

Abstract
Repeated administration of antipsychotic drugs to normal rats has been shown to induce a state of dopamine neuron inactivation known as depolarization block, which correlates with the ability of the drugs to exhibit antipsychotic efficacy and extrapyramidal side effects in schizophrenia patients. Nonetheless, in normal rats depolarization block requires weeks of antipsychotic drug administration, whereas schizophrenia patients exhibit initial effects soon after initiating antipsychotic drug treatment. We now report that, in a developmental disruption rat model of schizophrenia [methyl-azoxymethanol acetate (20 mg/kg, i.p.) injected into G17 pregnant female rats, with offspring tested as adults], the extant hyperdopaminergic state combines with the excitatory actions of a first- (haloperidol; 0.6 mg/kg, i.p.) and a second- (sertindole; 2.5 mg/kg, i.p.) generation antipsychotic drug to rapidly induce depolarization block in ventral tegmental area dopamine neurons. Acute injection of either antipsychotic drug induced an immediate reduction in the number of spontaneously active dopamine neurons (cells per electrode track; termed population activity). Repeated administration of either antipsychotic drug for 1, 3, 7, 15, and 21 d continued to reduce dopamine neuron population activity. Both acute and repeated effects on population activity were reversed by acute apomorphine injections, which is consistent with the reversal of dopamine neuron depolarization block. Although this action may account for the effects of D2 antagonist drugs on alleviating psychosis and the lack of development of tolerance in humans, the drugs appear to do so by inducing an offsetting deficit rather than attacking the primary pathology present in schizophrenia.
Introduction
All antipsychotic drugs (APDs) in use block dopamine (DA) D2 receptors (Carlsson and Lindqvist, 1963; Seeman et al., 1975; Kapur and Remington, 2001), and these drugs are most effective at alleviating the psychotic features of schizophrenia (Seeman, 1992; Kapur and Remington, 2001; Abi-Dargham, 2004; Cincotta and Rodefer, 2010). Moreover, in schizophrenia patients there is an elevated amphetamine-induced increase in raclopride displacement, which is a measure of DA release (Ginovart et al., 2004) in the striatum, and furthermore, this increased release is proportional to the amphetamine-induced exacerbation of psychosis (Laruelle et al., 1996; Abi-Dargham et al., 2004). These data are consistent with a hyperdopaminergic state in schizophrenia. However, evidence for an overactive DA system has not been forthcoming. Therefore, current theories suggest that the DA system is dysregulated, rendering it hyper-responsive to stimuli (Carlsson and Carlsson, 1990; Grace, 1991, 2000; Weinberger and Gallhofer, 1997). Data collected from the methyl-azoxymethanol acetate (MAM) developmental rat model of schizophrenia (Moore et al., 2006) suggest that this dysfunction derives from hippocampal overdrive of DA neurons in the ventral tegmental area (VTA), leading to increased numbers of DA neurons firing spontaneously (Lodge and Grace, 2007). Consequently, incoming stimuli would cause a greater phasic activation of the DA system (Lodge and Grace, 2006).
Nonetheless, how APDs achieve their therapeutic action is unclear. Thus, although APDs exert therapeutic benefit during the first days of administration (Kapur and Seeman, 2001; Kinon et al., 2010), the maximal antipsychotic action requires weeks to develop (Johnstone et al., 1978). In contrast, based solely on DA receptor antagonism, the maximal antipsychotic action should occur immediately, with tolerance developing over the ensuing weeks (Palmstierna and Wistedt, 1987; Baldessarini et al., 1988; Nyberg et al., 1995). One mechanism proposed for this delayed maximal action is the development of DA neuron depolarization block (Bunney and Grace, 1978; Chiodo and Bunney, 1983; Grace and Bunney, 1986; Grace et al., 1997). Thus, upon acute administration to normal rats, APDs block DA receptors and increase in the number of DA neurons firing (Bunney and Grace, 1978); however, after 3+ weeks of treatment, DA neurons undergo depolarization block, decreasing the number of DA neurons firing spontaneously due to overexcitation and spike inactivation. All drugs that are therapeutically effective cause depolarization block in the mesolimbic DA neurons, whereas those that cause extrapyramidal side effects induce depolarization block of the nigrostriatal DA neurons (Chiodo and Bunney, 1983; White and Wang, 1983b; Grace and Bunney, 1986; Hand et al., 1987; Skarsfeldt and Perregaard, 1990; Skarsfeldt, 1992). While this may account for some of the longer-term actions of APDs such as the lack of tolerance and delayed maximal efficacy, it does not account for the acute actions of these drugs in alleviating psychotic symptoms. On the other hand, these studies were done in normal rats. In the current study, we examined the effects of acute and repeated APD administration on DA neuron activity in the MAM model of schizophrenia, in which the DA neurons are already in an activated state.
Materials and Methods
Subjects and materials.
Rats used for the developmental rat model of schizophrenia used in this study were produced by injecting the DNA methylating agent MAM (Midwest Research Institute) to timed pregnant female rats at 17 d of gestation (GD17; 20 mg/kg, i.p., diluted in saline), as described previously (Moore et al., 2006). Female Sprague Dawley dams (Hilltop) arrived at GD15 and were housed individually on a normal 12 h light cycle (lights on at 7:00 A.M.). At GD17, pregnant females were randomly separated into two groups; one group received injection of 0.9% saline (i.p., 1 ml/kg), while the other group was injected with 20 mg/kg MAM in saline. P7 offspring were culled to 10 by removing females, and within 3 weeks from birth male offspring were weaned and housed in pairs or groups of three with littermates. A total of 142 adult rats, obtained from different litters, were used in this study, of which 84 were MAM and 58 saline offspring.
Two drugs were chosen for this study: haloperidol, a prototypical first-generation antipsychotic drug with potent D2 antagonistic properties, and sertindole, a second-generation drug that is also a D2 antagonist but is unique in not effectively blocking DA neuron cell body autoreceptors (Skarsfeldt, 1992; Valenti and Grace, 2010). Sertindole was generously provided by H. Lundbeck A/S; haloperidol, apomorphine hydrochloride hemihydrate, and all other drugs were obtained from Sigma-Aldrich. APDs were dissolved in 0.1 m acetic acid (Fisher Scientific) and the pH was adjusted with NaOH to reach pH ∼5.
All experiments and drug treatments performed in this study are in accordance with the guidelines outlined in the United States Public Health Service Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Acute and repeated APD treatments.
At 280–300 g, rats were transported to a separate facility and randomly assigned to a specific drug treatment. In rats tested for acute drug effects, 1.25 mg/kg sertindole or 0.6 mg/kg haloperidol were administered to either saline or MAM offspring through the tail or femoral vein under anesthesia (i.v.); and the effects on VTA DA neurons were tested starting at 1 h from injection (see below). Control rats were injected with 1 ml/kg drug solvent (0.1 m acetic acid, pH 5; VHC). A few rats in each group received systemic drug administration with recordings made 2 h (for sertindole) or 1 h (haloperidol) following the drug. As previous studies indicated that sertindole is poorly absorbed following systemic administration (Watanabe and Hagino, 1999; Dr. Arnt, personal communication), a higher dose of 2.5 mg/kg was used. Data obtained from rats that received either intravenous or intraperitoneal sertindole administration were consistent, and therefore these data were combined. The doses for both APDs were chosen based on previous experience of our laboratory for acute and repeated APD administration, to reproduce therapeutic blood level (Arnt, 1995; Grace et al., 1997); in addition, a daily administration of APD was opted to approximate the administration schedule in patients (Murray and Dean, 2008). It should be noted that previous studies showed that increasing the dose of antipsychotic drug threefold to fourfold does not alter the time course of induction of depolarization block (Jiang et al., 1988). To test the effect of repeated APD administration, the three treatment groups of MAM (vehicle, sertindole, or haloperidol) and two treatment groups of saline rats (haloperidol or sertindole) were separated into five subgroups for different durations of repeated drug administration: 1, 3, 7, 15, and 21 d. Sertindole (2.5 mg/kg), haloperidol (0.6 mg/kg), or vehicle (1 ml/kg) was administered by intraperitoneal injections, which occurred daily between 8:30 and 9:30 A.M. On the day of the experiment, rats were injected one additional time with the same compound (APD or VHC) administered during treatment, and the effect on VTA DA neuron activity was examined 2 h (for sertindole) or 1 h (for haloperidol) later.
