


Abstract
Controlling the patterns of splicing of specific genes is an important goal in the development of new therapies. We have shown that the splicing of a refractory exon, SMN2 exon 7, could be increased in fibroblasts derived from patients with spinal muscular atrophy by using bifunctional targeted oligonucleotide enhancers of splicing (TOES) oligonucleotides that anneal to the exon and contain a ‘tail’ of enhancer sequences that recruit activating proteins. We show here that there are striking agreements between the effects of oligonucleotides on splicing in vitro and on both splicing and SMN2 protein expression in patient-derived fibroblasts, indicating that the effects on splicing are the major determinant of success. Increased exon inclusion depends on the number, sequence and chemistry of the motifs that bind the activator protein SRSF1, but it is not improved by increasing the strength of annealing to the target site. The optimal oligonucleotide increases protein levels in transfected fibroblasts by a mean value of 2.6-fold (maximum 4.6-fold), and after two rounds of transfection the effect lasted for a month. Oligonucleotides targeted to the upstream exon (exon 6 in SMN) are also effective. We conclude that TOES oligonucleotides are highly effective reagents for restoring the splicing of refractory exons and can act across long introns.
INTRODUCTION
Pre-mRNA splicing is becoming a major target for the development of therapies. This situation has arisen for two reasons. First, it is evident that alternative splicing makes a major contribution to the regulation of gene expression. Almost all, 94%, of protein-coding genes contain introns and of these 90% show significant levels of alternative splicing; around 70% of these splices are tissue specific (1). Thus, a high proportion of mRNA sequences and proteins arise by alternative splicing and contribute to development. The second important factor is that the choice of splice sites depends on a number of very subtle aspects of the pre-mRNA sequence. These not only affect the use of bona fide sites but they are also required to distinguish between real sites and the very large number of sequences resembling splice sites that can be found in most genes (2,3). Indeed, it is estimated that between 12 and 19 distinct cues are involved in the selection of tissue-specific splice sites (4). The existence of alternative sites, the abundance of similar sequences and the activation or suppression of alternative sites in specific tissues all contribute to a system that appears to be vulnerable to mutations or to changes in the concentrations of the proteins that recognize these sequences. It is unsurprising that genetic diseases are commonly found to arise from mutations in sequences that affect splicing or that the progression of acquired diseases involves substantial changes in splicing patterns (5).
The first strategy used to redirect splicing for therapeutic purposes involved the use of oligonucleotides complementary to splice sites or, more recently, enhancer sequences (6–9). These were intended to block the binding of essential splicing factors at a specific exon, causing it to be skipped. This strategy has been proved to be very effective, and promising results have emerged from clinical trials of oligonucleotides that promote the skipping of a mutant dystrophin exon that causes Duchenne muscular dystrophy (10,11).
It was not quite so obvious how one might direct the inclusion of an exon. This is important for restitution of survival of motor neuron (SMN) protein expression in spinal muscular atrophy (SMA). SMA is a common autosomal recessive disorder caused by mutations in the gene SMN1. It is characterized by degeneration of the motor neurons in the anterior horn of the spinal cord, resulting in muscular atrophy and weakness, but the severity of the phenotype is modified by the copy number of a second SMN gene, SMN2, which is present as a result of an inverted duplication in chromosome 5q13. The SMN genes have eight exons, the first seven of which encode a 294 amino acid protein, but in SMN2 the majority of mRNA products lack exon 7 and the levels of functional full-length protein derived from SMN2 are very low as a result (12).
Two strategies have emerged for stimulating the inclusion of exon 7: the use of oligonucleotides that block silencer motifs or the use of bifunctional oligonucleotides. It is still difficult to predict the location of enhancer and silencer motifs within an exon (2,3), and the most effective route for finding appropriate oligonucleotides that block silencers is either to perform a systematic screen with a large number of candidate oligonucleotides (13,14) or to map the silencers by experiment. The other strategy is to increase the number of positively acting signals in an exon, which led us to invent bifunctional oligonucleotides (15). The oligonucleotides were designed with one domain that was intended to anneal to the target exon and a second (tail) domain that contained sequences to which activator proteins, such as the SR proteins, would bind (Figure 1). We demonstrated that one such oligonucleotide stimulated the splicing of SMN2 exon 7 in a model pre-mRNA in nuclear extracts and that it stimulated both splicing and SMN protein expression in fibroblasts derived from SMA patients. We designated this method as targeted oligonucleotide enhancers of splicing (TOES) (16). At the same time, peptide–PNA compounds were developed for the same purpose; in these, the PNA sequence annealed to the target exon and the peptide comprised repeats of an arginine–serine dipeptide that mimicked the activation (RS) domains of SR proteins. These are also effective in rescuing the splicing of a refractory exon in nuclear extracts (17).
Figure 1.

Design of TOES. The sequence of the SMN genes around exon 7 is shown, with the sequence of exon 7 in upper case and flanking intron sequences in lower case. Nucleotide 6 is shown as an asterisk, being C in SMN1 and T in SMN2. The shaded sequence is an enhancer that is recognized by Tra2β. Oligonucleotide 1 is shown annealed to nucleotides 2–16. The tail and annealing domains, shown in different colours, and the 5′-end of the tail (cap) may contain different nucleotide analogues in the oligonucleotides being tested. The line diagrams below the exon show oligonucleotides that anneal to different sites, the sites being shown by the nucleotide positions in the exon; the oligonucleotides are numbered as in Table 1.
The effectiveness of TOES as a potential therapy for SMA has been tested subsequently by two different strategies. A bifunctional oligonucleotide targeted to SMN2 exon 7 has been expressed in transgenic mice within a modified U7 snRNA gene. Expression of the TOES-U7 RNA in a mouse model of SMA produced a very substantial improvement in function and lifespan (18). Another variation of TOES involved targeting an intronic silencer upstream of SMN2 exon 7; this appeared to have the dual effect of blocking the silencer and recruiting activator proteins (19). Interestingly, this strategy resulted in increased inclusion of exon 7 when the oligonucleotides were injected into intracerebral ventricles, even though the recruitment of SR proteins to a site in an intron has been shown in other cases to inhibit splicing (20).
Optimizing the design of a TOES oligonucleotide requires consideration of a number of variables beyond those normally associated with a complementary silencing oligonucleotide. Obviously the sequence of the non-complementary tail has to represent an optimal binding site for an activator protein, but other factors include the best choice of protein, the number of protein molecules that need to be bound for maximum effectiveness, the ability of the protein to bind stable analogues of RNA and the optimal locations of the proteins relative to their sites of action.
The optimal location is difficult to predict. If enhancers could be identified reliably it might be thought best not to obstruct them, although a TOES oligonucleotide might have a dominant positive effect anyway. A second problem is that it is difficult to judge where an enhancer should be tethered because too little is known at a molecular level about where and how enhancer-bound SR proteins exert their effects. Following the identification of SR proteins as the main mediators of the effects of enhancer sequences (21), and evidence that they could mediate protein interactions via their RS domains (22), two very important mechanistic insights were made: (i) we showed that SR protein SRSF1 strengthened the binding of U1 snRNP complexes at 5′-splice sites (23), even though, as with the effects of SRFS1 on the selection of alternative 5′-splice sites (24,25), its RS domain is not required (26), and (ii) others showed that SR proteins enhanced the binding of the protein U2AF at upstream 3′-splice sites (27,28). These and subsequent observations led to the common picture that SR proteins bound to an exon stabilize complex formation at the flanking splice sites and thereby increase the probability of splicing to that exon, although the nature and number of the interactions involved is not known. However, recent investigations suggest that SR proteins bound to an exon stabilize RNA:RNA base pairing of U2 snRNA at the branchpoint and U6 snRNA at the 5′-splice site at later stages in spliceosome assembly (29,30). Thus, it is not yet clear which interactions should be targeted when designing a TOES oligonucleotide. We describe here an investigation into constraints on the design of the oligonucleotide, and suggest that this design has to reflect the rate-limiting steps in splicing to an exon. We have applied this to SMA as a model system and describe a highly effective TOES reagent inducing SMN2 exon 7 inclusion.
