

Abstract
Hepatitis B virus (HBV) infects humans and causes a wide range of clinical manifestations, from acute hepatitis to hepatocellular carcinoma (HCC). The HBV genome contains multiple promoters with gene expression regulated predominantly by the cellular transcription initiation machinery. Accordingly, the HBV-encoded pX, the only known viral regulator, is a potent transcription coactivator. We investigated the relationship between pX and cellular coactivators. We show that pX restores wild-type activity to inactive TBPAS mutants with poor TAFII250 and activator-binding activity. This pX-mediated recovery, however, is not obtained with inactive TBPAS mutants in binding of other general transcription factors. Remarkably, ts13, a cell line temperature sensitive for TAFII250 function, exhibiting growth arrest and apoptosis at the restrictive temperature, is rescued partially by pX expression, thus generating a pX-dependent cell growth. Collectively, our results suggest that pX suppresses some of the phenotypes of TBP and TAFII250 mutations, implying that pX circumvents the need for a holo-TFIID complex for transcription activation to proceed.
Keywords: Transcription coactivation, TAF independent transcription, cell cycle, apoptosis, BHK ts cells, HBxAg
Hepatitis B virus (HBV) is the prototype of the hepadnaviridae infecting human hepatocytes. Hepadnaviruses are closely associated with development of hepatocellular carcinoma (Wei et al. 1992). The 3.2-kbp genomic DNA of the virus contains multiple promoters replicating by self-encoded reverse-transcriptase. This compact array of promoters together with the viral enhancer generates a genetic entity with an unusually complex pattern of gene expression. Consequently, the synthesis of the viral transcripts is regulated on the transcription level and not RNA processing (for review, see Shaul 1991). To this end, HBV encodes a promiscuous regulator protein, pX, acting as a transcription coactivator (Haviv et al. 1995). The pX open reading frame is conserved among all the mammalian hepadnaviridae, and it was shown to be essential for woodchuck HBV (WHV) infectivity (Chen et al. 1993; Zoulim et al. 1994).
The X protein is a promiscuous activator of many cellular and viral promoters (Colgrove et al. 1989; Faktor and Shaul 1990; Cross et al. 1993). pX interacts with several preinitiation complex (PIC) components including the RNA Pol II enzyme (Haviv et al. 1996) probably through the RPB5 subunit (Cheong et al. 1995), TATA-binding protein (TBP) in an ATP-dependent manner (Qadri et al. 1995), transcription factor IIH (TFIIH) (Haviv et al. 1996; Qadri et al. 1996), and TFIIB (Haviv et al. 1996, 1998; Lin et al. 1997). Although pX interacts with the basal transcription factors, its transcriptional effect depends on the activator activation domain. An upstream DNA-binding domain is not sufficient (Unger and Shaul 1990; Haviv et al. 1995). It has been proposed that activation of promoters requires pX to interact with both the basal transcription complex and upstream bound activators (Haviv et al. 1996). Indeed, pX also interacts with some activators such as CREB/ATF and p53 (Maguire et al. 1991; Feitelson et al. 1993; Truant et al. 1995) and the potent activation domain of VP16 (Haviv et al. 1996).
The viral activators VP16 and E1a in the context of an artificial reporter system were shown to interfere competitively with each other (Martin et al. 1990). Interestingly, this interference, termed squelching, is alleviated in the presence of pX both in vitro and in vivo (Haviv et al. 1995, 1996). Addition of excess E1a to an in vitro transcription system responsive to the E1a activation domain, inhibited (squelched) both E1a-dependent and E1a-independent transcription by sequestering target factors required for E1a activation (Boyer and Berk 1993). From among the component activities reconstituting the E1a-responsive transcription, only holo-TFIID [TBP plus TBP-associated factors (TAFs); Dynlacht et al. 1991] was capable of reversing this inhibition, indicating that TAFs are the direct functional target of the E1a activation domain. As pX alone is capable of reversing the inhibition effect of excess E1a activation domain in vivo, a functional similarity between pX and TAFs can be proposed.
A remarkable feature of pX is its ability to stimulate reconstituted transcription in the absence of cellular coactivators and TAFs. A fully defined in vitro transcription reaction, using bacterially expressed recombinant general transcription factors (GTFs) together with homogenous RNA–Pol II does not support transcription activation. This is probably attributable to the absence of coactivators including TAFs (Parvin et al. 1992; Tyree et al. 1993; Chen et al. 1994). By including pX and recombinant Gal4–VP16 into the in vitro system, partial list of the cellular transcription machinery components including Pol II and recombinant TBP, TFIIF, and TFIIB, transcription activation commences (Haviv et al. 1996). Because VP16 was shown to require direct interaction with TAFs to support activated transcription (Goodrich et al. 1993), a mechanistic similarity between pX and TAFs again becomes apparent.
Here we used two different experimental systems to determine whether the TAF-independent effect of pX in vitro is maintained in vivo. The first system relies on ectopic expression of TBP mutants with reduced TAFII250-binding and activator response, which were used previously to study the role of TBP–GTF interactions in vivo (Tansey et al. 1994; Tansey and Herr 1995; Farmer et al. 1996; Bryant et al. 1996). The second system involves a cell line (ts13) with a conditional mutation in the TAFII250 gene that blocks cell proliferation at the restrictive temperature. We show that in both cases, transcription was enhanced with the addition of pX under the conditions in which TFIID was defective. Furthermore, expression of pX in the ts13 cell line conferred normal cell proliferation and viability at the restrictive temperature. Cumulatively, these results provide in vivo evidence that the HBV X protein circumvents the requirement for TAFs.