Single-unit extracellular recordings.
All electrophysiology recordings were performed as previously described (Valenti and Grace, 2010). In brief, rats were anesthetized with 8% chloral hydrate (i.p., 400 mg/kg) and placed in a stereotaxic apparatus (David Kopf Instruments). Body temperature and the level of anesthesia were constantly monitored, and anesthesia was adjusted by administration of additional chloral hydrate as needed. A burr hole was drilled in the region overlying the VTA (from bregma, AP: −5.3 mm, ML: −0.6 mm) on the right side of the rat brain. Glass microelectrodes (impedance, 8–12 MΩ) were lowered using a hydraulic microdrive, and sample regions of the VTA were examined by recording from spontaneously firing DA neurons. The electrode was passed through the VTA in 6–9 tracks in a predetermined grid pattern with each track separated by 200 μm, and proceeding from medial to lateral VTA (Valenti et al., 2011). The final count of all spontaneously firing DA neurons encountered in each rat divided by the number of tracks was defined as population activity. Estimating the population activity by counting the number of spontaneously firing DA neurons in each electrode track has proven to be an effective and reproducible means for estimating this parameter (West and Grace, 2000). DA neurons were distinguished from other VTA neurons with open filters (low cutoff, 50 Hz; high cutoff, 16 kHz), using well established criteria, including slow spontaneous firing rate, action potential shape, duration and location (Grace and Bunney, 1983). In particular, the long duration negative component, a distinguishing characteristic of these neurons (Grace and Bunney, 1983, 1984), was an important identification criterion. After allowing for a few minutes for the neuron to stabilize, 2–3 min of baseline activity was recorded to measure neuron firing rate and the proportion of action potentials occurring in bursts (percentage of burst firing) as defined previously (Grace and Bunney, 1984). Firing rate and percentage of burst firing were then averaged to give the average firing rate and average burst firing for the population of DA neurons recorded for each rat.
Reversal of depolarization block.
A subgroup of MAM rats that had been treated with either APD at different points of the treatment time course were injected with apomorphine (i.v.; Hollerman et al., 1992) after completing single-unit recordings from the VTA, to test for reversal of APD-induced depolarization block. Sertindole-treated MAM rats were injected with 30 μg/kg apomorphine, and 80 or 120 μg/kg apomorphine was used for rats treated with haloperidol. Higher doses of apomorphine were required in haloperidol-treated rats compared to sertindole-treated rats, since sertindole does not exert substantial autoreceptor antagonism (Valenti and Grace, 2010). Then a second hole was drilled on the left side of the brain overlying the contralateral VTA and a microelectrode was lowered to assess DA neuron firing and population activity as described above.
Histology and data analysis.
At the cessation of each experiment, recording sites were marked via electrophoretic ejection of Pontamine sky blue dye from the tip of the recording electrodes (approximately −20 μA constant current; Fintronics, bipolar constant current source). Rats were deeply anesthetized with chloral hydrate, decapitated and brains removed and fixed in 8% w/v paraformaldehyde in PBS for a minimum of 48 h. Following cryoprotection with 25% w/v sucrose in PBS, brains were sectioned and the 60 μm coronal sections obtained were mounted onto gelatin-chrom alum-coated slides, and stained with cresyl violet.
Analysis of DA neuron activity was performed off-line using the custom-design software Neuroscope (Brian Lowry). All data are reported as mean ± SEM. Statistics were calculated on raw data using SigmaStat 3.1 (Systat Software) for paired t test, two-way ANOVA, followed by all pairwise multiple comparison procedures (Holm–Sidak method). Two-sample Kolmogorov–Smirnov tests were performed on firing rate and burst distributions using MATLAB (The MathWorks).
Results
Acute administration of either sertindole or haloperidol to MAM rats rapidly reduced VTA DA neuron population activity
Previous reports have shown that APDs are present at target receptors within 1 h following acute administration to schizophrenia patients (Kapur and Remington, 2001; Agid et al., 2003; Kinon et al., 2010); thus, we first investigated whether acute administration of APD produces any change in the activity of MAM VTA DA neurons. In saline offspring injected with vehicle, the mean value of spontaneously active DA neurons per electrode track averaged 0.9 ± 0.1 (SAL + VHC: n = 8 rats, 53 DA neurons) (Floresco et al., 2003; Valenti and Grace, 2010). Following acute administration of 1.25 mg/kg sertindole (i.v., or 2.5 mg/kg, i.p.), there was a significant increase in population activity to 1.9 ± 0.1 DA neurons per track (SAL + SER: n = 4 rats, 53 DA neurons two-way ANOVA, F(1,2,259) = 36.6, p < 0.001; Fig. 1A), which is consistent with previous studies (Chiodo and Bunney, 1983; Skarsfeldt, 1992; Valenti and Grace, 2010). MAM offspring injected with vehicle exhibited high levels of population activity (MAM + VHC: 1.9 ± 0.1, n = 7 rats, 98 DA neurons), which has been suggested to correlate with the hyperdopaminergic condition of schizophrenia patients (Lodge and Grace, 2007). However, when MAM rats were injected acutely with sertindole, a significant reduction in population activity was observed (MAM + SER: 0.7 ± 0.03; n = 6 rats, 40 DA neurons; two-way ANOVA, F(1,2,259) = 36.6, p < 0.001). Thus, the resultant level of population activity was not different from that of untreated saline rats, indicating that a single dose of sertindole rapidly reversed the high levels of population activity in MAM rats (Fig. 1A). A similar result was produced upon testing the first-generation APD haloperidol. Acute administration of 0.6 mg/kg haloperidol induced a significant increase in population activity in saline offspring when compared to untreated saline rats (SAL + HAL: 1.8 ± 0.1, n = 4 rats, 48 DA neurons; two-way ANOVA, F(1,2,259) = 36.6, p < 0.001; Fig. 1B). Similar to sertindole, haloperidol also reversed the increased DA neuron population activity observed in the MAM rats, restoring the number of spontaneously firing DA neurons to control levels (MAM + HAL: 0.9 ± 0.1, n = 6 rats, 44 DA neurons; two-way ANOVA, F(1,2,259) = 36.6, p < 0.001) following drug administration (Fig. 1B). The change in population activity produced by sertindole versus haloperidol was not significantly different in the saline rats (two-way ANOVA, p > 0.05), nor was it significantly different in comparing sertindole versus haloperidol in the MAM rats (two-way ANOVA, p > 0.05).
Figure 1.
