

Abstract
The absence epilepsies are characterized by recurrent episodes of loss of consciousness associated with generalized spike-and-wave discharges, with an abrupt onset and offset, in the thalamocortical system. In the absence of detailed neurophysiological studies in humans, many of the concepts regarding the pathophysiological basis of absence seizures are based on studies in animal models. Each of these models has its particular strengths and limitations, and the validity of findings from these models for the human condition cannot be assumed. Consequently, studies in different models have produced some conflicting findings and conclusions. A long-standing concept, based primarily from studies in vivo in cats and in vitro brain slices, is that these paroxysmal electrical events develop suddenly from sleep-related spindle oscillations. More specifically, it is proposed that the initial mechanisms that underlie absence-related spike-and-wave discharges are located in the thalamus, involving especially the thalamic reticular nucleus. By contrast, more recent studies in well-established, genetic models of absence epilepsy in rats demonstrate that spike-and-wave discharges originate in a cortical focus and develop from a wake-related natural corticothalamic sensorimotor rhythm. In this review we integrate recent findings showing that, in both the thalamus and the neocortex, genetically-determined, absence-related spike-and-wave discharges are the manifestation of hypersynchronized, cellular, rhythmic excitations and inhibitions that result from a combination of complex, intrinsic, synaptic mechanisms. Arguments are put forward supporting the hypothesis that layer VI corticothalamic neurons act as ‘drivers’ in the generation of spike-and-wave discharges in the somatosensory thalamocortical system that result in corticothalamic resonances particularly initially involving the thalamic reticular nucleus. However an important unresolved question is: what are the cellular and network mechanisms responsible for the switch from physiological, wake-related, natural oscillations into pathological spike-and-wave discharges? We speculate on possible answers to this, building particularly on recent findings from genetic models in rats.
Keywords: epilepsy, EEG, electrophysiology, neocortex, thalamus
INTRODUCTION
Physiological oscillations in cortico-thalamo-cortical networks are involved in many normal neurocognitive functions including perception, cognition, attention, memory, wakefulness and sleep (Bouyer et al., 1981; Marczynski et al., 1984; Loring et al., 1985; Ribary et al., 1991; Pulvermuller et al., 1997; Tallon-Baudry et al., 1997; Keil et al., 1999; Steriade, 1999; Engel et al., 2001; Pinault, 2004). Additionally, it is increasingly evident that some natural oscillations develop into pathological oscillations (either by hypersynchronized natural oscillations or by evolving from a gradual transformation of natural oscillations). One of the most remarkable examples is the transformation of natural thalamocortical (TC) oscillations into spike-and-wave discharges (SWDs) that underlie absence seizures (Pinault et al., 2001; Pinault, 2003). In this review we focus on the cellular and network mechanisms that underlie the generation of SWDs, particularly on work from well-recognized, genetic models of absence epilepsy in rats.
Absence epilepsies are a group of idiopathic neurological disorders that are characterized by the occurrence of recurrent, unpredictable, non-convulsive (absence) seizures (Commission on Classification and Terminology of the International League Against Epilepsy, 1981). Clinically, absence seizures consist of a sudden loss of consciousness (awareness and responsiveness) without loss of either body tone or posture (Blumenfeld, 2005). They might be accompanied by minor motor clonic movements or automatisms, particularly if prolonged.
Absence seizures are brief, lasting 5–20 sec but, occasionally, are more prolonged and, rarely, non-convulsive status epilepticus (i.e. continuous absence seizure activity) occurs (Crunelli and Leresche, 2002). The clinical seizures are accompanied on a scalp electroencephalogram (EEG) by generalized SWDs with a frequency of 3Hz, which are recorded synchronously over both hemispheres and arise and cease abruptly out of an apparently normal background EEG activity.
Although it is generally believed that SWDs have sudden onset and offset, gradual modulations in alpha frequency were recorded in patients before the SWDs that accompany absence seizures (Inouye et al., 1990). This finding indicates that the period just preceding the occurrence of absence-related SWD represents a transition state from the background activity to the SWD.
On depth EEG recordings, SWDs are seen during absence seizures in both the thalamus and the cerebral cortex (Williams, 1953; Gloor and Fariello, 1988). Typical, well-organized, absence-related SWDs occur particularly during either immobile, inattentive wakefulness or drowsiness (Crunelli and Leresche, 2002). In contrast to focal onset, non-convulsive seizures (i.e. complex partial seizures), normal consciousness returns immediately following the cessation of the seizure activity with no period of post-ictal confusion.
Absence seizures also have a pharmacological response profile that distinguishes them from other, particularly focal, seizure types. Most (80%) patients will have control of their absence seizures with either valproate or ethosuximide (Avoli et al., 2001). However, several other anti-epileptic drugs that are effective in focal seizures, including carbamazepine, and drugs that enhance GABA activity in the brain, such as vigabatrin and tiagabine, are either ineffective or aggravate absence seizures, both in humans (Lehmann et al., 1999; Perucca et al., 1998) and in animal models (Micheletti et al., 1985; McLean et al., 2004; Wallengren et al., 2005; Liu et al., 2006).
Absence seizures occur either alone or in combination with other seizure types, particularly generalized tonic-clonic and myoclonic seizures, in a variety of generalized epilepsy syndromes, such as childhood absence epilepsy, juvenile absence epilepsy and in a proportion of patients with juvenile myoclonic epilepsy (Commission on Classification and Terminology of the International League Against Epilepsy, 1981). These syndromes are classified according to the ILAE system as primary (idiopathic) generalized epilepsies, in which it is presumed that seizures arise simultaneously in both hemispheres (i.e. generalized) and that, other than the tendency to have seizures, the brain is otherwise normal structurally and functionally. Both of these assumptions are being challenged by studies utilizing modern neurophysiological, neuroimaging and genetic techniques. In this review we concentrate predominantly on neurophysiological data, particularly from genetic rat models, that have revolutionized the concepts about the onset and generation of absence seizures.
ABSENCE EPILEPSIES HAVE A GENETIC BASIS
The epilepsy syndromes in which absence seizures occur are believed to be predominantly genetic in etiology (Scheffer and Berkovic, 2003). Twin studies have provided strong evidence for the importance of genetic influences, with a concordance for the presence of absence seizures of 70% and 33% in monozygotic twins and first-degree relatives, respectively (Berkovic et al., 1998). Several genetic defects have been found in rare families in whom the epilepsy strongly follows a monogenic inheritance pattern, with most of these affecting either ion channels or membrane receptors, including the g2-subunit of the GABA-A receptor complex (GABRG2) in a family in whom some members had childhood absence epilepsy (Wallace et al., 2001). However, the cause of the epilepsy in most patients with absence seizures is more complex, and it is almost certain that more than one gene is involved (i.e. polygenic) (Steinlein, 2004). Genome-wide scans have highlighted several loci as potentially carrying genetic polymorphisms rendering susceptibility to absence seizures, again mostly implicating ion channels and membrane receptors (reviewed in Crunelli and Leresche, 2002).
An important recent study found a variety of different mutations in the gene for the low threshold Ca2+ channel, CaV3.2 (CACNA1H), in 14 of 118 unrelated Han Chinese patients with idiopathic absence epilepsy, but in none of 230 control subjects (Chen et al., 2003). Several of these mutations alter the neurophysiological properties of the expressed T-type Ca2+ channels, facilitating increased Ca2+ influx with channel activation (Khosravani et al., 2004; Khosravani et al., 2005). Low threshold (T-type) Ca2+ channels appear to have a crucial role in the generation of the reciprocal TC burst firing that underlies absence seizures. Knock-out mice that lack the T-type channel Cav3.1 do not display TC burst firing and, unlike wild-type controls, are insensitive to SWDs induced by GABAB receptor agonists (Kim et al., 2001). Moreover, crossbreeding Cav3.1 knock-out mice with mutant mice with ataxia and absence epilepsy abolishes SWDs in the progeny (Song et al., 2004). Studies in Genetic Absence Epilepsy Rats from Strasbourg (GAERS) have demonstrated increased low threshold Ca2+ currents (Tsakiridou et al., 1995; Kuisle et al., 2006) and expression of mRNA for the 3.2 subunit (Talley et al., 2000) in the thalamic reticular nucleus (TRN) compared to non-epileptic control (NEC) rats. Recent work from our group has identified a homozygous, mis-sense, non-conservative, single nucleotide (G to C) mutation in the Cav3.2 gene in GAERS compared to NEC rats and the Rattus norvegicus genetic database in a region that is proposed to be involved in the regulation of channel inactivation (i.e. linker III–IV) (Kyi et al., 2006). Currently, in vitro and whole animal neurophysiological studies to correlate genotype-phenotype relationships are examining the importance of this mutation in the expression of absence seizures in GAERS. Genetic studies conducted in well-established genetic models of absence epilepsy (see below) support the polygenic nature of this idiopathic disorder (Gauguier et al., 2004; Rudolf et al., 2004).
OVERVIEW OF TC CIRCUITRY
The two crucial structures in the generation and maintenance of absence seizures are the thalamus and the cortex (Williams, 1953; Gloor and Fariello, 1988). A mapping study of SWDs in GAERS revealed that absence-related SWDs occur in both the dorsal thalamus (especially in the ventral and lateral regions, which correspond to the sensorimotor system) and the neocortex (Vergnes et al., 1990). Chemical ablation of either of these structures abolishes absence seizures in the GAERS model (Vergnes and Marescaux, 1992). Both the cerebral cortex and the thalamus are interconnected with other subcortical structures, forming highly distributed systems that are involved in different aspects of arousal and consciousness (attention, cognition, perception and sensorimotor processing) (Guillery and Sherman, 2002). The thalamus is connected reciprocally with the TRN and with the cerebral cortex. The TRN is a reservoir of GABAergic neurons that project exclusively into the dorsal thalamus (Fig. 1) (Pinault, 2004).
Fig. 1. Schematic of the thalamocortical system.
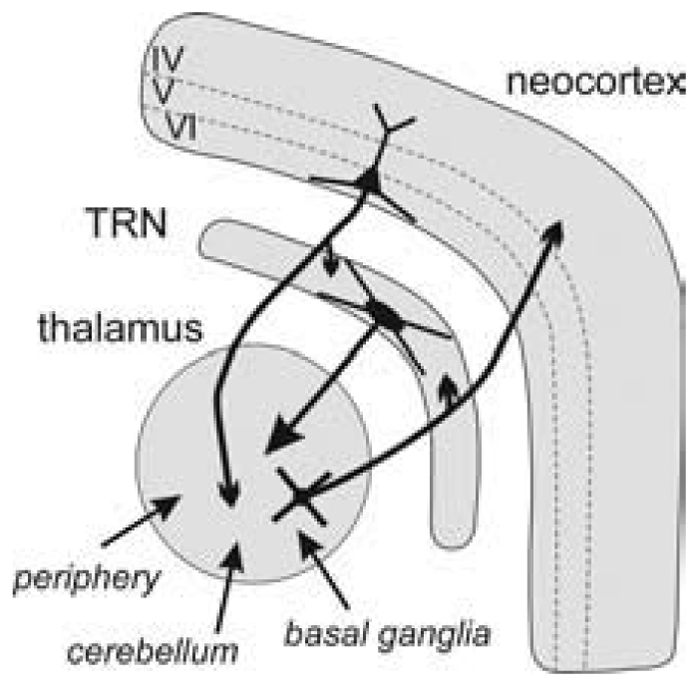
The thalamus is reciprocally connected with the cerebral cortex and with the thalamic reticular nucleus (TRN). It receives many inputs from the periphery (sensory), cerebellum and basal ganglia. The TRN is a reservoir of GABAergic neurons that project to the dorsal thalamus. Thalamocortical and corticothalamic neurons are glutamatergic and cross the TRN where they give off axon collaterals.
THE NEUROPHYSIOLOGICAL BASIS OF ABSENCE SEIZURES
In the middle of the 20th century, clinical and experimental findings supported the hypothesis that absence-related SWDs originate in subcortical structures, particularly the thalamus (the centrencephalic hypothesis) (Penfield and Jasper, 1954). Indeed, simultaneous EEG recordings in the thalamus, white matter and cerebral cortex during absence seizures in children seemed to indicate that SWDs started in the thalamus (Williams, 1953). Repetitive electrical stimulation at 3Hz of the midline and intralaminar nuclei of the cat’s thalamus produced an ‘arrest reaction’ and SWDs in the cerebral cortex (Jasper and Drooglever-Fortuyn, 1947).
At the same time, considering anatomical and electroclinical data, the centrencephalic hypothesis was proposed in opposition to the cortical theory (Gibbs and Gibbs, 1950). Over the past 1–2 decades the cortical theory of absence epilepsy has gained increasing prominence (Niedermeyer, 1996). This theory proposes that absence seizures result primarily from a cortical disorder that generates the discharges, and the thalamus is involved secondarily via normal TC interactions. Experimental support comes from studies in genetic rat models of absence epilepsy, such as GAERS and Wistar Albino Glaxo/Rijswijk (WAG/Rij) rats (Meeren et al., 2002; Pinault, 2003), and clinical (Bancaud et al., 1974; Lagae et al., 2001), EEG (Holmes et al., 2004) and imaging studies in patients (Lagae et al., 2001; Archer et al., 2003).