Results
TBPAS mutants regain activator response in the presence of pX
To examine in vivo the role of TBP in transcriptional activation, we used human TBP mutants with an altered DNA-binding specificity (TBPAS) that supports transcription of a reporter with an altered c-fos TATA box (TGTA box) linked to a UAS synthetic enhancer (Tansey et al. 1994; Tansey and Herr 1995). The TGTA reporter displays at least 25-fold less activity than the TATA reporter (Fig. 1A, bars 1,3), yet its activity is recovered by the cotransfected TBPAS (bar 5). pX coactivates transcription of the TGTA reporter only in the presence of TBPAS (bars 4,6), suggesting that pX-mediated transcription activation depends on TBP, and that pX cannot recruit the endogenous holo-TFIID to the TGTA reporter. The ability of TBP to respond to transcriptional activators in vivo is curiously resistant to clustered sets of alanine substitution mutations in different regions of the protein, including those that disrupt DNA-binding and basal transcription in vitro. Combined sets of these mutations, however, can attenuate the in vivo activity of TBP and can affect differentially response to different activation domains (Tansey et al. 1994). To investigate whether an intact TBP is required for pX effect, a double substitution mutant of TBPAS, termed H2+S3′/S4′, reported to have reduced binding to acidic activators and to TAFII250 and possibly other TBP targets (Tansey et al. 1994; Tansey and Herr 1995) was used. No recovery of the TGTA reporter by this TBP mutant was obtained (bar 7), reconfirming the previous reports that this mutant is not activator responsive in vivo. Remarkably, cotransfection of pX under this condition recovered TGTA reporter activity (bar 8). The mutated TBPAS is fivefold more pX responsive than the TBPAS (12.5- and 2.5-fold, respectively). pX was also capable of supporting transcription in the presence of otherwise inactive triple TBPAS mutants: H2+S3/S4+S3′/S4′ and H1′+H2+S3′/S4′ (bars 10,12). TBPAS was HA tagged, and α-HA immunoblotting was used to confirm that comparable amounts of the ectopic TBP protein were expressed (Fig. 1B). A panel of pX mutants was constructed by oligonucleotide-directed insertion of two codons (Arg Pro) at 10- or 20-amino-acid intervals along the entire gene. Analysis of these mutants revealed two separate internal functional domains of pX (Runkel et al. 1993). Interestingly the amino terminus domain overlaps with the Pol II-binding region and the carboxyl terminus domain with the TFIIB-binding region (Cheong et al. 1995; Haviv et al. 1998). Here we used two inactive pX mutants M6 and M13, within the amino and carboxyl termini domains, respectively. These pX mutants were inactive in TBPAS mutant rescue (Fig. 1C). Other inactive pX mutants M7 and M12 behaved similarly (data not shown). In these experiments we used the Gal4–p53 as activator but similar results were obtained with Gal4–VP16 (Fig, 2; data not shown).
Figure 1.

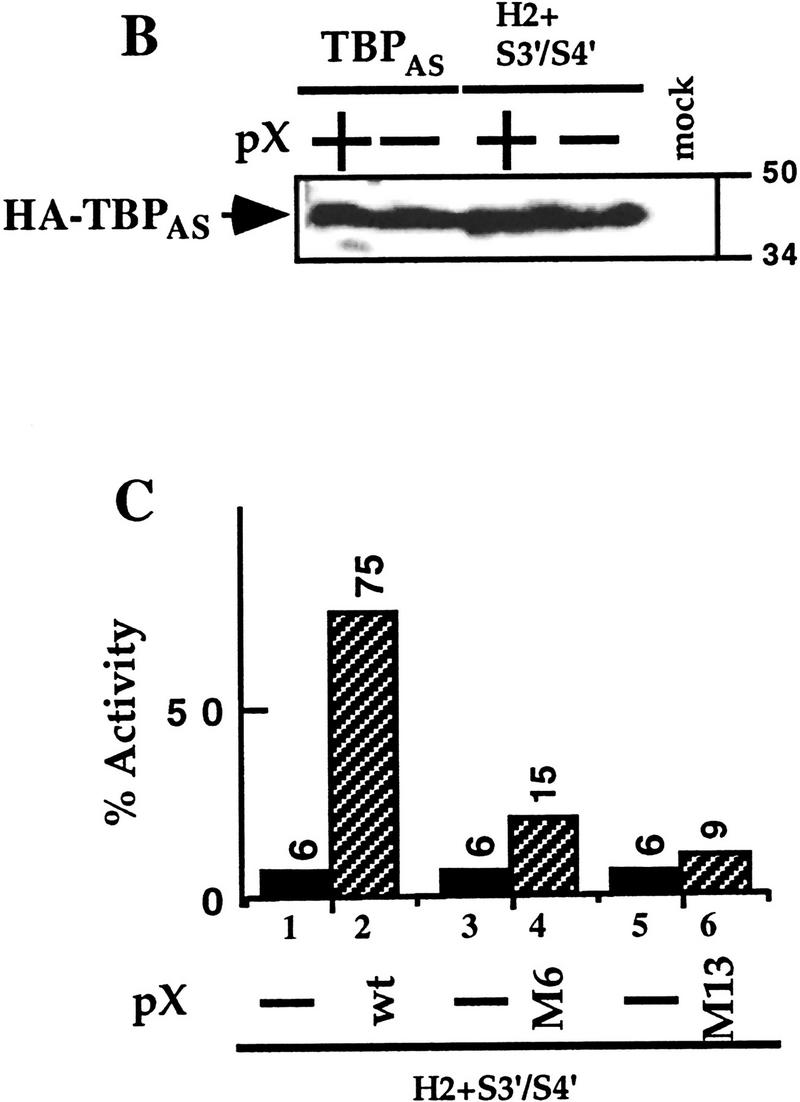
TBPAS mutants regain activator response in the presence of pX. (A) HepG2 cells were cotransfected with Gal4–p53 activator together with either a TATA or a TGTA reporter plasmids (0.2 μg/1.5-cm plates). These reporter plasmids contain an artificial enhancer that is activated by Gal4–p53 (0.02 μg), but not by Gal4 DNA-binding domain alone (Gal4–DBD). The TGTA reporter plasmid does not recognize the resident TBP protein, therefore is inactive (bars 3,4), unless a TBP with altered specificity (TBPAS) that recognizes the TGTA box is cotransfected (0.2 μg). Three different TBPAS mutants that show poor TAF-binding activity (Tansey et al. 1994; Tansey and Herr 1995; Bryant et al. 1996) were cotransfected (bars 7–12). Also cotransfected is a pX expressor plasmid (0.1 μg) where indicated (+). Luciferase activity relative to TATA-reporter plasmid alone is presented. (B) TBPAS is HA tagged, and the amount of the produced pHA–TBPAS in nuclear extracts of the transfected cells in A was analyzed by α-HA SDS-PAGE immunoblot. Migration of the molecular mass markers are shown (50 and 34 kD). (C) The ability of two pX mutants M6 and M13 to rescue the indicated TBPAS mutant is shown.