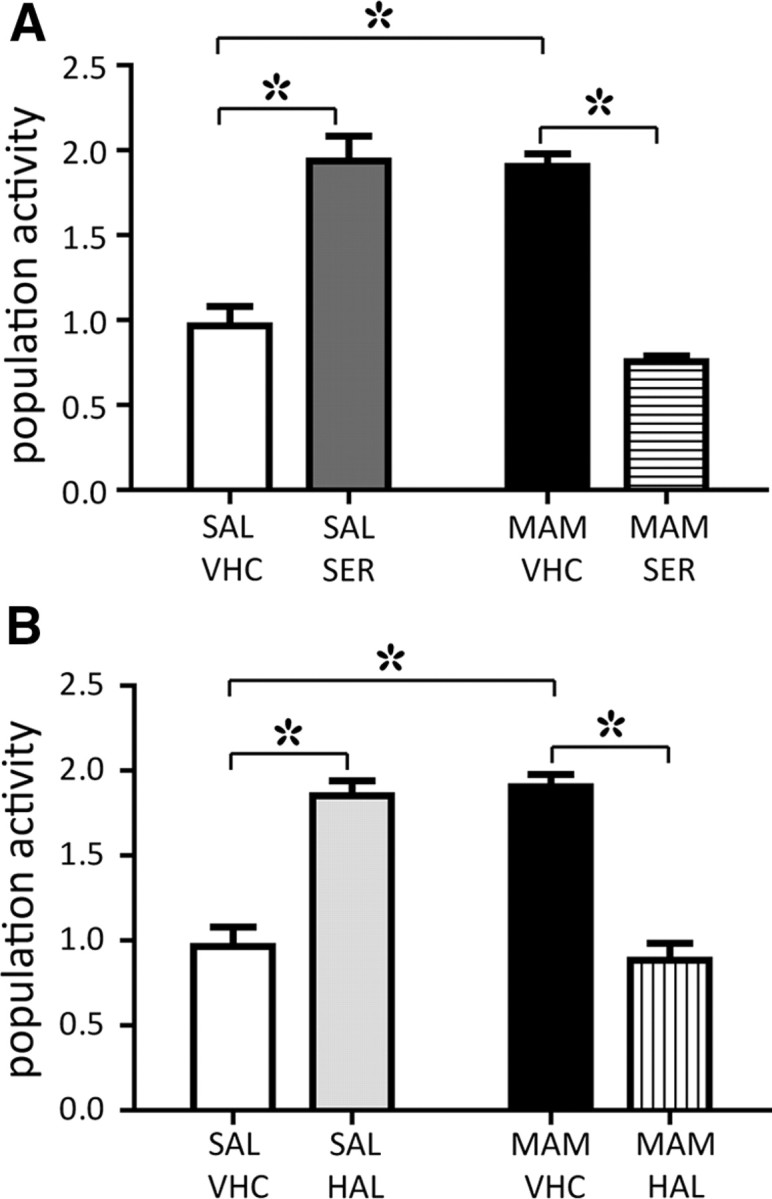
Acute sertindole and haloperidol administration induced a rapid decrease of VTA DA neuron population activity in MAM rats. A, Consistent with previous reports, a single, acute dose of sertindole induced a twofold increase in VTA DA neuron population activity of saline offspring (SAL+SER, 1.25 mg/kg, i.v., 2.5 mg/kg, i.p.; n = 4 rats, 53 DA neurons; dark gray bar) compared with saline rats injected with drug vehicle (SAL+VHC, 0.1 m AcA, pH ∼5, n = 8 rats, 53 DA neurons; white bar). MAM rats injected with vehicle exhibited elevated levels of spontaneously active DA neurons (MAM+VHC, n = 7 rats, 98 DA neurons; black bar); in contrast, administration of sertindole rapidly decreased population activity to control levels (MAM+SER, n = 6 rats, 40 DA neurons; horizontal line bar). B, Haloperidol administered at 0.6 mg/kg (i.p.) produced effects similar to sertindole on population activity in both SAL (SAL+HAL, n = 4 rats, 48 DA neurons; light gray bar) and MAM (MAM+HAL, n = 6 rats, 44 DA neurons; vertical line bar) rats. White and black bars as in A. *Two-way ANOVA followed by Holm–Sidak multiple-comparison test, p < 0.001.
Our previous study (Lodge and Grace, 2011) showed that in MAM rats, there was a preferential activation of DA neuron population activity in the lateral VTA. In the current study, we examined whether there was a difference in APD effect that was dependent on the medial–lateral location in the VTA. However, we found that sertindole and haloperidol produced equivalent increases across the medial–lateral extent of the VTA in saline rats (medial vs lateral in VHC, SER, HAL: two-way ANOVA, p = 1.0). Moreover, in MAM rats, sertindole and haloperidol produced equivalent decreases in population activity independent of location (medial vs lateral in VHC, SER, HAL: two-way ANOVA, p = 0.3).
Effects of acute APD administration on DA neuron firing rate and firing pattern
The effects of acute administration of either APD on DA neuron firing rate or percentage of action potentials occurring in bursts was examined. Sertindole did not significantly alter the average firing rate of VTA DA neurons when administered to either saline or MAM rats (SAL + VHC: 4.5 ± 0.1; SAL + SER: 4.0 ± 0.5; MAM + VHC: 3.7 ± 0.4 Hz; MAM + SER: 4.4 ± 0.3; two-way ANOVA, p = 0.2), and a similar result was observed with haloperidol administration (SAL + VHC: 4.5 ± 0.1, SAL + HAL: 4.0 ± 0.2, MAM + VHC: 3.7 ± 0.4, MAM + HAL: 4.3 ± 0.3; two-way ANOVA, p = 0.2). Although neither of the APDs tested produced any significant change in the average firing rate, further analysis revealed a shift in the firing rate distribution of DA neurons following acute sertindole administration. Thus, acute administration of sertindole did not significantly change DA firing rate distribution in saline rats (two-sample Kolmogorov-Smirnov test, p = 0.4; Fig. 2A), but induced a significant alteration in the firing rate distribution in MAM rats (two-sample Kolmogorov–Smirnov test, p = 0.02; Fig. 2B). In contrast, haloperidol administration did not significantly alter DA firing rate distribution either in saline or in MAM rats (two-sample Kolmogorov–Smirnov test; SAL + SER, p = 0.2, Fig. 2C; MAM + HAL, p = 0.4; Fig. 2D). Furthermore, there was no difference in firing rate distribution when comparing MAM and saline rats (two-sample Kolmogorov–Smirnov test, p = 0.2).
Figure 2.
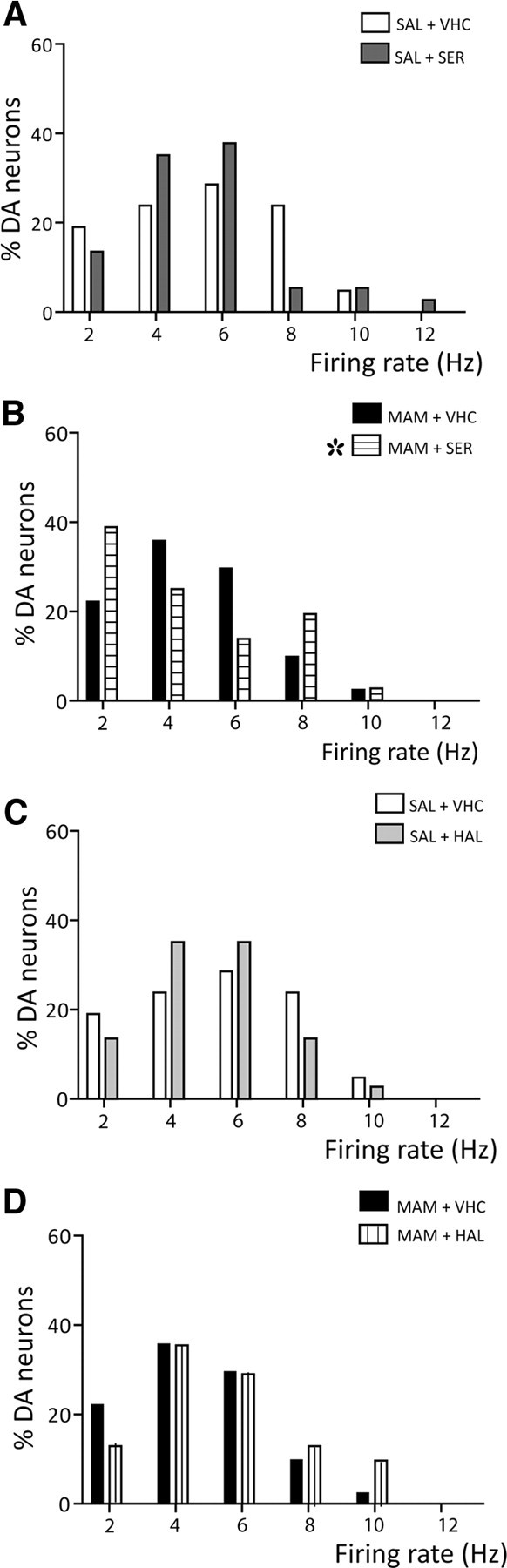
Acute administration of sertindole altered the distribution of firing rates in MAM-treated rats. A, Acute injection of sertindole (SAL+SER, dark gray bars) did not affect DA neuron firing rate distribution in untreated saline rats (SAL+VHC, white bars). B, In MAM rats, acute administration of 1.25 mg/kg sertindole (horizontal line bars) significantly altered the distribution of firing rates in MAM rats compared to the vehicle-treated MAM control (black bars) due to an increase in neurons firing at both low and high firing rates. In contrast, acute haloperidol did not alter firing rate distribution of DA neurons recorded in saline rats (C, SAL+HAL, light gray bars) or MAM rats (D, MAM+HAL, vertical line bars). White and black bars as in A and B. *Two-sample Kolmogorov–Smirnov test, p = 0.0161.
