

Abstract
The transcription factor IRF-1 has been implicated in tumor suppression: IRF-1 suppresses cell transformation and mediates apoptosis in vitro. Here we show that the loss of IRF-1 alleles per se has no effect on spontaneous tumor development in the mouse but dramatically exacerbates previous tumor predispositions caused by the c-Ha-ras transgene or by nullizygosity for p53. Grossly altered tumor spectrum, as compared to p53-null mice, was also observed in mice lacking both IRF-1 and p53, and cells from these mice show significantly higher mutation rate. Our results suggest that IRF-1 is a new member of the tumor susceptibility genes.
Keywords: IRF-1, c-Ha-ras, p53, tumor susceptibility gene, mutation frequency
The transcription factor IRF-1 (interferon regulatory factor-1) was originally identified as a regulator of the interferon (IFN) system (Miyamoto et al. 1988). More recent studies using IRF-1-null (IRF-1−/−) mice have revealed that IRF-1 has a crucial role in many aspects of host defense: It is essential in IFN-induced antiviral and antibacterial responses (Kamijo et al. 1994; Kimura et al. 1994), in the Th1-type adaptive immune response, and in the development of natural killer (NK) cells (Lohoff et al. 1997; Taki et al. 1997; Ogasawara et al. 1998).
Accumulating evidence has also suggested that IRF-1 controls tumor susceptibility. Transformed phenotypes of c-myc- or fosB-expressing cells, for example, can be suppressed by ectopic expression of IRF-1 (Tanaka et al. 1994b). Furthermore, unlike primary mouse embryonic fibroblasts (MEFs) from wild-type mice, MEFs from mutant mice homozygous for IRF-1 deficiency undergo transformation upon expression of an activated form of c-Ha-ras (Tanaka et al. 1994a). In addition, IRF-1 is essential to DNA damage-induced apoptosis in proliferating T lymphocytes and MEFs expressing an activated form of c-Ha-ras (Tanaka et al. 1994a; Tamura et al. 1995). Interestingly, IRF-1 also regulates DNA damage-induced cell cycle arrest in collaboration with the tumor suppressor p53 through transcriptional activation of the p21WAF1/CIP1 gene (Tanaka et al. 1996).
The human IRF-1 gene has been mapped to 5q31.1 (Willman et al. 1993). Genetic as well as epigenetic alterations in IRF-1 gene expression have been reported in human cancers. Defects in one or both IRF-1 alleles accompanied by deletion or translocation of 5q have been observed in acute leukemia (Willman et al. 1993). In addition, loss of functional IRF-1 mRNA expression due to skipping of specific exons has been reported in ∼20% of patients with myelodysplastic syndrome (MDS) or overt leukemia developing from MDS (Harada et al. 1994). More recently, frequent loss of heterozygosity at the IRF-1 locus has been reported in human gastric and esophageal cancer patients (Ogasawara et al. 1996; Tamura et al. 1996), among whom an inactivating point mutation in the IRF-1 gene was detected on the residual allele in at least one case of gastric cancer (Nozawa et al. 1998).
Although these observations lend support to the role of IRF-1 in tumor suppression, no systematic analysis has been carried out as to how the loss of IRF-1 affects tumor susceptibility in vivo. Moreover, subsequent to that in traditional tumor suppressor genes, interest has also grown in a class of tumor susceptibility genes that may suppress tumor development by indirect means (Demant 1992; Ghebranious and Donehower 1998; Kinzler and Vogelstein 1998). On this basis, we considered it valuable to examine to what extent loss-of-function mutation in IRF-1 alleles would affect tumor susceptibility. In this study we investigated the role of IRF-1 in tumor suppression in mice carrying null mutations in IRF-1 alleles (IRF-1−/− mice), with otherwise wild-type background and with backgrounds predisposed to tumor development owing to either expression of the c-Ha-ras transgene or null mutations in p53 alleles.
Results and Discussion
We first conducted a long-term investigation of spontaneous tumor development in a large cohort of IRF-1−/− mice. Only 2% of IRF-1−/− mice (6/315) developed tumors, characterized as malignant fibrous histiocytoma-like sarcoma, up to 200 days after birth (Figs. 1A and 2A). Although no tumor development was observed in wild-type littermates (0/625) during the same period (Fig. 1A), the difference between the two groups was not statistically significant (P = 0.12, Wilcoxon test). These findings thus indicated that the loss of IRF-1 expression per se provides little if any contribution to spontaneous tumor development. The question then arose as to whether the loss of IRF-1 affects tumor susceptibility when combined with other changes in oncogenes or tumor suppressor genes.
Figure 1.