The studies in the feline generalized penicillin epilepsy (FGPE) pharmacological model of absence seizures, which was developed in the 1960s (Prince and Farrell, 1969), altered the concepts regarding the neurophysiological basis of absence seizures. Gloor put forward the cortico-reticular hypothesis in which normal thalamic activity reaching hyper-excitable cortex triggers SWDs (Gloor, 1968). The FGPE model involves the administration of an intramuscular injection of a large dose of penicillin to cats, resulting in a transient epileptic state with electrographic, behavioural and pharmacological similarities with human absence seizures (Gloor and Fariello, 1988). In this feline model, SWDs were recorded to develop from a gradual transformation of sleep spindles (Kostopoulos, 2000). The occurrence of the SWDs coincided with a hypersynchronization of rhythmic cellular discharges in related cortical and thalamic regions (Avoli and Gloor, 1982; Avoli et al., 1983). More recently, comprehensive study in ferret thalamic slices revealed that spindle-like oscillations can be transformed into paroxysmal oscillations (von Krosigk et al., 1993). In thalamic relay neurons, these oscillations are underlain by enhanced GABA(B) receptor-mediated IPSPs, which occur following reduction of GABA(A) receptor-mediated inhibition. Together, these in vivo and in vitro neurophysiological studies yield data supporting Gloor’s cortico-reticular hypothesis (Gloor, 1968; Kostopoulos, 2001; McCormick and Contreras, 2001). Thus, these studies support the concept that sleep-related natural oscillations can give rise to absence-related SWDs. This has also been found in ketamine-xylazine anaesthetized cats (Steriade and Contreras, 1995).
ANIMAL MODELS OF ABSENCE SEIZURES AND EPILEPSY
Because absence epilepsy is not amenable to surgical treatment, there have been few invasive electrophysiological studies in patients for ethical reasons (Williams, 1953). Therefore, most neurophysiological data relating to absence seizures are derived from studies in animal models. It is important to recognize that the relevance of this data for the human condition cannot necessarily be assumed, and will depend on the validity and relevance of the animal model from which it is derived. Studies from different models produce contrasting findings, resulting in divergent conclusions regarding the pathophysiological basis of absence seizures. Examples that are discussed in this review include whether SWDs arise out of sleep spindles and whether the seizures have a focal cortical origin. Each of the conflicting conclusions might be valid for the particular model in which the observations were made, but judgments about which best applies to absence epilepsy in humans are difficult and require assumptions to be made. Criteria for an ideal animal model of absence epilepsy have been published (Danober et al., 1998; Snead et al., 1999). Currently, no available model is considered truly ‘ideal’, with each having its own specific advantages and limitations.
Nevertheless, studies in animal models are essential to gain insight and generate hypotheses regarding the pathophysiological processes that result in epilepsy because technical and ethical factors greatly limit what can be done experimentally in humans. Although in vitro studies are valuable, in vivo studies in whole animals remain the most important component of epilepsy research. Seizures derive from neuronal networks that are often topographically extensive, with many modulating inputs from nearby and remote areas. Therefore, to validly study seizures and epilepsy, an intact, functioning brain is required (Steriade, 2001a).
Many animal models of absence seizures and epilepsy have been described (Snead et al., 1999). These can be divided broadly into models of seizures and models of epilepsy. In the former, seizures are induced in otherwise non-epileptic animals by administering either a chemical or electrical stimulus that results in SWDs accompanied by behavioral arrest. Important examples of this are the FGPE model discussed previously (Prince and Farrell, 1969), and low-dose pentylenetetrazole (PTZ) (Marescaux et al., 1984a; Snead, 1998; McLean et al., 2004) and g-butyrolactone (GHB) models in rats (Snead, 1998). By contrast, in true models of epilepsy, animals have spontaneous, recurrent seizures and, therefore, more prima facie relevance to the human situation. Additionally, the true epilepsy models are less subject to biases and confounding effects related to the method of evoking the seizures. Most of the true epilepsy models have a genetic basis, but exposure to neurotoxic substances during neurodevelopment can also result in an epileptic animal (e.g. the AY-treated rat model of atypical absence epilepsy (Cortez et al., 2001). The genetic models can be divided into the monogenic mice models and the (presumed) polygenic rat models. However the former group, examples of which are tottering (Fletcher et al., 1996), lethargic (Burgess et al., 1997) and stargazer (Letts et al., 1998) mice, are generally considered less relevant to the human situation because of their monogenic mode of inheritance and the presence of other significant neurological deficits (e.g. ataxia). Increasingly, the presumed polygenic rat models, the most studied of which are GAERS (Marescaux et al., 1984b) and the WAG/Rij rats (Drinkenburg et al., 1991), are becoming models of choice for neurophysiological, neuropharmacological and genetic-expression studies of absence epilepsy.
There are many similarities between the two Wistar rat models. GAERS were derived originally from a Wistar rat colony in Strasbourg in whom it was observed that 20–30% of the animals had recurrent episodes resembling absence seizures with spontaneous, synchronous, bilateral SWDs on the EEG accompanied by behavioral arrest (Marescaux et al., 1984b). Selectively inbreeding according to the presence or absence of this epileptic phenotype resulted in a strain in which 100% of the animals have seizures by 14 weeks (i.e. GAERS) and another in whom 100% do not, even when followed for more than a year (i.e. NEC rats) (reviewed in Danober et al. 1998). Many studies from laboratories around the world have established that GAERS is an excellent model of absence epilepsy with an EEG, behavioral, etiological, pathophysiological and pharmacological profiles that are similar to that of human absence epilepsy. The availability of the NEC strain from the same original colony makes comparative studies very powerful for identifying neurobiological and neurobehavioural characteristics that are linked specifically to the epileptic phenotype (i.e. any feature that tracks with the phenotype has a high a-prior case for being either a cause of effect of the epileptic condition). The rats spend 10–15% of quiet wakefulness in seizures making them highly practical for pharmacological studies (Marescaux et al., 1984b; Stroud et al., 2005; Liu et al., 2006). The two most significant characteristics in which the model differs from the human condition is in the cycle frequency of the seizures (i.e. 7–11 versus 2.5–3.5 Hz) and their developmental ontogenesis (i.e. seizures commence in adolescent GAERS and become more frequent as they become older).
ABSENCE-RELATED SWDS ARE RELATED MORE TO WAKEFULNESS THAN TO SLEEP
Findings obtained from in vivo studies in the cat model (reviewed by Gloor and Fariello, 1988; Kostopoulos, 2000) and in vitro from brain slices (reviewed by McCormick and Contreras, 2001) have lead to the hypothesis that absence-related SWDs are related more to sleep than to wakefulness. This is an example of findings from animal models not being well assessed for their potential relevance to the human situation. In humans with primary generalized epilepsy SWDs occur during sleep and, on occasion, evolve from sleep spindles. Papers by Kellaway and colleagues (Kellaway, 1985; Kellaway et al., 1980) report that spike-and-wave bursts are more prevalent during the spindle stages of sleep, but they did not distinguish between well-organized and prolonged absences and irregular, short inter-ictal bursts or between convulsive and non-convulsive forms of generalized epilepsies.
In contrast, well-organized absence seizures clearly occur predominantly during wakefulness and drowsiness and rarely occur out of sleep (Niedermeyer, 1965; Mirsky et al., 1986; Halasz, 1991; Loiseau, 1992). Niedermeyer (Niedermeyer, 1965) reviewed 50 patients with absence epilepsy, recorded during wakefulness and sleep: in 39 cases absences were recorded only during wakefulness; in two cases during wakefulness and drowsiness; and in nine cases during wakefulness and sleep; in no case were absences recorded only during sleep. By contrast, inter-ictal discharges, which might not be seen during wakefulness, emerge during light sleep. Niedermeyer quoted ‘Petit mal absences or sub-ictal bursts with generalized synchronous 3/sec spike-wave complexes may occur in light sleep as well as during waking and drowsiness. More commonly, however, the well-organized complexes tend to disintegrate in sleep into subtle short sequences of spikes’ (Niedermeyer, 1982). Similarly, Pedley wrote ‘The well-formed 3-Hz spike-wave paroxysms seen with the patient awake give way during sleep to discharges of shorter duration composed of spikes or poly-spikes recurring as isolated bursts, with and without associated paroxysmal slow waves’ (Pedley, 1984). Furthermore, sleep spindles are a characteristic of deeper levels of sleep (Stage II and III), a time when true absences seizures occur rarely, if ever. Taken together, these clinical papers show that: (1) absence seizures usually peak after the morning awakening and before and after afternoon naps; (2) when absences occur at night it is usually during short, transient awakening from sleep; (3) the characteristic EEG features that are specific to absences disappeared during non-REM sleep but reverted to regular rhythmic 2–4Hz in REM sleep. However absences are rare during REM sleep.
The relationship of seizures in GAERS to the wake-sleep cycle parallels much more closely the human situation than those of the FGPE model. The group of Marescaux and Vergnes studied the occurrence of SWD in GAERS according to the vigilance state: 61% of SWD started and ended in wakefulness, 16% started in wakefulness and ended in light non-REM sleep, 9% started in light non-REM sleep and ended in wakefulness, and 14% started and ended in sleep (Lannes et al., 1988). Similarly, most SWDs occur during passive wakefulness or during transition from wake to light sleep in the WAG/Rij rat model (Drinkenburg et al., 1991).
On the basis of these clinical and experimental data, it seems clear that typical, well-organized, absence-related SWDs are related more to wakefulness than to sleep.
WAKE-RELATED SENSORIMOTOR OSCILLATIONS ARE MORE PRO-EPILEPTOGENIC THAN SLEEP SPINDLES IN GAERS
In GAERS, the seizure-related SWDs have been demonstrated to develop in the somatosensory TC system, either during quiet immobile wakefulness or under neuroleptanalgesia, from a natural, medium-voltage (< 0.4mV), 5–9Hz oscillation (Fig. 2) (Pinault et al., 2001). This natural rhythm, which emerges from a desynchronized EEG, is also recorded in NEC rats, the control inbred strain that is free of any spontaneous SWD, indicating that this physiological rhythm in itself is not sufficient to generate SWDs. It occurs in a highly coherent manner in the frontal and parietal cortices, and in the related thalamic regions (Pinault et al., 2006). In neuroleptanalgesied rats both the SWDs and the natural oscillations have an internal frequency that is lower by 2–3 Hz than that of both the SWDs and the physiological rhythm recorded in freely moving GAERS and NEC rats (Pinault, unpublished observations). The natural rhythm is, thus, probably identical to a previously identified rodent sensorimotor 7–12 Hz rhythm, which also occurs during body immobility and begins in the cortex before spreading to the thalamus and brainstem (Fanselow et al., 2001; Nicolelis and Fanselow, 2002). Therefore, the 5–9 Hz rhythm that is recorded in GAERS and NEC rats under neuroleptanalgesia is likely a wake-related sensorimotor (SM) rhythm (Pinault, 2003). The internal frequency of this rhythm is similar to that of spontaneously occurring SWDs (Pinault et al., 2001), which indicates that in GAERS the transition from SM oscillations to absence-related SWDs is a gradual process in cellular synchronization (Pinault et al., 2001). EEG recordings in patients with absence epilepsy revealed rapid changes in the EEG just preceding (,5sec) SWDs (Inouye et al., 1990), in which the principal wave components tended to approach gradually those of the SW complexes in amplitude and frequency.
Fig. 2. Sensorimotor (SM) oscillations give rise to absence-related SWDs.
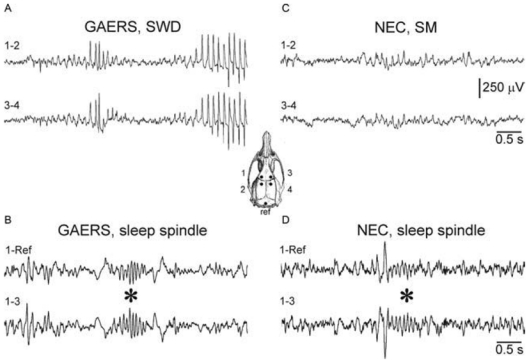
(A) In freely moving GAERS, SWDs spontaneously arise from bilaterally synchronous, medium-voltage SM (or 5–9Hz) oscillations in the surface EEG. (C) Similar synchronized rhythmic EEG activity, without SWDs, was recorded in non-epileptic control (NEC) rats. These SM oscillations are distinguishable from sleep spindles (waves at 7–15 Hz) (* in B), which were often preceded by a K-complex (* in D). The inset illustrates the location of the EEG electrodes (Ref = reference) on the frontoparietal cortex. Also note that, in contrast to sleep spindles (B and C), SM oscillations emerge from a desynchronized EEG (A and C). The scale bars are valid for each recording. Adapted from (Pinault et al., 2001).
In the frontoparietal cortex of rats, SM oscillations emerge from a desynchronized EEG whereas sleep-related spindles develop from a synchronized EEG (Fig. 2) (Pinault et al., 2001). In contrast to SM oscillations, spindle oscillations never last >2sec. Sensorimotor oscillations are often accompanied by small-amplitude (< 0.5mV), negative spike components. These might be embryo of the negative spike of the absence-related SW complex. Under neuroleptanalgesia, SM oscillations can co-exist with delta oscillations, whereas the incidence of spindle-like oscillations is low (Pinault et al., 2006). Systemic administration of barbiturates abolishes SWDs and SM oscillations, and increases the incidence of spindle-like oscillations (Fig. 3), which can contain spike components. Of importance, in GAERS spindle-like oscillations are never followed by SWDs.
Fig. 3. Pentobarbital abolishes SWDs and induces spindle-like oscillations.