With respect to firing pattern, the average percentage of burst firing was not significantly different following acute administration of either sertindole (SAL + VHC: 37.1 ± 6%, SAL + SER: 32.3 ± 7.6%, MAM + VHC: 29.6 ± 3.8%, MAM + SER: 42.6 ± 5.5; two-way ANOVA, p = 0.083) or haloperidol (SAL + VHC: 37.1 ± 6%, SAL + HAL: 34.5 ± 2.9%, MAM + VHC: 29.6 ± 3.8%, MAM + HAL: 39.3 ± 3.5%; two-way ANOVA, p = 0.08). In addition, neither acute administration of sertindole (SAL+VHC vs SAL+SER: two-sample Kolmogorov–Smirnov test, p = 0.9, Fig. 3A) or haloperidol (SAL+VHC vs SAL+HAL: two-sample Kolmogorov–Smirnov test, p = 0.8, Fig. 3C) affected the distribution of percentage burst firing in saline rats. In contrast, both acute sertindole and acute haloperidol induced a significant rightward shift in firing rate distribution in MAM rats (two-sample Kolmogorov–Smirnov test, MAM+VHC vs MAM+SER: p = 0.006, Fig. 3B; MAM+VHC vs MAM+HAL, p = 0.035, Fig. 3D). Such an increase in bursting in MAM rats may be indicative of neurons subthreshold for depolarization block.
Figure 3.
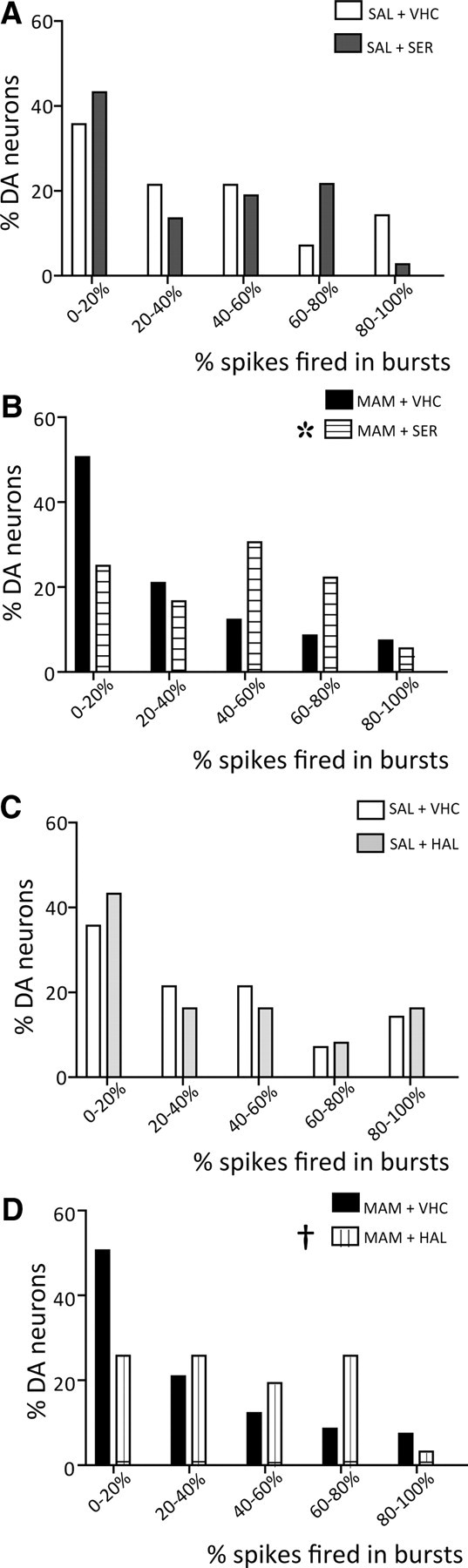
Acute administration of both APDs induced a rightward shift in the distribution of percentage burst firing of DA neurons recorded in MAM rats. A, C, Acute administration of either sertindole (A) or haloperidol (C) did not alter the distribution in percentage of burst firing of DA neurons recorded in saline rats (SAL+SER, dark gray bars; SAL+HAL, light gray bars). B, D, In contrast, when MAM rats were administered either sertindole (B, MAM+SER, horizontal line bars) or haloperidol (D, MAM+HAL, vertical line bars), there was a significant rightward shift in the distribution of percentage burst firing. Two-sample Kolmogorov–Smirnov test: *SER, p = 0.0063; †HAL, p = 0.0346.
Repeated administration of either sertindole or haloperidol decreased VTA DA neuron population activity in control and MAM-treated rats
Given that previous studies in control rats showed that APDs required 3+ weeks of administration to induce depolarization block (Bunney and Grace, 1978; Chiodo and Bunney, 1983; White and Wang, 1983a), the time course of the effects of repeated APD administration was tested in the MAM model. Sertindole or haloperidol was administered daily for up to 21 d to either saline or MAM offspring, and recordings of VTA DA neurons were performed at five distinct time points: after 1, 3, 7, 15, and 21 d of treatment.
Sertindole administration to MAM rats significantly decreased DA neuron population activity when compared to nontreated MAM rats (combined VHC: 1.7 ± 0.05, n = 20 rats, 261 DA neurons; acute SER: 0.8 ± 0.03, n = 6 rats, 40 DA neurons; 1 d: 0.7 ± 0.1, n = 6 rats, 36 DA neurons; 3 d: 0.6 ± 0.1, n = 4 rats, 21 DA neurons; 7 d: 0.5 ± 0.1, n = 5 rats, 21 DA neurons; 15 d: 0.5 ± 0.1, n = 5 rats, 18 DA neurons; 21 d: 0.4 ± 0.1, n = 5 rats, 18 DA neurons; two-way ANOVA, F(1,5,38) = 260.9, p < 0.001; Fig. 4A). In MAM rats treated repeatedly with sertindole, the number of DA neurons firing spontaneously was maintained across the first week of treatment (two-way ANOVA, p > 0.05); however, at 2 (two-way ANOVA, F(1,5,38) = 3.9, p = 0.003) and 3 (two-way ANOVA, F(1,5,38) = 3.9, p < 0.001) weeks, a further significant decrease was observed compared to acute administration (Fig. 4A). In saline rats, repeated systemic administration of sertindole did not significantly reduce population activity for the first 2 weeks (acute SER: 1.9 ± 0.03, n = 4 rats, 53 DA neurons; 1 d: 1.3 ± 0.3, n = 5 rats, 47 DA neurons; 3 d: 1.1 ± 0.3, n = 4 rats, 40 DA neurons; 7 d: 1.1 ± 0.2, n = 5 rats, 45 DA neurons; 15 d: 1.0 ± 0.1, n = 5 rats, 39 DA neurons; 21 d: 0.4 ± 0.1, n = 5 rats, 19 DA neurons; two-way ANOVA, p > 0.05; Fig. 4C), but did result in a significant decrease in population activity following 21 d of treatment (21 d SER vs all other SER treatments: two-way ANOVA, F(1,5,47) = 7.2, p < 0.019; Fig. 4B), which is consistent with previous studies (Skarsfeldt, 1992). The effect of sertindole on saline and MAM rats were significantly different following acute administration and during the first 2 weeks of treatment (two-way ANOVA, F(1,5,47) = 35.2, p < 0.001; Fig. 4B). However, at 21 d of repeated administration of sertindole in both saline and MAM rats, there was a further decrease in population activity which was not significantly different between the two groups (two-way ANOVA, p = 0.9; Fig. 4B).
Figure 4.