pX does not rescue TBPAS mutants at other GTF-interacting surfaces
To further characterize the mechanism of pX activity we used additional TBPAS mutants that were modified at the surfaces delineated to interact with other members of GTFs. For this purpose we used some of the radical TBP mutants generated by Bryant et al (1996) and the Gal4–VP16 activator. Here again the TBPAS H2+S3′/S4′ double mutant was active in the presence of pX (Fig. 2). This TBP mutant is ∼20-fold less active than the wild-type TBPAS; however, in the presence of pX, it became nearly as active as the wild-type TBPAS. Three other TBPAS point mutants, R235E, R239S, and P247E, found at the TBP surface predicted to be involved in activator/TAFs interactions, are pX responsive as well. In contrast, TBPAS mutants at regions on the surfaces predicted to bind either TFIIB or other GTFs; mutants E284R, E286R, G175R, and C176R were either inactive or only partially active in the presence of pX. The TBPAS point mutants are less active than the TBPAS H2+S3′/S4′ mutant possibly because the former produce ∼10-fold less protein as measured by Western blot analysis (data not shown). The majority of the pX-responsive TBPAS mutants have in vitro poor TAFII250 and activator interacting activity (Tansey et al. 1994). Thus, the fact that in the presence of pX these defective TBP mutants can support transcription activation suggests the interesting possibility that pX can substitute for some TAFII250 functions. No activation was obtained when the Gal4–pX fusion protein was used instead of the activator, ruling out the possibility that in these experiments pX complemented the activator function.
Figure 2.

pX does not rescue TBPAS mutants at another GTF-interacting surface. Cells were cotransfected with either Gal4–VP16 activator or Gal4–pX under the conditions described in Fig. 1. The Gal4–pX plasmid was constructed by cloning the entire X ORF at the carboxyl terminus end of the first 147 amino acids (DNA-binding domain) of Gal4. Two different reporter plasmids were used: TATA and TGTA. The TBPAS mutants are listed under the corresponding lanes. The obtained units of luciferase were compared to that obtained with TATA reporter in the absence of pX (taken as 100%). The experiments with the TGTA reporter plasmid were repeated three times and the average values with the calculated standard deviations are shown. The amount of protein produced by the TBPAS mutant H2+S3′/S4′ was fivefold higher than with single amino acid substitution mutants (data not shown).
A cell line with a TAFII250 temperature-sensitive mutant regains cyclin A expression in the presence of pX
The BHK21-derived thermosensitive cell lines ts13 and tsBN462 are defective in transcriptional activation of certain genes involved in cell cycle progression. These two cell lines arose from independent screens for thermosensitivity, yet carry the same G690D mutation in the TAFII250 gene (Talavera and Basilico 1977; Hayashida et al. 1994). A thermosensitive transcription defect in ts13 cells was observed on a cyclin A promoter (Wang and Tjian 1994). We transfected transiently ts13 cells with a cyclin A reporter together with a plasmid that expresses either wild-type or mutant pX and monitored activity at permissive (32.5°C), and restrictive (39.5°C) temperatures. A wild-type TAFII250 expressor plasmid was used as a positive control. As expected, under the restrictive temperature a 10-fold decrease of cyclin A reporter activity was obtained (Fig. 3A), which was recovered by cotransfected TAFII250. Interestingly, pX was also able to support cyclin A transcription under restrictive conditions. This cyclin A reporter activation was not obtained with transcriptionally inactive pX M6 and M13 mutants. At 39.5°C, the reporter is 15-fold more pX responsive than at 32.5°C (Fig. 3B). This differential temperature-sensitive pX response was not seen with the cell cycle-regulated E2F-responsive reporter (Fig. 3B), and with both SV40- and CMV-based reporters (not shown). The effect of pX, therefore, cannot be explained simply by changing the cell cycle. Furthermore, pX stimulates down-regulated cyclin A transcription 5 hr after the temperature upshift long before the cell cycle block is noticed (not shown). These results again support the possibility that pX can substitute for some TAFII250 functions. Moreover, they suggest that in vivo, pX can suppress the phenotype of cells bearing mutations in TAFII250.
Figure 3.


pX enables expression of cyclin A promoter under restrictive temperature in ts13 cells. (A) Luciferase activity of the cyclin A (cycA) reporter plasmid in ts13 cells after transient cotransfection, with increasing amounts (0.02, 0.08, and 0.2 μg) of the expressor plasmids of wild-type pX and pX mutants [M6 and M13 (Runkel et al. 1993)], as indicated. TAFII250 expressor plasmid was used as a control. Luciferase activity of the cyclin A reporter is presented in arbitrary units. (B) The fold of pX effect on the activity of cyclin A promoter was compared to that of E2F promoter. Hatched bars represent activity at 39.5°C; black bars at 32.5°C.
pX confers ts13 viability at the restrictive temperature
To test further the ability of pX to circumvent the need for TAFs in vivo, we performed genetic rescue experiments. We transfected ts13 cells with different pX expression vectors and directly selected thermoresistant cells at 39.5°C. Interestingly, pX-transfected plates produced a significant number of thermoresistant colonies, as compared to the control vectors (Fig. 4A, plate I; Table 1A). Two pX mutants, M6 and M13, which failed to rescue TBPAS mutants and activate the cyclin A promoter, are inactive (M13) or poorly active (M6) in rescuing the ts13 cells (Fig. 4A, plates II and III; Table 1C).
Figure 4.

pX suppresses the temperature-sensitive phenotype of ts13 cells. (A) ts13 cells were transfected with the HsX expressor plasmids (Runkel et al. 1993); (I) wild-type X; (II) M7 HsXm; (III) M13 HsXm and with the tetracycline-sensitive expression system that include (IV) pLPD450-fX + pUHD15-1; (V) pLPD450-fX; and (VI) pUHD15-1 plasmids. pUHD15-1 expresses the production of the tTA transactivator that activates the LPD450-fX plasmid to express the Flag-tagged X; therefore, pX is produced only in plate IV. Colonies obtained after 3 weeks of growth at 39.5°C were stained. (B) Proteins from representative colonies isolated from a plate that was transfected as in plate IV were analyzed by SDS-polyacrylamide gel immunoblot with α-HA monoclonal antibody for pHA-X detection. Note that some colonies (4 and 8) shut down pX expression in response to tetracycline incubation for 24 hr.