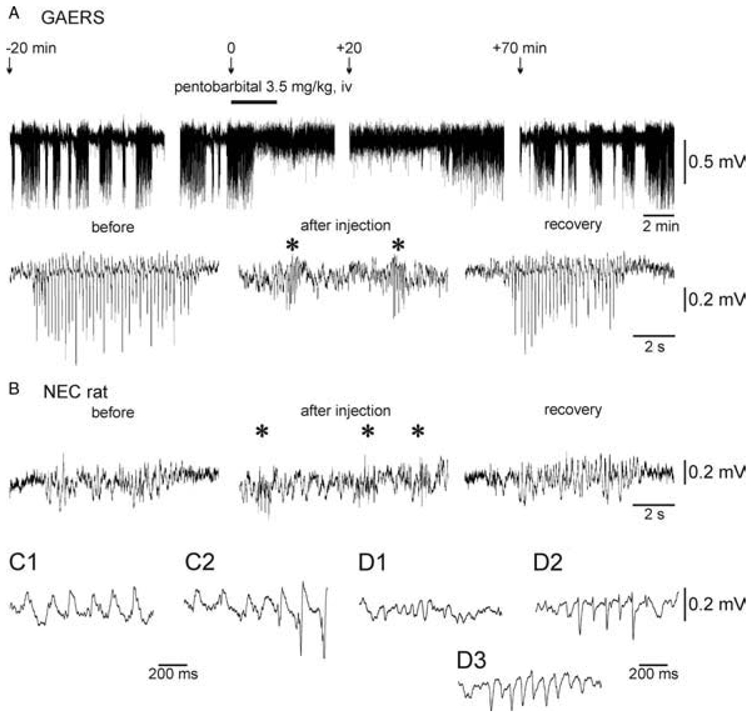
(A) Top: Surface EEG recordings (truncated) over the primary somatosensory cortex in a GAERS under neuroleptic analgesia, which received a subanaesthetic dose of pentobarbital. Bottom: Expanded EEG traces. Before pentobarbital injection and during the recovery period, the EEG displays recurrent high-voltage SWDs of variable duration. Note that pentobarbital suppresses SWDs and induces the occurrence of spindle-like episodes (*) in a GAERS (A) and in a non-epileptic control (NEC) rat (B). (B) In a NEC rat, SM oscillations occur spontaneously before and at least 20–30min after pentobarbital injection (recovery). (C) Expanded traces of a typical SM oscillation recorded in a NEC rat (C1) and a SM oscillation that gives rise to a SWD in a GAERS (C2). (D) Three types of spindle-like oscillations recorded in GAERS and NEC rats after barbiturate administration: (D1) contains only sinusoid-like waves; (D2) biphasic spikes; and (D3) mainly monophasic spikes. Adapted from (Pinault et al., 2006).
Within the somatosensory TC system SM oscillations occur in a more coherent manner than spindle-like oscillations (Pinault et al., 2006). As discussed later, the intracellular events underlying SM and spindle-like oscillations are different in thalamic relay and reticular neurons.
DISTINCT INTRACELLULAR CORRELATES OF SENSORIMOTOR AND SPINDLE-LIKE OSCILLATIONS
Sensorimotor and spindle-like oscillations share a common frequency band (~5–15Hz) and might contribute to the generation of SWDs in the rat (Pinault, 2003) and in FGPE model (Kostopoulos et al., 1981) respectively, so it is important to compare in vivo their respective underlying thalamic cellular mechanisms. In both thalamic relay and reticular neurons, SM oscillations emerge from a significantly more depolarized membrane potential (> −65mV) than do spindle-like oscillations (Fig. 4A–D) (Pinault et al., 2006). Also, the membrane input resistance of both cell types is significantly lower just before the occurrence of SM oscillations than before spindle-like oscillations. Furthermore, spectral analysis has revealed that spontaneously occurring membrane potential oscillations of these thalamic neurons include beta and gamma activities, which are more powerful just before SM oscillations than those occurring just before spindle-like oscillations (Fig. 5) (Pinault et al., 2006). Together, these in vivo findings indicate that TRN and TC neurons receive more incoming tonic excitatory inputs when they are going to generate SM oscillations than when they are going to generate spindle-like oscillations.
Fig. 4. Thalamic relay (A,C,E,F) and reticular (B,D,G,H) intracellular rhythmic activities associated with either spontaneously occurring SM or spindle-like oscillations in the related surface EEG.
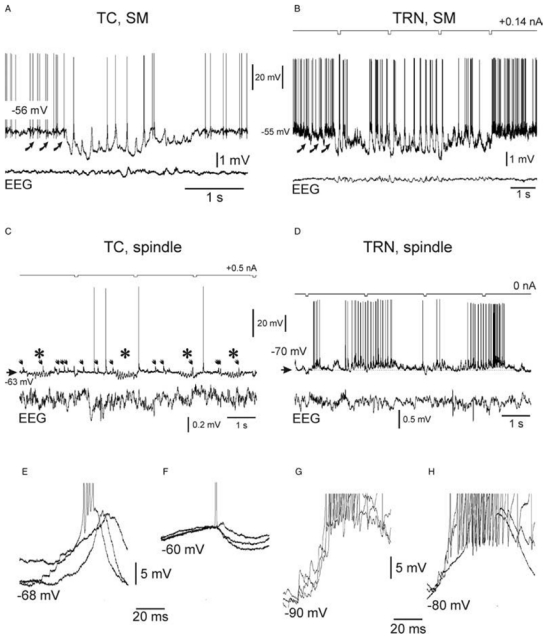
Intracellular recordings are from non-epileptic control (NEC) rats. (A) Intracellular rhythmic activity of a thalamocortical (TC) neuron associated with spontaneously occurring SM oscillations in the simultaneous surface EEG recording. (B) Intracellular rhythmic activity of a thalamic reticular nucleus (TRN) neuron associated with spontaneously occurring SM oscillations in the related surface EEG. A current square pulse of −0.2 nA was delivered every 2sec. The curved arrows in A and B indicate barrages of EPSPs. (C) Barbiturate-induced intracellular spindle-like oscillations (7–15Hz; *) in a TC neuron. Note that individual synaptic potentials of presumably lemniscal origin (arrows) occur during and in between the spindle-like episodes. Note that these postsynaptic potentials do not trigger a low-threshold Ca2+ potential. (D) Barbiturate-induced intracellular spindle-like oscillations (7–15 Hz) in a TRN neuron. (E,F) Superimposition of three successive rhythmic depolarizations, which occur during SM (E) and during spindle-like oscillations (F) in a TC cell. Note the occurrence of small synaptic and/or intrinsic unitary events at the beginning of the depolarizing waves in E. In E, one of the three depolarizations triggers an apparent low-threshold Ca2+ spike topped by a high-frequency burst of action potentials, whereas in F only a single action potential was evoked during one of the events. (G,H) Superimposition of three successive rhythmic depolarizations, which occur during SM (G) and spindle-like oscillations (H) in a TRN cell. Note the occurrence of synaptic and/or intrinsic unitary events at the beginning of the depolarizing waves in G and H. The action potentials are truncated in E–H. Adapted from (Pinault, 2003; Pinault et al., 2006).
Fig. 5. The membrane potential of thalamic neurons displays more powerful fast rhythmic activities between SM oscillations and SWDs than between spindle-like oscillations.
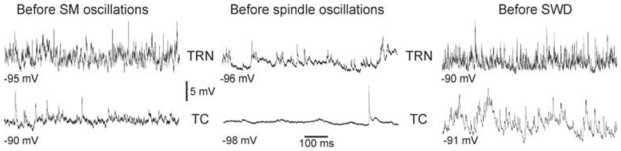
Intracellular recordings of spontaneously occurring membrane potential oscillations in thalamic reticular nucleus (TRN) and thalamocortical (TC) neurons, which occur before the occurrence of SM oscillations (in a NEC rat under neuroleptic analgesia), the occurrence of absence-related SWDs (in GAERS, under neuroleptic analgesia), and the occurrence of spindle-like oscillations (in a NEC rat after barbiturate treatment). These recordings were obtained while applying DC hyperpolarizing current of at least −1 nA. Adapted from (Pinault et al. 2006).
Most excitatory inputs of thalamic relay and reticular neurons arise in layer VI of the neocortex (Bourassa et al., 1995). Thus, layer VI corticothalamic (CT) neurons might have a leading role in determining the up and down states of the membrane potential in thalamic relay and reticular neurons (Pinault et al., 2006). Moreover, irregular, but tonic, firing has been recorded in layer VI CT neurons in between SM oscillations or absence-related SWDs (Pinault, 2003). Our data, thus, indicate that desynchronized cortical activity (steady up state) might be a prerequisite in GAERS for the initiation of SM oscillations and SWDs but is not necessary for the generation of sleep spindles, which usually emerge from a synchronized EEG.
In relay neurons, SM oscillations are characterized mainly by rhythmic subthreshold/threshold depolarizations that occur during a tonic hyperpolarization (Fig. 4A) and which can trigger a low-threshold Ca2+ potential crowned by a high-frequency burst of APs (Fig. 4E) (Pinault, 2003). By contrast, spindle-like oscillations are characterized mostly by a rhythmic, GABA(A) receptor-mediated, short-lasting hyperpolarization (Fig. 4C, F) (Pinault et al., 2006). In TRN cells, SM oscillations occur during a tonic hyperpolarization (Fig. 4B), whereas spindle-like oscillations occur during a slow depolarizing wave (Fig. 4D). In these GABAergic neurons, both oscillation types usually produce rhythmic high-frequency bursts of APs (Fig. 4G, H), which are commonly underlain by at least a low-threshold Ca2+ potential (Pinault, 2003).
The low-threshold Ca2+ potential is important in determining the internal pattern of the high-frequency burst of APs. The acceleration-deceleration pattern of SM oscillation-related TRN bursts is less pronounced than the well-known acceleration-deceleration pattern of TRN spindle bursts (Pinault et al., 2006), which are caused by the underlying low-threshold Ca2+ potential (Mulle et al., 1986; Spreafico et al.,1988; Avanzini et al., 1989; Bal and McCormick, 1993; Contreras et al., 1993). In addition, the instantaneous frequency within SM bursts is significantly lower than that in spindle bursts (Pinault et al., 2006). Furthermore, SWD-related bursts in the TRN have the highest internal frequency (Pinault et al., 2006), which indicates that the epilepsy-related TRN bursts are underlain by at least a low-threshold Ca2+ potential (Pinault, 2003; Slaght et al., 2002) that is, on average, more powerful than that underlying the SM-related and spindle-related TRN bursts. This might be the manifestation of cellular hyper-synchronization associated with absence-related SWDs and is consistent with the finding that GAERS have increased low-threshold Ca2+ potentials (Tsakiridou et al., 1995) and expression of the Cav3.2 subunit in the TRN compared with NEC rats (Talley et al., 2000).
The difference between the intracellular events underlying SM oscillations and spindle-like oscillations, and the similarities between those underlying SM oscillations and generalized SWDs (see below), indicate that, in GAERS, SM oscillations are more pro-epileptogenic than sleep spindles (Pinault et al, 2006).
SIMILAR ELECTROPHYSIOLOGICAL CHARACTERISTICS OF SENSORIMOTOR OSCILLATIONS AND ABSENCE-RELATED SWDS
On the surface EEG over the frontoparietal cortex in GAERS, absence-related generalized SWDs are significantly more persistent (usually several tens of seconds) and more stereotyped (because of the regular, rhythmic occurrence of the epileptiform event, such as the SW complex) than SM oscillations (3.4 ± 0.9sec, mean ± S.E.M., 0.5–20sec). In thalamic relay and reticular neurons, the probability of having an AP discharge on every cycle tends to be higher during SWDs than during SM oscillations (Pinault et al., 2001; Pinault, 2003). However, whatever the type of oscillations, this probability is usually much lower in TC than in TRN neurons mainly because of a coincidence in TC neurons of a TRN-induced GABA(A) IPSP with the recurrent SW-related depolarization (Pinault et al., 1998; Pinault, 2003; also see below).
Furthermore, in vivo, intracellular recordings have revealed that the cellular mechanisms underlying SM oscillations and generalized SWDs in GAERS are qualitatively similar (Pinault, 2003; see below). However, in TRN cells the SW-related depolarization is, on average, shorter in duration than that associated with natural SM oscillations (up to 110msec versus 180msec) (Fig. 6A–D). The relatively short duration of the SW-related depolarization is presumably conditioned by the occurrence of a Ca2+-dependent K+ hyperpolarization as a result of cellular hypersynchronization (Fig. 6D) (Pinault, 2003).
Fig. 6. Normal- and absence-related intracellular SM oscillations in thalamic reticular nucleus neurons are qualitatively similar.
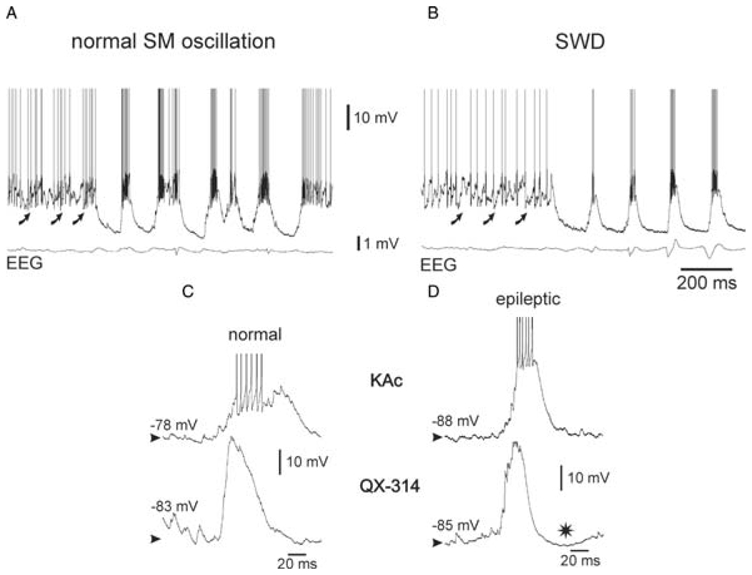
Intracellular recordings of thalamic reticular nucleus neurons during a spontaneously occurring SM oscillation (A) and a spike-and-wave discharge (SWD) (B) in the EEG in a NEC rat and in a GAERS, respectively. The curved arrows in A and B indicate rhythmic barrages of EPSPs. (C,D) Comparison of two typical recurring threshold depolarizing waves, which were recorded during normal SM oscillations in a NEC rat (C) and during SWDs in a GAERS (D), respectively. The intracellular recordings were made with a KAc-(upper traces) or QX-314-filled (lower traces) micropipette. * in D indicates a late hyperpolarization that probably results from a Ca2+-activated K+ current. The action potentials are truncated in C and D for clarity. Adapted from (Pinault, 2003).