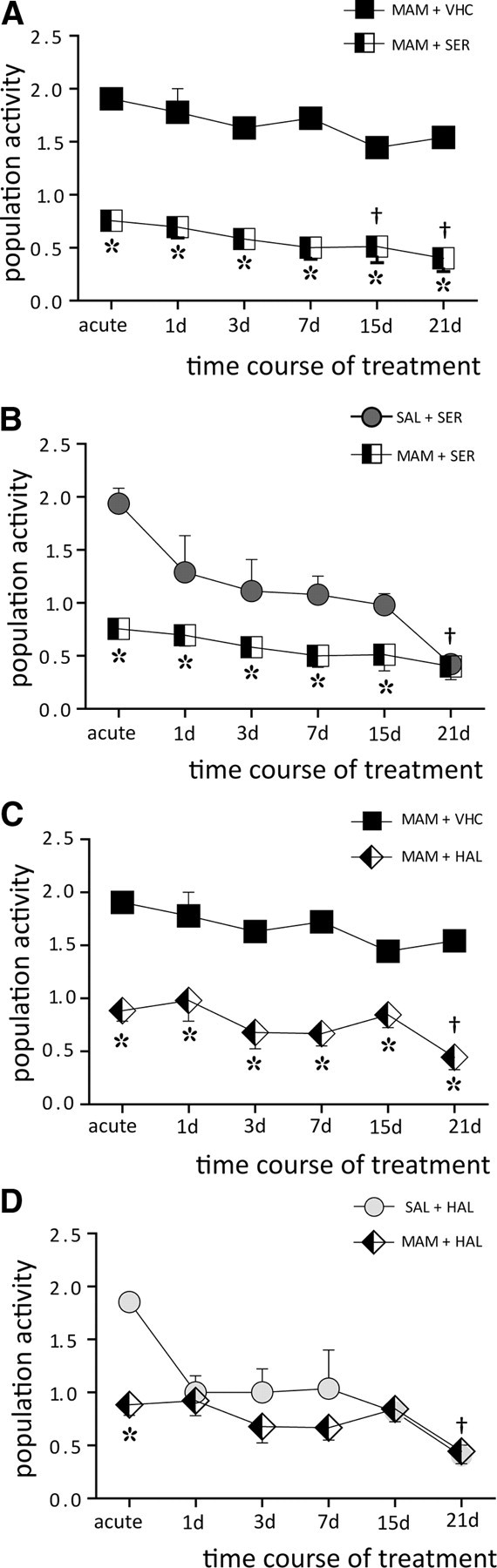
Repeated administration of antipsychotic drugs further reduced MAM VTA DA neuron population activity. Time courses of the effects of repeated antipsychotic drug treatment on saline and MAM rat VTA DA neuron population activity tested at six time points (see Materials and Methods). A, Repeated administration of sertindole to MAM rats (n = 31 rats, 154 DA neurons; black and white squares) induced a pronounced decrease in VTA DA neuron population activity compared to MAM rats injected with vehicle (n = 20 rats, 261 DA neurons; black squares) at each of the time points examined (*MAM + VHC vs MAM + SER, two-way ANOVA followed by Holm–Sidak multiple-comparison test, p < 0.001). Within the first 2 weeks of treatment, the effect of sertindole on population activity was not different from acute administration; however, at 2 and 3 weeks a further significant decrease was observed (†acute SER vs 15 d and acute SER vs 21 d: two-way ANOVA followed by Holm–Sidak multiple-comparison test, p = 0.003 and p < 0.001). B, As previously reported, treatment with the second-generation antipsychotic drug, sertindole, significantly reduced DA neuron population activity in saline rats (dark gray circles) only at 21 d (†significant difference between SAL + 21 d SER vs all other SAL + SER treatments: n = 28 rats, 243 DA neurons; two-way ANOVA followed by Holm–Sidak multiple-comparison test, p < 0.05). In MAM rats, the sertindole-induced decrease in population activity (black and white squares) was significantly different from the sertindole-induced decrease in saline (dark gray circles) for the first 2 weeks of treatment (*SAL + SER vs MAM + SER, two-way ANOVA followed by Holm–Sidak multiple-comparison test, p < 0.05). In contrast, at 21 d of sertindole administration both groups showed a significant decrease in DA neuron population activity compared to predrug controls (two-way ANOVA, p < 0.001). C, Similar to sertindole, repeated administration of haloperidol in MAM rats (black and white diamonds) significantly reduced DA neuron population activity compared to untreated MAM rats (black squares; two-way ANOVA followed by Holm–Sidak multiple-comparison test; n = 33 rats, 202 DA neurons; *MAM+VHC vs MAM+HAL, p < 0.001); however, a further decrease was observed in this case only at 21 d (two-way ANOVA followed by Holm–Sidak multiple-comparison test; †acute HAL vs 21 d, p = 0.011). D, Consistent with previous reports, haloperidol induced a significant decrease in population activity in saline rats (light gray circle) at 21 d of treatment (†SAL + 21 d HAL vs all other SAL + HAL treatments, n = 22 rats, 174 DA neurons: two-way ANOVA followed by Holm–Sidak multiple-comparison test, p < 0.05). In contrast to sertindole, haloperidol induced a significant difference in population activity of MAM vs SAL only following acute administration (*two-way ANOVA, p < 0.001), whereas the effect of repeated haloperidol treatment in MAM rats was not significantly different from controls for all of the other time points of treatment (two-way ANOVA, p > 0.05).
Repeated administration of haloperidol to MAM rats significantly decrease VTA DA neuron population activity compared to vehicle (acute HAL: 0.8 ± 0.1, n = 6 rats, 43 DA neurons; 1 d: 0.9 ± 0.1, n = 7 rats, 53 DA neurons; 3 d: 0.7 ± 0.1, n = 5 rats, 29 DA neurons; 7 d: 0.7 ± 0.1, n = 5 rats, 26 DA neurons; 15 d: 0.8 ± 0.1, n = 5 rats, 32 DA neurons; 21 d: 0.4 ± 0.1, n = 5 rats, 19 DA neurons; two-way ANOVA, F(1,5,41) = 133.5, p < 0.001; Fig. 4C). In addition, in MAM rats haloperidol induced a further change in population activity at 21 d of treatment compared to acute administration (two-way ANOVA, F(1,5,41) = 2.6, p = 0.011; Fig. 4C). Thus, repeated haloperidol administration in saline rats induced a pronounced decrease in population activity only at 21 d of treatment (acute HAL: 1.8 ± 0.1, n = 4 rats, 48 DA neurons; 1 d: 1.0 ± 0.2, n = 4 rats, 35 DA neurons; 3 d: 1.0 ± 0.2, n = 3 rats, 27 DA neurons; 7 d: 1.0 ± 0.3, n = 3 rats, 27 DA neurons; 15 d: 0.8 ± 0.1, n = 3 rats, 19 DA neurons; 21 d: 0.4 ± 0.1, n = 5 rats, 18 DA neurons; 21 d HAL vs all other HAL treatments: two-way ANOVA, F(1,5,44) = 9.9, p < 0.012; Fig. 4D) as reported previously (Bunney and Grace, 1978; Chiodo and Bunney, 1983; White and Wang, 1983b). Similar to sertindole, this decrease has been suggested to occur via the induction of depolarization block (Grace and Bunney, 1986; Grace et al., 1997). However, the number of neurons remaining spontaneously active following repeated haloperidol administration in MAM rats was not significantly different from saline across the time course of treatment (two-way ANOVA, p ≫ 0.05).
In contrast to the changes in population activity, there was no effect of either APD on DA neuron firing rate (two-way ANOVA, p > 0.05), burst firing (two-way ANOVA, p > 0.05), or distributions of these parameters (two-sample Kolmogorov–Smirnov test, p > 0.05) during the course of treatment in saline rats or in MAM rats. Furthermore, there were no differences when comparing MAM and saline rats (SAL + repeated APD vs MAM + repeated APD: two-way ANOVA, p > 0.05).
