibition on the ability of Bcr–Abl to inhibit apoptosis, we monitored 32D/p185/SR cell viability following IL-3 withdrawal. Cell death was not observed upon IL-3 withdrawal of 32D/p185/SR cells because Bcr–Abl continued to render the cells IL-3-independent even in the presence of the IκBα-SR (Fig. 4D). Similar results were obtained with 32D/p210/SR cells (data not shown). Significant cell death was observed in 32D parental control cells following IL-3 withdrawal (Fig. 4D). It is important to note that 32D/Bcr–Abl/SR cells can survive in the absence of IL-3 whether the cell lines are obtained by infecting 32D/Bcr–Abl cells with the IκBα-SR or by introducing Bcr–Abl into 32D/SR cells (data not shown). Identical results were also obtained in DAGM/p210/SR myeloid cells (data not shown). In addition, 32D/Bcr–Abl cells expressing the IκBα-SR do not exhibit measurable growth defects compared with controls as shown through growth curve analysis and flow cytometric profiles (data not shown). Bcr–Abl also inhibits apoptosis in response to exposure to DNA damaging agents (Laneuville et al. 1994; McGahon et al. 1994; Cortez et al. 1995). However, 32D/Bcr–Abl and 32D/Bcr–Abl/SR cells respond identically to DNA damage induced by etoposide treatment or exposure to ionizing radiation (data not shown). Therefore, NF-κB activation is not required for Bcr–Abl to inhibit apoptosis of 32D cells following IL-3 withdrawal or in response to various DNA damaging agents (see also Discussion).
32D/Bcr–Abl cells expressing the IκBα-SR are deficient in their ability to cause tumor formation in nude mice
Whereas parental 32D cells are nontumorigenic, expression of Bcr–Abl in these cells generates a cell line capable of forming tumors in nude mice (Daley and Baltimore 1988; Pendergast et al. 1993b; Afar et al. 1994; Cortez et al. 1995). Recent evidence indicates that NF-κB may be involved in oncogenic transformation (Kitajima et al. 1992; Beauparlant et al. 1994; La Rosa et al. 1994; Lee et al. 1995; Baldwin 1996; Finco et al. 1997; Mayo et al. 1997). Therefore, our observations that Bcr–Abl activates NF-κB functional activity led us to examine whether NF-κB is involved in the tumorigenic potential of Bcr–Abl. 32D/Bcr–Abl cells and 32D/Bcr–Abl/SR-mp cells expressing the IκBα-SR were injected subcutaneously into 5- to 6-week-old athymic nude mice and tumor formation was monitored. 32D/p185 and 32D/p210 cells formed tumors which were first observed ∼9 days postinjection. The appearance of 32D/Bcr–Abl/SR–mp tumors occurred ∼12 days postinjection. Interestingly, at 18 days postinjection, 32D/p185/SR–mp or 32D/p210/SR–mp cells had formed tumors of greatly reduced size as compared with wild-type Bcr–Abl tumors (Table 1, Fig. 5A). Western blot analysis of tumors from 32D/p185/SR–mp cell injections indicate that the cells in these tumors no longer express the IκBα-SR and endogenous IκBα levels are equivalent to those in tumors formed by 32D/p185 (Fig. 5B). Bcr–Abl is still expressed in these tumors (data not shown). Similar results were obtained with 32D/p210/SR–mp cells (data not shown). In fact, no tumor examined from mass population 32D/Bcr–Abl/SR cells retained the IκBα-SR and all showed expression of endogenous IκBα. This result suggested that the mass populations used for injections were likely populations representing cells that express the IκBα-SR and a smaller population that does not (see above). On injection of the mass populations, the cells that contain the IκBα-SR may be deficient in tumor formation, but the remaining cells that lack IκBα-SR expression grow to form tumors. This would explain the tumorigenesis data observed with the 32D/Bcr–Abl/IκBα-SR–mp cell lines and would explain the delayed appearance of tumors by these cells. In order to address this point, clonal cell lines were injected that coexpressed Bcr–Abl and the IκBα-SR. Two 32D/p185/SR and two 32D/p210/SR clonal cell lines were injected into nude mice and monitored for tumor appearance. Although 32D/Bcr–Abl cells and the mass population 32D/Bcr–Abl/SR cells had formed sizeable tumors by 18 days postinjection, the 32D/Bcr–Abl/SR clones showed no evidence of tumor formation at this time (Table 1). Continued observation of the mice injected with 32D/Bcr–Abl/SR clones revealed that 4 of 12 injections eventually formed tumors ∼32 days postinjection, which is ∼21–23 days slower than the appearance of wild-type tumors. These clonal cell line-derived tumors were removed 41 days postinjection and cell extracts were prepared for Western analysis. The cells from these tumors express the IκBα-SR, but more importantly, they contain elevated levels of endogenous IκBα (data not shown). Therefore, these tumors may have formed from a population of cells that was able to partially overcome NF-κB inhibition by the IκBα-SR. Taken together, these data strongly indicate that the activation of NF-κB by Bcr–Abl is required for tumorigenesis of Bcr–Abl-expressing cells.
Table 1.
Tumorigenicity of 32D cells expressing Bcr–Abl or Bcr–Abl/IκBα–SR
| 32D cell line
|
Observation of tumors formed 18 days postinjection
|
|
|---|---|---|
| tumors/injection
|
avg. massa
|
|
| p210 | 5/5 | 510 ± 103 |
| p210/SR | 12/13 | 118 ± 26 |
| p210/SR clonesb | 0/8 | — |
| p185 | 5/5 | 1073 ± 323 |
| p185/SR | 13/13 | 209 ± 47 |
| p185/SR clonesb | 0/4 | — |
Animals with tumors were sacrificed 18 days postinjection, the tumors were removed and weighed; mass of tumors is represented in mg ± s.e.m.
Bcr–Abl/SR clonal injections represent four clonal isolates: two p210/SR clones and two p185/SR clones. Tumors eventually appeared in 4 of 12 clonal injections (see text).
Figure 5.