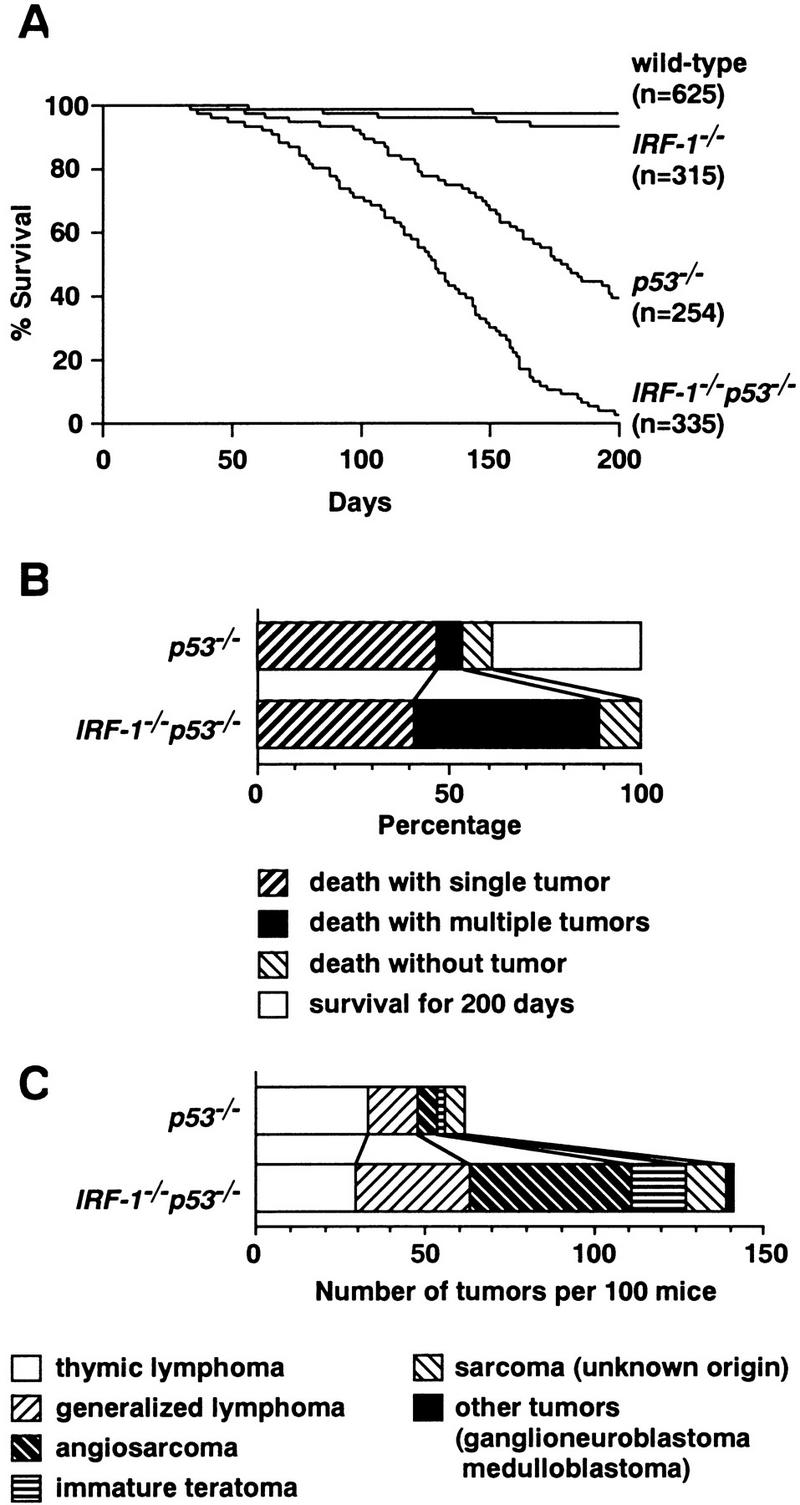
Survival rate, cause of death, and histology of tumors in subject mice. (A) Survival curves of wild-type, IRF-1−/−, p53−/−, and IRF-1−/−p53−/− mice followed up to 200 days by the Kaplan–Meier method. Mice were sacrificed upon becoming apparently moribund. (n) Total number of mice in each genotype. At 200 days, 2% of IRF-1−/− mice, 56% of p53−/− mice, and 96% of IRF-1−/−p53−/− mice developed tumors; 0.2% of wild-type mice died before 200 days, but no tumors were found. (B) The first 100 mice of each genotype were monitored for up to 200 days and autopsied to assess for the presence and number of tumors. (C) Raw numbers of histological types of tumors in p53−/− and IRF-1−/−p53−/− mice. Data shown are from the same cohort of 100 mice as in B. The bar patterns for tumor types are shown in B and C.
Figure 2.