APDs reduced DA neuron population activity via induction of depolarization block
Our data indicate that acute and repeated APD administration induced a significant decrease in DA neuron population activity in the MAM-treated rats; however, whether this was due to inhibition or to depolarization block was not evident. Therefore, we examined whether the proposed overexcitation leading to depolarization block could be reversed by administering doses of the DA agonist apomorphine that had been shown previously to be effective in reversing depolarization block (Bunney and Grace, 1978). At the termination of recordings, apomorphine was injected intravenously and recordings were performed in the contralateral VTA. In rats treated with sertindole, 30 μg/kg apomorphine reversed the sertindole-induced decrease in population activity (acute: VTA(right) = 0.7, VTA(left) = 2.2, n = 1 rat; 1 d: VTA(right) = 1.1, VTA(left) = 1.8, n = 1 rat; 3 d: VTA(right) = 0.8, VTA(left) = 1.8, n = 1 rat; 7 d: VTA(right) = 0.8, VTA(left) = 1.8, n = 1 rat; 15 d: VTA(right) = 0.6, VTA(left) = 2.1, n = 2 rats; 21 d: VTA(right) = 0, VTA(left) = 2.0, n = 1 rat; t test, t = −8.3, p < 0.001; Fig. 5A). Administration of 80 or 120 μg/kg apomorphine in rats treated with haloperidol produced analogous effects, and similarly reversed the haloperidol-induced decrease in population activity at each of the time points examined (acute: VTA(right) = 0.7, VTA(left) = 2, n = 1 rat; 1 d: VTA(right) = 1.1, VTA(left) = 1.9, n = 2 rat; 3 d: VTA(right) = 0.4, VTA(left) = 1.7, n = 1 rats; 7 d: VTA(right) = 0.8, VTA(left) = 1.8; 15 d: VTA(right) = 1.0, VTA(left) = 1.6, n = 1 rat; 21 d: VTA(right) = 0.7, VTA(left) = 2.0, n = 1 rat; t test, t = −7.765, p < 0.001; Fig. 5B).
Figure 5.
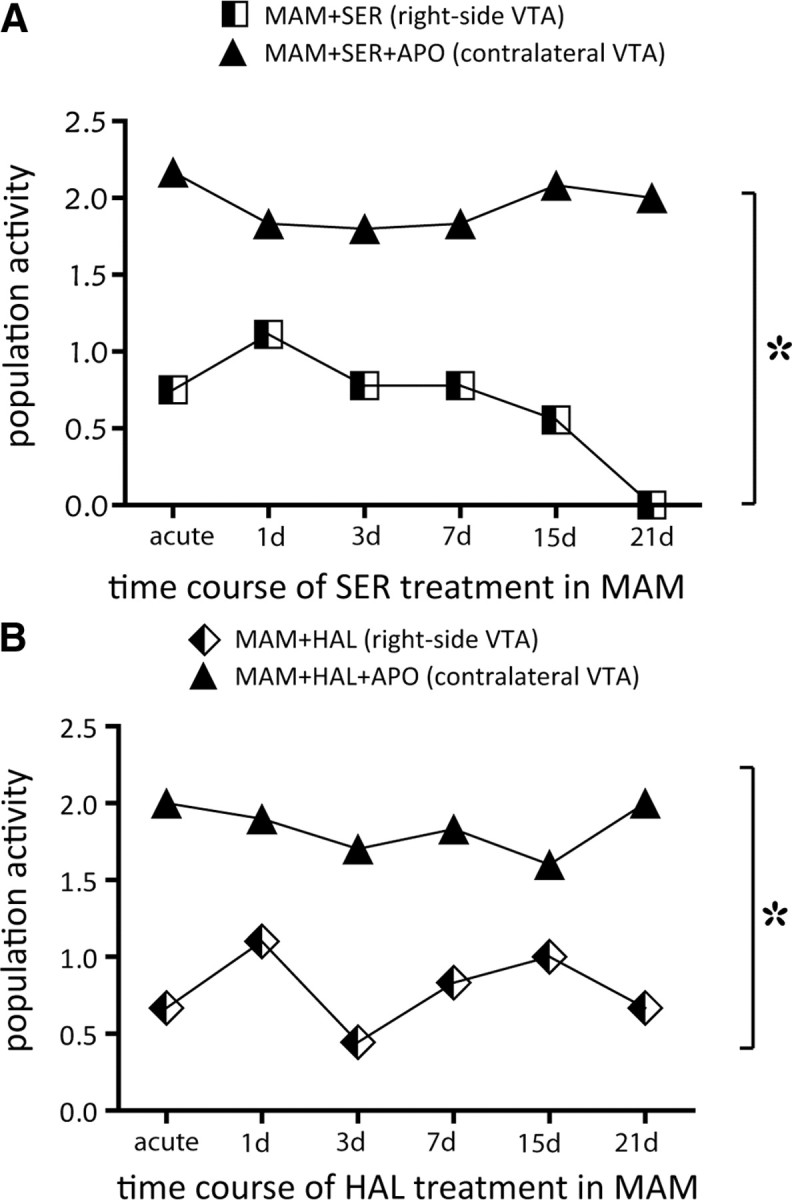
: Antipsychotic drug-induced decrease in VTA DA neuron population activity was due to the induction of depolarization block. A, In MAM rats, sertindole induced a decrease in DA neuron population activity at each of the six time points of treatment examined (MAM+SER, black and white squares). Following intravenous injection of 30 μg/kg apomorphine, DA neuron population activity on the contralateral side of the same rats was restored to predrug levels (MAM+SER+apomorphine, black triangles). B, Same as in A, but for haloperidol treatment (black and white diamonds) and haloperidol + 80–120 μg/kg apomorphine (black triangles). t test: *each point of the time course MAM + APD vs MAM + APD + apomorphine, p < 0.001.
Discussion
DA antagonist APDs have been in use for nearly 60 years; however, the mechanism by which they alleviate psychosis is still not completely understood. Research in animal models has to date been examined only in normal rats. Nonetheless, it is known that schizophrenia patients respond much differently to APD administration than the control population. In this study, we show that a developmental animal model of schizophrenia that has a clear hyperdopaminergic phenotype (Lodge and Grace, 2007) demonstrates electrophysiological responses to both first- and second-generation APDs that are qualitatively different from that observed in normal animals. Indeed, it is the presence of the hyper-responsive DA system, in the form of increased DA neuron population activity, that enables antipsychotic medications to restore baseline DA neuron activity levels, albeit not by restoring the system to a normal condition.
In control rats, acute APD administration leads to compensatory changes in activity, including increased firing rate and burst firing. However, the most robust change is in the number of DA neurons firing. Thus, following acute administration of either first-generation (Bunney and Grace, 1978) or second-generation (Chiodo and Bunney, 1983; White and Wang, 1983a; Skarsfeldt, 1992; Valenti and Grace, 2010) APDs, there is a twofold increase in the number of spontaneously firing DA neurons. However, if the APDs are given repeatedly over a 3+ week period, a different phenomenon ensues—i.e., one of depolarization block. In this state, the DA neurons are not firing due to overdepolarization and inactivation of spike generation (Grace and Bunney, 1986). This phenomenon has been repeated across a number of laboratories and drugs (Chiodo and Bunney, 1983; White and Wang, 1983a; Grace, 1991; Skarsfeldt, 1992). This delayed onset of depolarization block was proposed to explain the delayed development of therapeutic actions and parkinsonian side effects associated with these medications (Grace et al., 1997).
On the other hand, recent studies have pointed out that, although the parkinsonian side effects of first-generation APDs are delayed in development, the onset of antipsychotic efficacy occurs with a rapid time course. Thus, Agid et al. (2003) showed that the onset of antipsychotic action can occur within the first few doses of APD treatment. Although this does not parallel what one observes in normal rats, it is important to note that in the normal rat APDs induce a large increase in DA neuron population activity that can act to offset the effects of receptor blockade (Bunney and Grace, 1978; Chiodo and Bunney, 1983; White and Wang, 1983a). As a result, one would predict that in the normal individual, the effects of APDs administered acutely would be blunted. This contrasts with the condition in the MAM model of schizophrenia, in which DA neuron population activity is already near maximal, with 2× the number of DA neurons firing spontaneously due to a ventral hippocampal overdrive (Lodge and Grace, 2007). In this condition, the ability to increase DA neuron population activity is blunted, leading to the rapid induction of depolarization block. This is similar in nature to the effects of first-generation APDs in rats with partial lesions of the nigrostriatal DA system, in which doses of DA antagonists that do not substantially affect locomotion in control rats produce immediate depolarization block and akinesia in rats in which the DA system has already undergone compensation for the lesion (Bunney and Grace, 1978; Hollerman et al., 1992). In both lesion-induced depolarization block in the substantia nigra and APD-induced depolarization block produced acutely in the MAM-treated rat and following 21 d of drug administration in normal rats, the depolarization block is reversed by administering an agent that normally inhibits DA neuron firing—i.e., the direct-acting DA agonist apomorphine (Hollerman et al., 1992). This has consistently been an accepted method for distinguishing decreases in DA neuron population activity that are due to depolarization block from that resulting from inhibition of firing (e.g., maintained stress; Moore et al., 2001).