MATERIALS AND METHODS
Oligonucleotides
Oligonucleotides were synthesized and purified by Eurogentec. They were dissolved to standard concentrations and stored according to the manufacturer’s instructions.
Splicing in vitro
Radiolabelled transcripts were prepared as described previously from a construct comprising SMN2 exon 7 and flanking intron sequences (∼150 nt from intron 6 and 300 from intron 7) inserted into a shortened version of rabbit β-globin intron 2 and flanking exon sequences (15). A mixture of oligonucleotide and transcript was heated at 70°C for 30 s and then allowed to cool to ambient temperature over 30 min. A mixture of nuclear extract, buffer, salts and ATP was added, producing final concentrations of 2 mM MgCl2, 38.5 mM KCl, 2 mM ATP, 10 mM creatine phosphate and 35% nuclear extract (Cilbiotech). Reactions were incubated at 30°C for 2 h. After treatment, samples were analysed on 6% denaturing polyacrylamide gels. The radiation present in the two isoforms of mRNA and the remaining pre-mRNA was quantified with a phosphor imager (Packard), adjusted for the content of radiolabelled nucleotides and the proportion of exon inclusion mRNA calculated. This figure was expressed as a ratio relative to the level of inclusion of RNA spliced in parallel in the absence of oligonucleotides. Splicing assays to test the effect of the mutation in the ESE (Figure 7) were done in 50% nuclear extract, 3.2 mM MgCl2, 50 mM KCl, 50 mM K glutamate. For ribonuclease H cleavage assays, the TOES oligonucleotides were annealed to pre-mRNA in 100 mM K glutamate, 50 mM Hepes.KOH, and then incubated in 40% nuclear extract as for splicing but in the absence of ATP and creatine phosphate. After 5 min at 30°C, the DNA oligonucleotide was added to 500 nM and ribonuclease H to 0.5 U/µl, and incubation was continued for 30 min.
Figure 7.

Rescue of splicing to SMN exon 7 by oligonucleotide 1 in vitro after mutation of GAA motifs. SMN2 is the normal substrate used for in vitro assays; ESE-M is an otherwise identical transcript in which the GAA repeats in exon 7 had been mutated and nucleotide 6 had been converted to the SMN1 nucleotide C. The pre-mRNA is the upper band in the triplet adjacent to the diagram of the pre-mRNA; the inclusion and exclusion mRNA isoforms are also marked. The oligonucleotides shown were used at 100 and 250 nM, as shown. Increased splicing to exon 7 is associated with increased intensities of a higher band corresponding to mRNA in which the second but not the first intron has been spliced, as noted previously (15).
Transfection of SMA fibroblasts
Skin fibroblast cell line 7384 was derived from a patient with type II SMA who has a homozygous deletion of the SMN1 gene and three copies of SMN2. Fibroblasts were cultured in DMEM containing 10% foetal calf serum. Cells were seeded into six-well plates with a concentration of ∼2 × 105 cells per well, which gives 90% confluence when transfected the next day. Oligonucleotides were complexed with Lipofectamine 2000 (Invitrogen) in Opti-MEM (Invitrogen) according to the manufacturer’s instructions, and added to the cells. Cells were harvested 12 h post-transfection for RNA extraction, and 24 h post-transfection for protein extraction. All transfections have been repeated at least three times throughout the entire study.
Reverse transcription PCR and quantitative real-time PCR
Total RNA from the cultured cells was extracted with the Qiagen RNeasy kit. Almost 500 ng of each RNA sample was used for first-strand cDNA synthesis with a Superscript III reverse transcription kit (Invitrogen). Primers (Forward: 5′- CTC CCA TAT GTC CAG ATT CTC TT-3′ and Reverse: 5′-CTA CAA CAC CCT TCT CAC AG-3′) were used to amplify full-length (505 bp) and Δ7 SMN2 (451 bp) from cDNA. The products were amplified semi-quantitatively using 25 PCR cycles (94°C for 30 s, 55°C for 30 s and 72°C for 30 s). All PCR products were checked by running on 1% agarose gels.
Quantitative real-time PCR was done with the Eurogentec MESA Blue qPCR kit. Samples were incubated in a 25 µl reaction mix according to manufacturer’s instructions. The same cDNA products as above were used as template for real-time PCR. The specific full-length SMN2 primers, forward 5′-ATA CTG GCT ATT ATA TGG GTT TT-3′ and reverse 5′-TCC AGA TCT GTC TGA TCG TTT C-3′ (133 bp); and specific SMN Δ7 primers, forward 5′-TGG ACC ACC AAT AAT TCC CC-3′ and reverse 5′-ATG CCA GCA TTT CCA TAT AAT AGC C-3′ (125 bp) were used. Quantitative real-time PCR was performed using Applied Biosystem fast 7500 Real Time PCR System using the recommended programme: activation at 95°C for 5 min, 40 cycles of 95°C for 3 s and 60°C for 1 min. Quantification was based on concurrent standard curves produced from serial dilutions of cDNA from untreated 7384 SMA fibroblasts. The cycle at which the amount of fluorescence was above the threshold (Ct) was detected. The ratios of full-length SMN2 to Δ7 SMN2 of the treated samples were normalized taking the ratio of the untreated sample as 1.0.
Western blotting
Fibroblasts were lysed in buffer containing 75 mM Tris–HCl (pH 6.8) and 0.25% SDS supplemented with protease inhibitor cocktail (Roche Diagnostics). The lysates were centrifuged at 13 000 g for 15 min at 4°C to collect the supernatants. The protein concentration was analysed using a BCA kit (Pierce) according to the manufacturer’s instructions and 5 µg protein was loaded into NuPAGE pre-cast gels (10% Bis–Tris, Invitrogen). Separated proteins were then transferred electrophoretically to nitrocellulose membrane (GE Healthcare). Membranes were blocked overnight in 10% semi-skimmed milk in PBS-0.1% Tween, then incubated for 1 h at room temperature with mouse anti-SMN monoclonal antibody (1:3000, BD Transduction laboratories) and mouse anti-β-tubulin monoclonal antibody (Clone TUB2.1, 1:5000, Sigma) in 5% Milk–PBS–Tween. Membranes were incubated for 1 h at room temperature followed with secondary HRP-conjugated anti-mouse IgG antibody (Amersham) at a dilution of 1:50 000 in PBS–Tween. Blots were developed using an enhanced chemiluminescence detection kit (GE Healthcare). Quantification was done with ImageJ software.
Protein–RNA interactions
To assay the binding of recombinant SRSF1 by native gel electrophoresis, 1.5 pmol of 5′-end-labelled oligonucleotide 27 were incubated on ice for 15 min with 20 pmol SRSF1 in 5 µl of 20 mM Tris–HCl, pH 7.5, 100 mM KCl, 10 mM MgCl2. A 25 pmol of competitor oligonucleotide was added, as labelled in Figure 7, and incubation continued for an hour. Subsequently, 3 µl of 80% glycerol were added, and 5 µl portions loaded on a pre-cooled native gel (10% polyacrylamide, 25 mM Tris base, 200 mM glycine, 5% glycerol). Electrophoresis was for 1 h at 4°C at 20 V/cm.