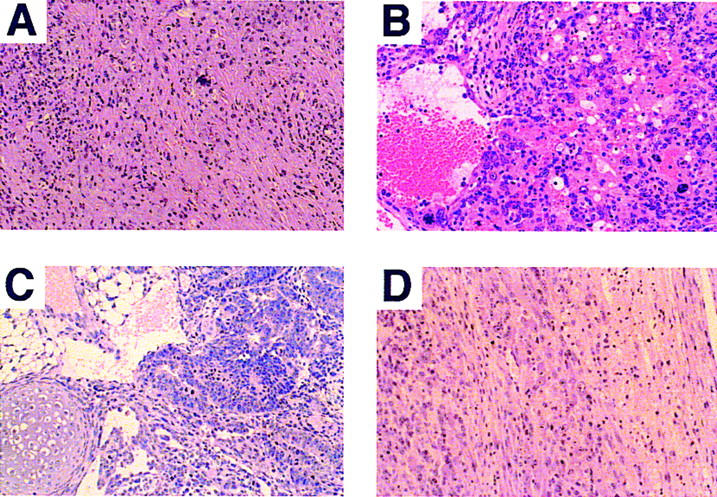
Histological analysis of representative tumors obtained from IRF-1−/− and IRF-1−/−p53−/− mice (hematoxylin and eosin staining). (A) Malignant fibrous histiocytoma-like sarcoma characteristically found in IRF-1−/− mice. Tumor chiefly consists of atypical spindle-shaped cells. (B) Angiosarcoma in an IRF-1−/−p53−/− mouse. Atypical tumor cells surround individual or groups of erythrocytes and also form neoplastic blood vessels. (C) Immature teratoma in testis of a male IRF-1−/−p53−/− mouse. Tumor consists of immature adenomatous, adipose-like, and chondroid tissue components. (D) Ganglioneuroblastoma in an IRF-1−/−p53−/− mouse. The spinal cord (right) is invaded by neuron-like tumor cells with enlarged nuclei and prominent nucleoli (left). This type of tumor was not observed in singly null mice. Original magnifications: (A,C,D) 500×; (B) 600×.
In view of our previous finding that IRF-1-deficient MEFs expressing activated c-Ha-ras undergo transformation and become resistant to DNA damage-induced apoptosis (Tanaka et al. 1994a), we examined the effect of loss-of-function mutation in the IRF-1 gene on tumor development in mice carrying the human c-Ha-ras gene. IRF-1−/− mice were crossed with mice carrying five to six copies of normal human c-Ha-ras gene (rasH2 mice; Saitoh et al. 1990) to generate rasH2 mice with an IRF-1-null background. All mice were sacrificed at 6 months after birth. Whereas only 7% (2/30) of rasH2 mice heterozygous for the IRF-1 mutation (IRF-1+/−/rasH2 mice) developed tumors during this period, a total of 44% (12/27) of IRF-1−/−/rasH2 mice developed tumors in various organs, among which angiosarcomas were found most frequently (76%; 19 of 25 tumors; Table 1). This tumor spectrum is similar to that described originally in rasH2 mice (Saitoh et al. 1990). These results suggest that loss of IRF-1 contributes to tumor development in conjunction with the c-Ha-ras gene in vivo.
Table 1.
Tumors observed in IRF-1−/−/rasH2 and IRF-1+/−/rasH2 mice by 6 months of age
|
Mouse line (tumor incidence)a
|
Case
|
Sex
|
Tumor type
|
Organ (no. of sites)
|
|---|---|---|---|---|
| IRF-1−/−/rasH2 | 1 | M | angiosarcoma | skin |
| (12/27; 44%) | sarcoma (NOS)b | abdomen (4) | ||
| 2 | M | angiosarcoma | skin | |
| papilloma | skin | |||
| 3 | M | angiosarcoma | skin (2), spleen, liver | |
| 4 | M | angiosarcoma | skin (2) | |
| 5 | M | angiosarcoma | skin (2) | |
| 6 | M | angiosarcoma | skin (2) | |
| 7 | M | angiosarcoma | spleen | |
| 8 | F | angiosarcoma | skin | |
| 9 | F | angiosarcoma | skin | |
| 10 | F | angiosarcoma | skin | |
| 11 | F | angiosarcoma | skin | |
| 12 | F | angiosarcoma | kidney | |
| IRF-1+/−/rasH2 | 1 | M | papilloma | skin |
| (2/30; 7%) | 2 | M | angiosarcoma | skin |
Number of tumor-bearing mice/total number of mice. Tumors larger than 5 mm in diameter were diagnosed as positive.
Not otherwise specified.