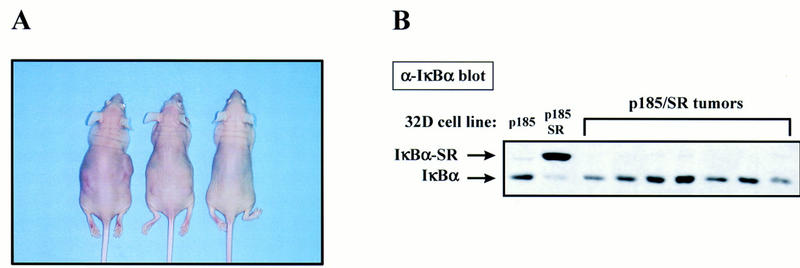
NF-κB is required for tumor formation by Bcr–Abl. (A) Cells (1 × 106) were injected subcutaneously into each flank of 5- to 6-week-old athymic nu/nu mice. Pictured left to right are mice injected with 32D/p210, a 32D/p210/IκBα-SR mass population, and a 32D/p210/IκBα-SR clonal cell line (E2). These mice were sacrificed 18 days postinjection and photographed. (B) Tumors formed from 32D/p185/IκBα-SR–mp cells do not express the IκBα-SR.Tumors formed from 32D/p185 and 32D/p185/IκBα-SR–mp cell injections were removed from the animals and equal protein from whole cell extracts were analyzed by Western blotting with a carboxy-terminal IκBα polyclonal antibody. Equal protein from cell extracts prepared from 32D/p185 and 32D/p185/IκBα-SR cell lines were analyzed as above. Mobility of endogenous IκBα and the IκBα-SR is indicated with arrows.
Expression of the IκBα-SR inhibits transformation of primary bone marrow cells by Bcr–Abl
Although NF-κB is required for Bcr–Abl-mediated tumorigenicity in an established 32D cell line (Table 1), we were interested in determining whether NF-κB is also required for Bcr–Abl-mediated transformation of primary pre-B lymphocytes. Expression of Bcr–Abl from a retroviral vector causes transformation of primary mouse bone marrow cells resulting in outgrowth of pre-B lymphocytes in vitro (Rosenberg and Baltimore 1978; Whitlock and Witte 1982; McLaughlin et al. 1987). To determine whether NF-κB is necessary for Bcr–Abl-mediated transformation of hematopoietic cells, primary bone marrow transformation assays (McLaughlin et al. 1987, 1989) were performed to determine whether the IκBα-SR is capable of suppressing transformation by Bcr–Abl. Primary bone marrow transformation assays have been used successfully in previous studies investigating downstream requirements of Bcr–Abl-mediated transformation (Sawyers et al. 1992; Dickens et al. 1997; Skorski et al. 1997). To ensure that both p185 Bcr–Abl and the IκBα-SR were simultaneously expressed in each infected cell, we constructed bicistronic retroviral vectors that allow the simultaneous expression of two genes from a single retrovirus. Infection of primary bone marrow cells with control bicistronic retrovirus (empty–empty) does not lead to transformation of the cultures (Table 2). Infection of primary bone marrow cells with a bicistronic retrovirus that contains the gene to express only p185 Bcr–Abl (p185–empty) or a retrovirus that encodes for both p185 Bcr–Abl and the antisense sequence to the IκBα-SR (p185–AS–IκBα-SR) leads to the growth of transformed bone marrow cell cultures (Table 2). However, infection of bone marrow cells with a retrovirus that encodes both p185 Bcr–Abl and the IκBα-SR (p185–IκBα-SR) results in transformation of only three of nine bone marrow cell culture samples (Table 2). Importantly, Western blot analysis of these three transformed samples reveals that the IκBα-SR is not expressed in these cells (data not shown). These data resemble results obtained in the tumor formation assays (Table 1), in which successful tumor growth caused by Bcr–Abl/IκBα-SR cells was associated with loss of expression of the IκBα-SR (Fig. 5B). Similar results to those generated with the Bcr–Abl/IκBα-SR bicistronic virus were obtained when primary bone marrow cells were infected with two retroviruses separately encoding p185 Bcr–Abl and the IκBα-SR (data not shown). It should be noted that we were able to sustain growth of bone marrow cultures that express the IκBα-SR under the condition of G418 selection and in the presence of exogenously added growth factors that allow growth of primary bone marrow cells without the requirement for transformation (Skorski et al. 1996) (data not shown). This indicates that the IκBα-SR is not toxic to the growth of primary bone marrow cells. Together, this data strongly indicates a positive role for NF-κB in leukemic transformation by Bcr–Abl.
Table 2.
Growth of primary bone marrow cell cultures following infection with Bcr–Abl or Bcr–Abl/IκBα–SR retroviruses
| Virus
|
Number of infected cell cultures that have reached a density >1 × 106 cells/mla
|
||
|---|---|---|---|
| day 15
|
day 20
|
day 26
|
|
| empty–empty | 0/9 | 0/9 | 0/9 |
| p185–empty | 4/9 | 8/9 | 8/9 |
| p185–Sense–IκBα–SR | 1/9 | 3/9 | 3/9b |
| p185–Antisense–IκBα–SR | 0/3 | 2/3 | 3/3 |
Cell counts were monitored on a daily basis; cultures reaching >1 × 106 cells/ml are considered transformed.
Western analysis reveals that these three transformed cultures do not express the IκBα–SR.
Discussion
In this study we identify NF-κB as a downstream component of a Bcr–Abl-initiated signaling pathway. Utilizing electrophoretic mobility shift analyses and transcriptional luciferase reporter assays we show that Bcr–Abl activates NF-κB functional activity in 32D myeloid and Baf3 pro-B cells. NF-κB activity is not necessary for normal IL-3 signaling in 32D cells or in DAGM myeloid cells (data not shown) and is not necessary for the ability of Bcr–Abl to inhibit apoptosis in response to IL-3 withdrawal. However, NF-κB activity is required for tumorigenesis and transformation initiated by Bcr–Abl.
Whereas the role for NF-κB in tumorigenesis has yet to be firmly established, recent work has suggested that this transcription factor may play a role in this process. The inhibition of NF-κB activity through the use of antisense oligonucleotides to p65 was shown to inhibit HTLV-1 Tax-induced tumorigenesis (Kitajima et al. 1992). More recently, NF-κB activation has been shown to be necessary for tumor formation by Hodgkin’s lymphoma cells in mice (Bargou et al. 1997). Also, an increase in NF-κB levels were identified in breast cancer cell lines, primary human breast cancer tumors and in primary rat mammary tumors when compared with nontransformed controls (Sovak et al. 1997). In addition, NF-κB transcriptional activity is required for oncogenic Ras-induced cellular transformation (Finco et al. 1997), which likely occurs through the inhibition of transformation-associated apoptosis (Mayo et al. 1997). The role of NF-κB in Bcr–Abl-mediated transformation is unknown and is currently under investigation.