For immunoprecipitation, protein G agarose bead slurry (50%) was preincubated with mAB96 antibody (Zymed) in PBS containing 0.1 mg/ml BSA for 2 h at 4°C with rotation. Antibody-bound protein G was washed by pelleting at 12 000g at 4°C in PBS. 5′-end-labelled oligonucleotide (2 pmol/µl) was incubated with 10 µl antibody-bead complexes, 0.8 µg SRSF1 and 400 µl IPP200 Buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.05% NP40 and 200 mM NaCl) for 2 h at 4°C. Protein-antibody bead complexes were pelleted at 12 000 g and washed for 15 min at 4°C in IPP200 three times. Final pelleted samples were resuspended in 10 µl 2 × SDS sample buffer and separated by electrophoresis in SDS on 15% polyacrylamide gels. Gels were dried on 3 MM Whatman paper and exposed to a phosphor screen.
RESULTS
TOES oligonucleotides contain a protein binding domain (tail) comprising repeated motifs believed to be optimal binding sites for specific proteins. In the first exemplars, the annealing domain was complementary to nucleotides 2–16 of SMN2 exon 7. Motifs that were known binding sites for an activator, SRSF1, stimulated inclusion and those for two inhibitors, polypyrimidine tract binding protein (PTB) and hnRNP A1, reduced the efficiency of splicing in vitro (15).
To establish principles for designing TOES oligonucleotides, we varied the chemical structure of the nucleotide analogues in the tail and the annealing domain, the number of potential binding sites in the tail, the sequences of these sites and the sites of annealing of the oligonucleotide. The efficacy of the oligonucleotides was assessed by measuring the splicing of SMN exon 7 in nuclear extracts and by transfection of 7384 fibroblasts derived from a SMA patient, measuring both changes in SMN2 splicing by real time PCR and SMN protein expression. Oligonucleotides were used at concentrations of 250, 500, 1000 and 5000 nM for splicing in vitro and at 50, 100 and 250 nM for the transfection of cells. Table 1 summarizes the results obtained with the oligonucleotides at 250 nM. It is important to note that the values stated for splicing in vitro show the relative increase in the level of exon inclusion mRNA as a proportion of the sum of the unspliced pre-mRNA and the two isoforms of spliced mRNA. The purpose of this is to avoid using the ratio of inclusion and exclusion isoforms alone, which might produce an apparent increase in inclusion if skipping had been inhibited. The values stated for splicing in fibroblasts cannot be readily presented in the same way, and they show the ratio of inclusion mRNA to exclusion mRNA, normalized against untreated cells. However, these results can be compared with measurements of the relative levels of SMN protein in treated and untreated cells, which would allow aberrant measurements resulting from the inhibition of skipping to be detected.
Table 1.
Summary of properties and activities of oligonucleotides
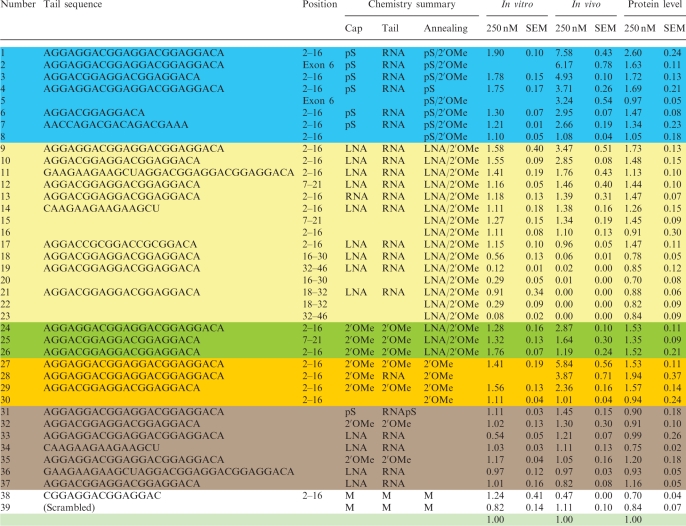 |
Number is the number assigned to each oligonucleotide; Position shows the nucleotides in the exon, numbered from the 5′-end, to which the oligonucleotide anneals; In vitro refers to splicing of RNA in nuclear extract and shows the proportion of mRNA including exon 7 as [inc/(inc + exc + pre-mRNA)] in the presence of oligonucleotide at 250 nM relative to the same ratio in the absence of oligonucleotide in the same experiment, with the standard error of the mean shown (SEM); In vivo refers to splicing of the endogenous SMN2 gene in 7384 fibroblasts transfected with oligonucleotide at 250 nM and shows the ratio of products including exon 7 to the products without, as determined by real-time PCR, relative to the levels measured for parallel untransfected samples; protein level refers to the ratio of SMN protein to tubulin protein signals on western blotting of extracts from 7384 fibroblasts transfected with oligonucleotides at 250 nM, relative to the same ratio in untransfected cells. The chemistry is designated as: pS, phosphorothioate; 2′OMe, 2′-O-methyl; LNA, locked nucleic acid; M, morpholino. In all cases, the reference value in the absence of oligonucleotide is taken as 1.00 (green shading).
The effects of oligonucleotides on splicing in vitro and in SMA fibroblasts are compared in Figure 2. These effects show a striking agreement for most of the oligonucleotides that stimulate inclusion, which suggests that their different effects on exon 7 inclusion in SMA fibroblasts result from their effects on splicing as opposed to differences in the stability or uptake of the oligonucleotides. Moreover, an increase in the proportion of exon 7 inclusion in vitro is generally associated with increased levels of SMN protein expression (Figure 3), which suggests that there are no significant detrimental effects of the oligonucleotide on translation. We conclude that the efficiency of splicing to exon 7 is the main determinant of the levels of SMN protein expression after treatment with TOES oligonucleotides.
Figure 2.

Comparison of the activities of bifunctional oligonucleotides on splicing in transfected SMA fibroblasts and in vitro. The ratio of inclusion to exclusion isoforms from fibroblasts transfected with oligonucleotides at 250 nM, determined by quantitative real-time PCR, is plotted against the relative increase in efficiency of splicing to exon 7 in vitro in the presence of oligonucleotides at the same concentration (values from Table 1). The results have been grouped according to the chemistries of the oligonucleotides used. The results for oligonucleotides 6, 3 and 1 (left to right) have been circled.
Figure 3.

Comparison of the activities of bifunctional oligonucleotides on SMN protein expression and splicing in transfected SMA fibroblasts. The level of SMN protein expression relative to that of tubulin, and expressed relative to the corresponding level in untransfected cells, is plotted against the ratios of the mRNA isoforms determined by real-time PCR. As in Figure 3, the results are derived from experiments with the oligonucleotides at 250 nM, and they are grouped according to the chemistries of the oligonucleotides used. The results for oligonucleotides 6, 3 and 1 (left to right) have been circled.