Table 1.
Efficiency of ts13 colony formation at 39.5°C.
| Constructs
|
No. of colonies (± s.d.)a
|
No. of plates
|
Constructs
|
No. of colonies (± s.d.)a
|
No. of plates
|
|---|---|---|---|---|---|
| A. Different X-containing plasmid vectors confer ts13 viability at 39.5°C | |||||
| Virgin vectors | X-containing vectors | ||||
| pUHD10−3+tTA | 0 ± 0 | 4 | pUHD10−3−fX+tTA | 26 ± 16 | 4 |
| SBC+tTA | 0 ± 0 | 5 | SBC−HAX+tTA | 63 ± 15 | 5 |
| LPD450+tTA | 0 ± 0 | 4 | LPD450−fX+tTA | 36 ± 16 | 4 |
| pBABE−puro | 0 ± 0 | 2 | pBABE−puro−fX | 51* | 2 |
| B. tTA-regulated X-plasmid vectors confer ts13 viability at 39.5°C by tTA | |||||
| Without tTA | with tTA | ||||
| pUHD10−3−fX | 0 ± 0 | 4 | pUHD10−3−fX+tTA | 26 ± 16 | 4 |
| LPD450−fX | 0 ± 0 | 4 | LPD450−fX+tTA | 36 ± 16 | 4 |
| SBC−HAX | 0 ± 0 | 5 | SBC−HAX+tTA | 63 ± 15 | 5 |
| C. Wild-type but not mutant pX confes ts13 viability at 39.5°C | |||||
| Mutant pX | wild type pX | ||||
| M6 HsXm | 0 ± 2 | 3 | HsX | 21 ± 12 | 3 |
| M13 HsXm | 0 ± 0 | 3 | |||
| Total | 0 ± 2 | 21 | 42 ± 17 | 18 | |
(*) All colonies were also puromycin resistant.
Number of colonies per 10-cm plates.
To link pX directly to the selective advantage of ts13 cells, we used tetracycline (Tet)-inducible expression of pX. An efficient rescue was observed only when the Tet-regulated pX expressor plasmids were activated by tTA, the activator of Tet-inducible vectors (Fig. 4A, plate IV; Table 1B). Neither the tTA activator (plate VI; Table 1A) nor the Tet-regulated pX expression vector (plate V; Table 1A) alone could rescue the cells. Twenty-four hours after incubation at 39.5°C with and without Tet, individual colonies were picked (150 colonies) and tested for pX expression either by PCR (data not shown) or by anti-X immunoblot (see representative samples in Fig. 4B). Every pX-transfected colony, growing at 39.5°C expressed detectable levels of pX substantiating its role in ts13 rescue. Furthermore, when a plasmid coexpressing pX and the puromycin resistance gene was used, all thermoresistant colonies were also puromycin resistant (Table 1A). Although unlikely, the possibility that during ts selection the cells have acquired mutations that affect ts phenotype was not ruled out. To address this point, cells were first drug selected at permissive temperature and then transferred to the restrictive 39°C. Despite the fact that equal number of drug-resistant colonies were obtained in the presence and the absence of pX, only the latter survived the temperature challenge (Fig. 5). These results implicated expression of pX as the factor, maintaining viability of the ts13 cells at restrictive temperature.
Figure 5.

pX-containing ts13 cells are thermoresistant. ts13 cells were transfected with the indicated plasmids and exposed to puromycin selection for 2 weeks. Plates with equal number of colonies were further incubated at the restrictive 39°C temperature for 3 days before staining.
pX promotes cell cycle progression of ts13 cells at restrictive temperature
The transcription defect in ts13 leads to growth arrest at G1 and eventual cell death (Sekiguchi et al. 1991, 1995). To address directly the ability of pX to rescue the cell cycle block in these cells, we followed entry into the cell cycle from G0 by flow cytometry analysis (Resnitzky and Reed 1995). Various ts13-derived cell lines were serum starved at 32.5°C, and then serum stimulated at either 32.5°C or 39.5°C. Flow cytometry analysis indicated that the starved cells were arrested in G0/G1 (data not shown). Consistent with their inability to grow at the restrictive temperature ts13 cells, 8 hr after serum addition, contained 18% and 2% of the cells in S-phase, at 32.5°C and 39.5°C, respectively (Fig. 6I,II). As expected expression of wild-type TAFII250 rendered these cells thermoresistant for DNA replication, and 29% of these cells were in S-phase at 39.5°C (Fig. 6III). Expression of pX also rendered 25% of the cells at S-phase at 39.5°C (Fig. 6IV).
Figure 6.

pX rescues the ts13 cell cycle defect. G0/G1-arrested cells were serum stimulated and replated for 8 hr at either 32.5°C (I), or 39.5°C (II,III,IV), as indicated. The analyzed ts13 cells were either parental (I,II), or stable transformants expressing wild-type TAFII250 (III), or pX (IV). Cell cycle distribution is presented as a flow cytometry graph of BrdU vs. propidium iodide incorporation. S-phase population, with high BrdU, was calculated by cell-quest gating.
The pX effect on cell cycle progression of ts13 cells at restrictive temperature is direct
The use of tetracycline-regulated pX expression enabled us to link directly the cell cycle state of the cells with pX expression. Whereas TAFII250 colonies were unaffected by Tet (data not shown), the thermoresistant phenotype is reversed when pX expression is shut off. A threefold reduction in the S-phase index of the X-expressing cells occurred when serum starvation proceeded in the presence of Tet, before the temperature challenge (Fig. 7A). This reduction in the S-phase index correlated with the detected amount of pX (Fig. 7B). No significant difference in the cell cycle profile was noticed at permissive temperature in the presence of Tet (Fig. 7C). Thus, pX is directly responsible for the ability of these cells to grow under restrictive temperature.
Figure 7.

Tet-inducible pX links pX expression to cell growth. (A) A pX-expressing clone of ts13 (as in Fig. 6IV) was incubated either with or without Tet (during serum starvation), before analysis as described in Fig 6. (B) α-X immunoblot demonstrates that the tetracycline treatment in A efficiently shuts off pX expression. (C) The same cell line under permissive temperature either with or without Tet was analyzed by FACS for cell cycle profile.