In conclusion, in GAERS absence-related SWDs correspond to the sudden transformation of natural, medium-voltage SM oscillations, which are moderately synchronized (Pinault et al., 2001; Pinault, 2004; Pinault et al., 2006) into hyper-synchronous, stereotyped, high-voltage oscillations (or SWDs). However, the cellular and network mechanisms that are responsible for the conversion of physiological into pathological oscillations are unknown (see below).
LAYER VI CORTICOTHALAMIC NEURONS ACT AS ‘DRIVERS’ DURING ABSENCE SEIZURES
In vivo, paired, single-unit, extracellular recordings of TC and TRN neurons of the somatosensory system were performed in GAERS during spontaneously occurring bilateral SWDs (Pinault et al., 2001). Cross-correlation analyses of related TC and TRN discharges have demonstrated that they occur in a much more synchronous and phase-locked manner during SWDs than during natural medium-voltage SM oscillations (Fig. 7). Three-dimensional reconstruction of the recorded neurons has revealed that these high-degree SWD-related synchronizations are not related to direct synaptic connections between the recorded TC and TRN neurons (Fig. 7E) (Pinault, 2003).
Fig. 7. Paired, single-cell, extracellular recordings of TC and TRN neurons of the somatosensory system during a spontaneously occurring, medium-voltage, SM oscillation and an absence-related spike-and-wave discharge in a GAERS.
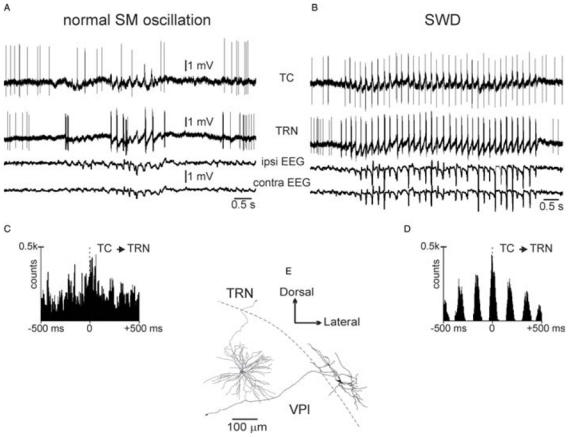
(A,B) Recordings are from the same TC–TRN pair in a GAERS. (C,D) Superimposition of four representative cross-correlograms (2 msec resolution) computed from four paired recordings obtained during normal (C) or epileptic (D) SM oscillations. (E) Three-dimensional reconstruction of the two recorded neurons. Note that they are not directly connected. Abbreviations: ipsi, ipsilateral; contra, contralateral.
Because CT neurons of layer VI are glutamatergic and innervate both TC and TRN neurons, paired extracellular recordings of histologically identified CT neurons with related TC or TRN neurons were performed during spontaneously occurring SWDs (Pinault, 2003). These demonstrate that CT AP discharges are also phase-locked with the SW complex, but they phase-lead related TC and TRN discharges by 7msec on average (Fig. 8).
Fig. 8. Dual extracellular recordings of a CT neuron with either a TC (A1–A3,D) or a TRN (B1–B3,E) cell during spontaneous SWDs.
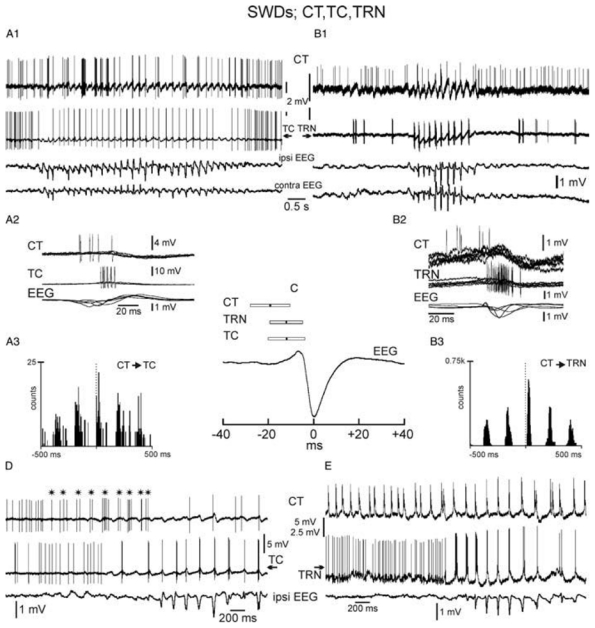
(A2,B2) Five superimposed, successive, recordings of SW-related action potential discharges. (A3,B3) Cross-correlograms (2 msec resolution) computed from pair recordings. (C) Means and S.D. of the time relationship between the CT, TC and TRN action potential discharges and the SW complex (CT: −19.4 ± 8.8 msec, 88 SW complexes from four rats: TRN: −11.9 ± 7.3 msec, 108 SW complexes from six rats: TC: −12.6 ± 8.2 msec, 133 SW complexes, seven rats. (D) * indicates the beginning of the CT rhythmic discharge, which starts before both the TC rhythmic firing and the occurrence of the seizure. (E) The CT cell discharges in a rhythmic manner well before the related TRN neuron and before the beginning of the SWD.
To determine the membrane mechanisms that are responsible for the generation of synchronized TC and TRN discharges, in vivo, intracellular recordings of TC and TRN neurons were performed during spontaneously occurring SWDs (Pinault, 2003). In both TC and TRN cells, a rhythmic depolarization was recorded concurrent with the SW complex (Fig. 9). This depolarization occurs during a tonic hyperpolarization and can be topped by an AP discharge, as previously noted. The SW-related depolarization has an indented time course, its amplitude is abolished when setting the membrane potential close to AP threshold, and its frequency of occurrence is not affected by injection of sustained hyperpolarizing or depolarizing currents. Together, these electrophysiological features indicate that, in both TC and TRN neurons, the SW-related depolarization is the summation of numerous EPSPs, which further trigger voltagedependent components, especially a low-threshold Ca2+ potential crowned by a high-frequency burst of action potentials (APs) (Pinault, 2003). Both the SW-related depolarization and the low-threshold Ca2+ potential can concur with some cycles but, as a rule, the depolarization occurs systematically earlier that the intrinsic potential (Pinault, 2003).
Fig. 9. Intracellular activities of TC (A,C) and TRN (B,D) neurons during spontaneously occurring SWDs in GAERS.
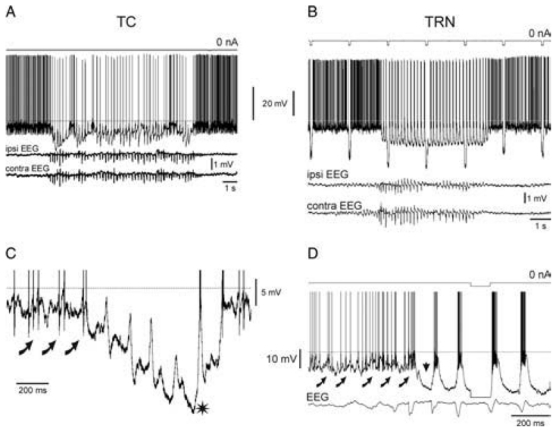
The traces in A and B are expanded in C and D, respectively, to show the first intracellular events that are associated with the ‘onset’ of SWDs. Curved arrows indicate rhythmic barrages of EPSPs, which occur just before the beginning of spontaneously occurring SWDs. The horizontal dotted line indicates the action potential threshold (− 58mV) at rest. The action potentials are clipped in C and D. Adapted from (Pinault, 2003).
Thus, our in vivo anatomo-electrophysiological data from GAERS demonstrate that TC and TRN discharges, which occur in a synchronous and phase-locked manner during every SW complex, are synaptically triggered by a common excitatory input. Because CT neurons of layer VI are glutamatergic, innervate both TC and TRN neurons (Bourassa et al., 1995) and fire a few msec before them, the absence-related rhythmic synchronous EPSPs are probably the result of the firing of these CT neurons (Fig. 10). This absence-related CT firing cannot originate in layer V pyramidal neurons because they do not innervate the TRN and the ventral posterior nucleus (see below).
Fig. 10. Likely spatio-temporal cellular interactions between related CT, TC and TRN neurons during absence-related-SWD in GAERS.
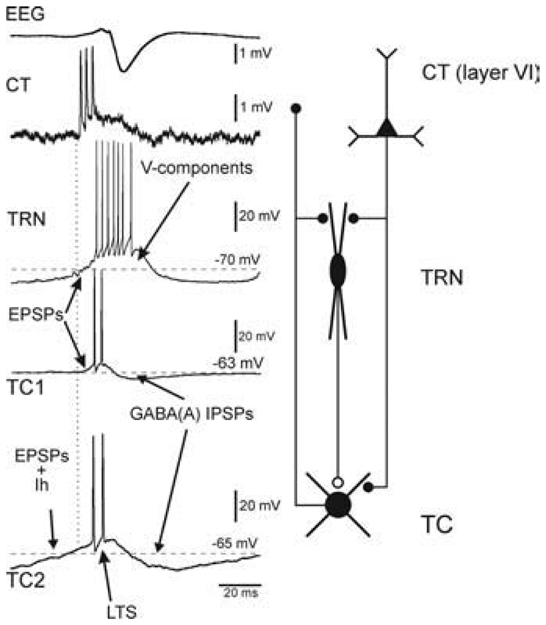
In both non-epileptic control rats and GAERS, at least two types of TC neurons coexist, one of which (TC2) is endowed with a presumed H-current. Note that thalamic, relay and reticular, discharges occur in synchronous, phase-locked manners during the occurrence of an absence-related rhythmic spike-and-wave complex in the cortical surface EEG. Left: SW-related extracellular CT and intracellular TC and TRN activities. From top to bottom: A SW complex (EEG), an extracellular CT discharge, an intracellular TRN discharge, and two typical intracellular TC1 and TC2 discharges. The second TC cell (TC2) exhibits a presumed H-current that coincides with an EPSP barrage. The ramp-shaped depolarization, which includes a presumed Ih, can trigger a low-threshold Ca2+ spike (LTS). In TRN cells, the EPSP barrage can trigger voltage-dependent components (V-components, including a low-threshold Ca2+ potential). Right: Schematic of the anatomical relationships between the three main elements that make up the TC system. Adapted from (Pinault, 2003).
Several lines of evidence strongly indicate that layer VI neurons behave more like ‘drivers’ than ‘modulators’ during absence seizures. The dorsal thalamus includes first-order (FO) and higher-order (HO) nuclei (Guillery, 1995; Guillery and Sherman, 2002). HO thalamic nuclei receive two extrareticular GABAergic inputs, one from the zona incerta and the other from the anterior pretectal nucleus (Bartho et al., 2002; Bokor et al., 2005). Glutamatergic TC axons cross the TRN where they give off collaterals (Jones, 1985). Corticothalamic axons, which are also glutamatergic, originate from cortical layers V and VI. Layer V CT axons do not innervate TRN cells but innervate only HO thalamic nuclei (Deschênes et al., 1994; Feig and Harting, 1998), whereas layer VI axons innervate the TRN and both FO and HO nuclei (Bourassa et al., 1995). Layer V CT neurons are considered to play a key relay role in large-scale cortico-thalamo-cortical network communication.
On the basis of morphological characteristics of their terminations, Layer V CT axons are considered as drivers and layer VI CT axons as modulators (Guillery, 1995; Guillery and Sherman, 2002). Layer V axons are thick and give rise to clusters of large terminals (like prethalamic axons ending in FO nuclei), whereas layer VI axons are thin and give rise to a greater number of smaller terminals, which are dispersed in larger thalamic territories and in slabs in the TRN. Thus, the finding that layer VI CT neurons that project to FO somatosensory (ventral posterior) nuclei exert a leading role in the absence-related rhythmic hypersynchronization of related TC and TRN neurons strongly indicates that layer VI neurons, at least those of the somatosensory system, can behave rather like ‘drivers’. Because layer VI cells innervate large thalamic territories, this electrophysiological property would be efficient to recruit large numbers of TC neurons on each cycle in FO and HO thalamic nuclei. The hypothesis that CT neurons behave like drivers during the generation of absence seizures is also supported indirectly by electrophysiological studies in ferret thalamic slices (Bal et al., 2000; Blumenfeld and McCormick, 2000).
However, knowing that layer VI of the neocortex contains at least two major types of CT neurons, one innervating only FO nuclei, and the other FO and HO nuclei (Zhang and Deschênes, 1997; Deschênes et al., 1994; Guillery and Sherman, 2002), it is important to know whether all types of layer VI CT neurons operate similarly and in a synchronous manner during the generation of absence-related generalized SWDs. Indeed, if all layer VI CT neurons behave like drivers during absence seizures, those that project to certain HO nuclei would be expected to fire after those innervating FO nuclei (in particular ventral posterior nuclei) because it has been demonstrated that, in at least some HO nuclei, such as the intralaminar centrolateral and paracentral nuclei (Fig. 11), TC neurons fire after FO TC neurons (Seidenbecher and Pape, 2001). By contrast, in our own recordings we have found that in other HO thalamic neurons (medial dorsal and dorsal posterior nuclei), TC neurons sometimes fire well before FO ventral posterior TC neurons and the surface EEG spike (Pinault and O’Brien, unpublished findings). The most striking finding in these recordings is that the time relationship between the TC firing and the EEG spike is considerably more variable from discharge-to-discharge in HO than in FO nuclei. This indicates that the firing of HO thalamic nuclei TC neurons are less synchronized with the SWDs than FO nuclei TC neurons, at least in the somatosensory system.
Fig. 11. Temporal relationship between an absence-related spike-and-wave complex and associated first-order and higher-order thalamic cellular activities in GAERS.