The signaling pathway(s) employed by Bcr–Abl to activate NF-κB remains to be elucidated. Classically, NF-κB is activated by extracellular stimulation that utilizes a signaling pathway that leads to IκBα phosphorylation, ubiquitination, and degradation by the 26S proteasome (Finco et al. 1995; Verma et al. 1995). Free NF-κB is then capable of entering the nucleus to increase the transcription of target genes. 32D cells expressing Bcr–Abl show an increase in nuclear NF-κB (see Fig. 1). Therefore, Bcr–Abl may utilize a similar signaling pathway in 32D cells to target IκBα degradation and subsequent NF-κB nuclear translocation. A hallmark event of this pathway is IκBα phosphorylation, however, we have not determined whether IκBα phosphorylation plays a role in Bcr–Abl-mediated activation of NF-κB in these cells. Also, Bcr–Abl activates NF-κB-dependent gene expression in Baf3 cells without causing nuclear translocation of NF-κB (see Figs. 1 and 2), implicating alternative pathways to NF-κB activation by Bcr–Abl. Recent data from our laboratory and others have suggested that the activation of NF-κB-dependent gene expression is not entirely regulated by the nuclear translocation of NF-κB (Yoza et al. 1996; Finco et al. 1997; Wesselborg et al. 1997). Expression of oncogenic Ras in NIH-3T3 cells results in the activation of NF-κB-dependent gene expression in the absence of nuclear translocation. Further investigation revealed that NF-κB-dependent gene expression by Ras is induced by activating the transactivation function of p65 (Finco et al. 1997). Similarly, Bcr–Abl activates RelA/p65 in a Ras-dependent manner. Bcr–Abl activation of NF-κB in 32D cells may be, in part, caused by the modest increase in nuclear translocation as well as an increase in transactivation potential. Therefore, Bcr–Abl may signal via multiple pathways to activate NF-κB.
Bcr–Abl signals to NF-κB activation through a Ras-dependent pathway. Ras has been shown to play a critical role in the antiapoptotic and transforming signal elicited by Bcr–Abl (Pendergast et al. 1993b; Cortez et al. 1995; Gishizky et al. 1995; Goga et al. 1995). Whereas the mechanism of Ras-mediated activation of NF-κB is not known, Bcr–Abl activates PI3K and JNK (Raitano et al. 1995; Cortez et al. 1997; Skorski et al. 1997), both of which can function downstream of Ras and may be capable of stimulating NF-κB transcriptional activity independent of inducing nuclear translocation. Inhibition of Ras function blocks NF-κB activation and also inhibits Bcr–Abl-mediated transformation.
Although our data clearly show that Bcr–Abl activates NF-κB, additional data show that Bcr–Abl is able to delay and reduce NF-κB activation that occurs in response to TNFα stimulation by negatively regulating the degradation of IκBα (J.Y. Reuther and A.S. Baldwin, unpubl.). It is possible that Bcr–Abl is simply affecting signaling downstream of the tumor necrosis factor receptor (TNFR). However, this data may reveal a dual function for Bcr–Abl that also appears to occur with v-Abl. v-Abl activates NF-κB (data not shown) but has also been shown to block immunoglobulin light-chain gene rearrangement and the constitutive NF-κB binding activity present in IL-7 expanded pre-B cells by negatively regulating IκBα degradation (Chen et al. 1994; Klug et al. 1994). It is possible that v-Abl activates the transactivation function of RelA/p65 as does Bcr–Abl and that the inhibition of NF-κB DNA binding by these activated tyrosine kinases is a distinct and separate pathway from that which leads to an increase in the transactivation function of RelA/p65. These different methods of regulating NF-κB may reflect a need to tightly control the expression of genes regulated by NF-κB and may indicate distinct functions for increased nuclear accumulation of NF-κB versus the activation of the transactivation function of RelA/p65 by Bcr–Abl.
Because our laboratory and others have shown a role for NF-κB in the inhibition of apoptosis (Arsura et al. 1996; Beg and Baltimore 1996; Van Antwerp et al. 1996; Liu et al. 1996; Wang et al. 1996) we were surprised to find that NF-κB was not required by Bcr–Abl to inhibit apoptosis in response to IL-3 withdrawal or upon exposure to etoposide and ionizing radiation (data not shown). However, it is possible that Bcr–Abl elicits multiple pathways to inhibit apoptosis (including NF-κB), and the inhibition of any one signal may not induce apoptotic conditions in culture. Bcr–Abl activates phosphatidylinositol-3 kinase (PI 3-kinase) and Akt, which have been shown to mediate the inhibition of apoptosis in hematopoietic cells in response to IL-3 (Skorski et al. 1995, 1997; Kennedy et al. 1997; Songyang et al. 1997). In addition, the antiapoptotic function of Bcr–Abl has been linked to its ability to activate Ras and in some cells to increase Bcl-2 expression (Sanchez-Garcia and Grutz 1995; Cortez et al. 1996). Bcr–Abl may use multiple pathways to block apoptosis and cause deregulated proliferation. Therefore, the inhibition of NF-κB by the IκBα-SR may not be sufficient to demonstrate a potential role for NF-κB as an antiapoptotic factor, at least in 32D cells. In addition, although the inhibition of NF-κB does not lead to apoptosis of Bcr–Abl-expressing cells upon IL-3 withdrawal in vitro, it is possible that the antiapoptotic function of NF-κB may be critical in primary bone marrow cell transformation or during the development of tumors in mice.