The length of the tail domain
The original oligonucleotide (number 1 in Table 1) contained six repeats of the motif GGA, within three repeats of the sequence GGAGGAC. We tested whether it would be possible to use oligonucleotides with shorter tails by designing variants that lacked one or three of the GGA motifs. Table 1 shows the oligonucleotides arranged in blocks according to the chemistries of their three parts, the ranking within each block being determined by the effectiveness with which they stimulated exon 7 inclusion in transfected fibroblasts. Comparison of oligonucleotides 1, 3 and 6 shows that their effectiveness both in vitro and in vivo depends directly upon the number of GGA repeats (6, 5 and 3, respectively), and oligonucleotides lacking either a tail or an annealing domain are inactive (Figures 2 and 3; representative western blots and a qualitative PCR analysis of splicing from transfected cells are shown in Figure 4A and B).
Figure 4.

Importance of the length and sequence of the tail domain. (A) Western blots showing SMN and tubulin proteins after transfection of SMA fibroblasts with oligonucleotides containing different lengths of tail, as labelled above the lanes (oligonucleotide 1, 6 GGA motifs; oligonucleotide 3, 5 GGA motifs; oligonucleotide 6, 3 GGA motifs). The figures below the lanes show the normalized ratios of SMN:tubulin. (B) Qualitative PCR of the spliced isoforms of endogenous SMN2 mRNA after transfection of SMA fibroblasts by the designated oligonucleotides. The site annealed refers to the 5′-most nucleotide in the exon to which the oligonucleotide is complementary. (C) Qualitative PCR illustrating the effectiveness of different tail sequences.
Sequence of the tail domain
The GGA motifs in oligonucleotide 1 were chosen on the basis that they appeared to be core elements of the binding site for the SR protein SRSF1. We tested two other sequences that have been identified as functional binding sites for SRSF1: oligonucleotide 7, which has two copies of CAGACG (31) in its tail of 19 nt, and oligonucleotide 17, which has a tandem repeat of CCGCGGA (32) in a tail of 20 nt. In comparison with oligonucleotides 3 and 10, which have five GGA motifs in RNA tails of 20 nt with the appropriate pS/2′-O-methyl and LNA/2′-O-methyl annealing domains, respectively, both alternative tail sequences perform relatively poorly in splicing assays in vitro and in vivo (Table 1 and Figure 4C). An important natural enhancer sequence in the middle of exon 7 contains two GAA motifs (33), and there is evidence that they form the binding site for the activator protein Tra2β (34). We tested three GAA motifs in oligonucleotide 14. Although this performed better than oligonucleotide 17, it fell far short of oligonucleotide 10 (Table 1 and Figure 4C). For their length and in a given chemical background, it appears that the GGA-based motifs are most potent.
Chemistry of the tail domain
The sugars in the tail of oligonucleotide 1 are all ribose groups. These were used because it was not clear whether RNA binding proteins would tolerate alterations to the ribose groups, despite the likelihood that an RNA moiety would compromise the stability of the oligonucleotide in a cell. A comparison of oligonucleotide 1 with oligonucleotide 27, which is fully 2′-O-methyl substituted and significantly less efficient in all assays (Table 1), suggested that RNA binding might be compromised. However, oligonucleotides differing only in their tail and cap chemistries did not show consistent differences (oligonucleotides 10 and 13 versus 26 and 27 versus 28; Table 1 and Figure 5A). We infer that 2′-O-methyl tails do not impede recognition of the tail's enhancer sequences by activator proteins, and that the difference in performance between oligonucleotides 1 and 27 might be attributed to the properties of the annealing region.
Figure 5.

Importance of nucleotide modifications in the annealing domain and their effects on the stability of the duplex. (A) Analysis of splicing in vitro. Transcripts containing SMN2 exon 7 and flanking β-globin exons were incubated with oligonucleotides, as designated by the numbers above the lanes, and incubated in HeLa nuclear extract before analysis by polyacrylamide gel electrophoresis. The concentrations of the oligonucleotides used were 250 or 500 nM, as shown. The spliced mRNA isoforms with inclusion or exclusion of exon 7 are shown alongside the image. Splicing intermediates are as illustrated previously (15). (B) Identification of the bands between the mRNA isoforms. Splicing reactions were done as above in the presence (+) or absence (−) of oligonucleotide 1 and then incubated with ribonuclease H and DNA oligonucleotides complementary to the exon or intron marked. (C) Pre-mRNA was incubated with oligonucleotides 1, 27 and 9, which contain the same sequence but with pS/2′-O-methyl, 2′-O-methyl and LNA/2′-O-methyl annealing domains, respectively, and then incubated in nuclear extract under conditions promoting formation of committed splicing complexes (−ATP). DNA oligonucleotides with the same sequence as the annealing domain and ribonuclease H were added to produce cleavage of any exposed pre-mRNA.
Chemistry of the annealing domain
The original annealing domain was completely substituted with 2′-O-methyl groups and partially with phosphorothioate. The principal determinants of a successful annealing domain were expected to be the stability of the duplex formed, the specificity of duplex formation and the resistance of the analogue to nucleases. Since the annealing site is rich in (A+U), it would be predicted that increasing the stability of the hybrid by incorporating LNA derivatives (35) into the annealing region would increase activity.
The results in Table 1 and examples in Figure 5A show that this expectation was not borne out, even for splicing in vitro. Oligonucleotides 10, 13, 26 and 29, which have tails containing 5 GGA motifs but annealing regions comprising LNA/2′-O-methyl or 2′-O-methyl modifications, are all less active overall than oligonucleotide 3, which has a 2′-O-methyl/phosphorothioate annealing region; similarly, oligonucleotide 9 is less active than oligonucleotide 1. The band in between the mRNA isoforms that is induced by addition of the most active oligonucleotides (1 and 3) was shown by ribonuclease H cleavage to comprise intron 2 (Figure 5B).
Since the LNA/2′-O-methyl oligonucleotides stimulated inclusion in vitro, rather than skipping or even a failure to splice, we can exclude any generic inhibition of splicing by off-target annealing as a plausible explanation of their relatively poor effects. An alternative explanation might be that the LNA/2′-O-methyl domain formed stable secondary structures that would prevent the oligonucleotide annealing to pre-mRNA. We tested whether the oligonucleotide had annealed in nuclear extracts using ribonuclease H protection (Figure 5C). The LNA/2′-O-methyl oligonucleotide provided complete protection at all concentrations tested. We have shown previously that the annealing of a DNA oligonucleotide and ribonuclease H cleavage can be treated as an irreversible pseudo-first order reaction such that the extent of cleavage depends on the level of binding or rate of dissociation by other components such as the TOES oligonucleotide (26). The results suggest that increased stability of the duplex with the target improves binding but reduces TOES activity.
Position of annealing
The extrinsic enhancer activity of the TOES oligonucleotides may depend on the location of the tail within the exon, and it is possible that a sequence that acts as an enhancer in one position might act as a silencer in another. We compared the effects of location for oligonucleotides with LNA/2′-O-methyl annealing domains of equal lengths and RNA tails containing five GGA motifs (Figure 6A). The results are shown in Table 1 and illustrative in vitro splicing and qualitative PCR reactions are shown in Figure 6B and C. The activities of the oligonucleotides decreased as the 5′-end of the annealing portion shifted from position +2 in the exon to +7, +18 and +32. Interestingly, the equivalent untailed oligonucleotides at positions +7 to +32 produced similar outcomes, so that at position +7 the oligonucleotide stimulated splicing slightly while at position +32 it inhibited completely. This means that the tail contributed significantly only for oligonucleotides beginning at nucleotide +2. To test whether this success depended on the site of annealing or the tail itself, we tested another oligonucleotide that annealed to positions 16–30 but was fused to the tail at its 3′-end. This oligonucleotide, number 18, also inhibited inclusion of exon 7 and comparison with oligonucleotide 20 shows that the tail had little effect (Figure 6 and Table 1). We conclude that the site of annealing has a major effect on activity, as might be expected, but that the tail only modulates this significantly with the oligonucleotides in which the tail is attached at position 16 and the annealing domain is on its 3′ side, i.e. to the upstream side of the exon.