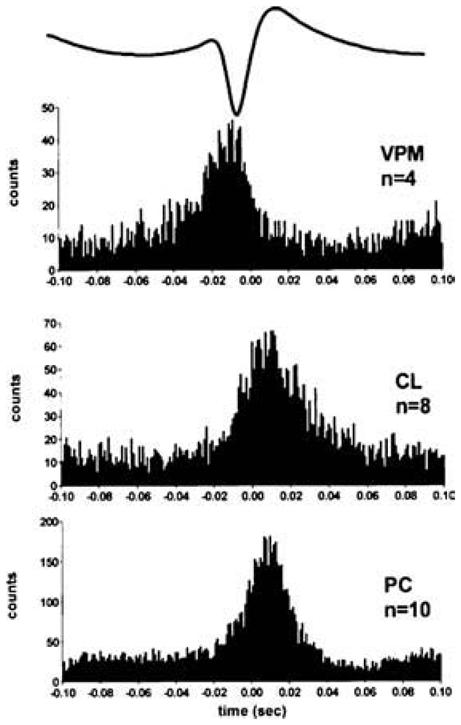
Peri-event time histograms of unit activity (bin width 1msec) associated with the spike component of the SW complex on the EEG demonstrate activity in first-order (VPM) and higher-order (PC and CL) thalamic nuclei. Note that higher-order nuclei fire after first order thalamic nuclei during the SW complex. Trace above histograms shows an averaged spike-wave complex. Abbreviations: CL, centrolateral; PC, paracentral VPM; medial part of the ventral posterior nucleus. Adapted from (Seidenbecher and Pape, 2001).
Of importance, in vivo single-cell anatomical studies using the juxtacellular technique (Pinault, 1996) demonstrate that the rat somatosensory cortex contains another important type of projection neurons, the cortico-cortical cells (Zhang and Deschênes, 1997). These neurons have two important features: (1) they innervate local and distant cortical areas, including the second somatosensory cortex and the motor cortex. Axonal branches have also been observed in the corpus callosum (Zhang and Deschênes, 1997); and (2) an in vitro electrophysiological study demonstrates that corticocortical neurons are endowed with pronounced phasic firing that is characterized by a brief high-frequency burst of APs in response to a current pulse of depolarization (Mercer et al., 2005). These anatomo-functional properties provide further support for the hypothesis that layer VI corticocortical neurons might also behave like drivers. Together these findings indicate that, at least in GAERS, layer VI of the somatosensory cortex includes all the ingredients for generation, synchronization, and spreading of genetically-determined SWDs for one to other cortical areas (especially the sensorimotor system) and to the thalamus.
RHYTHMIC AND TONIC HYPERPOLARIZATIONS IN THALAMIC RELAY AND RETICULAR NEURONS DURING ABSENCE-RELATED SWDS
In GAERS during absence seizures, in both TC and TRN neurons, the SW-related CT-induced depolarization (see above) is followed by a short-lasting hyperpolarization, forming a depolarizing wave-hyperpolarizing wave sequence during a tonic hyperpolarization (Fig 9) (Pinault et al., 1998; Slaght et al., 2002; Pinault, 2003). In TC neurons the short-lasting hyperpolarization is mediated by a Cl− conductance due to TRN-induced activation of GABA(A) receptors (Pinault et al., 1998; Pinault, 2003). Moreover, microiontophoretic application of bicuculline, a GABA(A) receptor antagonist, on TC neurons during absence seizures significantly increases the duration of SW-related burst firing in WAG/Rij rats (Staak and Pape, 2001).
By contrast, GABA(B) receptors do not contribute significantly to the generation of absence-related rhythmic and tonic hyperpolarization in GAERS (Feig and Harting, 1998; Staak and pape, 2001). Together these findings do not support the hypothesis that rhythmic GABA(B) receptor-mediated IPSPs and low-threshold Ca2+ potentials topped by high-frequency bursts of APs are essential in the generation of absence-related intracellular activity in TC neurons, which is an hypothesis that was born from an in vitro study of thalamic slices of the visual system of ferrets (von Krosigk et al., 1993).
In TRN neurons the short-lasting hyperpolarization appears to be mediated by a Ca2+-dependent K+ current (Pinault, 2003). In these neurons, this current is likely to potentiate the de-inactivation of T-type Ca2+ channels, which, once activated by incoming EPSPs from synapsing TC neurons, would lead to the generation of rhythmic robust burst firing during absence seizures (Tsakiridou et al., 1995). In both TC and TRN neurons, the SWD-related tonic hyperpolarization is likely to be mediated by a disfacilitation induced by the sudden arrest of firing simultaneously in a large number of layer VI CT neurons (Pinault et al., 2006). Furthermore, in both TC and TRN neurons the tonic hyperpolarization is abolished when their membrane potential is brought to AP threshold but not by applying strong hyperpolarizing currents. In addition, the membrane potential of both cell types contains beta and gamma oscillations, which are significantly more powerful just before the occurrence of absence seizures (Pinault et al., 2006). These fast membrane potential oscillations are associated with irregular, but tonic, firing in layer VI CT neurons (Pinault, 2003). Therefore, layer VI CT neurons have a major role not only during absence-related rhythmic hypersynchronized depolarizations (or rhythmic short-lasting up states) but also in maintaining a depolarizing pressure (or steady up state) in thalamic relay and reticular neurons in between SWDs.
The tonic hyperpolarization during SWDs is usually deeper in TRN neurons than in TC neurons (Fig. 9A, B) (Pinault, 2003). This difference might be accounted for by the fact that, in contrast to TC neurons, TRN neurons receive glutamatergic afferents from both the neocortex and the dorsal thalamus. However, the proposed disfacilitation mechanism does not exclude the contribution of K+ currents in the tonic hyperpolarization (Pinault et al., 1998; Polack and Charpier, 2006). It is worth mentioning that, during absence seizures, the intrinsic and synaptic mechanisms underlying the rhythmic and tonic hyperpolarizations interact with each other in CT, TC and TRN neurons.
THALAMIC RELAY AND RETICULAR NEURONS HAVE OPPOSITE BEHAVIOUR DURING ABSENCE-RELATED SWDS
As stated above, in GAERS during SWDs, TRN and TC neurons have a propensity to fire in a synchronous, phase-locked manner during every SW complex. However, both the probability of firing and the number of APs during the SW complex are much lower in a majority (75%) of TC neurons than in TRN cells (Pinault et al., 1998; Pinault, 2003). It is worth renoting that, in contrast to TC neurons, nearby TRN neurons tend to fire a high-frequency burst of APs during every SW complex (Slaght et al., 2002; Pinault, 2003). A lower probability of firing in TC neurons has also been recorded in ketamine-xylazine anaesthetized cats in most (60%) TC neurons during the evolution of sleep-related oscillations into SWDs (Steriade and Contreras, 1995). In GAERS, the lower probability of firing of TC neurons during the SW-related depolarization is likely to be due mainly to the coincident TRN-induced GABA(A) receptor-mediated rhythmic hyperpolarization and the tonic hyperpolarization, which persists as long as SWDs (Pinault et al., 1998; Pinault, 2003). Another mechanism that might also contribute to the contrasting behavior of TC and TRN neurons during absence-related SWDs is that CT synapses, which are glutamatergic and excitatory, are functionally more efficient in TRN than in TC neurons, as demonstrated in vitro in CT slices of mice (Golshani et al., 2001; Steriade, 2001b).
WHAT ARE THE RELATIVE CONTRIBUTIONS OF FO AND HO THALAMIC NUCLEI DURING ABSENCE SEIZURES?
The SWD-related inhibitions of TC neurons are thought to be important neuronal substrates for the loss of consciousness experienced by the patients during absence seizures (Kostopoulos, 2001). During absence-related inhibitions TC neurons (at least those of FO nuclei) are not responsive to pre-thalamic signals, and so cannot relay peripheral signals to the cerebral cortex (Pinault, 2003; Steriade and Contreras, 1995). This raises an important issue as to whether HO nuclei TC neurons relay information from one neocortical area to others during absence seizures. An in vivo study in GAERS shows that most TC neurons in intralaminar nuclei display synchronized high-frequency bursts of APs, which are time-locked with the SW complex (Seidenbecher and Pape, 2001). Our own recordings have extended these to other HO nuclei (medial dorsal, posterior group, central median and lateral habenular nuclei and in addition have shown that other HO TC neurons) show a rhythmic inhibition of their tonic firing during SWDs, which is phasically related to the hyperpolarizing wave (Pinault and O’Brien, unpublished findings). These findings indicate that in HO nuclei normal TC neuronal firing is altered during absence seizures so that they are unable to reliably relay information from one cortical area to another, as is required for conscious operations. Therefore these functional interruptions of normal cortico-thalamo-cortical signaling might be an important neuronal substrate of the loss of consciousness that occurs during absence seizures.
As stated previously, layer V CT neurons have a key relay role in large-scale cortico-cortical network communication, which are the major neuronal substrates of HO processes, (i.e. consciousness, cognition, attention and perception). Paired cellular recordings in vivo in the somatosensory cortex of GAERS reveal that during absence seizures layer V neurons usually fire after layer VI neurons during the SW complex (Pinault, 2003). Because layer V CT neurons innervate HO nuclei but not FO nuclei, HO nuclei TC neurons (at least in some HO nuclei) would be expected to fire after FO TC nuclei neurons (Pinault, 2003). This hypothesis is supported by findings from an independent, in vivo study in GAERS, which demonstrates that TC neurons of intralaminar nuclei (HO nuclei) fire after TC neurons of the ventral posterior nucleus (FO nucleus) during the SW complex (Fig. 11) (Seidenbecher and Pape, 2001). As discussed above, our recordings indicate that this time relationship might differ somewhat for other HO nuclei, but that the discharge-to-discharge variability is consistently greater than in the FO nuclei. These findings indicate that CT neurons innervating HO nuclei show a less tight relationship to the SWDs that those innervating FO nuclei.
Spontaneous, high-voltage, rhythmic spike (HVRS) discharges at 6–12Hz have been recorded in the EEG of the frontoparietal cortex of Long-Evans rats (Semba et al., 1980; Nicolelis and Fanselow, 2002; Shaw and Liao, 2005). It has recently been reported that there are behavioral and EEG similarities between Long-Evans rats and the genetic rat models of absence epilepsy, such as GAERS (Shaw and Liao, 2005). In vivo intracellular recordings of thalamic neurons of the ventral lateral nucleus, from which neurons project to layer V neurons of the motor cortex (whose axons can be activated antidromically from the ventral lateral nucleus), have demonstrated that thalamic neurons fire, on average, 12msec before layer V neurons during every cycle of HVRS activity (Polack and Charpier, 2006). This study provides strong experimental evidence to support the hypothesis that HVRS discharges in Long-Evans rats are similar to absence-related SWDs. Also, knowing that layer V pyramidal cells project mainly to HO thalamic nuclei, this study supports the hypothesis that, in general, HO nuclei TC neurons fire after FO nuclei TC neurons.
It is worth emphasizing that HO thalamic nuclei also receive inputs from layer VI pyramidal neurons, and that layer V CT neurons also innervate GABAergic neurons of the zona incerta and anterior pretectal nucleus, which, in turn, innervate HO thalamic nuclei (Bartho et al., 2002; Bokor et al., 2005). These anatomical connections highlight two important questions: (1) what are the relative contributions of inputs from layer V and layer VI neurons projecting to HO nuclei during absence seizures; and (2) what are the relative contributions of TRN and extra-TRN GABAergic inputs in HO thalamic nuclei during the generation of absence-related SWDs. Addressing these issues will help understanding of the neuronal mechanisms underlying conscious operations.
DIVERSE ROLES OF PACEMAKER CURRENTS IN THE GENERATION OF ABSENCE-RELATED SWDS
Hyperpolarization-activated cation non-selective (HCN) channels have important roles in determining the resting membrane potential, in the excitability of neuronal networks, and in the timing of neuronal oscillations in cortical and subcortical structures, including in the TC system (Pape, 1996; Luthi and McCormick, 1998; Pape et al., 2007). HCN channels give rise to somatodendritic inward currents (Ih) that are sensitive to voltage (activated by hyperpolarization) and to intracellular cAMP levels (Pape, 1996; Robinson and Siegelbaum, 2003). The synthesis of cAMP is triggered, in great part, by Ca2+ entry through low-threshold Ca2+ potentials, for example, during persistent TC oscillations (Luthi and McCormick, 1998; Wang et al., 2002). Properties of Ih are deeply disturbed in neurological diseases, including during epileptogenesis and epilepsy-related abnormal oscillations (Chen et al., 2001; Strauss et al., 2004; Budde et al., 2005; Kuisle et al., 2006).
In vivo intracellular recordings have revealed that, in GAERS and NEC rats, most (75%) TC neurons of the somatosensory thalamus (medial and lateral parts of the ventral posterior nucleus) display an apparent hyperpolarization-activated depolarizing sag potential (Pinault, 2003; Kuisle et al., 2006). The depolarizing sag potential is the physiological correlate of Ih voltage gating. During absence seizures, TC neurons that display the depolarizing sag generate a ramp-shaped depolarization just before the SW-related depolarizing wave (barrage of EPSPs), which can trigger a low-threshold Ca2+ potential that is crowned by a high-frequency burst of APs (Pinault, 2003). It is known that Ih contributes to the level of the resting membrane potential and to intrinsic pacemaker activities (reviewed by Pape, 1996; Luthi and McCormick, 1998). Additionally, TC neurons that display the depolarizing sag potential have a relatively high membrane input resistance, making them hyperexcitable.
No significant differences in passive and active membrane properties, including the amplitude of the sag potential (in relation to the degree of hyperpolarization), have been found between TC neurons of GAERS and NEC rats in vivo (Kuisle et al., 2006). By contrast, a diminished cAMP-dependent Ih regulation has been demonstrated in vitro in TC neurons of the somatosensory thalamus of GAERS before and after the onset of their epilepsy (Fig. 12) (Kuisle et al., 2006). A lowered sensitivity of Ih to cAMP has also been identified in pre-epileptic WAG/Rij rats (Budde et al., 2005), supporting the hypothesis that abnormal cAMP sensitivity of HCN channels in TC neurons contribute in the epileptogenesis in the two (GAERS and WAG/Rij rats) genetic models of absence epilepsy. For example, low sensitivity of HCN channels to cAMP might facilitate the occurrence of burst discharges in TC neurons during epileptogenesis, thereby increasing their excitability (Budde et al., 2005).