Because Bcr–Abl activates NF-κB and because NF-κB is required for Bcr–Abl-mediated tumorigenesis and transformation, it is likely that the products of NF-κB responsive genes play a role in Bcr–Abl mediated leukemogenesis. In hematopoietic cells, growth factor deprivation results in a decrease in c-myc mRNA and protein. However, cells expressing Bcr–Abl maintain elevated levels of c-myc mRNA and protein following growth factor deprivation. In addition, c-myc is required for transformation by Bcr–Abl (Sawyers et al. 1992). These results may reflect the ability of Bcr–Abl to activate transcription factors that positively regulate the c-myc promoter. The identification of two NF-κB sites in the c-myc promoter/enhancer and the realization that activation of c-myc by IL-1 requires functional NF-κB binding sites led to the discovery that NF-κB regulates c-myc transcription (La Rosa et al. 1994). The regulation of c-myc by NF-κB provides evidence of a role for NF-κB in growth control, and, therefore, Bcr–Abl may, in part, use NF-κB to maintain c-myc expression. However, it has been shown recently that v-Abl can activate the c-myc promoter through E2F in a Ras-dependent manner (Wong et al. 1995; Zou et al. 1997). It is possible that E2F as well as NF-κB play a role in the regulation of the c-myc promoter downstream of activated Abelson tyrosine kinases. The loss of tumorigenesis may also result from a loss in expression of cell surface proteins involved in cell adhesion that are regulated by NF-κB (Baldwin 1996). These cell surface proteins may be required to provide proper cellular interactions for tumor formation. In addition, it still remains that NF-κB may play a role in the inhibition of apoptosis and that these effects can only be realized in tumor formation in nude mice or in transformation of primary bone marrow cells . Therefore, Bcr–Abl/SR cells may be deficient in tumorigenesis or transformation because of an increase in apoptosis.
Future studies will be directed toward addressing the role NF-κB plays in Bcr–Abl-mediated tumorigenesis and transformation. Although the precise mechanism(s) by which inhibition of NF-κB impairs Bcr–Abl-mediated transformation is unclear, the finding that NF-κB is activated by Bcr–Abl and is required for oncogenesis provides new insights into Bcr–Abl signaling. It will be important to conduct further experiments to determine whether NF-κB inhibitors will have therapeutic potential for Ph1+ ALL and CML.
Materials and methods
Cell culture
Cells stably expressing Bcr–Abl (p185tkneo or p210tkneo) were obtained by retroviral infection of 32D and DAGM cells or by electroporation of Bcr–Abl (p185pSRα or p210pSRα) followed by IL-3 deprivation. Cells expressing Flag–IκBαS32/36A (IκBα-SR) were obtained by retroviral infection of 32D, 32D/p185pSRα, and 32D/p210pSRα cells growing in the presence of WEHI-conditioned medium (WEHI-cm) as a source of IL-3 (32D/SR, 32D/p185, or p210/SR). Helper-free retroviral stocks were produced with the pSRαMSVTkneo vector as described previously (Muller et al. 1991). Retroviruses were produced in Bosc-23 cells or in 293T cells (Pear et al. 1993). Retroviral infections were performed as described previously (Pendergast et al. 1993a). G418 (0.5 mg/ml, GIBCO-BRL, Gaithersburg, MD) was added 48 hr postinfection to select for populations expressing the genes. Clonal cell lines were obtained by limiting dilution. Expression of Bcr–Abl and the IκBα-SR was confirmed by Western blotting.
32D myeloid, DAGM myeloid, and Baf3 pro-B cells were maintained in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS, GIBCO-BRL), 10% WEHI-cm, and 100 μg/ml penicillin and 100 μg/ml streptomycin (P/S, Sigma Chemical Co.). 32D/p185tkneo, 32D/p210tkneo, 32D/p185/SR, and 32D/p210/SR cells were maintained in RPMI-1640 medium supplemented with 10% FCS, P/S, and G418. 32D/SR cells were maintained in RPMI-1640 supplemented with 10% FCS, 10% WEHI-cm, P/S, and G418. 32D/p185pSRα, 32D/p210pSRα, and Baf3/p185pSRα cells were maintained in RPMI-1640, 10% FCS, and P/S unless otherwise indicated.
Plasmids
The Flag–IκBα-SR expression vector is a gift from D. Ballard (Vanderbilt University, Nashville, TN) and has been described previously (Brockman et al. 1995). 3XMHC WTluc (WT κB luc) and 3XMHC mutantluc (mutant κB luc) are gifts from B. Sugden (University of Wisconsin, Madison) (Mitchell and Sugden 1995). Gal4–SP1-B is a gift from X. Wang (Duke University Medical Center, Durham, NC). RasA17 is a gift from C. Der (University of North Carolina, Chapel Hill). To construct the retroviral pSRαMSVTkneo–Flag–IκBα-SR vector, HindIII linkers were introduced at the Not1 site of Flag–IκBα-SR and the cDNA was then cloned as a HindIII fragment into pSRαMSVTkneo. The bicistronic retroviral vector (pSRαMSVCMV) is a modified version of the one described previously in Sawyers et al. (1992). p185-empty was constructed by subcloning p185 as an EcoRI fragment into the EcoRI site 3′ of the MSV-LTR. XbaI sites were introduced into Flag–IκBα-SR by PCR and this product was subcloned into the XbaI site 3′ of the CMV promoter. Confirmation of the retroviral constructs was confirmed by Western blotting following transfection of 293T cells.
Transient transfection reporter assays
For NF-κB luciferase reporter assays, 9.9 × 106 cells/0.3 ml were mixed with 5 μg of luciferase reporter (WT κB luc or mut κB luc) and 2 μg of cmv–LacZ expression vector for assays performed with stable cell lines or with 5 μg of luciferase reporter and 5 μg of Bcr–Abl or v-Abl expression vectors for assays performed in parental 32D and Baf3 cells. For Gal4 luciferase assays, cells were mixed with 1 μg of Gal–luc, 100 ng–5 μg Bcr–Abl expression vector, and 100 ng–200 ng Gal4 or Gal4 fusion expression vector. Samples were electroporated at 260 V, 960 μFD, and incubated at 37°C in 10 ml of appropriate growth medium. Cells were collected 48 hr post-transfection and washed in phosphate-buffered saline, cells were pelleted and resuspended in 0.25 m Tris at pH 7.6. Cells were lysed by three freeze/thaw cycles and lysate was clarified by centrifugation. Supernatant was collected and protein concentration determined with Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA). Luciferase assays were performed by mixing 50–200 μg of lysate with 0.25 m Tris at pH 7.6 to a total volume of 100 μl. Assay reagents were ATP reagent (1 mm DTT, 2 mm ATP, 25 mm glycylglycine, 4 mm EGTA, 15 mm MgSO4, 15 mm K2HPO4) and Luciferin reagent [200 μm luciferin (Sigma Chemical Co.), 25 mm glycylglycine]. Luciferase assays were performed in a AutoLumat LB953 (E G & G Berthold, Aliquippa, PA) as per manufacturer’s instructions. For normalization of luciferase data, 5–10 μl of lysate was incubatedwith 100 μl of Z buffer at pH 7.0 (60 mm Na2HPO4 · 7H2O, 40 mm NaH2PO4 · H2O, 10 mm KCl, 1 mm MgSO4 · 7H2O, 39 mm β-mercaptoethanol) containing chlorophenolred β-d-galactopyranoside (CPRG, Boehringer Mannheim Corp., Indianapolis, IN) at 37°C. On color change, assays were read at 595 nm.