We next investigated the relationship between IRF-1 and p53 on tumor expression. One of the best characterized mouse models for tumor suppression, which are notable for spontaneous tumor development, is the p53-deficient mouse (Jacks 1996; Ghebranious and Donehower 1998). Because IRF-1 cooperates with p53 in regulation of the cell cycle (Tanaka et al. 1996) and both IRF-1 and p53 are essential to oncogene-induced apoptosis (Tanaka et al. 1994a), we were particularly interested to determine if combined loss-of-function mutations in the IRF-1 and p53 alleles would affect tumor development in any way. That is, if IRF-1 functions only as a mediator in some of the p53 pathways, we would not expect superimposition of IRF-1 null mutations to alter tumor susceptibility of p53−/− mice. To address these issues, we generated mice carrying null mutations for both IRF-1 and p53 alleles (IRF-1−/−p53−/− mice). Results showed that whereas only 56% (137/254) of p53−/− mice showed tumor development within 200 days, tumor incidence increased to 96% (322/335) in IRF-1−/−p53−/− mice (Fig. 1A). Furthermore, death due to tumors was observed at a much earlier age in IRF-1−/−p53−/− mice (Fig. 1A), and the frequency of multiple tumors in individual mice was increased approximately sevenfold (Fig. 1B). Moreover, the spectrum of developed tumors was also significantly altered in IRF-1−/−p53−/− mice; the incidence of generalized lymphoma, angiosarcoma, and immature teratoma were notably increased, whereas that of thymic lymphoma was decreased (Fig. 1C). It is noteworthy that the doubly deficient mice developed tumors that were not observed in singly null mice, namely ganglioneuroblastoma and medulloblastoma (Fig. 1C). Histopathological data for some of these characteristic tumors are presented in Figure 2B–D.
Taken together, the early onset of tumorigenesis, increased tumor incidence, enhanced multiplicity, and notable alteration of the tumor spectrum in IRF-1−/−p53−/− mice suggested again that the loss of IRF-1 affects tumor susceptibility in mice. Moreover, the results suggest that IRF-1 manifests tumor suppressor activity in vivo through a mechanism(s) distinct from those for p53; that is, IRF-1 is not hypostatic to p53 in affecting tumor predisposition.
It has been reported that IRF-1−/− mice show several immunological disorders, most notably a severe defect in the development of NK cells (Ogasawara et al. 1998). It was therefore conceivable that the accelerated tumor development observed in IRF-1−/− p53−/− mice may be due to combination of the loss of tumor suppression by p53 and impairment of the IRF-1-controlled tumor surveillance system. To test this possibility, we next generated IRF-1−/−p53−/− ↔ p53−/− chimeric mice by aggregation of respective embryos (see Materials and Methods for details), in which the immunological disorders due to IRF-1 deficiency were no longer detectable due to the contribution of cells containing wild-type IRF-1 genes (i.e., p53−/− cells; data not shown). In these mice, cells more susceptible to tumorigenesis could be assessed by examination of the genotype of the developed tumors. PCR was done to confirm ∼50% chimerism of the two genotypes in peripheral blood leukocytes and tail tissues isolated from these chimeric mice at a stage before any tumor development was detectable, and Southern blot analysis was done at sacrifice to confirm this chimerism in non-tumor-bearing tissues (data not shown). As summarized in Table 2, of 12 tumors developed in 10 chimeric mice, 9 were found to originate in IRF-1−/− p53−/− cells and only 3 in p53−/− cells. We also compared tumor incidence between IRF-1−/−p53−/− ↔ wild-type and p53−/− ↔ wild-type aggregation chimeric mice. As expected, IRF-1−/−p53−/− ↔ wild-type chimeric mice developed more tumors (mostly lymphomas) and died earlier than p53−/− ↔ wild-type chimeric mice (data not shown). Thus, IRF-1−/−p53−/− cells appear to be intrinsically more susceptible to tumorigenesis than p53−/− cells in vivo, and the enhanced tumor-prone phenotype of IRF-1−/−p53−/− mice may be directly attributable to the profound oncogenic potential of IRF-1−/−p53−/− cells.
Table 2.
Origin of tumors observed in IRF-1−/−p53−/− ↔ p53−/− chimeric mice
|
Case
|
Sex
|
Survival (days)
|
Tumor type
|
Genotype of tumora
|
|---|---|---|---|---|
| 1 | M | 71 | thymic lymphoma | IRF-1−/−p53−/− |
| 2 | M | 90 | thymic lymphoma | IRF-1−/−p53−/− |
| 3 | F | 95 | thymic lymphoma | p53−/− |
| angiosarcoma | p53−/− | |||
| 4 | M | 96 | angiosarcoma | IRF-1−/−p53−/− |
| 5 | M | 98 | lipoma | IRF-1−/−p53−/− |
| 6 | M | 110 | sarcoma (NOSb) | p53−/− |
| 7 | M | 113 | angiosarcoma | IRF-1−/−p53−/− |
| 8 | M | 131 | angiosarcoma | IRF-1−/−p53−/− |
| 9 | M | 153 | generalized lymphoma | IRF-1−/−p53−/− |
| 10 | M | 190 | thymic lymphoma | IRF-1−/−p53−/− |
| generalized lymphoma | IRF-1−/−p53−/− |
The genotype of each tumor was determined by PCR or Southern blot analysis of the IRF-1 and p53 alleles as described (Matsuyama et al. 1993; Tsukada et al. 1993).
Not otherwise specified.