Fig. 12. The Ih of GAERS TC cells has a diminished sensitivity to submaximal, near-physiological cAMP pulses, but not to saturating concentrations of cAMP.
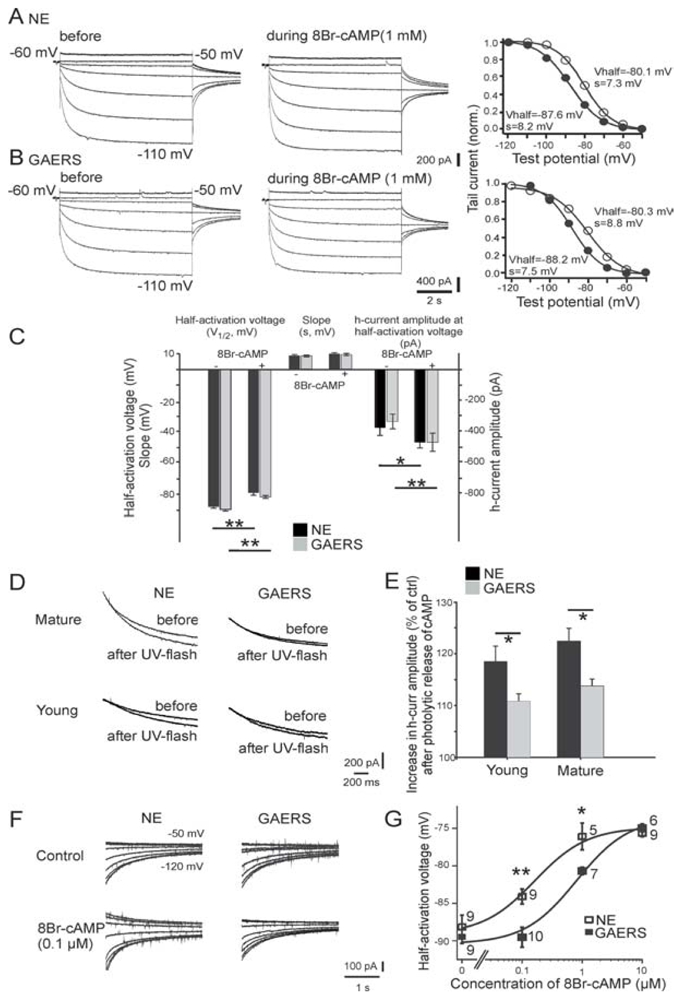
(A,B) Current responses of mature TC cells from a non-epileptic control (NEC) rat (A) and a GAERS (B) to increasing negative test voltages (test voltages −50mV and −110mV are indicated next to the traces) before, and in the continuous presence of, 1mM 8Br-cAMP. Corresponding activation curves, constructed from tail currents evoked at −80mV (see Methods), are shown to the right. Thick lines represent the optimal fit of a Boltzmann curve, with the resulting values for the half-activation voltage (Vhalf) and the slope (s) indicated next to them. Filled and open circles represent values before and during 8Br-cAMP application, respectively. (C) Pooled data for V1/2, for s, and for the current amplitude at −90mV before, and in the continuous presence of 8Br-cAMP (1mM). Except for s, 8Br-cAMP significantly altered all control values. The changes in all three parameters were indistinguishable between mature NEC rat (n = 8) and GAERS (n = 7) TC cells. (D) Responses of Ih to photolytic release of caged cAMP in mature (top row) and young (bottom row) NEC animals and GAERS. Overlay of current responses to 30mV hyperpolarizing voltage steps before and after application of a UV flash in cells perfused with caged cAMP. Holding potential was − 60mV. For clarity, only current relaxations during the hyperpolarizing voltage step are shown; passive responses to the step voltage were blanked. (E) The percentage increase in current response in young and mature NEC animals (young, n = 12; mature, n = 8) and GAERS (young, n = 12; mature, n = 12). (F) Representative tail currents obtained from TC cells in the absence of (control) or during perfusion of the cellular interior with 0.1mM 8Br-cAMP. The same voltage protocol as in A was used, voltage steps applied prior to evoking tail currents are indicated next to the traces, and tails were evoked at −75mV. (G) Concentration-response curve for the effect of 8Br-cAMP on the half-activation voltage. The number of recorded cells is indicated next to the symbols. Fitting of the Hill equation was achieved by fixing the Hill coefficient to 1 (see Methods in Kuisle, 2006), yielding a ~5-fold increase of half maximal concentration of 8Br-cAMP in GAERS. *P < 0.05, ** P <0.01. From (Kuisle et al., 2006).
The decreased sensitivity of Ih to cAMP in adult GAERS is associated with a significant increase (59%) in expression of the relatively cAMP-insensitive HCN1 channel isoform in the medial part of the ventral posterior nucleus and the somatosensory sector of the TRN, compared with age-matched NEC rats (Kuisle et al., 2006). This region of the thalamus is related functionally to the epileptic focus of absence seizures, which has been demonstrated in the somatosensory cortex of WAG/Rij rats (Meeren et al., 2002; see below). The concentration of mRNA encoding the cAMP-sensitive HCN2 channel isoform, which is the most abundant in the thalamus (Santoro et al., 2000), is the same in GAERS and NEC rats. Furthermore, the concentration of mRNA encoding the HCN1 isoform is similar in the somatosensory cortex of GAERS and NEC rats (Kuisle et al., 2006). Western blot analyses have revealed similar levels of the HCN1 and HCN2 isoforms in the thalamus of adult GAERS and NEC rats.
Upregulation of Ih is induced in vitro following repetitive low-threshold Ca2+ potentials in TC neurons of GAERS (Kuisle et al., 2006). This upregulation manifests itself in an after-depolarization, which attenuates the rhythmic occurrence of a low-threshold Ca2+ potential topped by a high-frequency burst of APs. A small, long-lasting depolarization is recorded in vivo in TC neurons at the end of spontaneously occurring absence-related SWDs (Pinault et al., 1998). This slow depolarization, which involves enhanced intracellular Ca2+ accumulation from repetitive low-threshold Ca2+ discharges, might reflect upregulation of Ih channels, at least in TC neurons.
Recordings from the somatosensory cortex on coronal brain slices from WAG/Rij rats have revealed a smaller Ih conductance compared to control rats (Strauss et al., 2004). The decrease in Ih corresponds to 34% reduction of HCN1 protein, although expression of HCN1 mRNA remains stable. The other three Ih subunit proteins (HCN2–4) and mRNA expression remain unchanged. Using ZD7288, a selective Ih antagonist, it has been shown in cortical slices that changes in Ih might be involved in temporal summation of EPSPs, thereby contributing to the development of hyperexcitability in the cortex of WAG/Rij rats. However, molecular studies in GAERS did not reveal significant changes in the content of Ih subunit proteins and mRNA in the neocortex (Kuisle et al., 2006).
Together these findings indicate that HCN channels have a dual role in absence epilepsy: (1) diminished cAMP-dependent Ih regulation of HCN channels precedes and is likely to promote epileptogenesis in genetic models of absence epilepsy; and (2) adaptive responses stabilizing Ih function contribute to the cessation of absence-related SWDs.
ABSENCE-RELATED SWDS CORRESPOND TO CORTICOTHALAMIC HYPER-RESONANCES
Simultaneous multi-site recordings of field potentials at the three levels (thalamus, TRN and layer VI) of the somatosensory TC system of GAERS have shown that SM oscillations often start in layer VI of the somatosensory cortex before thalamic oscillations and before the occurrence of SWDs on the cortical surface (Fig. 13A, B) (Pinault, 2003). In addition, paired recordings of identified layer VI CT neurons and of related thalamic neurons have revealed that CT cells often start to fire in a rhythmic manner before thalamic relay and reticular neurons and before the occurrence of absence-related SWDs (Fig. 8D, E). Also, paired recordings of thalamic relay and reticular neurons show that TRN cells display rhythmic burst activity almost always before TC neurons (Fig. 14A, B) (Pinault et al., 2001), and, in contrast to TC neurons, TRN cells fire systematically in the burst mode on each cycle of the CT-induced rhythmic depolarization (Fig. 9C, D) (Pinault et al., 2001; Pinault, 2003). This means that TRN cells are more electro-responsive to CT inputs than TC neurons and, thus, start to resonate before TC neurons. Therefore, it is tempting to put forward the hypothesis that absence-related SWDs correspond to a CT-induced hyperresonance phenomenon (Pinault, 2003).
Fig. 13. Triple extracellular field potential recordings of somatosensory-related thalamic and cortical sites along with spontaneously occurring SWDs in the surface EEG.
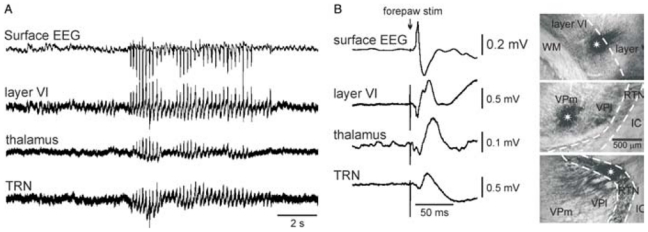
The photomicrographs in B (up, dorsal; right, lateral) show large extracellular applications (epicentre indicated by a white asterisk) of dextran biotin amine where extracellular field potential recordings were carried out simultaneously in the somatosensory system, that is, from top to bottom, in layer VI, in the thalamus, and in the thalamic reticular nucleus (TRN). The bandpass was 0.1–6 kHz. (B) Left: evoked potentials following electrical stimulation of the contralateral forepaw. Abbreviations: IC, internal capsule; VPl, ventral posterolateral thalamic nucleus; VPm, ventral posteromedial thalamic nucleus; WM, white matter. Adapted from (Pinault, 2003).
Fig. 14. TRN neurons discharge in the burst mode almost always before TC neurons during the spontaneous generation of SWD in GAERS.
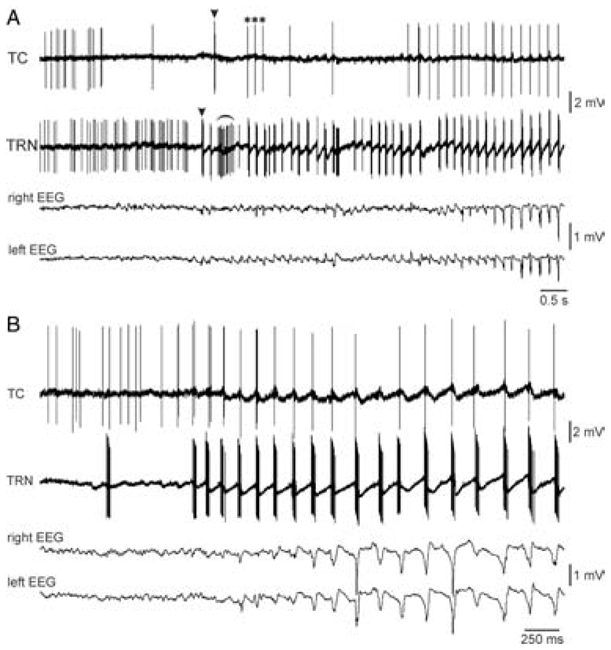
(A,B) Two experiments in which related TC and TRN extracellular activities were simultaneously recorded during the development of a spontaneous SWD in the surface EEG of the related cortex. (A) Arrowheads and * indicate the first TC and TRN bursts and the first three AP occurring at 6Hz, respectively. The curved line indicates a short-lasting episode during which he TRN cell fired rhythmic bursts at ~15Hz. Adapted from (Pinault et al., 2001).
This CT-induced resonance phenomenon might also involve reciprocal interactions between TC and TRN neurons, with complex interactions between their synaptic and intrinsic membrane properties. In the scenario proposed (Fig. 10) (Pinault, 2003) (1) through an unknown mechanism, layer VI CT cells start to oscillate at 5–9Hz (SM rhythm), phase-locking threshold and subthreshold oscillations in cortical and thalamic relay and reticular neurons; (2) TRN cells start to resonate massively before TC neurons, displaying rhythmic robust high-frequency burst of APs (Pinault et al., 2001) underlain by at least a low-threshold Ca2+ potential (Slaght et al., 2002; Pinault, 2003); (3) these TRN bursts generate GABA(A) receptor-mediate IPSPs in target TC neurons; (4) sudden rhythmic interruption of CT firing induces a rhythmic hyperpolarization and contributes to the tonic hyperpolarization in TC and TRN neurons (Pinault et al., 2006); (5) in a majority of TC neurons, the recurrent hyperpolarization activate Ih strengthening functional interactions between TC, CT and TRN neurons and launching, after a few cycles, cellular hypersynchronization, as mentioned above. The synaptic build-up of cellular synchronization might also involve gap junction couplings (Hughes et al., 2002; Landisman et al., 2002; Landisman and Connors, 2005; Gigout et al., 2006; Proulx et al., 2006). Thus, TC neurons massively recruit new active units at the cortical level, particularly in layers IV/V. The recruitment of layer V cells through this mechanism might explain why HO nuclei TC neurons fire in the burst mode after some HO nuclei TC neurons during absence-related absence seizures (see above).
This supports the concept that both the thalamus and the neocortex form a unified functional system (Steriade, 1999), running at the same frequency during absence-related SWDs. However, the time relationship of the firing of the CT, TC and TRN neurons obeys to anatomical rules and to intrinsic properties of these neurons.