Cell extracts and Western blotting
Nuclear (NE) and cytoplasmic (CE) extracts were prepared as described previously (Beg et al. 1993). For Bcr–Abl expressing cells, CE and NE were made from cells growing in the presence and absence of IL-3. For TNFα timecourse, 2 × 105 cells/ml were plated 18–24 hr prior to treatment. TNFα (Promega Corp., Madison, WI) was added to 10 ng/ml and samples were incubated at 37°C for the indicated times. Whole cell extracts of tumor samples were prepared by lysing crushed, frozen tumors in 2–4 pellet volumes of lysis buffer (20 mm Tris at pH 8.0, 10% glycerol, 137 mm NaCl, 5 μg/ml each aprotinin, leupeptin, and pepstatin, and 1 mm PMSF) for 20–30 min on ice. Lysates were clarified by centrifugation, the supernatant was collected and protein concentrations were determined. For Western blotting, equal protein per sample was separated in 8% SDS-polyacrylamide gels and transferred to nitrocellulose. Blots were blocked in 5% milk and probed with an anti-ABL monoclonal antibody or an anti-carboxy-terminal IκBα polyclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Blots were probed with a secondary Ab conjugated to horseradish peroxidase (Promega Corp.) and protein bands were visualized with enhanced chemiluminescence detection (Amersham Life Science, Arlington Heights, IL).
EMSA
EMSAs were performed as described previously (Haskill et al. 1991). The DNA probe used contains an NF-κB site (underlined) from the H-2κb gene (5′-CAGGGCTGGGGATTCCCATCTCCACAGTTTCACTTC-3′) (Baldwin and Sharp 1987, 1988; Scheinman et al. 1993). Supershift assays were performed on an anti-p50 polyclonal antibody (Santa Cruz Biotechnology, Inc.) and an anti-p65 polyclonal antibody (Rockland, Boyertown, PA)
Cell viability and apoptosis
Cells were treated with 10 ng/ml mouse TNFα (Boehringer Mannheim Corp.) for the indicated times and cell viability was determined by trypan blue exclusion. Apoptosis was determined by flow cytometry as described previously (Cortez et al. 1995).
Tumor challenges
Cells were washed twice in RPMI-1640 only and resuspended at 1 × 107 cells/ml in RPMI-1640 supplemented with 0.1% FCS. Cells (1 × 106) were injected subcutaneously into each flank of 5- to 6-week-old athymic nu/nu female mice (Taconic Farms, German Town, NY).
Bone marrow transformation assay
Titers of the bicistronic retroviruses were determined by infection of Rat1 fibroblasts followed by indirect immunofluorescence to detect Bcr–Abl. Bone marrow transformation assays were performed as described (McLaughlin et al. 1987, 1989; Sawyers et al. 1992). Briefly, 5 × 106 freshly isolated bone marrow cells from BALB/c mice (Charles River Laboratories) were infected with equivalent titers of the bicistronic retroviruses encoding Bcr–Abl and the IκBα-SR in the presence of 4 μg/ml Polybrene for 6 hr. The cells were then plated in 4 ml of IMDM containing 10% FBS, 55 μm β-mercaptoethanol, glutamine, and P/S. Cultures were fed twice weekly and nonadherent cell counts were determined. Cultures reaching a nonadherent cell density of 1 × 106 cells/ml were considered transformed.
Acknowledgments
We thank Patricia C. Cogswell for assistance with removing tumors from the animals and for advice on transfections. We also thank Robert C. Quackenbush and Patricia Zipfel for assistance with the bone marrow transformation assay. We thank Dean Ballard for Flag–IκBα-SR expression vector; Bill Sugden for WT and mutant κB luc reporter constructs; Xiao-Fan Wang for Gal4–SP1-B expression vector; and Channing Der for Ras A17 expression vector. J.Y.R. was supported by a predoctoral National Institutes of Health (NIH) training grant. G.W.R. was supported by an Environmental Protection Agency Fellowship. D.C. was a Howard Hughes Medical Institute predoctoral fellow. A.M.P. is a Whitehead Scholar and a Scholar of the Leukemia Society of America. This work was supported in part by a NIH grants CA61033 (to A.M.P.) and CA72771 (to A.S.B.) and Leukemia Society of America grant (to A.S.B.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jhall@med.unc.edu; FAX (919) 966-0444.