Although the APDs apparently were effective in the MAM-treated rat in inducing excitation-mediated inactivation of DA neuron firing, they did not produce a significant change in the average firing rate of the DA neurons. This was likely due to the effects produced across the population of neurons recorded. Thus, although there was not an average increase in firing rate, sertindole did cause a significant shift in the firing rate distribution in MAM rats (haloperidol showed a trend to cause a similar shift, but did not reach significance). This suggests that the change in firing rate was masked by the change in distribution—i.e., the activation of nonfiring neurons into slow firing balancing out the shift of firing neurons to faster rates. With respect to burst firing, both sertindole and haloperidol shifted the distribution significantly to the right; this increase in neurons showing a high degree of burst firing was likely indicative of neurons that were approaching depolarization block (Grace et al., 1997).
With continued treatment, additional differences between the saline and MAM rats emerged. Thus, in the MAM rats 2 weeks of sertindole treatment resulted in a maintained lower level of DA neuron population activity compared to the baseline state; this contrasted with the control rats in which baseline activity was reinstated with treatment. Although haloperidol only decreased population activity below saline with acute administration, it nonetheless did induce depolarization block across all time points. However, at 21 d of APD treatment, all groups of rat demonstrated equivalent levels of population activity corresponding to induction of depolarization block in saline animals and a small accentuation of depolarization block in the MAM-treated rat. Therefore, with continued treatment, depolarization block tended to become more prominent. Such a condition may be consistent with the propensity of APDs to show maximal efficacy with >3 weeks of treatment, without showing tolerance (Johnstone et al., 1978; Palmstierna and Wistedt, 1987).
Of course, these studies were performed on either control rats or MAM-treated rats with nearly identical prenatal disruption. In contrast, schizophrenia patients show a wide range of premorbid states and responses to APDs. Interestingly, studies have shown that the level of tonic striatal DA D2 occupancy predicts faster responses of psychotic symptoms to APDs (Abi-Dargham et al., 2000). Furthermore, it is generally accepted that patients with the most prominent psychotic signs typically show the best response to antipsychotic medication (Murray and Dean, 2008). By analogy to the MAM-treated rat, this would be consistent with a condition in which the patients showing the greatest DA neuron population activity would also be more susceptible to the rapid induction of depolarization block. Indeed, Kinon et al. (2010) have shown that the response of the patient to initial APD treatment best predicts their eventual therapeutic efficacy, which would again be consistent with a DA system that is most readily overdriven by the APD.
Of course much of this argument depends on the validity of the MAM model. The model has shown substantial face and construct validity with respect to behavior, pharmacology, and neuroanatomical changes (Moore et al., 2006). Furthermore, the model does demonstrate consistency with respect to the behavioral hyper-responsivity to amphetamine (Moore et al., 2006; Lodge and Grace, 2007), which appears to be a good correlate of DA neuron population activity across models (Lodge and Grace, 2007; Cifelli and Grace, 2011; Valenti et al., 2011). Therefore, although DA neuron population activity cannot be evaluated in human schizophrenia patients, the correspondence to increased response to amphetamine in imaging studies (Laruelle et al., 1996; Abi-Dargham et al., 2000) and with respect to exacerbation of psychosis (Janowsky et al., 1973; Laruelle et al., 1999) lends credence to this association.
In summary, the rapid induction of depolarization block by therapeutically effective APDs is consistent with the dopaminergic nature of psychosis, the rapid response of schizophrenia patients to antipsychotic medication, and the lack of substantial development of tolerance. Physiologically, this appears to be produced by reversing what we propose to underlie psychosis—i.e., a pathologically high number of DA neurons firing. Thus, the primary effect of induction of partial depolarization block is to restore the basal condition of DA neuron population activity. However, this is not to say that the DA system is restored to normal, since partial depolarization block is not likely to be the “normal” state of the DA system. Instead, it is inducing an offsetting pathological condition, one in which DA neuron population activity is attenuated, but also where increases in population activity cannot be produced when environmental conditions warrant. A much more effective approach would be to attack the disorder at the site of pathology, which we and others have proposed may be the limbic hippocampal region (Heckers et al., 1998; Malaspina et al., 1999; Medoff et al., 2001; Lodge and Grace, 2008; Grace, 2010; Tamminga et al., 2010). If a therapeutic approach that reverses the disrupted systems that are responsible for overdriving DA neuron population activity is developed, it is highly likely that this will be reflected in a more positive patient outcome. Nonetheless, this study provides direct evidence that the current generations of APDs, via overdrive of DA neurons, can restore somewhat normal responses. However, by doing so, the ability of the DA system to respond to normal activating stimuli by increasing DA neuron population activity is also blunted.
Footnotes
This work was supported in part by a research grant from H. Lundbeck H/S and United States Public Health Service Grant MH57440 to A.A.G. and the Ri.MED Foundation to P.C. We thank Nicole MacMurdo and Sandra Lolic for technical assistance and Brian Lowry for the development of the custom-designed computer software Neuroscope. Brian Lowry was affiliated with the Department of Neuroscience, University of Pittsburgh (Pittsburgh, PA), and employed by A.A.G. at the time this work was completed.
A.A.G. has the following conflicts to disclose: consultant for Johnson & Johnson; research grants from Lundbeck, GSK, EMD Serrono, pharmaceutical company talks (Abbott, Roche, Lilly); advisory boards (as nonmember invited speaker, including Merck).