GLOBAL AND CELLULAR FEATURES ASSOCIATED WITH THE ‘ONSET’ OF ABSENCE-RELATED SWDS
It is worth first noting that, using surface EEG recordings, it is difficult to detect the beginning of an absence-related generalized SWD. Surface electro-corticographic recordings of the somatosensory cortex of GAERS reveal that the first rhythmic waves of SWDs are medium-voltage SM oscillations (Fig. 2) (Pinault et al., 2001). This transformation corresponds mainly to progressive synaptic build-up of rhythmic cellular hypersynchronizations via short-and long-range projection pathways. Indeed a medium-voltage negative spike can accompany any cycle of SM oscillations, which can then suddenly reach maximal amplitude with the onset of the absence seizures. Multi-site recordings of the extracellular field potentials in the somatosensory cortex and related thalamus show that layer VI cortical cells begin to display rhythmic oscillatory firing before any oscillations or SWDs are recorded at the cortical surface or in the related thalamic regions (Seidenbecher and Pape, 2001; Pinault, 2003). The cellular mechanisms underlying the generation of SM oscillations in layer VI remain unknown.
Of importance, intracellular recordings show that in most TC and TRN neurons recorded the rhythmic depolarization, which corresponds to the summation of CT-induced EPSPs, occurs a few times just before the ‘onset’ of absence-related SWDs (Fig. 9C,D) (Pinault, 2003). In addition, their amplitude increases progressively in parallel with the development of SWDs. By contrast, Slaght et al. (Slaght et al., 2002) report that, in GAERS, SWDs start with a large hyperpolarization in TRN neurons. However, this statement is questionable considering that several rhythmic depolarizations (in the same frequency range as that of the SW complex in absence-related SWDs) are the first event visible just before the appearance of SWDs in the cortex (Figs 5A1, 5B1 and 7A, Slaght et al., 2002). In our laboratory, the first large hyperpolarization has also been recorded in most TRN cells in GAERS after the occurrence of several rhythmic CT-induced depolarizations at the beginning of SWDs and the proceeding SM oscillations (Fig. 9C, D) (Pinault, 2003). Thus, the first large hyperpolarization in TRN neurons might correspond to the ‘onset’ of the tonic or steady hyperpolarization, which lasts as long as absence-related generalized SWDs. Taken together, these findings support the hypothesis that the cellular and network mechanisms underlying the onset of generalized SWDs in the somatosensory TC system are located in the cortex. However, this conclusion does not exclude an initial contribution of a subcortical (extrathalamic or intrathalamic) structure in the evolution of the normal rhythmic waves (SM oscillations) into SWDs in the TC somatosensory system, which SM oscillations then finally become generalized to other systems.
DO ABSENCE SEIZURES ARISE IN A LOCALIZED CORTICAL FOCUS?
A growing body of experimental evidence indicates that absence seizures commence in the neocortex, and that they might arise from a topographically localized (focal) region (Meeren et al., 2002). This is in contrast to the conventional view of generalized seizures, in which the seizures are supposed to arise simultaneously in a wide area of brain in both hemispheres (Commission on Classification and Terminology of the International League Against Epilepsy, 1981). The most convincing demonstration of this is by Meeren and colleagues in WAG/Rij rats (Meeren et al., 2002). Using multisite EEG recordings from a grid of electrodes placed on the cortical surface, and depth electrodes in the thalamus, the authors demonstrated that seizures commence focally in the somatosensory cortex, specifically the perioral area, which includes the nose and vibrissae (Fig. 15) (Meeren et al., 2002). Of importance, these body areas display tremors during absence-related SWDs (Vergnes et al., 1982; Meeren et al., 2002). Further studies by the same group demonstrated that injection of the local anesthetic, lignocaine, into this ‘focus’ bilaterally suppressed the occurrence of SWDs (compared with saline) in all brain regions recorded (Sitnikova and van Luijtelaar, 2004). Our and others studies in GAERS have confirmed that seizures in this model also commence in the somatosensory cortex (Pinault, 2003). Consistent with this, focal cortical injections in GAERS of the anti-absence drug, ethosuximide, results in a major suppression of SWDs, whereas injection into the thalamus has little effect on the seizures (Manning et al., 2004).
Fig. 15. Overview of the corticocortical (black arrows) and corticothalamic (gray arrows) interdependencies during spontaneous absence seizures in WAG/Rij rats as established by nonlinear association analyses.
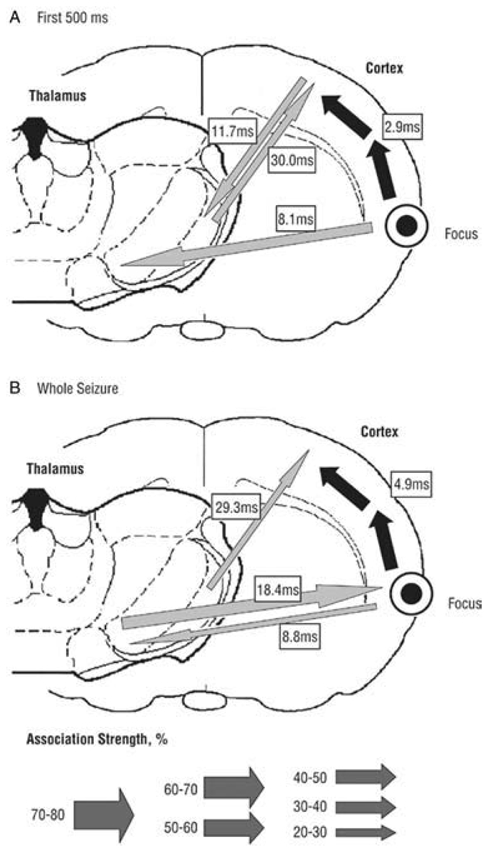
The thickness of the arrows represents the strength of the association, while the direction of the arrowhead points toward the lagging site. (A) The first 500 msec of the seizure. A cortical focus was found in the upper lip and nose area (perioral region, parietal 2) of the somatosensory cortex, as this site consistently led the other cortical recording sites. The hind paw area was found to lag by 2.9 msec with respect to this focal site. Concerning the corticothalamic interrelationships, the cortical focus led the thalamic ventroposterior medial nucleus with a delay of 8.1msec. (B) The whole seizure. The same cortical focus as during the first 500msec was found consistently. Compared with the first 500msec, the time delay from the cortical focus to the nonfocal cortical sites increases and the direction of the corticothalamic couplings changes. Adapted from (Meeren et al., 2005).
Clinical studies and observations also support the proposition that absence seizures might arise from a focal region of the cortex. Generalised SWDs, which resemble those seen with absence seizures, might be generated from either focal lesions or electrical stimulation in the frontal lobe (Bancaud et al., 1974; Lagae et al., 2001). Dipole source analysis of EEG recordings acquired with a dense electrode array indicate a discrete frontal focus in patients with primary generalized epilepsy, including two patients with typical childhood absence epilepsy (Holmes et al., 2004). fMRI studies of patients during SWDs have suggested a focus of ictal neuronal activity in the precentral sulcus bilaterally (Archer et al., 2003). Additionally, a recently published C-11-flumazenil PET study performed on members of a family with genetic generalized epilepsy (including several with absence epilepsy) caused by a mutation in the g2-subunit of the GABA-A receptor complex, demonstrates decreases in binding maximally in specific focal regions of the frontal lobes (i.e. insular and anterior cingulate) (Fedi et al., 2006).
The intracortical location of the generator of the seizures is currently unknown. A study conducted in vitro in slices of rat neocortex demonstrates that layer V neurons are endowed with the singular intrinsic property that allows them to generate prolonged synchronized 5–9Hz rhythmic activities (Silva et al., 1991). These intrinsic oscillations involve Na+ and Ca2+ currents, at least. Furthermore, layer V cells are characterized by a bistable state around firing threshold, and can switch their regular firing pattern from one (burst mode) to another (single-AP mode) in an abrupt manner. Based on this it is tempting to hypothesize that the pacemaker generating the SM oscillations that subsequently evolve into seizures might be located in cortical layer V. However, against this is the demonstration that layer V neurons fire after layer VI cells in the somatosensory cortex (parietal 1 region) during absence seizures in GAERS (Pinault, 2003). It is worth noting that layer VI CT cells are the unique source of dense excitatory inputs to the TRN and in ventral posterior nucleus (see above). Thus, a subpopulation of layer VI CT neurons might have similar intrinsic properties to layer V pyramidal neurons, an issue that deserves further experiments. Whatever the location of the ‘seizure generator’ in the neocortex of GAERS and WAG/Rij rats, these particular neurons would have a singular emergent property to suddenly transmit their synchronized intrinsic oscillations simultaneously to cortical and to thalamic neurons via short-and long-range projections pathways.
POSSIBLE MECHANISMS RESPONSIBLE FOR GENERATING NATURAL SENSORIMOTOR OSCILLATIONS AND THEIR SWITCH INTO GENERALIZED SWDS
As mentioned previously, the natural SM rhythm itself is not sufficient to give rise to SWDs in GAERS because it is also recorded in NEC rats who do not express SWDs. So, the key question concerns the molecular, cellular and networks mechanisms that suddenly switch the normal, moderately synchronized, SM oscillations into hypersynchronized oscillations (or SWDs) in genetic models of absence epilepsy. This important issue prompted us to speculate.
At least two major processes are required for generation of the SWDs: (1) a cellular or network pacemaker that generates the physiological SM rhythm (see above); and (2) a cellular or network trigger that switches the physiological SM oscillations into pathological SWDs.
(1) The expression of the pacemaker activity might involve tonic activation of NMDA receptors in layer V pyramidal neurons, which have been demonstrated to be required to allow them to generate intrinsic 5–9Hz oscillations (Silva et al., 1991). It is, thus, worth noting that NMDA-related activity is significantly more important in middle and deep layers of the neocortex of GAERS than NEC rats (Pumain et al., 1992). Consistent with this, a recent study in in vitro brain slices demonstrates increased NMDA-mediated synaptic excitability in the deep layers of the somatosensory cortex of WAG/Rij rats compared to NEC rats (D’Antuono et al., 2006). Also, in vivo and in vitro studies demonstrate that neocortical disinhibition generates synchronized 7–14Hz oscillations, which are driven by deep cortical layers and spread to the thalamus via layer VI projection neurons (Castro-Alamancos, 2000; Castro-Alamancos and Rigas, 2002).
Regional changes in ion channel expression might underlie the generation of the oscillatory neuronal activity in the cortical focus. In WAG/Rij rats mRNA and proteins of the Na+ channels Nav1.1 and 1.6 are focally over-expressed in the cortical layers II–IV in a region that corresponds to the focus identified by Meeren et al. (2002), and increasing expression of these channels with age correlates with the increase in amount of SWDs seen in these rats (Klein et al., 2004).
Changes in neuronal morphology might also contribute to an increased propensity to generate and/or propagate oscillatory activity in the cortical focus. A quantitative and qualitative morphometric Golgi-staining study demonstrated a disordered pattern of dendritic arborization in cortical layers I–III focally in the epileptogenic region in WAG/Rij rats (Karpova et al., 2005). Do these morphological changes correlate with the upregulation of Na+ channels?
Another possible mechanism for the generation of focal oscillatory activity in the neocortex might result from complex interactions between an increase of extracellular concentration of K+, deep neuronal hyperpolarization, and Ih in a subpopulation of neurons (Timofeev et al., 2002). If so, focal paroxysmal activity would spread rapidly and simultaneously in the cortex and thalamus using synaptic pathways.
(2) The transformation of normal SM oscillations into the pathological SWDs might result from the presence of hyperexcitable TC or cortico-thalamo-cortical networks in the epileptic animals. This theory supposes that normal, physiological SM oscillations are generated in the somatosensory cortex and spread to engage related thalamic regions, but in epileptic animals certain properties of their TC network facilitate the development of a hyper-resonant state with rapid, pathological recruitment of many thalamocortical neurons, which results in a widespread, self-sustaining, hypersynchronous, oscillatory firing pattern. Such a hyperexcitable TC system might result from electrochemical changes in the cortex, the thalamus or both. It is likely that several changes contribute, and that these are more topographically widespread that those responsible for generation of the focal pacemaker rhythm, given rapid propagation of the synchronous SWD activity to large areas of the cortex in both hemispheres.
Experimental observations indicate a number of factors that might contribute to this hyperexcitable state. These include: the previously mentioned alterations in Ih function, as demonstrated in the thalamus of GAERS (Kuisle et al., 2006), and in the cortex (Strauss et al., 2004) and thalamus (Budde et al., 2005) of WAG/Rij rats; enhanced NMDA-related excitability in cortical neurons, as demonstrated in GAERS (Pumain et al., 1992) and in the deep layers of the cortex in WAG/Rij rats; and deficits in cortical (D’Antuono et al., 2006; Luhmann et al., 1995) and/or possibly intra-TRN inhibition (Slaght et al., 2002) that result in reduced dampening of rhythmic firing within these highly distributed systems. Also, impaired glutamate-related and GABA-related metabolic links between astrocytes and glutamatergic neurons in the neocortex might be a causal factor in the generation of generalized SWDs in genetically determined SWDs (Melo et al., 2006).
CONCLUSIONS
Absence epilepsy is a genetically-based human disease, the pathophysiological basis of which is incompletely understood. Findings from studies in animal models form the basis of many of the proposed theories for the generation and underlying neurophysiology of absence seizures. However, studies in different models result in conflicting conclusions. In-bred Wistar rat strains that display spontaneous absence-like seizures associated with generalized SWDs on the EEG and in TC structures, that are genetically determined, are increasingly being validated as models of many aspects of the human condition. Recent studies in these models have challenged a number of previously held concepts regarding the neurophysiology of absence seizures, which were based on investigations in other models, particularly cats. It is clear that in rat models, at least, that absence-related seizures are generated in focal regions of the cortex, and less direct evidence for this is also accumulating from human studies. In rat models, seizures arise out of a physiological SM rhythm that is seen particularly during relaxed wakefulness. The mechanisms that transform this normal rhythm into epileptic SWDs are unknown, but might involve widespread neurochemical changes in the TC system in epileptic individuals that predispose them to develop hyper-resonance phenomena with these TC oscillations. Characterizing the molecular, cellular and network mechanisms of genetically-determined absence seizures will provide helpful indications to better understand impaired consciousness and develop innovative therapeutic strategies.