References
- Afar DEH, Goga A, McLaughlin J, Witte ON, Sawyers CL. Differential complementation of Bcr-Abl point mutants with c-Myc. Science. 1994;264:424–426. doi: 10.1126/science.8153630. [DOI] [PubMed] [Google Scholar]
- Alkalay I, Yaron A, Hatzubai A, Jung S, Avraham A, Gerlitz O, Pashut-Lavon I, Ben-Neriah Y. In vivo stimulation of I kappa B phosphorylation is not sufficient to activate NF- kappa B. Mol Cell Biol. 1995;15:1294–1301. doi: 10.1128/mcb.15.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsura M, Wu M, Sonenshein GE. TGFβ1 inhibits NF-κB/Rel activity inducing apoptosis of B cells: Transcriptional activation of IκBα. Immunity. 1996;5:31–40. doi: 10.1016/s1074-7613(00)80307-6. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- ————— A 65-kD subunit of active NF-κB is required for inhibition of NF-κ B by IκB. Genes & Dev. 1989;3:1689–1698. doi: 10.1101/gad.3.11.1689. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Baldwin AS. The NF-kappa B and I-kappa B proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baldwin AS, Sharp PA. Binding of a nuclear factor to a regulatory sequence in the promoter of the mouse H-2Kb class I major histocompatibility gene. Mol Cell Biol. 1987;7:303–313. doi: 10.1128/mcb.7.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Two transcription factors, NF-kappa B and H2TF1, interact with a single regulatory sequence in the class I major histocompatibility complex promoter. Proc Natl Acad Sci. 1988;85:723–727. doi: 10.1073/pnas.85.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dorken B. Constitutive nuclear factor-κB-RelA activation is required for proliferation and survival of Hodgkin’s Disease tumor cells. J Clin Invest. 1997;100:2961–2969. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauparlant P, Kwan I, Bitar R, Chou P, Koromilas AE, Sonenberg N, Hiscott J. Disruption of I kappa B alpha regulation by antisense RNA expression leads to malignant transformation. Oncogene. 1994;9:3189–3197. [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κB in preventing TNFα-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr I κB interacts with the nuclear localization sequences of the subunits of NF-κB: A mechanism for cytoplasmic retention. Genes & Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- Beg AA, Finco TS, Nantermet PV, Baldwin JAS. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκBα: A mechansim for NF-κB activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours V, Burd PR, Brown K, Villalobos J, Park S, Ryseck RP, Bravo R, Kelly K, Siebenlist U. A novel mitogen-inducible gene product related to p50/p105-NF-kappa B participates in transactivation through a kappa B site. Mol Cell Biol. 1992;12:685–695. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Coupling of a signal response domain in IκBα to multiple pathways for NF-κB activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Park S, Kanno T, Franzoso G, Siebenlist U. Mutual regulation of the transcriptional activator NF-kappa B and its inhibitor, I kappa B-alpha. Proc Natl Acad Sci. 1993;90:2532–2536. doi: 10.1073/pnas.90.6.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of IκB-α proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- Carlesso N, Griffin JD, Druker BJ. Use of a temperature-sensitive mutant to define the biological effects of the p210 BCR-ABL tyrosine kinase on proliferation of a factor-dependent murine myeloid cell line. Oncogene. 1994;9:149–156. [PubMed] [Google Scholar]
- Chen Y-Y, Wang LC, Huang MS, Rosenberg N. An active v-abl protein tyrosine kinase blocks immunoglobulin light-chain gene rearrangement. Genes & Dev. 1994;8:688–697. doi: 10.1101/gad.8.6.688. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets I κB alpha to the ubiquitin-proteasome pathway. Genes & Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- Chiao PJ, Miyamoto S, Verma IM. Autoregulation of I kappa B alpha activity. Proc Natl Acad Sci. 1994;91:28–32. doi: 10.1073/pnas.91.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson B, Strife A. Linkage of proliferative and maturational abnormalities in chronic myelogenous leukemia and relevance to treatment. Leukemia. 1993;7:1683–1721. [PubMed] [Google Scholar]
- Cortez D, Kadlec L, Pendergast AM. Structural and signaling requirements for BCR-ABL-mediated transformation and inhibition of apoptosis. Mol Cell Biol. 1995;15:5531–5541. doi: 10.1128/mcb.15.10.5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Stoica G, Pierce JH, Pendergast AM. The BCR-ABL tyrosine kinase inhibits apoptosis by activating a Ras-dependent signaling pathway. Oncogene. 1996;13:2589–2594. [PubMed] [Google Scholar]
- Cortez D, Reuther GW, Pendergast AM. The Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and stimulates G1 to S phase transition in hematopoietic cells. Oncogene. 1997;15:2333–2342. doi: 10.1038/sj.onc.1201400. [DOI] [PubMed] [Google Scholar]
- Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia specific p210 BCR/ABL protein. Proc Natl Acad Sci. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, VanEtten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210 bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. Phosphorylation of I kappa B alpha precedes but is not sufficient for its dissociation from NF-kappa B. Mol Cell Biol. 1995;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Finco TS, Baldwin AS., Jr Mechanistic aspects of NF-kappa B regulation: The emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS., Jr Oncogenic Ha-Ras-induced signaling activates NF-κB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997;272:24113–24116. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- Ganchi PA, Sun SC, Greene WC, Ballard DW. I kappa B/MAD-3 masks the nuclear localization signal of NF-kappa B p65 and requires the transactivation domain to inhibit NF-kappa B p65 DNA binding. Mol Cell Biol. 1992;3:1339–1352. doi: 10.1091/mbc.3.12.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore TD. NF-kappa B, KBF1, dorsal, and related matters. Cell. 1990;62:841–843. doi: 10.1016/0092-8674(90)90257-f. [DOI] [PubMed] [Google Scholar]
- Gishizky ML, Cortez D, Pendergast AM. Mutant forms of growth factor-binding protein-2 reverse BCR-ABL-induced transformation. Proc Natl Acad Sci. 1995;92:10889–10893. doi: 10.1073/pnas.92.24.10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goga A, McLaughlin J, Afar DEH, Saffran DC, Witte ON. Alternative signals to RAS for hematopoietic transformation by the BCR-ABL oncogene. Cell. 1995;8:981–988. doi: 10.1016/0092-8674(95)90277-5. [DOI] [PubMed] [Google Scholar]