Figure 6.

Effects of the site of annealing on the efficiency of TOES oligonucleotides. (A) Diagram showing the positions of annealing of the oligonucleotides to exon 7. Numbers are the designations of the oligonucleotides. Oligonucleotides 1 and 8 anneal to exon 7 positions 2–16, 18 and 20 to 16–30, 21 and 22 to 18–32, 19 and 23 to 32–46, and 12, 25 and 15 to 7–21. Of these, oligonucleotides 8, 20, 22, 23 and 15 lack tails. (B) In vitro splicing assays with the oligoribonucleotides shown above the lanes. The oligonucleotides labelled as bGt and bG are complementary to β-globin exon 2; bG lacks a tail. The diagrams on the right show the pre-mRNA and the two mRNA product isoforms; the diagrams in grey show intermediates. As noted for Figure 6, the presence of the RNA in which intron 2 has been spliced is an indication of use of the inclusion pathway of splicing. The bands above the pre-mRNA in lanes 19 and bGt, and above the inclusion mRNA in bGt, result from persistent annealing of the oligonucleotide in the denaturing gel. This is a feature of the oligonucleotides in which the annealing portion contains LNA/2′-O-methyl analogues and 40% or more (G+C). (C) Qualitative RT–PCR analysing splicing of endogenous SMN2 in fibroblasts transfected with some of the same oligonucleotides.
The inability of the tails on oligonucleotides 18 and 21 to rescue the inhibition caused by the corresponding annealing domains (oligonucleotides 20 and 22) might be caused by the inability of the tail to recruit proteins that compensate for the functions of proteins that would normally bind to this part of the exon. These oligonucleotides overlap the core Tra2β-dependent enhancer (Figure 1). We tested whether the tail could compensate for defective binding of Tra2β by mutating the binding site from AGAAGGAAGG to CGCACGATTA, which reduced splicing to exon 7 substantially; the 5′-end of the exon was mutated to match SMN1 to ensure a minimal level of inclusion. Splicing in vitro (Figure 7) showed that oligonucleotide 1 restored splicing efficiently, whereas an oligonucleotide containing GAA motifs (number 14) was ineffectual and one containing both GGA and GAA motifs (number 11) was marginally active. This result confirms that the failure of oligonucleotides 18 and 21 is not the result of a failure by the tail to compensate for the loss of Tra2β activity but is consistent with the possibility that the tail is inactive at positions 30 and 32.
Stimulation of splicing by bifunctional oligonucleotides targeted to an upstream exon
We observed previously that oligonucleotide 1 appeared to stimulate splicing in vitro of the downstream intron preferentially. This effect can be seen also in the in vitro splicing reactions in Figures 5A, 6B and 7, where predominance of the exon 7 inclusion mRNA is associated with increased levels of the intermediate RNA in which the downstream intron had spliced but the upstream one was retained. If TOES oligonucleotides have a polar effect, acting primarily on the downstream intron, then splicing of the upstream intron might be facilitated by a TOES oligonucleotide that annealed to the upstream exon. An oligonucleotide was tested that annealed to β-globin exon 2, which is the upstream exon in our in vitro substrate. This oligonucleotide contains five GGA motifs in an RNA tail with an LNA cap and an LNA/2′-O-methyl annealing domain. This proved to be effective at 250 nM; at 500 nM it was the most effective oligonucleotide (bGt, Figure 6B). This principle was tested on the endogenous SMN2 gene with an oligonucleotide similar to oligonucleotide 1 but complementary to exon 6. Even though this oligonucleotide annealed ∼5800 nt upstream of exon 7, it stimulated inclusion of exon 7 very substantially (oligonucleotide 2, Table 1).
Binding of the tail to SRSF1
If bifunctional oligonucleotides act by recruiting enhancer binding proteins such as SRSF1, then the activity of the tail should depend on SRSF1 binding. The formation of complexes between the oligonucleotides and recombinant SRSFS1 was measured by a competition assay: phosphorylated protein was incubated with labelled oligonucleotide 27 (six GGA motifs, fully 2′-O-methyl-substituted) and the complexes challenged with excess competitor oligonucleotides. After electrophoresis on a native gel, a successful competitor will be seen to have displaced the labelled oligonucleotide from the complex. Figure 8A shows that a 17-fold excess of oligonucleotides with six GGA motifs in the tail successfully displaced the labelled oligonucleotide. The thin bands above the complex and in the middle of the gel are multimers of the labelled oligonucleotide; smeary bands in the middle of the gel may represent partial dissociation of protein. In contrast, oligonucleotides lacking tails do not compete. This was confirmed by measuring binding at a range of protein concentrations in the presence of an 1800-fold excess of untailed competitor (Figure 8B). The competitor had no effect. Unsurprisingly, the morpholino oligo (38) shows little evidence of binding to protein.
Figure 8.

Dependence of binding by SRSF1 on oligonucleotide tail sequences. (A) Native gel electrophoresis showing displacement of SRSF1 protein from oligonucleotide 27 by competing bifunctional oligonucleotides. Reactions containing 5′-end-labelled oligonucleotide 27 (six GGA motifs, fully 2′O-methyl modified) were incubated with excess recombinant SRSF1 protein and then mixed with an excess of the oligonucleotide designated above the lanes. The displaced and bound labelled oligonucleotides are marked. (B) Competition between oligonucleotide 27 and untailed oligonucleotide 8 present at an 1800-fold higher concentration. (C) Immunoprecipitation of labelled oligonucleotides with anti-SRSF1 antibody after incubation with recombinant SRSF1. Oligonucleotides are numbered above each lane. The immunoprecipitates and input were analysed by gel electrophoresis and exposed to a phosphor screen simultaneously.
The importance of the tail for binding to SRSF1 was confirmed by direct immunoprecipitation of labelled oligonucleotides after incubation at a concentration of 8 nM with SRSF1 and immunoprecipitation buffer (Figure 8C). All oligonucleotides with a tail-bound SRSF1, irrespective of the presence of an annealing domain, except for oligonucleotide 21. The latter has a pyrimidine-rich annealing domain that could form a secondary structure with the tail and thus prevent binding of SRSF1 to the free oligonucleotide. We conclude that the tail sequence is able to mediate SRSF1 binding.
Effects of tail chemistry on persistence of effects
Oligonucleotides containing an RNA tail are clearly effective reagents. However, the RNA domain might allow rapid degradation of the oligonucleotide. To test whether the chemistry of the tail or the annealing domain affects the persistence of the response, oligonucleotides 1, 9 and 27 were transfected into fibroblasts from a patient with type II SMA as before and samples were removed at intervals up to 10 days after transfection. Figure 9A shows that in all three cases there was a significant increase in the levels of SMN protein until ∼7 days after transfection (the relative increase in levels of SMN is shown under each lane). Surprisingly, the shortest duration was seen with 2′-O-methyl modification (oligonucleotide 27). If transfection with oligonucleotide 1 is followed after 5 days by a second transfection and protein expression is followed at intervals thereafter, it can be seen that SMN protein levels remain high after all periods tested, i.e. even up to 28 days after the second transfection (Figure 9B).
Figure 9.