Discussion
HBV gene expression relies on multiple transcription initiation and reintiation events. It has been proposed that pX, the only known nonstructural HBV protein, functions in the absence of some of the components of the cellular transcription machinery. In vitro pX supports activator-dependent transcription with recombinant, bacterially expressed general transcription factors together with homogenous RNA Pol-II, but in the absence of TFIID components other than TBP (Haviv et al. 1996). In this report we describe observations suggesting that this ability of pX to function in the absence of wild-type TFIID is maintained in vivo. pX recovers transcription in the presence of overexpressed defective TBP mutants and suppresses the phenotype of the ts13 TAFII250 temperature-sensitive mutation.
We used a human TBP mutant with an altered DNA-binding specificity (TBPAS) to examine the role of TBP in pX-mediated transcriptional activation in vivo. The ability of TBP to respond to transcriptional activators in vivo is attenuated by substitution mutations in different regions of the protein with distinctive surface interactions (Tansey et al. 1994; Bryant et al. 1996). We show here that the pX recovery of activity is specific to activator-refractory TBPAS mutants but not TBPAS mutants with defective interactions with other components of the transcription machinery. The H2+S3′/S4′ TBPAS mutant displayed very poor TAFII250 interaction activity and it was concluded that mis-assembled TFIID complexes are responsible for the reduced activator response (Tansey et al. 1994). Although it is not clear whether this is the only molecular defect, our work confirms that these TBPAS mutants are indeed activator refractory, yet activity is rescued by pX. It is possible that suppression of these TBPAS mutants by pX requires TBP interactions, other than with TAFII250. However, the fact that pX can stimulate the activity of the otherwise inactive cyclin A promoter in ts13 cells under conditions inactivating TAFII250, further supports the hypothesis that in vivo, the assembly of TAFs into the TFIID complex is not an absolute requirement for pX effect on transcription.
The TAFII250-substituted mutant in ts13 cells affects transcription at 39.5°C leading to cell cycle block (Talavera and Basilico 1977). This defect is specific to certain genes, such as cyclin A, in which expression drops at the restrictive temperature. However, despite intensive attempts, cyclin A alone could not rescue these cells (Talavera and Basilico 1977; Sekiguchi et al. 1991, 1995; Hayashida et al. 1994), indicating that the altered expression is not limited to cyclin A. pX expression, however, is sufficient to elicit temperature-resistant cell growth and colony formation (Fig. 4 and Table 1), indicating that pX is capable of stimulating the expression of the set of genes required for ts13 cell growth. Our experiments with reporter plasmids have demonstrated that pX is not sensitive to the TAFII250 mutation, and the induction of these genes by pX can be explained by its capacity to circumvent TAFII250 requirement. However, the effect of pX on transcription in ts13 cells is not identical to that of TAFII250. Unlike the latter, pX stimulated cyclin A expression at permissive temperature as well. Also, as compared to the wild-type TAFII250 rescue, the efficiency of rescue by pX was significantly lower and required longer incubation time.
The selective advantage of the rescued ts13 cells is dependent on pX expression, as implied by the detection of pX mRNA and protein in every colony formed. Also, we found the ts13 cell cycle, in colonies bearing the X-gene under the regulation of Tet, to be Tet sensitive. Cell cycle blocks in mammals and yeast (obtained either by mutations or environmental conditions) can be bypassed by mitogens or genetic means that are not related directly to the cause of the block. We do not think that pX acts through this mechanism for a number of reasons. First, 5 hr after temperature upshift, pX stimulates down-regulated cyclin A transcription (data not shown). Second, the fold of pX effect on E2F, another cell cycle-regulated promoter, did not change at different temperatures (Fig. 3B) suggesting that pX activates specifically the cyclin A promoter whose expression was selectively down-regulated in the absence of TAFII250. Third, pX was unable to promote cell cycle progression when the cell cycle block was induced by mitogen depletion, or by lovastatin (data not shown). Therefore, we found it likely that pX overcomes ts13 cell cycle block at the restrictive temperature by directly suppressing the TAFII250 ts phenotype.
An alternative model assumes pX affects cellular signal transduction leading to indirect transcription modulation. It has been reported that pX transactivates AP-1 and other protein kinase C (PKC) dependent transcription factors by direct activation of PKC (Kekule et al. 1993). However, use of physiological concentrations of PKC inhibitors H7 and staurosporin uncoupled PKC from pX activity (Murakami et al. 1994). Others have reported that pX activates AP-1 through the Ras–Raf pathway (Benn and Schneider 1994; Natoli et al. 1994). However, neither Ras nor Raf dominant-negative mutants could block the induction of AP-1 by pX (Y. Shaul et al., unpubl.). In fact, no protein–protein interactions were reported to occur between pX and any protein that is involved in signaling, whereas pX binds to many components of the transcription machinery. These include TFIIB (Haviv et al. 1996, 1998; Lin et al. 1997), TFIIH (Haviv et al. 1996; Qadri et al. 1996), Pol II (Cheong et al. 1995; Haviv et al. 1996), and activators (Maguire et al. 1991; Wang et al. 1994; Haviv et al. 1996). However, the effect of pX on signaling might be indirect and obtained through transcription activation of yet unidentified cytokines or growth factors. In this regard, the fact that pX resembles the enzyme nucleoside-diphosphote kinase (NDPK) structurally and functionally (De-Medina and Shaul 1994) is intriguing. This may suggest that pX modulates small G-proteins by converting the bound GDP to GTP.
A central GTF component is TFIIB that interacts with TBP and Pol II–TFIIF. We have recently studied the role of TFIIB–pX interaction in transcription coactivation by pX by using TFIIB mutants that are inactive in either Pol II–TFIIF or TBP binding. Neither of these TFIIB mutants supports transcription. Remarkably, TFIIB mutants with disrupted Pol II–TFIIF binding, but with pX binding, acquire wild-type phenotype in the presence of pX both in vivo and in vitro (Haviv et al. 1998). These results not only substantiate the fact that TFIIB is the in vivo target of pX but also suggest that pX may establish the otherwise inefficient TFIIBm–Pol II–TFIIF interaction by acting as a molecular bridge.