Given that tumors develop from cells that have acquired genetic alterations in critical tumor suppressor genes and oncogenes, increased susceptibility to mutations is an important factor in tumorigenesis (Strauss 1998). To gain further understanding of the tumor-prone phenotype of IRF-1−/−p53−/− cells, we next examined the frequency of mutations induced by DNA-damaging agents that lead to ouabain resistance (Ouar) in MEFs. Whereas wild-type and IRF-1−/− MEFs showed no Ouar colony formation when treated with cisplatin, p53−/− MEFs formed a significant number of Ouar colonies. Interestingly, the number of Ouar colonies increased approximately fourfold in IRF-1−/−p53−/− MEFs (Fig. 3A). IRF-1−/−p53−/− MEFs also showed a high frequency of Ouar colony formation when treated with N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), a DNA alkylating mutagen, whereas wild-type and singly null MEFs showed no significant colony formation (Fig. 3B). Furthermore, whereas p53−/− MEFs showed sensitivity to cisplatin in a dose-dependent manner, as reported previously (Hawkins et al. 1996), this sensitivity was increased further in the IRF-1−/−p53−/− MEFs (data not shown). This increase in Ouar colony numbers by cisplatin and MNNG treatment and hypersensitivity to cisplatin in IRF-1−/−p53−/− MEFs suggest that IRF-1 may be involved in DNA repair systems in combination with p53, such as nucleotide excision repair, base excision repair, and other repair mechanism by O6-methylguanine-DNA methyltransferase (Tanaka and Wood 1994; Sekiguchi et al. 1996). However, we found no gross alterations in the expression of genes known to be involved in these repair systems by loss of IRF-1 (data not shown); hence, the mechanism by which IRF1 mediates regulation of DNA repair remains to be characterized.
Figure 3.
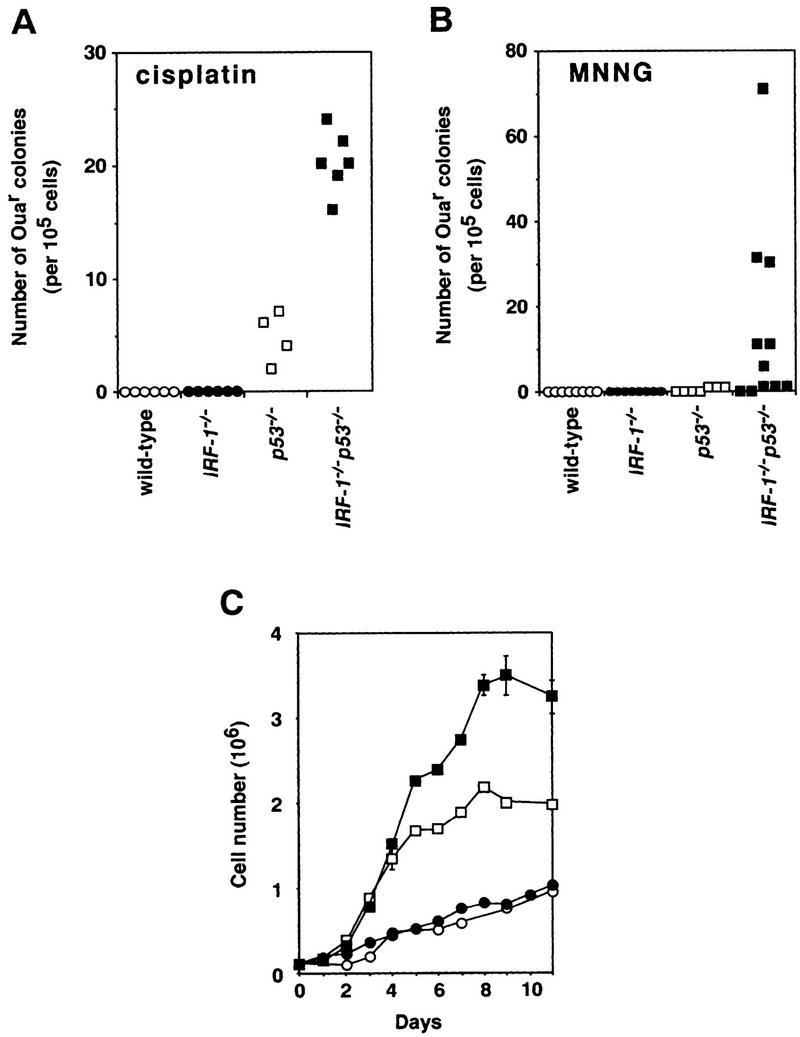
Cellular abnormalities in IRF-1−/−p53−/− MEFs. (A,B) Mutation frequency in MEFs treated with cisplatin (0.05 μg/ml for 72 hr; A) or MNNG (5 μm for 3 hr; B). The numbers of Ouar colonies/105 MEF cells were plotted. Plating efficiencies of p53−/− and IRF-1−/−p53−/− MEFs were similar (∼90%). (C) Representative growth curves of MEFs. Cells were plated at a density of 105 cells/35-mm dish at passage 4, and cell number counted (bars indicate s.d.). Experiments performed on at least four clones of each genotype showed the results to be essentially reproducible. Mean doubling times in log phase for wild-type and IRF-1−/− MEFs were similar (66.6 ± 39.8 and 56.9 ± 18.6 hr, respectively). p53−/− and IRF-1−/−p53−/− MEFs showed similar growth rates (mean doubling time, 22.9 ± 2.5 and 25.0 ± 4.3 hr, respectively). Saturation density of MEFs of each genotype was 11.0 ± 2.5 × 105 (wild-type, ○), 8.6 ± 1.9 × 105 (IRF-1−/−, ●), 26.8 ± 4.4 × 105 (p53−/−, □), and 35.4 ± 3.4 × 105 cells (IRF-1−/− p53−/−, █).