Time course of SMN protein expression after transfection with oligonucleotides 1, 9 and 27. (A) SMA fibroblasts were transfected and samples taken at the intervals shown (d, days) for analysis by western blotting. (B) Persistence of elevated SMN protein levels after a second transfection with oligonucleotide 1. SMA fibroblasts were transfected and then re-transfected after 5 days. Samples were taken at the number of days indicated after the second transfection and analysed by western blotting. Numbers under the lanes indicate the fold increases in the ratio of SMN to tubulin protein.
DISCUSSION
The purposes of this research were to optimize the design of the bifunctional oligonucleotide used to rescue the splicing of SMN2 exon 7 and to establish principles that could be used to design oligonucleotides to rescue the splicing of other exons. The efficiencies of the oligonucleotides were assessed by three main assays, i.e. splicing in vitro, measurements of the relative levels of the endogenous isoforms expressed in transfected fibroblasts and measurements of the level of protein expressed in these fibroblasts. The assays of splicing in nuclear extracts use transcripts containing exon 7 and flanking intron portions fused to flanking globin sequences. The endogenous sequence from exons 6–8 of SMN is about 7 kb, which is impracticable for this purpose because splicing in vitro becomes very inefficient with transcripts longer than around 1 kb. However, the short substrate contains most of the sequences identified to modulate exon 7 inclusion and responds appropriately to mutations in them, including sensitivity to exon nucleotide 6, and to the TOES oligonucleotide (15). It had been anticipated that differences in lipofectamine-mediated uptake, stability within the cell or delivery to the nucleus would produce substantial differences between the in vitro and cell-based assays, but Figures 2 and 3 show that there was a surprising level of agreement for most oligonucleotides. Thus, splicing is the main determinant of efficiency. The comparisons among oligonucleotides showed in addition that: (i) the effectiveness of the oligonucleotides was determined by the number of GGA motifs in the tail, (ii) GGA motifs were the most successful sequence tested and could bind SRSF1, (iii) 2′-O-methyl substitutions in the tail did not reduce activity, (iv) modifications increasing the stability of annealing reduced activity, (v) position is an important determinant, (vi) annealing to an upstream exon is effective and (vii) activity persists for at least 28 days after a second transfection and that RNA-containing tails confer a more enduring response than 2′-O-methyl analogues.
The finding that the length of the tail, or the number of GGA repeats, has such a strong influence on the efficiency of a TOES oligonucleotide and has interesting mechanistic implications. A systematic study of the effects of multiple doublesex enhancers showed that their effects were additive (36), from which it was inferred that the limiting factor in their actions was the probability that a bound activator protein would interact with constitutive splicing components. Our results are consistent with those. However, a recent analysis of responses to the transcription factor NF-KB has provided a formal description of other possible models (37). We envisage four possible mechanisms for the effects of tail length, shown in Figure 10. Model (i) suggests that there is a requirement for a threshold number of proteins bound; models (ii) and (iii) suggest that the multiple repeats of GGA increase either the probability of occupancy by a protein or the probability that a bound protein will interact subsequently with other components, depending on which step is limiting, whereas model (iv) suggests that bound proteins can participate in several independent slow reactions. Model (i) can be excluded by the graded response of tails with different numbers of GGA motifs. Recently, we determined the numbers of molecules of PTB bound to polypyrimidine tracts by single molecule methods (38). Based on our examination of possible arrangements of the proteins on to the tracts, we suggested that proteins containing multiple RNA binding domains with similar specificities, connected by flexible linkers, would have higher apparent affinities for sequences with repeated target motifs as a result of the multiplicity of possible binding sites and the multiplicity of arrangements possible for the domains within each site; any inhibition of the binding of multiple proteins might be attenuated if the binding of each domain were weak and therefore permitted shuffling of the domains (38). Such a mechanism might be consistent with the effects of tail length according to model (ii). However, the lack of detailed structures of the RRM domains of SRSF1 with RNA limits our ability to interpret the results. We are currently using single molecule methods to investigate the mechanism.
Figure 10.

Possible models for the effects of the length of the tail on the efficiency of splicing rescue. The diagram shows the bifunctional oligonucleotide and protein SRSF1, which has two RRM domains (ovals) and a RS-rich C-terminal region (shown unstructured). (i), a threshold number of molecules of protein are required to bind before activity is possible, possibly because proteins interact and binding is cooperative; (ii), the greater number of repeated motifs in the longer tails increases an otherwise extremely low affinity or probability of binding, (iii), the probability that a bound protein makes productive contacts is low, but occupancy by multiple molecules increases this probability, and (iv), bound proteins can make a number of different contacts and the presence of multiple proteins allows a number of concurrent interactions with other splicing components.
The substantial differences in performance between different tail sequences were surprising. The tails based on GAC, GGACCG and GAA motifs (oligonucleotides 7, 17 and 14, respectively) were not very effective at all, even though the GAC and GGACGG motifs in oligonucleotides 7 and 17 were based on sequences identified as very effective sites mediating activation by SRSF1, based on functional selection (31,32), GAA motifs have been reported to be optimal binding sites for SRSF1 in SELEX experiments with recombinant protein (39), and recent transcriptomic analyses by CLIP-SEQ confirm that the motif GAAGAR is enriched in transcripts bound in vivo by SRSF1 (40). However, GAA and particularly AGAA motifs are also associated with binding by the activator Tra2β (41,42), which is known to bind such motifs in the ESE of SMN2 exon 7 (34) and to stimulate the recruitment of U2 snRNP to the 3′-splice site (43). Thus, if oligonucleotide 14 bound Tra2β it should stimulate splicing. Preliminary results confirm that the oligonucleotide can bind purified Tra2β (N. Owen, unpublished data). Our results may indicate that the level of Tra2β binding to SMN2 exon 7 is not limiting, even though it has been reported that transfection of plasmids expressing Tra2β increases inclusion (34). The more important limitation may be the loss of an SRSF1 site in SMN2 exon 7 where the SMN1 and SMN2 exons differ at nucleotide 6 (44,45).
Different tail sequences in TOES oligonucleotides have been tested previously by expressing the oligonucleotide from a snRNA promoter in transfected or infected cells. Three sequences were expressed from a U6 snRNA promoter (46). These were rich in (A+C), (G+C) and GGA motifs, with the stated intentions of recruiting SRSF1, SRSF2 and Tra2β, respectively. They all worked, although they annealed (with a mismatch) to positions 6–26 in exon 7 and, in the absence of a control untailed sequence in parallel experiments, it was not clear to what extent the success was the result simply of annealing. Several other sequences have been expressed within a U7 minigene and compared with the original tail domain of our oligonucleotide 1 (47). Two of the sequences were largely identical to the (A+C)-rich and (G+C)-rich sequences tested by Baughan et al. (46), but they were inactive when targeted to nucleotides 31–48. Of the six tail sequences tested, the original sequence of oligonucleotide 1 was by far the most active. We conclude that GGA motifs confer the highest activity.