pX mutants at either Pol II- or TFIIB-binding regions, M6 and M13 mutants, respectively (Cheong et al. 1995; Haviv et al. 1998), are inactive in suppressing TBP mutants. This raises the possibility that pX interaction with Pol II and TFIIB is required for pX to suppress the phenotype of the TBP and TAFII250 mutants. Therefore, the different roles of pX (i.e., TAF substitution and bridging Pol II to TFIIB) might be related. This possibility is in accordance with the fact that TAFs themselves interact with TFIIB and other GTFs. It has been reported that TAFII40 binds both TFIIB and acidic activators (Goodrich et al. 1993; Hori et al. 1995). In yeast TAFII30 is associated with TFIIF, Pol II, and the SWI/SNF coactivator complex (Henry et al. 1994; Cairns et al. 1996). Some of these interactions are functionally significant. For example, anti-TAFII100 antibodies that were raised against the domain that binds RAP30 (TFIIF small subunit) inhibited in vitro transcriptional activity of TFIID (Dubrovskaya et al. 1996). Furthermore, TAFII250 binds RAP74 (TFIIF large subunit) and the RAP74-binding domain was required for ts13 cell line complementation assays (Ruppert and Tjian 1995). Collectively, these molecular events support the possibility that TAFs play a role in establishing an active transcription complex not only by direct interaction with the activators but also with the components of GTFs. Thus, in this regard the mode of pX interactions with the transcription apparatus resembles partially that of components of holo-TFIID.
The pX activity described here is novel. Different viruses express proteins that bind and regulate TFIID (Zhou et al. 1992; Boyer and Berk 1993; Goodrich et al. 1993; Mazzarelli et al. 1995; Tong et al. 1995; Zhou and Sharp 1995; Carrozza and DeLuca 1996; Damania and Alwine 1996). A transient rescue of the temperature-sensitive transcription of cyclin A reporter in ts13 cells was described for the SV40 large T-antigen (Damania and Alwine 1996). Large T-antigen incorporates into the holo-TFIID complex and directly interacts with some of TAFs. Unlike pX, T antigen does not circumvent the need for TAFs, and functions in a TAF-dependent manner. Also, in contrast to pX, stably expressing large T-antigen actually accelerates ts13 cell death (Sekiguchi et al. 1991, 1995).
A unique feature of HBV is its mode of gene expression. The synthesis of all the viral transcripts is regulated on the transcriptional level and not during RNA processing. HBV bears multiple (about four) promoters despite its small genome size (3.2 kb). The ability of pX to suppress the phenotype of mutations at TBP and TAFII250 implies that pX circumvents the requirement for a holo-TFIID complex for transcription activation. This activity of pX should serve the virus needs in effectively using the cellular transcription machinery. Furthermore, the circular nature of the HBV genome might impose repeated assembly and disassembly of the transcription machinery on the HBV genome while the transcription bubble runs through the promoter regions. The ability to support transcription in the absence of some of the GTFs might very well be an important mechanism to support this compact gene expression program.
Finally, despite the likelihood that pX is involved in induction of hepatocellular carcinoma in some of the long-term HBV carriers, we still lack experimental evidence and a molecular explanation. Here we demonstrate that under certain conditions pX may rescue cells from apoptosis and drive arrested cells to divide. To our knowledge this is the first example in which cell growth depended on pX expression.
Materials and methods
Transient transfections
The TGTA reporter, TBPAS, Gal4–p53 and Gal4–VP16 expressors were used for transient cotransfection with or without a pX expressor plasmid (pSV2X), into HepG2, HtTA, or COS cells, and analyzed as previously described (Haviv et al. 1995). The nature of the different TBPAS mutants was described (Tansey et al. 1994; Tansey and Herr 1995; Bryant et al. 1996). Transient cotransfection of the cyclin A reporter (0.2 μg in a total of 1.5 μg of DNA into 1.5-cm plates), with expressors of TAFII250 or pX as indicated into ts13 cells, was performed in duplicates as in Wang and Tjian (1994). Cells were grown for 24 hr and then duplicates were transferred for an additional 5 hr to either 32.5°C or 39.5°C and harvested for luciferase assays.
ts13 rescue assay and the plasmids
Ten-centimeter plates of ts13 cells were transfected with 10 μg of plasmids in a total of 20 μg of DNA by the CaPi method as in Haviv et al. (1995). LPD450, SBC, and pUHD10-3 are various tTA (Tet-controlled transactivator)-dependent expression vectors and their expression is turned off by Tet (Resnitzky and Reed 1995). LPD450-fX is an EBV-based episomal expressor plasmid in which the X ORF with a flag tag was cloned under Tet-regulated promoter that contains the tTA-binding site. fX was also cloned in the UHD10-3 plasmid under its Tet-responsive promoter. HA-tagged X ORF was cloned in the SBC plasmid to construct the SBC–HAX plasmid. pUHD15-1 is an expressor plasmid that contains the tTA transactivator gene. pBABE–puro-fX is a retroviral vector that contains both the puromycin resistant and the Flag-tagged pX genes. The colonies that were obtained 3 weeks after transfection and growth at 39.5°C (medium was changed every 3 days) were either stained with 1% methylene blue or selected for further studies. Six-centimeter plates (with or without 2 μg/ml Tet) of each clone, were extracted with Tri-Reagent (MRC) for preparation of DNA, RNA, and proteins. Genomic DNA and RNA were used for PCR and RT–PCR analysis, respectively, with the X gene and the TAFII250 cDNA specific primers (Sekiguchi et al. 1991). Proteins were analyzed by SDS-polyacrylamide gel immunoblots (Haviv et al. 1996).
FACS analysis of the cell cycle
Cells were serum starved for 1 week, then serum stimulated, and replated for 8 hr, at either 32.5°C, or 39.5°C (Resnitzky and Reed 1995). Cells were then labeled (30 min) with 5 mm BrdU, fixed, double stained with FITC-conjugated α-BrdU (Becton-Dickinson) and propidium iodide, and analyzed by flow cytometry (FACScan Becton-Dickinson).
Acknowledgments
We thank Drs. W.P. Tansey and W. Herr for the TBPAS expression vectors; A.J. Berk for the TGTA reporter and TBPAS expression vectors; B. Henglein and P. Jansen-Duerr for the cyclin A reporter; S. Ruppert, E.H. Wang, and R. Tjian for the ts13 cells and the wild-type TAFII250 expression vector; and L. Runkel and H. Schaller for the X two-codon insertion mutants. We thank Drs. D. Resnitzki and R. Dikstein for critical reading of the manuscript and O. Barak for his linguistics comments. We thank S. Budilovski for her technical assistance. This research was supported by a grant from the national council for research and development of Israel and the Deutsches Krebsforschunszentrum (DKFZ) and Israel’s Ministry of Science.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lvshaul@weizmann.weizmann.ac.il; FAX 972-8 9344108.