The accumulation of genetic alterations can be augmented by inappropriate regulation of apoptosis and cell growth (Sherr 1996; Evan and Littlewood 1998). In this context, it is already known that activated IRF-1−/− T lymphocytes (splenocytes) are resistant to apoptosis upon γ-irradiation (Tamura et al. 1995). As expected, IRF-1−/−p53−/− splenocytes were also resistant to radiation-induced apoptosis, which occurred normally in p53−/− splenocytes (data not shown). Next we compared the growth profiles of MEFs among the four genotypes. MEFs grew better in the absence of p53, but loss of IRF-1 showed no significant effect on growth rate in log phase. Interestingly, p53−/− MEFs showed even higher saturation density when IRF-1 was additionally absent (Fig. 3C). Thus, the combined loss of IRF-1 and p53 in MEFs results in acquisition of abnormal growth capacity, suggesting that the impairment in cell cycle machinery in p53−/− MEFs is further affected by the additional loss of IRF-1. The p16INK4a/retinoblastoma (Rb)-linked pathway has been proposed as a major mechanism of cell cycle regulation that is distinct from the p53-dependent pathway (Sherr 1996; Haber 1997). However, the expression of p16INK4a, CDK4, cyclins D1, D2, and D3, and Rb proteins in MEFs were not significantly altered by loss of IRF-1 (H. Nozawa, unpubl.), suggesting that IRF-1 regulates the cell cycle through an as yet unknown mechanism(s) distinct from the p16INK4a/Rb pathways. Obviously, further work will be required to elucidate the mechanism by which IRF-1 deficiency affects tumor susceptibility, by identifying the critical target gene(s) of this transcription factor.
Efforts have been made to investigate genes involved in tumor susceptibility by analyzing their genetic changes in various human cancers. Phenotypes in mice mutated for tumor suppressor genes provide further understanding of their roles in tumor development in vivo (Jacks 1996; Ghebranious and Donehower 1998). Moreover, multiple loss-of-function mutations in these genes can facilitate tumor development, as demonstrated through the generation of mice with compound mutations of genes such as p53, Rb, ataxia–telangiectasia mutated (atm), and adenomatous polyposis coli (Apc) (Williams et al. 1994; Reitmair et al. 1996; Westphal et al. 1997). In these cases, however, mutation in a single gene (heterozygous or homozygous) induces a cancer-predisposing phenotype (Donehower et al. 1992; Jacks et al. 1992; Barlow et al. 1996). In addition, many of the tumor suppressor genes appear to be essential for development, as introduction of nullizygosity in these genes causes embryonic lethality (Jacks 1996; Ghebranious and Donehower 1998). In contrast, the loss of IRF-1 per se does not have an immediate visible effect on tumor development in vivo. Rather, its effect becomes apparent when combined with other genetic abnormalities. It has been demonstrated that a tumor modifier, Mom1, affects the susceptibility of intestinal tumors in mice carrying the ApcMin mutation (Dietrich et al. 1993; Gould and Dove 1997); whereas loss of IRF-1 increases tumor incidence of many organs in mice carrying the c-Ha-ras transgene or nullizygosity for p53. In this context IRF-1 may belong to a new class of tumor susceptibility gene. Given that the risk of tumor incidence is increased significantly when IRF-1 is functionally inactivated in combination with other genetic alterations, it is conceivable that the loss of IRF-1 may also be involved in the process of development of human cancers.
Materials and methods
Generation of mutant mice
To generate IRF-1−/− mice carrying human c-Ha-ras transgenes, IRF-1−/− mice (Matsuyama et al. 1993) were mated with rasH2 mice (Saitoh et al. 1990) to produce IRF-1-null (IRF-1−/−/rasH2) or heterozygous (IRF-1+/−/rasH2) mice carrying ras transgenes.
Murine IRF-1 and p53 genes are both located on chromosome 11 (Rotter et al. 1984; Buckwalter et al. 1992). To generate IRF-1−/−p53−/− mice, IRF-1−/− mice and p53−/− mice (Tsukada et al. 1993) were crossed, and offspring were mated with C57BL/6 mice to obtain mice carrying a chromosome 11 containing both the targeted IRF-1 and p53 loci in cis-configuration by meiotic recombination. Heterozygous animals were inbred to obtain mice homozygous for the null IRF-1 and p53 alleles. Wild-type, IRF-1−/− and p53−/− mice of the same background were used for comparisons.