- Grilli M, Chiu JJ, Lenardo MJ. NF-kappa B and Rel: Participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- Haskill S, Beg AA, Tompkins SM, Morris JS, Yurochko AD, Sampson-Johannes A, Mondal K, Ralph P, Baldwin AS., Jr Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell. 1991;65:1281–1289. doi: 10.1016/0092-8674(91)90022-q. [DOI] [PubMed] [Google Scholar]
- Heisterkamp N, Jenster G, Ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukemia in bcr/abl transgenic mice. Nature. 1990;344:252–253. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- Henkel T, Zabel U, Van Zee K, Muller JM, Fanning E, Baeuerle PA. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell. 1992;68:1121–1133. doi: 10.1016/0092-8674(92)90083-o. [DOI] [PubMed] [Google Scholar]
- Kawakami K, Scheidereit C, Roeder RG. Identification and purification of a human immunoglobulin-enhancer- binding protein (NF-kappa B) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc Natl Acad Sci. 1988;85:4700–4704. doi: 10.1073/pnas.85.13.4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher MA, Knott A, McLaughlin J, Rosenberg N, Witte ON. Differences in oncogenic potency but not target cell specificity distinguish the two forms of the Bcr-Abl oncogene. Mol Cell Biol. 1991;11:4710–4716. doi: 10.1128/mcb.11.9.4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes & Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Kieran M, Blank V, Logeat F, Vandekerckhove J, Lottspeich F, Le Bail O, Urban MB, Kourilsky P, Baeuerle PA, Israel A. The DNA binding subunit of NF-kappa B is identical to factor KBF1 and homologous to the rel oncogene product. Cell. 1990;62:1007–1018. doi: 10.1016/0092-8674(90)90275-j. [DOI] [PubMed] [Google Scholar]
- Kitajima I, Shinohara T, Bilakovics J, Brown DA, Xu X, Nerenberg M. Ablation of transplanted HTLV-I Tax-transformed tumors in mice by antisense inhibition of NF-kappa B. Science. 1992;258:1792–1795. doi: 10.1126/science.1299224. [DOI] [PubMed] [Google Scholar]
- Klug CA, Gerety SJ, Shah PC, Chen Y-Y, Rice NR, Rosenberg N, Singh H. The v-Abl tyrosine kinase negatively regulates NF-κB/Rel factors and blocks κ gene transcription in pre-B lymphocytes. Genes & Dev. 1994;8:678–687. doi: 10.1101/gad.8.6.678. [DOI] [PubMed] [Google Scholar]
- Kurzrock R, Gutterman JU, Talpaz M. The molecular genetics of Philadelphia chromosome-positive leukemias. New Eng J Med. 1988;319:990–998. doi: 10.1056/NEJM198810133191506. [DOI] [PubMed] [Google Scholar]
- La Rosa FA, Pierce JW, Sonenshein GE. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol Cell Biol. 1994;14:1039–1044. doi: 10.1128/mcb.14.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laneuville P, Timm M, Hudson AT. BCR-ABL expression in 32D c13(G) cells inhibits apoptosis induced by protein tyrosine kinase inhibitors. Cancer Res. 1994;54:1360–1366. [PubMed] [Google Scholar]
- Lee H, Arsura M, Wu M, Duyao M, Buckler AJ, Sonenshein GE. Role of Rel-related factors in control of c-myc gene transcription in receptor-mediated apoptosis of the murine B cell WEHI 231 line. J Exp Med. 1995;181:1169–1177. doi: 10.1084/jem.181.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Brown K, Siebenlist U. Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha: Signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B. Proc Natl Acad Sci. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-KappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Mayo MW, Wang C-Y, Cogswell PC, Rogers-Graham KS, Lowe SW, Der CJ, Baldwin AS., Jr Requirement for NF-κB activation to suppress p53-independent apoptosis induced by oncogenic ras. Science. 1997;278:1812–1815. doi: 10.1126/science.278.5344.1812. [DOI] [PubMed] [Google Scholar]
- McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- McLaughlin J, Chianese E, Witte ON. In vitro transformation of immature hematopoietic cells by the p210 BCR/ABL oncogene product of the Philadelphia chromosome. Proc Natl Acad Sci. 1987;84:6558–6562. doi: 10.1073/pnas.84.18.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Alternative forms of the BCR–ABL oncogene have quantitatively different potencies for stimulation of immature lymphoid cells. Mol Cell Biol. 1989;9:1866–1874. doi: 10.1128/mcb.9.5.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JV. The diversity of Bcr–Abl fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–2384. [PubMed] [Google Scholar]
- Mitchell T, Sugden B. Stimulation of NF-kappa B-mediated transcription by mutant derivatives of the latent membrane protein of Epstein-Barr virus. J Virol. 1995;69:2968–2976. doi: 10.1128/jvi.69.5.2968-2976.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AJ, Young JC, Pendergast AM, Pondel M, Landau NR, Littman DR, Witte ON. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol Cell Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri A, Chang CC, Lombardi L, Salina M, Corradini P, Maiolo AT, Chaganti RS, Dalla-Favera R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-kappa B p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- Nolan GP, Ghosh S, Liou HC, Tempst P, Baltimore D. DNA binding and I kappa B inhibition of the cloned p65 subunit of NF-kappa B, a rel-related polypeptide. Cell. 1991;64:961–969. doi: 10.1016/0092-8674(91)90320-x. [DOI] [PubMed] [Google Scholar]
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497–1500. [Google Scholar]
- Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- Pane F, Frigeri F, Sindona M, Luciano L, Ferrara F, Cimino R, Meloni G, Saglio G, Salvatore F, Rotoli B. Neutrophilic-chronic myeloid leukemia: A distinct disease with a specific molecular marker (BCR/ABL with C3/A2 junction) Blood. 1996;88:2410–2414. [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast AM, Gishizky ML, Havlik MH, Witte ON. SH1 domain autophosphorylation of p210 BCR/ABL is required for transformation but not growth factor independence. Mol Cell Biol. 1993a;13:1728–1736. doi: 10.1128/mcb.13.3.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, Gishizky ML. BCR–ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993b;75:175–185. [PubMed] [Google Scholar]
- Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci. 1995;92:11746–11750. doi: 10.1073/pnas.92.25.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan L, Rosenberg N. abl genes. Biochim Biophys Acta. 1989;989:209–224. doi: 10.1016/0304-419x(89)90043-7. [DOI] [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Ribeiro RC, Abromowitch M, Raimondi SC, Murphy SB, Behm F, Williams DL. Clinical and biological hallmarks of Philadelphia chromosome in childhood acute lymphoblastic leukemia. Blood. 1987;70:948–953. [PubMed] [Google Scholar]