References
- Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci. 1994;91:10350–10354. doi: 10.1073/pnas.91.22.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer TG, Berk AJ. Functional interaction of adenovirus E1A with holo-TFIID. Genes & Dev. 1993;7:1810–1823. doi: 10.1101/gad.7.9.1810. [DOI] [PubMed] [Google Scholar]
- Bryant GO, Martel LS, Burley SK, Berk AJ. Radical mutations reveal TATA-box binding protein surfaces required for activated transcription in vivo. Genes & Dev. 1996;10:2491–2504. doi: 10.1101/gad.10.19.2491. [DOI] [PubMed] [Google Scholar]
- Cairns BR, Henry NL, Kornberg RD. TFG/TAF30/ANC1, a component of the yeast SWI/SNF complex that is similar to the leukemogenic proteins ENL and AF-9. Mol Cell Biol. 1996;16:3308–3316. doi: 10.1128/mcb.16.7.3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, DeLuca NA. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol Cell Biol. 1996;16:3085–3093. doi: 10.1128/mcb.16.6.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Kaneko S, Girones R, Anderson RW, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol. 1993;67:1218–1226. doi: 10.1128/jvi.67.3.1218-1226.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Attardi LD, Verrijzer CP, Yokomori K, Tjian R. Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell. 1994;79:93–105. doi: 10.1016/0092-8674(94)90403-0. [DOI] [PubMed] [Google Scholar]
- Cheong J, Yi M, Lin Y, Murakami S. Human RPB5, a subunit shared by eukaryotic nuclear RNA polymerases, binds human hepatitis B virus X protein and may play a role in X transactivation. EMBO J. 1995;14:143–150. doi: 10.1002/j.1460-2075.1995.tb06984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrove R, Simon G, Ganem D. Transcriptional activation of homologous and heterologous genes by the hepatitis B virus X gene product in cells permissive for viral replication. J Virol. 1989;63:4019–4026. doi: 10.1128/jvi.63.9.4019-4026.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross JC, Wen P, Rutter WJ. Transactivation by hepatitis B virus X protein is promiscuous and dependent on mitogen-activated cellular serine/threonine kinases. Proc Natl Acad Sci. 1993;90:8078–8082. doi: 10.1073/pnas.90.17.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damania B, Alwine JC. TAF-like function of SV40 large T antigen. Genes & Dev. 1996;10:1369–1381. doi: 10.1101/gad.10.11.1369. [DOI] [PubMed] [Google Scholar]
- De-Medina T, Shaul Y. Functional and structural similarity between the X protein of hepatitis B virus and nucleoside diphosphate kinases. FEBS Lett. 1994;351:423–426. doi: 10.1016/0014-5793(94)00900-7. [DOI] [PubMed] [Google Scholar]
- Dubrovskaya V, Lavigne AC, Davidson I, Acker J, Staub A, Tora L. Distinct domains of hTAFII100 are required for functional interaction with transcription factor TFIIF beta (RAP30) and incorporation into the TFIID complex. EMBO J. 1996;15:3702–3712. [PMC free article] [PubMed] [Google Scholar]
- Dynlacht BD, Hoey T, Tjian R. Isolation of coactivators associated with the TATA-binding protein that mediate transcriptional activation. Cell. 1991;66:563–576. doi: 10.1016/0092-8674(81)90019-2. [DOI] [PubMed] [Google Scholar]
- Faktor O, Shaul Y. The identification of hepatitis B virus X gene responsive elements reveals functional similarity of X and HTLV-I tax. Oncogene. 1990;5:867–872. [PubMed] [Google Scholar]
- Farmer G, Bargonetti J, Zhu H, Friedman P, Prywes R, Prives C. Functional interaction between p53, the TATA-binding protein (TBP), and TBP-associated factors in vivo. Mol Cell Biol. 1996;16:4295–4304. doi: 10.1128/mcb.16.8.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feitelson MA, Zhu M, Duan LX, London WT. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene. 1993;8:1109–1117. [PubMed] [Google Scholar]
- Goodrich JA, Hoey T, Thut CJ, Admon A, Tjian R. Drosophila TAFII40 interacts with both a VP16 activation domain and the basal transcription factor TFIIB. Cell. 1993;75:519–550. doi: 10.1016/0092-8674(93)90386-5. [DOI] [PubMed] [Google Scholar]
- Haviv I, Vaizel D, Shaul Y. The X protein of hepatitis B virus coactivates potent activation domains. Mol Cell Biol. 1995;15:1079–1085. doi: 10.1128/mcb.15.2.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— pX, the HBV-encoded coactivator, interacts with components of the transcription machinery and stimulates transcription in a TAF-independent manner. EMBO J. 1996;15:3413–3420. [PMC free article] [PubMed] [Google Scholar]
- Haviv, I., M. Shamay, and Y. Shaul. 1998. Hepatitis B virus pX targets TFIIB in transcription coactivation. Mol. Cell. Biol. (in press). [DOI] [PMC free article] [PubMed]
- Hayashida T, Sekiguchi T, Noguchi E, Sunamoto H, Ohba T, Nishimoto T. The CCG1/TAFII250 gene is mutated in thermosensitive G1 mutants of the BHK21 cell line derived from golden hamster. Gene. 1994;141:267–270. doi: 10.1016/0378-1119(94)90583-5. [DOI] [PubMed] [Google Scholar]
- Henry NL, Campbell AM, Feaver WJ, Poon D, Weil PA, Kornberg RD. TFIIF-TAF-RNA polymerase II connection. Genes & Dev. 1994;8:2868–2878. doi: 10.1101/gad.8.23.2868. [DOI] [PubMed] [Google Scholar]
- Hori R, Pyo S, Carey M. Protease footprinting reveals a surface on transcription factor TFIIB that serves as an interface for activators and coactivators. Proc Natl Acad Sci. 1995;92:6047–6051. doi: 10.1073/pnas.92.13.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway [see comments] Nature. 1993;361:742–725. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- Lin Y, Nomura T, Cheong J, Dorjsuren D, Iida K, Murakami S. Hepatitis B virus X protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J Biol Chem. 1997;272:7132–7139. doi: 10.1074/jbc.272.11.7132. [DOI] [PubMed] [Google Scholar]