Generation of chimeric mice
Aggregation chimeric mice were produced according to methods described elsewhere (Nagy and Rossant 1993). Briefly, eight-cell stage embryos derived from IRF-1−/−p53−/−, p53−/− and wild-type mice cryopreserved by the vitrification method were collected and the zonae pellucidae was removed by acid Tyrode’s solution. To generate IRF-1−/−p53−/− ↔ p53−/− chimeric mice, IRF-1−/−p53−/− and p53−/− embryos were aggregated at a ratio of 1:1 and at the blastocyst stage transferred into the uteri of pseudopregnant recipients. Similarly, IRF-1−/−p53−/− ↔ wild-type and p53−/− ↔ wild-type chimeric mice were generated by aggregation of IRF-1−/−p53−/− and wild-type embryos, and p53−/− and wild-type embryos, respectively.
Histology
Tissue specimens were fixed in 10% buffered formalin, blocked in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin.
Cell culture and cell proliferation assay
Primary MEFs were isolated from embryos at 12–14 days of gestation and maintained as described previously (Tanaka et al. 1994a). MEFs were plated on 35-mm dishes at passage 4 (1 × 105 cells per dish) and cultured. Saturation density was determined as the maximum cell number during 3 weeks of culture.
Mutation frequencies of MEFs treated with mutagens
MEFs were treated continuously with 0.05 μg/ml cisplatin (Sigma, St. Louis, MO) for 72 hr, or 5 μm MNNG (Sigma), for 3 hr and incubated in mutagen-free medium for 6 days. Cells (1 × 105) were then replated on 100-mm dishes and cultured in DMEM supplemented with 10% FCS containing 3 mm ouabain (Sigma) for 8 weeks.
Acknowledgments
We thank Shinsuke Taki, David M. Livingston, Charles J. Sherr, Yoichi Taya, Toshio Mori, Fumio Hanaoka, Chikahide Masutani, Mutsuo Sekiguchi, Yusaku Nakabeppu, and Joji Inazawa for their invaluable advice and helpful discussion. We are also grateful to Shinsuke Saito and Naoki Hata for technical advice and to Guy Harris for revision of the manuscript. This work was supported in part by a special grant for Advanced Research on Cancer from the Ministry of Education, Science, and Culture of Japan, and a Research Grant of the Princess Takamatsu Cancer Research Fund. H.N. is a Research Fellow of the Japan Society for the Promotion of Science.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL nobtanak@m.u-tokyo.ac.jp; FAX 81-3-5841-3450.
References
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Buckwalter MS, Lossie AC, Scarlett LM, Camper SA. Localization of the human chromosome 5q genes Gabra-1, Gabrg-2, Il-4, Il-5, and Irf-1 on mouse chromosome 11. Mamm Genome. 1992;3:604–607. doi: 10.1007/BF00350629. [DOI] [PubMed] [Google Scholar]
- Demant P. Genetic resolution of susceptibility to cancer—New perspectives. Semin Cancer Biol. 1992;3:159–166. [PubMed] [Google Scholar]
- Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- Ghebranious N, Donehower LA. Mouse models in tumor suppression. Oncogene. 1998;17:3385–3400. doi: 10.1038/sj.onc.1202573. [DOI] [PubMed] [Google Scholar]
- Gould KA, Dove WF. Localized gene action controlling intestinal neoplasia in mice. Proc Natl Acad Sci. 1997;94:5848–5853. doi: 10.1073/pnas.94.11.5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber DA. Splicing into senescence: The curious case of p16 and p19ARF. Cell. 1997;91:555–558. doi: 10.1016/s0092-8674(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Harada H, Kondo T, Ogawa S, Tamura T, Kitagawa M, Tanaka N, Lamphier MS, Hirai H, Taniguchi T. Accelerated exon skipping of IRF-1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene. 1994;9:3313–3320. [PubMed] [Google Scholar]
- Hawkins DS, Demers GW, Galloway DA. Inactivation of p53 enhances sensitivity to multiple chemotherapeutic agents. Cancer Res. 1996;56:892–898. [PubMed] [Google Scholar]
- Jacks T. Lessons from the p53 mutant mouse. J Cancer Res Clin Oncol. 1996;122:319–327. doi: 10.1007/BF01220798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, Le J, Koh SI, Kimura T, Green SJ, Mak TW, Taniguchi T, Vlicek J. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263:1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, Taniguchi T. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science. 1994;264:1921–1924. doi: 10.1126/science.8009222. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- Lohoff M, Ferrick D, Mittrucker HW, Duncan GS, Bischof S, Rollinghoff M, Mak TW. Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. Immunity. 1997;6:681–689. doi: 10.1016/s1074-7613(00)80444-6. [DOI] [PubMed] [Google Scholar]
- Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kündig TM, Amakawa R, Kishihara K, Wakeham A, Potter J, Furlonger CL, Narendran A, Suzuki H, Ohashi PS, Paige CJ, Taniguchi T, Mak TW. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-β gene regulatory elements. Cell. 1988;54:903–913. doi: 10.1016/s0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J. Production of completely ES cell-derived fetus. In: Joyner AL, editor. Gene targeting, a practical approach. Oxford, UK: IRL Press; 1993. pp. 147–180. [Google Scholar]
- Nozawa H, Oda E, Ueda S, Tamura G, Maesawa C, Muto T, Taniguchi T, Tanaka N. Functionally inactivating point mutation in the tumor-suppressor IRF-1 gene identified in human gastric cancer. Int J Cancer. 1998;77:522–527. doi: 10.1002/(sici)1097-0215(19980812)77:4<522::aid-ijc8>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Ogasawara S, Tamura G, Maesawa C, Suzuki Y, Ishida K, Satoh N, Uesugi N, Saito K, Satodate R. Common deleted region on the long arm of chromosome 5 in esophageal carcinoma. Gastroenterology. 1996;110:52–57. doi: 10.1053/gast.1996.v110.pm8536888. [DOI] [PubMed] [Google Scholar]
- Ogasawara K, Hida S, Azimi N, Tagaya Y, Sato T, Yokochi-Fukuda T, Waldmann TA, Taniguchi T, Taki S. Requirement for IRF-1 in the microenvironment supporting development of natural killer cells. Nature. 1998;391:700–703. doi: 10.1038/35636. [DOI] [PubMed] [Google Scholar]
- Reitmair AH, Cai JC, Bjerknes M, Redston M, Cheng H, Pind MT, Hay K, Mitri A, Bapat BV, Mak TW, Gallinger S. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–2926. [PubMed] [Google Scholar]
- Rotter V, Wolf D, Pravtcheva D, Ruddle FH. Chromosomal assignment of the murine gene encoding the transformation-related protein p53. Mol Cell Biol. 1984;4:383–385. doi: 10.1128/mcb.4.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh A, Kimura M, Takahashi R, Yokoyama M, Nomura T, Izawa M, Sekiya T, Nishimura S, Katsuki M. Most tumors in transgenic mice with human c-Ha-ras gene contained somatically activated transgenes. Oncogene. 1990;5:1195–1200. [PubMed] [Google Scholar]
- Sekiguchi M, Nakabeppu Y, Sakumi K, Tuzuki T. DNA-repair methyltransferase as a molecular device for preventing mutation and cancer. J Cancer Res Clin Oncol. 1996;122:199–206. doi: 10.1007/BF01209646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Strauss BS. Hypermutability in carcinogenesis. Genetics. 1998;148:1619–1626. doi: 10.1093/genetics/148.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki S, Sato T, Ogasawara K, Fukuda T, Sato M, Hida S, Suzuki G, Mitsuyama M, Shin EH, Kojima S, Taniguchi T, Asano Y. Multistage regulation of Th1-type immune responses by the transcription factor IRF-1. Immunity. 1997;6:673–679. doi: 10.1016/s1074-7613(00)80443-4. [DOI] [PubMed] [Google Scholar]
- Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S, Taniguchi T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 1995;376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- Tamura G, Ogasawara S, Nishizuka S, Sakata K, Maesawa C, Suzuki Y, Terashima M, Saito K, Satodate R. Two distinct regions of deletion on the long arm of chromosome 5 in differentiated adenocarcinomas of the stomach. Cancer Res. 1996;56:612–615. [PubMed] [Google Scholar]
- Tanaka K, Wood RD. Xeroderma pigmentosum and nucleotide excision repair of DNA. Trends Biochem Sci. 1994;19:83–86. doi: 10.1016/0968-0004(94)90040-X. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell. 1994a;77:829–839. doi: 10.1016/0092-8674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Taniguchi T. Suppression of c-myc or fosB-induced cell transformation by the transcription factor IRF-1. Cancer Lett. 1994b;83:191–196. doi: 10.1016/0304-3835(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, Aizawa S, Tokino T, Oren M, Taniguchi T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature. 1996;382:816–818. doi: 10.1038/382816a0. [DOI] [PubMed] [Google Scholar]
- Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S, Matsuo I, Ikawa Y, Aizawa S. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene. 1993;8:3313–3322. [PubMed] [Google Scholar]
- Westphal CH, Rowan S, Schmaltz C, Elson A, Fisher DE, Leder P. atm and p53 cooperate in apoptosis and suppression of tumorigenesis, but not in resistance to acute radiation toxicity. Nat Genet. 1997;16:397–401. doi: 10.1038/ng0897-397. [DOI] [PubMed] [Google Scholar]
- Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994;7:480–484. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- Willman CL, Sever CE, Pallavicini MG, Harada H, Tanaka N, Slovak ML, Yamamoto H, Harada K, Meeker TC, List AF, Taniguchi T. Deletion of IRF-1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science. 1993;259:968–971. doi: 10.1126/science.8438156. [DOI] [PubMed] [Google Scholar]