References
- Abi-Dargham A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol. 2004;7(Suppl 1):S1–S5. doi: 10.1017/S1461145704004110. [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia.[see comment] Proc Natl Acad Sci U S A. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Dargham A, Kegeles LS, Zea-Ponce Y, Mawlawi O, Martinez D, Mitropoulou V, O'Flynn K, Koenigsberg HW, Van Heertum R, Cooper T, Laruelle M, Siever LJ. Striatal amphetamine-induced dopamine release in patients with schizotypal personality disorder studied with single photon emission computed tomography and [123I]iodobenzamide. Biol Psychiatry. 2004;55:1001–1006. doi: 10.1016/j.biopsych.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Agid O, Kapur S, Arenovich T, Zipursky RB. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry. 2003;60:1228–1235. doi: 10.1001/archpsyc.60.12.1228. [DOI] [PubMed] [Google Scholar]
- Arnt J. Differential effects of classical and newer antipsychotics on the hypermotility induced by two dose levels of D-amphetamine. Eur J Pharmacol. 1995;283:55–62. doi: 10.1016/0014-2999(95)00292-s. [DOI] [PubMed] [Google Scholar]
- Baldessarini RJ, Cohen BM, Teicher MH. Significance of neuroleptic dose and plasma level in the pharmacological treatment of psychoses. Arch Gen Psychiatry. 1988;45:79–91. doi: 10.1001/archpsyc.1988.01800250095013. [DOI] [PubMed] [Google Scholar]
- Bunney BS, Grace AA. Acute and chronic haloperidol treatment: comparison of effects on nigral dopaminergic cell activity. Life Sci. 1978;23:1715–1727. doi: 10.1016/0024-3205(78)90471-x. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963;20:140–144. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia—implications for schizophrenia and Parkinson's disease. Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- Chiodo LA, Bunney BS. Typical and atypical neuroleptics: differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. J Neurosci. 1983;3:1607–1619. doi: 10.1523/JNEUROSCI.03-08-01607.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifelli P, Grace AA. Pilocarpine induced temporal lobe epilepsy in the rat is associated with increased dopamine neuron activity. Int J Neuropsychopharmacol. 2011 doi: 10.1017/S1461145711001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cincotta SL, Rodefer JS. Emerging role of sertindole in the management of schizophrenia. Neuropsychiatr Dis Treat. 2010;6:429–441. doi: 10.2147/ndt.s7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Wilson AA, Houle S, Kapur S. Amphetamine pretreatment induces a change in both D2-Receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol Psychiatry. 2004;55:1188–1194. doi: 10.1016/j.biopsych.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31:330–341. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Grace AA. Dopamine system dysregulation by the ventral subiculum as the common pathophysiological basis for schizophrenia psychosis, psychostimulant abuse, and stress. Neurotox Res. 2010;18:367–376. doi: 10.1007/s12640-010-9154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons—1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: analysis using in vivo intracellular recording. J Pharmacol Exp Ther. 1986;238:1092–1100. [PubMed] [Google Scholar]
- Grace AA, Bunney BS, Moore H, Todd CL. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997;20:31–37. doi: 10.1016/S0166-2236(96)10064-3. [DOI] [PubMed] [Google Scholar]
- Hand TH, Hu XT, Wang RY. Differential effects of acute clozapine and haloperidol on the activity of ventral tegmental (A10) and nigrostriatal (A9) dopamine neurons. Brain Res. 1987;415:257–269. doi: 10.1016/0006-8993(87)90207-1. [DOI] [PubMed] [Google Scholar]
- Heckers S, Rauch SL, Goff D, Savage CR, Schacter DL, Fischman AJ, Alpert NM. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci. 1998;1:318–323. doi: 10.1038/1137. [DOI] [PubMed] [Google Scholar]
- Hollerman JR, Abercrombie ED, Grace AA. Electrophysiological, biochemical, and behavioral studies of acute haloperidol-induced depolarization block of nigral dopamine neurons. Neuroscience. 1992;47:589–601. doi: 10.1016/0306-4522(92)90168-2. [DOI] [PubMed] [Google Scholar]
- Janowsky DS, el-Yousel MK, Davis JM, Sekerke HJ. Provocation of schizophrenic symptoms by intravenous administration of methylphenidate. Arch Gen Psychiatry. 1973;28:185–191. doi: 10.1001/archpsyc.1973.01750320023004. [DOI] [PubMed] [Google Scholar]
- Jiang LH, Tsai M, Wang RY. Chronic treatment with high doses of haloperidol fails to decrease the time course for the development of depolarization inactivation of midbrain dopamine neurons. Life Sci. 1988;43:75–81. doi: 10.1016/0024-3205(88)90239-1. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Crow TJ, Frith CD, Carney MW, Price JS. Mechanism of the antipsychotic effect in the treatment of acute schizophrenia. Lancet. 1978;1:848–851. doi: 10.1016/s0140-6736(78)90193-9. [DOI] [PubMed] [Google Scholar]
- Kapur S, Remington G. Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry. 2001;50:873–883. doi: 10.1016/s0006-3223(01)01251-3. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: A new hypothesis. Am J Psychiatry. 2001;158:360–369. doi: 10.1176/appi.ajp.158.3.360. [DOI] [PubMed] [Google Scholar]
- Kinon BJ, Chen L, Stauffer VL, Sniadecki J, Ascher-Svanum H, Kollack-Walker S, Jacob J, Kapur S. Early onset of antipsychotic action in schizophrenia: evaluating the possibility of shorter acute efficacy trials. J Clin Psychopharmacol. 2010;30:286–289. doi: 10.1097/JCP.0b013e3181dcb7c3. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006;31:1356–1361. doi: 10.1038/sj.npp.1300963. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Amphetamine activation of hippocampal drive of mesolimbic dopamine neurons: a mechanism of behavioral sensitization. J Neurosci. 2008;28:7876–7882. doi: 10.1523/JNEUROSCI.1582-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Divergent activation of ventromedial and ventrolateral dopamine systems in animal models of amphetamine sensitization and schizophrenia. Int J Neuropsychopharmacol. 2011 doi: 10.1017/S1461145711000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina D, Storer S, Furman V, Esser P, Printz D, Berman A, Lignelli A, Gorman J, Van Heertum R. SPECT study of visual fixation in schizophrenia and comparison subjects. Biol Psychiatry. 1999;46:89–93. doi: 10.1016/s0006-3223(98)00306-0. [DOI] [PubMed] [Google Scholar]
- Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11:543–550. doi: 10.1002/hipo.1070. [DOI] [PubMed] [Google Scholar]
- Moore H, Rose HJ, Grace AA. Chronic cold stress reduces the spontaneous activity of ventral tegmental dopamine neurons. Neuropsychopharmacology. 2001;24:410–419. doi: 10.1016/S0893-133X(00)00188-3. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60:253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray RM, Dean K. Schizophrenia and related disorders. In: Murray RM, Kendler KS, McGuffin P, Wesseley S, Castle DJ, editors. Essential psychiatry. Ed 4. Cambridge: Cambridge UP; 2008. pp. 284–319. [Google Scholar]
- Nyberg S, Nordstrom AL, Halldin C, Farde L. Positron emission tomography studies on D2 dopamine receptor occupancy and plasma antipsychotic drug levels in man. Int Clin Psychopharmacol. 1995;10(Suppl 3):81–85. [PubMed] [Google Scholar]
- Palmstierna T, Wistedt B. Absence of acquired tolerance to neuroleptics in schizophrenic patients. Am J Psychiatry. 1987;144:1084–1085. doi: 10.1176/ajp.144.8.1084. [DOI] [PubMed] [Google Scholar]
- Seeman P. Dopamine receptor sequences. Therapeutic levels of neuroleptics occupy D2 receptors, clozapine occupies D4. Neuropsychopharmacology. 1992;7:261–284. [PubMed] [Google Scholar]
- Seeman P, Chau-Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci U S A. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarsfeldt T. Electrophysiological profile of the new atypical neuroleptic, sertindole, on midbrain dopamine neurones in rats: acute and repeated treatment. Synapse. 1992;10:25–33. doi: 10.1002/syn.890100105. [DOI] [PubMed] [Google Scholar]
- Skarsfeldt T, Perregaard J. Sertindole, a new neuroleptic with extreme selectivity on A10 versus A9 dopamine neurones in the rat. Eur J Pharmacol. 1990;182:613–614. doi: 10.1016/0014-2999(90)90067-g. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167:1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- Valenti O, Grace AA. Antipsychotic drug-induced increases in ventral tegmental area dopamine neuron population activity via activation of the nucleus accumbens-ventral pallidum pathway. Int J Neuropsychopharmacol. 2010;13:845–860. doi: 10.1017/S1461145709990599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Lodge DJ, Grace AA. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci. 2011;31:4280–4289. doi: 10.1523/JNEUROSCI.5310-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Hagino Y. The atypical antipsychotic sertindole enhances efflux of dopamine and its metabolites in the rat cortex and striatum. Eur J Pharmacol. 1999;367:19–23. doi: 10.1016/s0014-2999(98)00936-4. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Gallhofer B. Cognitive function in schizophrenia. Int Clin Psychopharmacol. 1997;12(Suppl 4):S29–S36. doi: 10.1097/00004850-199709004-00006. [DOI] [PubMed] [Google Scholar]
- West AR, Grace AA. Striatal nitric oxide signaling regulates the neuronal activity of midbrain dopamine neurons in vivo. J Neurophysiol. 2000;83:1796–1808. doi: 10.1152/jn.2000.83.4.1796. [DOI] [PubMed] [Google Scholar]
- White FJ, Wang RY. Differential effects of classical and atypical antipsychotic drugs on A9 and A10 dopamine neurons. Science. 1983a;221:1054–1057. doi: 10.1126/science.6136093. [DOI] [PubMed] [Google Scholar]
- White FJ, Wang RY. Comparison of the effects of chronic haloperidol treatment on A9 and A10 dopamine neurons in the rat. Life Sci. 1983b;32:983–993. doi: 10.1016/0024-3205(83)90929-3. [DOI] [PubMed] [Google Scholar]