- Maguire HF, Hoeffler JP, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein–protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- Martin KJ, Lillie JW, Green MR. Evidence for interaction of different eukaryotic transcriptional activators with distinct cellular targets. Nature. 1990;346:147–152. doi: 10.1038/346147a0. [DOI] [PubMed] [Google Scholar]
- Mazzarelli JM, Atkins GB, Geisberg JV, Ricciardi RP. The viral oncoproteins Ad5 E1A, HPV16 E7 and SV40 TAg bind a common region of the TBP-associated factor-110. Oncogene. 1995;11:1859–1864. [PubMed] [Google Scholar]
- Murakami S, Cheong J, Ohno S, Matsushima K, Kaneko S. Transactivation of human hepatitis B virus X protein, HBx, operates through a mechanism distinct from protein kinase C and okadaic acid activation pathways. Virology. 1994;199:243–246. doi: 10.1006/viro.1994.1119. [DOI] [PubMed] [Google Scholar]
- Natoli G, Avantaggiati ML, Chirillo P, Puri PL, Ianni A, Balsano C, Levrero M. Ras- and Raf-dependent activation of c-jun transcriptional activity by the hepatitis B virus transactivator pX. Oncogene. 1994;9:2837–2843. [PubMed] [Google Scholar]
- Parvin JD, Timmers HT, Sharp PA. Promoter specificity of basal transcription factors. Cell. 1992;68:1135–1144. doi: 10.1016/0092-8674(92)90084-p. [DOI] [PubMed] [Google Scholar]
- Qadri I, Maguire HF, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc Natl Acad Sci. 1995;92:1003–1007. doi: 10.1073/pnas.92.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadri I, Ferrari ME, Siddiqui A. The hepatitis B virus transactivator protein, HBx, interacts with single- stranded DNA (ssDNA). Biochemical characterizations of the HBx-ssDNA interactions. J Biol Chem. 1996;271:15443–15450. doi: 10.1074/jbc.271.26.15443. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkel L, Fischer M, Schaller H. Two-codon insertion mutations of the HBx define two separate regions necessary for its trans-activation function. Virology. 1993;197:529–536. doi: 10.1006/viro.1993.1626. [DOI] [PubMed] [Google Scholar]
- Ruppert S, Tjian R. Human TAFII250 interacts with RAP74: Implications for RNA polymerase II initiation. Genes & Dev. 1995;9:2747–2755. doi: 10.1101/gad.9.22.2747. [DOI] [PubMed] [Google Scholar]
- Sekiguchi T, Nohiro Y, Nakamura Y, Hisamoto N, Nishimoto T. The human CCG1 gene, essential for progression of the G1 phase, encodes a 210-kilodalton nuclear DNA-binding protein. Mol Cell Biol. 1991;11:3317–3325. doi: 10.1128/mcb.11.6.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi T, Nakashima T, Hayashida T, Kuraoka A, Hashimoto S, Tsuchida N, Shibata Y, Hunter T, Nishimoto T. Apoptosis is induced in BHK cells by the tsBN462/13 mutation in the CCG1/TAFII250 subunit of the TFIID basal transcription factor. Exp Cell Res. 1995;218:490–498. doi: 10.1006/excr.1995.1183. [DOI] [PubMed] [Google Scholar]
- Shaul Y. Regulation of HBV Transcription. In: McLachlan A, editor. Molecular biology of the hepatitis B virus. Boca Raton, FL: CRC Press; 1991. pp. 193–212. [Google Scholar]
- Talavera A, Basilico C. Temperature sensitive mutants of BHK cells affected in cell cycle progression. J Cell Physiol. 1977;92:425–436. doi: 10.1002/jcp.1040920310. [DOI] [PubMed] [Google Scholar]
- Tansey WP, Herr W. The ability to associate with activation domains in vitro is not required for the TATA box-binding protein to support activated transcription in vivo. Proc Natl Acad Sci. 1995;92:10550–10554. doi: 10.1073/pnas.92.23.10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey WP, Ruppert S, Tjian R, Herr W. Multiple regions of TBP participate in the response to transcriptional activators in vivo. Genes & Dev. 1994;8:2756–2769. doi: 10.1101/gad.8.22.2756. [DOI] [PubMed] [Google Scholar]
- Tong X, Wang F, Thut CJ, Kieff E. The Epstein-Barr virus nuclear protein 2 acidic domain can interact with TFIIB, TAF40, and RPA70 but not with TATA-binding protein. J Virol. 1995;69:585–588. doi: 10.1128/jvi.69.1.585-588.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol. 1995;69:1851–1859. doi: 10.1128/jvi.69.3.1851-1859.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyree CM, George CP, Lira DL, Wampler SL, Dahmus ME, Zawel L, Kadonaga JT. Identification of a minimal set of proteins that is sufficient for accurate initiation of transcription by RNA polymerase II. Genes & Dev. 1993;7:1254–1265. doi: 10.1101/gad.7.7a.1254. [DOI] [PubMed] [Google Scholar]
- Unger T, Shaul Y. The X protein of the hepatitis B virus acts as a transcription factor when targeted to its responsive element. EMBO J. 1990;9:1889–1895. doi: 10.1002/j.1460-2075.1990.tb08315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang EH, Tjian R. Promoter-selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250. Science. 1994;263:811–814. doi: 10.1126/science.8303298. [DOI] [PubMed] [Google Scholar]
- Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Fourel G, Ponzetto A, Silvestro M, Tiollais P, Buendia MA. Hepadnavirus integration: Mechanisms of activation of the N-myc2 retrotransposon in woodchuck liver tumors. J Virol. 1992;66:5265–5276. doi: 10.1128/jvi.66.9.5265-5276.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Sharp PA. Novel mechanism and factor for regulation by HIV-1 Tat. EMBO J. 1995;14:321–328. doi: 10.1002/j.1460-2075.1995.tb07006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lieberman PM, Boyer TG, Berk AJ. Holo-TFIID supports transcriptional stimulation by diverse activators and from a TATA-less promoter. Genes & Dev. 1992;6:1964–1974. doi: 10.1101/gad.6.10.1964. [DOI] [PubMed] [Google Scholar]
- Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68:2026–2030. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]