- Rice NR, Ernst MK. In vivo control of NF-κB activation by IκBα. EMBO J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Baltimore D. The effect of helper virus on Abelson virus-induced transformation of lymphoid cells. J Exp Med. 1978;147:1126–1141. doi: 10.1084/jem.147.4.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Witte ON. The viral and cellular forms of the Abelson (abl) oncogene. Adv Virus Res. 1988;35:39–81. doi: 10.1016/s0065-3527(08)60708-3. [DOI] [PubMed] [Google Scholar]
- Ruben SM, Dillon PJ, Schreck R, Henkel T, Chen CH, Maher M, Baeuerle PA, Rosen CA. Isolation of a rel-related human cDNA that potentially encodes the 65-kD subunit of NF-kappa B. Science. 1991;251:1490–1493. doi: 10.1126/science.2006423. [DOI] [PubMed] [Google Scholar]
- Ryseck RP, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, Bravo R. RelB, a new Rel family transcription activator that can interact with p50-NF-kappa B. Mol Cell Biol. 1992;12:674–684. doi: 10.1128/mcb.12.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia L, Grutz G. Tumorigenic activity of the BCR-ABL oncogenes is mediated by BCL2. Proc Natl Acad Sci. 1995;92:5287–5291. doi: 10.1073/pnas.92.12.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992;70:901–910. doi: 10.1016/0092-8674(92)90241-4. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Beg AA, Baldwin AS., Jr NF-kappa B p100 (Lyt-10) is a component of H2TF1 and can function as an I kappa B-like molecule. Mol Cell Biol. 1993;13:6089–6101. doi: 10.1128/mcb.13.10.6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RM, Perkins ND, Duckett CS, Andrews PC, Nabel GJ. Cloning of an NF-kappa B subunit which stimulates HIV transcription in synergy with p65. Nature. 1991;352:733–736. doi: 10.1038/352733a0. [DOI] [PubMed] [Google Scholar]
- Schmitz M, dos Santos Silva M, Baeuerle P. Transactivation domain 2 (TA2) of p65 NF-Kappa B. Similarity to TA1 and phorbol ester-stimulated activity and phosphorylation in intact cells. J Biol Chem. 1995;270:15576–15584. doi: 10.1074/jbc.270.26.15576. [DOI] [PubMed] [Google Scholar]
- Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-κB regulates IκB by two distinct mechanisms. Genes & Dev. 1993;7:1266–1276. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen S-C, Zon G, Gerwitz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 Kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia Chromosome-positive cells. Blood. 1995;86:726–736. [PubMed] [Google Scholar]
- Skorski T, Nieborowska-Skorska M, Wlodarski P, Perrotti D, Martinez R, Wasik MA, Calabretta B. Blastic transformation of p53-deficient bone marrow cells by p210bcr-abl tyrosine kinase. Proc Natl Acad Sci. 1996;93:13137–13142. doi: 10.1073/pnas.93.23.13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R, Wlodarksi P, Perrotti D, Chan OT, Wasik MA, Tsichlis PN, Calabretta B. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3κ/Akt-dependent pathway. EMBO J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF. Interleukin 3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sovak MA, Bellas R E, Kin DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S-C, Ganchi PA, Ballard DW, Greene WC. NF-κB controls expression of inhibitor IκBα: Evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Traenckner EB-M, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF kappa B. EMBO J. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traencker EB-M, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human IκB-α on serines 32 and 36 controls IκB-α proteolysis and NF-κB activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtieri M, Tweardy DJ, Caracciolo D, Johnson K, Mavilio F, Altmann S, Santoli D, Rovera G. Cytokine-dependent granulocytic differentiation: Regulation of proliferative and differentiative responses in a murine progenitor cell line. J Immunol. 1987;138:3829–3835. [PubMed] [Google Scholar]
- Van Antwerp DJV, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: Intimate tales of association and dissociation. Genes & Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Voncken JW, Kaartinen V, Pattengale PK, Germeraad WTV, Groffen J, Heisterkamp N. BCR/ABL P210 and P190 cause distinct leukemia in transgenic mice. Blood. 1995;86:4606–4611. [PubMed] [Google Scholar]
- Wada H, Mizutani S, Nishimura J, Usuki Y, Kohsaki M, Komai M, Kaneko H, Sakamoto S, Delia D, Kanamaru A. Establishment and molecular characterization of a novel leukemic cell line with Philadelphia chromosome expressing p230 BCR/ABL fusion protein. Cancer Res. 1995;55:3192–3196. [PubMed] [Google Scholar]
- Wang C-Y, Mayo MW, Baldwin JAS. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wesselborg S, Bauer MKA, Vogt M, Schmitz ML, Schulze-Osthoff K. Activation of transcription factor NF-kappa B and p38 mitogen-activated protein kinase is mediated by distinct and separate stress effector pathways. J Biol Chem. 1997;272:12422–12429. doi: 10.1074/jbc.272.19.12422. [DOI] [PubMed] [Google Scholar]
- Whiteside ST, Ernst MK, LeBail O, Laurent-Winter C, Rice N, Israel A. N- and C-terminal sequences control degradation of MAD3/IκBα in response to inducers of NF-κB activity. Mol Cell Biol. 1995;15:5339–5345. doi: 10.1128/mcb.15.10.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock CA, Witte ON. Long term culture of B lymphocytes and their precursors from murine bone marrow. Proc Natl Acad Sci. 1982;79:3608–3612. doi: 10.1073/pnas.79.11.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, Zou X, Merrell KT, Patel AJ, Marcu KB, Chellappan S, Calame K. v-Abl activates c-myc transcription through the E2F site. Mol Cell Biol. 1995;15:6535–6544. doi: 10.1128/mcb.15.12.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoza BK, Hu JYQ, McCall CE. Protein-tyrosine kinase activation is required for lipopolysaccharide induction of interleukin 1β and NF-κB activation, but not NF-κB nuclear translocation. J Biol Chem. 1996;271:18306–18309. doi: 10.1074/jbc.271.31.18306. [DOI] [PubMed] [Google Scholar]
- Zabel U, Baeuerle PA. Purified human IκB can rapidly disocciate the complex of the NF-κB transcription factor with its cognate DNA. Cell. 1990;61:255–265. doi: 10.1016/0092-8674(90)90806-p. [DOI] [PubMed] [Google Scholar]
- Zabel U, Henkel T, Silva MS, Baeuerle PA. Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus. EMBO J. 1993;12:201–211. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Rudchenko S, Wong K, Calame K. Induction of c-myc transcription by the v-Abl tyrosine kinase requires Ras, Raf1, and cyclin-dependent kinases. Genes & Dev. 1997;11:654–662. doi: 10.1101/gad.11.5.654. [DOI] [PubMed] [Google Scholar]