The position of annealing of a bifunctional oligonucleotide was very important. There are two separate considerations: the effects of annealing, which might obstruct enhancer or silencer motifs or alter secondary structures, and limitations on the position of the tail within the exon. The effects of annealing were distinguished by using untailed control oligonucleotides. A number of proteins are associated with the exon, including Tra2β (34), hnRNP A1 (48,49), Sam68 (50), hnRNP G (51), hnRNP Q (52) and SRSF9 (53). Where the binding sites of the proteins have been mapped, they appear to be either around nucleotide 6 or associated with the GAA motifs around the enhancer between nucleotides 19 and 27. In contrast, systematic surveys by saturating mutagenesis (54) or oligonucleotide scanning (13) show a much more complicated pattern. Oligonucleotides 18, 20, 21 and 22 inhibit inclusion; since they anneal across GAA motif nucleotides 19–27, we infer that they inhibit essential binding by Tra2β. Surprisingly, the successful TOES sequences expressed from a U6 snRNA promoter annealed to this region (46). Since mutating nucleotides 21–30 did not prevent splicing being rescued by oligonucleotide 1, we inferred that the tail can compensate for the absence of Tra2β binding but that its position is critical.
Oligonucleotides 19 and 23 inhibited, even though they do not obstruct any known binding site, but they overlap a region close to the GAA motifs that has previously been shown to be a target for inhibition by oligonucleotides (nucleotides 29–34). The same region has been targeted by a TOES sequence fused to U7 snRNA, but in this case exon 7 inclusion was stimulated (47). The slight positive effect of oligonucleotides 12 and 17 (annealing to positions 7–21) is consistent with the results from oligonucleotide scanning across the exon, which showed that an oligonucleotide annealing to this site produced the highest positive response among the oligonucleotides tested (13). In this case, the failure of the tail to improve the outcome is quite striking.
A possible explanation for the strong dependence of tail activity on its location and position relative to the annealing domain was that the TOES oligonucleotides act on the downstream 5′-splice site, which we inferred from their preferential stimulation of splicing of the downstream intron. The tail may need to be positioned on the 5′ rather than the 3′-end of the oligonucleotide, i.e., on the 5′ splice site side of the double-stranded region, so that RNA flexibility permits interactions. However, the oligonucleotide cannot be too close to the 5′-splice site, since U1 snRNPs form interactions up to 18 nt 5′- of the 5′-splice site (55) and the possibility of inhibition by proximity has been shown by the mutual interference of 5′-splice sites 25 nt apart (23,56). With this in mind, we designed the oligonucleotides complementary to the upstream globin exon and to SMN2 exon 6, both of which stimulated exon 7 inclusion efficiently, such that they were, respectively, 51 and 66 nt upstream of the 5′-splice sites. Similarly, the site we selected for stimulating inclusion of Ron exon 11 (57) was 55 nt upstream of the 5′-splice site. We infer that oligonucleotides annealing further downstream than nucleotides 2–16 in SMN2 exon 7 might be too close to the 5′-splice site.
The observation that TOES oligos targeted to upstream exons is effective and raises interesting mechanistic questions. In the short substrate used for splicing in vitro, it is likely that the formation of cross-intron interactions is rate-limiting. Increasing the level of binding of U1 snRNPs at the 5′-splice site of the upstream β-globin exon might increase the rate of splicing in vitro, although it is not obvious why it would increase the probability of splicing to the 3′-splice site of SMN2 exon 7 rather than the distal β-globin exon. The effects on the endogenous SMN2 gene in fibroblasts are very striking. Exon 6 is so far away (5.8 kb) from exon 7 that interactions across each exon would be expected to precede cross-intron interactions and to determine the splicing outcome (58). Based on this model, any increase in the efficiency of binding of splicing factors to exon 6 mediated by the TOES oligo would be expected to ensure exon 6 inclusion rather than exon 7 inclusion. However, the results might be explained by a kinetic model. If the TOES oligo annealed to exon 6 increased the stability or rate of U1 snRNP binding to the 5′-splice site during transcription it might increase the chance that the U1 was present on exon 6 at the moment when the 3′-splice site of exon 7 was transcribed. This might result in commitment to splicing between exons 6 and 7. Under normal circumstances, if U1 binding were weaker, then perhaps U1 snRNP would be more likely to bind exon 6 after both 3′-splice sites (7 and 8) have been transcribed and exon 7 would not have a kinetic advantage. It will be important for applications in other genes to identify the mechanisms involved.
The therapeutic value of oligonucleotides depends on the availability of convenient methods for delivering them to target tissues and their safety, and the cost-effectiveness of any such therapy depends in part on the frequency and dosage of administration. It has been demonstrated in mice that oligonucleotides that anneal to a silencer in SMN2 intron 7 can rescue necrosis in a mouse model of SMA when injected into cerebral ventricles (59) and that TOES oligonucleotides can rescue the phenotype of SMA mice when expressed from a transgene (18). Although ensuring the delivery of exogenous oligonucleotides to the central nervous system will require major advances in the methods of delivery, the success of oligonucleotides stimulating skipping of a dystrophin exon in clinical trials with systemic administration bodes well for the future. The oligonucleotides targeted to a silencer in intron 7 were shown to persist for at least 2 months in neuronal tissue and to produce an effect for at least half a year (59). These oligonucleotides were fully modified. We were surprised to find that the effects of a second transfection of a TOES oligonucleotide persisted for at least a month in fibroblasts, not least because in oligonucleotide 1 the majority of the tail sequence comprises of unmodified RNA. A likely explanation of this persistence is that the tail domain of the oligonucleotide becomes incorporated into discrete and well-defined RNP complexes, as we have observed in nuclear extracts (R. Dickinson, unpublished data).
We conclude that the very high levels of recovery of SMN2 exon 7 inclusion and SMN protein expression associated with oligonucleotide 1 and the persistence of its effects suggest that it is an appropriate candidate for further investigation as a potential therapeutic reagent in SMA. The levels of enhancement of SMN protein expression exceed any other quantitative data yet published. We suggest the following design principles for TOES oligonucleotides: (i) the tail should comprise GGA motifs, (ii) as many as possible should be included, (iii) the tail should be unmodified, apart from a cap, (iv) annealing should not be too stable, (v) the oligonucleotide should anneal more than 40 nt upstream of a 5′-splice site and (vi) splicing of the target exon should be limited by its 5′-splice site strength. It is likely that even more effective reagents could be made if more was known about the mechanisms by which enhancers and the SR proteins bound to them act to stimulate splicing.
FUNDING
Wellcome Trust (grant 074984); the European Commission (grant EURASNET-LSHG-CT-2005-518238, European Network of Excellence in Alternative Splicing to I.C.E.); Great Ormond Street Hospital Children’s Charity (to F.M.); Biotechnology and Biological Sciences Research Council (A. Malygin, Underwood Fellowship); Wellcome VIP award (to H.Z.); MRC (L.D.S., postgraduate studentship). Funding for open access charge: Wellcome Trust.
ACKNOWLEDGEMENTS
The authors thank Dr B. Yue for pure recombinant SRSF1.
REFERENCES
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chasin LA. Searching for splicing motifs. Adv. Exp. Med. Biol. 2007;623:85–106. doi: 10.1007/978-0-387-77374-2_6. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Burge B. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 5.Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim. Biophys. Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl Acad. Sci. USA. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunckley MG, Manoharan M, Villiet P, Eperon IC, Dickson G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 1998;7:1083–1090. doi: 10.1093/hmg/7.7.1083. [DOI] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A, Houlleberghs H, van Deutekom JC, van Ommen GJ, t Hoen PA. Exonic sequences provide better targets for antisense oligonucleotides than splice site sequences in the modulation of Duchenne muscular dystrophy splicing. Oligonucleotides. 2010;20:69–77. doi: 10.1089/oli.2009.0215. [DOI] [PubMed] [Google Scholar]
- 9.Popplewell LJ, Trollet C, Dickson G, Graham IR. Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol. Ther. 2009;17:554–561. doi: 10.1038/mt.2008.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 12.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 Exon 7 Inclusion by Antisense Oligonucleotides Targeting the Exon. PLoS Biol. 2007;5:e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc. Natl Acad. Sci. USA. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eperon IC, Muntoni F. Response to Buratti et al.: Can a ‘patch’ in a skipped exon make the pre-mRNA splicing machine run better? Trends Mol. Med. 2003;9:233–234. doi: 10.1016/s1471-4914(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 17.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 18.Meyer K, Marquis J, Trub J, Nlend Nlend R, Verp S, Ruepp MD, Imboden H, Barde I, Trono D, Schumperli D. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum. Mol. Genet. 2009;18:546–555. doi: 10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 19.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum. Mol. Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibrahim EC, Schaal TD, Hertel KJ, Reed R, Maniatis T. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc. Natl Acad. Sci. USA. 2005;102:5002–5007. doi: 10.1073/pnas.0500543102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian M, Maniatis T. A splicing enhancer complex controls alternative splicing of doublesex pre-mRNA. Cell. 1993;74:105–114. doi: 10.1016/0092-8674(93)90298-5. [DOI] [PubMed] [Google Scholar]
- 22.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 23.Eperon IC, Ireland DC, Smith RA, Mayeda A, Krainer AR. Pathways for selection of 5′ splice sites by U1 snRNPs and SF2/ASF. EMBO J. 1993;12:3607–3617. doi: 10.1002/j.1460-2075.1993.tb06034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caceres JF, Krainer AR. Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. EMBO J. 1993;12:4715–4726. doi: 10.1002/j.1460-2075.1993.tb06160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo P, Manley JL. Functional domains of the human splicing factor ASF/SF2. EMBO J. 1993;12:4727–4737. doi: 10.1002/j.1460-2075.1993.tb06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eperon IC, Makarova OV, Mayeda A, Munroe SH, Caceres JF, Hayward DG, Krainer AR. Selection of alternative 5′ splice sites: role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol. Cell. Biol. 2000;20:8303–8318. doi: 10.1128/mcb.20.22.8303-8318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Hoffmann HM, Grabowski PJ. Intrinsic U2AF binding is modulated by exon enhancer signals in parallel with changes in splicing activity. RNA. 1995;1:21–35. [PMC free article] [PubMed] [Google Scholar]
- 28.Zuo P, Maniatis T. The splicing factor U2AF35 mediates critical protein-protein interactions in constitutive and enhancer-dependent splicing. Genes Dev. 1996;10:1356–1368. doi: 10.1101/gad.10.11.1356. [DOI] [PubMed] [Google Scholar]
- 29.Shen H, Green MR. A pathway of sequential arginine-serine-rich domain-splicing signal interactions during mammalian spliceosome assembly. Mol. Cell. 2004;16:363–373. doi: 10.1016/j.molcel.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 30.Shen H, Green MR. RS domain-splicing signal interactions in splicing of U12-type and U2-type introns. Nat. Struct. Mol. Biol. 2007;14:597–603. doi: 10.1038/nsmb1263. [DOI] [PubMed] [Google Scholar]
- 31.Liu HX, Zhang M, Krainer AR. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998;12:1998–2012. doi: 10.1101/gad.12.13.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 33.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 34.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc. Natl Acad. Sci USA. 2000;97:9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen M, Nielsen CB, Nielsen KE, Jensen GA, Bondensgaard K, Singh SK, Rajwanshi VK, Koshkin AA, Dahl BM, Wengel J, et al. The conformations of locked nucleic acids (LNA) J. Mol. Recognit. 2000;13:44–53. doi: 10.1002/(SICI)1099-1352(200001/02)13:1<44::AID-JMR486>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 36.Hertel KJ, Maniatis T. The function of multisite splicing enhancers. Mol. Cell. 1998;1:449–455. doi: 10.1016/s1097-2765(00)80045-3. [DOI] [PubMed] [Google Scholar]
- 37.Giorgetti L, Siggers T, Tiana G, Caprara G, Notarbartolo S, Corona T, Pasparakis M, Milani P, Bulyk ML, Natoli G. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell. 37:418–428. doi: 10.1016/j.molcel.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Cherny D, Gooding C, Eperon GE, Coelho MB, Bagshaw CR, Smith CW, Eperon IC. Stoichiometry of a regulatory splicing complex revealed by single-molecule analyses. EMBO J. 2010;29:2161–2172. doi: 10.1038/emboj.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tacke R, Manley JL. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995;14:3540–3551. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanford JR, Wang X, Mort M, Vanduyn N, Cooper DN, Mooney SD, Edenberg HJ, Liu Y. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tacke R, Tohyama M, Ogawa S, Manley JL. Human Tra2 proteins are sequence-specific activators of pre-mRNA splicing. Cell. 1998;93:139–148. doi: 10.1016/s0092-8674(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 42.Tsuda K, Someya T, Kuwasako K, Takahashi M, He F, Unzai S, Inoue M, Harada T, Watanabe S, Terada T, et al. Structural basis for the dual RNA-recognition modes of human Tra2-β RRM. Nucleic Acids Res. 2011;39:1538–1553. doi: 10.1093/nar/gkq854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martins de Araujo M, Bonnal S, Hastings ML, Krainer AR, Valcarcel J. Differential 3′ splice site recognition of SMN1 and SMN2 transcripts by U2AF and U2 snRNP. RNA. 2009;15:515–523. doi: 10.1261/rna.1273209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 46.Baughan T, Shababi M, Coady TH, Dickson AM, Tullis GE, Lorson CL. Stimulating full-length SMN2 expression by delivering bifunctional RNAs via a viral vector. Mol. Ther. 2006;14:54–62. doi: 10.1016/j.ymthe.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen-Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol. Ther. 2007;15:1479–1486. doi: 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- 48.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 49.Vezain M, Saugier-Veber P, Goina E, Touraine R, Manel V, Toutain A, Fehrenbach S, Frebourg T, Pagani F, Tosi M, et al. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum. Mutat. 2010;31:E1110–E1125. doi: 10.1002/humu.21173. [DOI] [PubMed] [Google Scholar]
- 50.Pedrotti S, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Stamm S, Manley JL, Sette C. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010;29:1235–1247. doi: 10.1038/emboj.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum. Mol. Genet. 2002;11:2037–2049. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 52.Chen HH, Chang JG, Lu RM, Peng TY, Tarn WY. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol. Cell. Biol. 2008;28:6929–6938. doi: 10.1128/MCB.01332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Young PJ, DiDonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2beta1. Hum. Mol. Genet. 2002;11:577–587. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 54.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10:1291–1305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chabot B, Steitz JA. Multiple interactions between the splicing substrate and small nuclear ribonucleoproteins in spliceosomes. Mol. Cell. Biol. 1987;7:281–293. doi: 10.1128/mcb.7.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cunningham SA, Else AJ, Potter B, Eperon IC. Influences of separation and adjacent sequences on the use of alternative 5′ splice sites. J. Mol. Biol. 1991;217:265–281. doi: 10.1016/0022-2836(91)90541-d. [DOI] [PubMed] [Google Scholar]
- 57.Ghigna C, De Toledo M, Bonomi S, Valacca C, Gallo S, Apicella M, Eperon I, Tazi J, Biamonti G. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: Therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol. 2010;7:495–503. doi: 10.4161/rna.7.4.12744. [DOI] [PubMed] [Google Scholar]
- 58.Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc. Natl Acad. Sci. USA. 2005;102:16176–16181. doi: 10.1073/pnas.0508489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]