Acknowledgments
We thank Any Boehrer for breeding and selection of GAERS, Christian Marescaux and collaborators for the validation of GAERS as a model of absence epilepsy and for previous findings obtained in GAERS, and also Marguerite Vergnes for many discussions. D.P. is supported by the French Institute of Health and Medical Research (INSERM) and by the University of Louis Pasteur (faculté de médecine), the Fondation Française pour la Recherche sur l’Epilepsie, and the Electricité de France. T.O’B. is supported by the Australian National Health and Medical Research Council (NH & MRC), project grants 400088, 400106 and 406640, The National Alliance for Research on Schizophrenia and Depression (USA), The Royal Melbourne Hospital Neuroscience Foundation and The University of Melbourne.
Contributor Information
Didier Pinault, Physiopathologie clinique et expérimentale de la schizophrénie INSERM : U666, IFR37, Université Louis Pasteur - Strasbourg I, FR.
Terence J. O'Brien, Department of Medicine Royal Melbourne Hospital, University of Melbourne, AU
References
- Archer JS, Abbott DF, Waites AB, Jackson GD. fMRI “deactivation” of the osterior cingulate during generalized spike and wave. Neuroimage. 2003;20:1951–1922. doi: 10.1016/s1053-8119(03)00294-5. [DOI] [PubMed] [Google Scholar]
- Avanzini G, de Curtis M, Panzica F, Spreafico R. Intrinsic properties of nucleus reticularis thalami neurones of the rat studied in vitro. Journal of Physiology (London) 1989;416:111–122. doi: 10.1113/jphysiol.1989.sp017752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Gloor P. Role of the thalamus in generalized penicillin epilepsy: observations on decorticated cats. Experimental Neurology. 1982;77:386–402. doi: 10.1016/0014-4886(82)90252-7. [DOI] [PubMed] [Google Scholar]
- Avoli M, Gloor P, Kostopoulos G, Gotman J. An analysis of penicillin-induced generalized spike and wave discharges using simultaneous recordings of cortical and thalamic single neurons. Journal of Neurophysiology. 1983;50:819–837. doi: 10.1152/jn.1983.50.4.819. [DOI] [PubMed] [Google Scholar]
- Avoli M, Rogawski MA, Avanzini G. Generalized epileptic disorders: an update. Epilepsia. 2001;42:445–457. doi: 10.1046/j.1528-1157.2001.39800.x. [DOI] [PubMed] [Google Scholar]
- Bal T, McCormick DA. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: a mammalian pacemaker. Journal of Physiology (London) 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancaud J, Talairach J, Morel P, Bresson M, Bonis A, Geier S, et al. “Generalized” epileptic seizures elicited by electrical stimulation of the frontal lobe in man. Electroencephalography and Clinical Neurophysiology. 1974;37:275–282. doi: 10.1016/0013-4694(74)90031-5. [DOI] [PubMed] [Google Scholar]
- Bartho P, Freund TF, Acsady L. Selective GABAergic innervation of thalamic nuclei from zona incerta. European Journal of Neuroscience. 2002;16:999–1014. doi: 10.1046/j.1460-9568.2002.02157.x. [DOI] [PubMed] [Google Scholar]
- Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Annals of Neurology. 1998;43:435–445. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- Bokor H, Frere SG, Eyre MD, Slezia A, Ulbert I, Luthi A, et al. Selective GABAergic control of higher-order thalamic relays. Neuron. 2005;45:929–940. doi: 10.1016/j.neuron.2005.01.048. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, McCormick DA. Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci. 2000;20:5153–5162. doi: 10.1523/JNEUROSCI.20-13-05153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumendfeld H. Consciousness and epilepsy: why are patients with absence seizures absent? Progress in Brain Research. 2005;150:271–289. doi: 10.1016/S0079-6123(05)50020-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa J, Pinault D, Deschênes M. Corticothalamic projections from the cortical barrel field to the somatosensory thalamus in rats: a single-fibre study using biocytin as an anterograde tracer. European Journal of Neuroscience. 1995;7:19–30. doi: 10.1111/j.1460-9568.1995.tb01016.x. [DOI] [PubMed] [Google Scholar]
- Bouyer JJ, Montaron MF, Rougeul A. Fast fronto-parietal rhythms during combined focused attentive behaviour and immobility in cat: cortical and thalamic localizations. Electroencephalography and Clinical Neurophysiology. 1981;51:244–252. doi: 10.1016/0013-4694(81)90138-3. [DOI] [PubMed] [Google Scholar]
- Budde T, Caputi L, Kanyshkova T, Staak R, Abrahamczik C, Munsch T, et al. Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy. Journal of Neuroscience. 2005;25:9871–9882. doi: 10.1523/JNEUROSCI.2590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA. Origin of synchronized oscillations induced by neocortical disinhibition in vivo. Journal of Neuroscience. 2000;20:9195–9206. doi: 10.1523/JNEUROSCI.20-24-09195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Calcagnotto ME. Presynaptic long-term potentiation in corticothalamic synapses. Journal of Neuroscience. 1999;19:9090–9097. doi: 10.1523/JNEUROSCI.19-20-09090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Rigas P. Synchronized oscillations caused by disinhibition in rodent neocortex are generated by recurrent synaptic activity mediated by AMPA receptors. Journal of Physiology. 2002;542:567–581. doi: 10.1113/jphysiol.2002.019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nature Medicine. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lu J, Pan H, Zhang Y, Wu H, Xu K, et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Annals of Neurology. 2003;54:239–243. doi: 10.1002/ana.10607. [DOI] [PubMed] [Google Scholar]
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electrographic classification of epileptic seizures. Epilepsia. 1981;22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- Contreras D, Curro DR, Steriade M. Electrophysiological properties of cat reticular thalamic neurones in vivo. Journal of Physiology (London) 1993;470:273–294. doi: 10.1113/jphysiol.1993.sp019858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, McKerlie C, Snead OC., III A model of atypical absence seizures: EEG, pharmacology, and developmental characterization. Neurology. 2001;56:341–349. doi: 10.1212/wnl.56.3.341. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nature Reviews Neuroscience. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Progress in Neurobiology. 1998;55:27–57. doi: 10.1016/s0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- D’Antuono M, Inaba Y, Biagini G, D’Arcangelo G, Tancredi V, Avoli M. Synaptic hyperexcitability of deep layer neocortical cells in a genetic model of absence seizures. Genes, Brain and Behavior. 2006;5:73–84. doi: 10.1111/j.1601-183X.2005.00146.x. [DOI] [PubMed] [Google Scholar]
- Deschênes M, Bourassa J, Pinault D. Corticothalamic projections from layer V cells in rat are collaterals of long-range corticofugal axons. Brain Research. 1994;664:215–219. doi: 10.1016/0006-8993(94)91974-7. [DOI] [PubMed] [Google Scholar]
- Drinkenburg WH, Coenen AM, Vossen JM, Van Luijtelaar EL. Spike-wave discharges and sleep-wake states in rats with absence epilepsy. Epilepsy Research. 1991;9:218–224. doi: 10.1016/0920-1211(91)90055-k. [DOI] [PubMed] [Google Scholar]
- Engel AK, Fries P, Singer W. Dynamic predictions: oscillations and synchrony in top-down processing. Nature Reviews Neuroscience. 2001;2:704–716. doi: 10.1038/35094565. [DOI] [PubMed] [Google Scholar]
- Fanselow EE, Sameshima K, Baccala LA, Nicolelis MA. Thalamic bursting in rats during different awake behavioral states. Proceedings of the National Academy of Sciences of the USA. 2001;98:15330–15335. doi: 10.1073/pnas.261273898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedi M, Berkovic SF, Marini C, Mulligan R, Tochon-Danguy H, Reutens DC. A GABAA receptor mutation causing generalized epilepsy reduces benzodiazepine receptor binding. Neuroimage. 2006;32:995–1000. doi: 10.1016/j.neuroimage.2006.05.059. [DOI] [PubMed] [Google Scholar]
- Feig S, Harting JK. Corticocortical communication via the thalamus: ultrastructural studies of corticothalamic projections from area 17 to the lateral posterior nucleus of the cat and inferior pulvinar nucleus of the owl monkey. Journal of Comparative Neurology. 1998;395:281–295. doi: 10.1002/(sici)1096-9861(19980808)395:3<281::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O’Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Gauguier D, van Luijtelaar G, Bihoreau MT, Wilder SP, Godfrey RF, Vossen J, et al. Chromosomal mapping of genetic loci controlling absence epilepsy phenotypes in the WAG/Rij rat. Epilepsia. 2004;45:908–915. doi: 10.1111/j.0013-9580.2004.13104.x. [DOI] [PubMed] [Google Scholar]
- Gigout S, Louvel J, Pumain R. Effects in vitro and in vivo of a gap junction blocker on epileptiform activities in a genetic model of absence epilepsy. Epilepsy Research. 2006;69:15–29. doi: 10.1016/j.eplepsyres.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Gloor P. Generalized cortico-reticular epilepsies. Some considerations on the pathophysiology of generalized bilaterally synchronous spike and wave discharge. Epilepsia. 1968;9:249–263. doi: 10.1111/j.1528-1157.1968.tb04624.x. [DOI] [PubMed] [Google Scholar]
- Gloor P, Fariello RG. Generalized epilepsy: some of its cellular mechanisms differ from those of focal epilepsy. Trends in Neurosciences. 1988;11:63–68. doi: 10.1016/0166-2236(88)90166-x. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4-subunit number at corticothalamic synapses on two populations of thalamic neurons. Proceedings of the National Academy of Sciences of the USA. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW. Anatomical evidence concerning the role of the thalamus in corticocortical communication: a brief review. Journal of Anatomy. 1995;187:583–592. [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, Sherman SM. Thalamic relay functions and their role in corticocortical communication: generalizations from the visual system. Neuron. 2002;33:163–175. doi: 10.1016/s0896-6273(01)00582-7. [DOI] [PubMed] [Google Scholar]
- Halasz P. Sleep, arousal and electroclinical manifestations of generalized epilepsy with spike wave pattern. Epilepsy Research (Suppl) 1991;2:43–48. [PubMed] [Google Scholar]
- Holmes M, Brown M, Tucker D. Are generalized seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence epilepsy. Epilepsia. 2004;45:1568–1579. doi: 10.1111/j.0013-9580.2004.23204.x. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Blethyn KL, Cope DW, Crunelli V. Properties and origin of spikelets in thalamocortical neurones in vitro. Neuroscience. 2002;110:395–401. doi: 10.1016/s0306-4522(01)00577-2. [DOI] [PubMed] [Google Scholar]
- Inouye T, Sakamoto H, Shinosaki K, Toi S, Ukai S. Analysis of rapidly changing EEGs before generalized spike and wave complexes. Electroencephalogr Clin Neurophysiol. 1990;76:205–221. doi: 10.1016/0013-4694(90)90016-d. [DOI] [PubMed] [Google Scholar]
- Jasper HH, Drooglever-Fortuyn J. Experimental studies on the functional anatomy of petit mal epilepsy. Research publications Association for Research in Nervous and Mental Disease. 1947;26:272–298. [Google Scholar]
- Jones EG. The Thalamus. Plenum Press; 1985. [Google Scholar]
- Karpova A, Bikbaev A, Coenen A, van Luijtelaar G. Morphometric Golgi study of cortical locations in WAG/Rij rats: the cortical focus theory. Neuroscience Research. 2005;51:119–128. doi: 10.1016/j.neures.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Keil A, Muller MM, Ray WJ, Gruber T, Elbert T. Human gamma band activity and perception of a gestalt. Journal of Neuroscience. 1999;19:7152–7161. doi: 10.1523/JNEUROSCI.19-16-07152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellaway P. Sleep and epilepsy. Epilepsia. 1985;26(Suppl 1):S15–30. doi: 10.1111/j.1528-1157.1985.tb05720.x. [DOI] [PubMed] [Google Scholar]
- Kellaway P, Frost JD, Jr, Crawley JW. Time modulation of spike-and-wave activity in generalized epilepsy. Annals of Neurology. 1980;8:491–500. doi: 10.1002/ana.410080506. [DOI] [PubMed] [Google Scholar]
- Khosravani H, Altier C, Simms B, Hamming KS, Snutch TP, Mezeyova J, et al. Gating effects of mutations in the Cav3.2 T-type calcium channel associated with childhood absence epilepsy. Journal of Biological Chemistry. 2004;279:9681–9684. doi: 10.1074/jbc.C400006200. [DOI] [PubMed] [Google Scholar]
- Khosravani H, Bladen C, Parker DB, Snutch TP, McRory JE, Zamponi GW. Effects of Ca(v)3.2 channel mutations linked to idiopathic generalized epilepsy. Annals of Neurology. 2005;57:745–749. doi: 10.1002/ana.20458. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, et al. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- Klein J, Khera D, Nersesyan H, Kimchi E, Waxman S, Blumenfeld H. Dysregulation of sodium channel expression in cortical neurons in a rodent model of absence epilepsy. Brain Research. 2004;1000:102–109. doi: 10.1016/j.brainres.2003.11.051. [DOI] [PubMed] [Google Scholar]
- Kostopoulos G, Gloor P, Pellegrini A, Siatitsas I. A study of the transition from spindles to spike and wave discharge in feline generalized penicillin epilepsy: EEG features. Experimental Neurology. 1981;73:43–54. doi: 10.1016/0014-4886(81)90044-3. [DOI] [PubMed] [Google Scholar]
- Kostopoulos GK. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clinical Neurophysiology. 2000;111(Suppl 2):S27–S38. doi: 10.1016/s1388-2457(00)00399-0. [DOI] [PubMed] [Google Scholar]
- Kostopoulos GK. Involvement of the thalamocortical system in epileptic loss of consciousness. Epilepsia. 2001;42(Suppl 3):13–19. doi: 10.1046/j.1528-1157.2001.042suppl.3013.x. [DOI] [PubMed] [Google Scholar]
- Kuisle M, Wanaverbecq N, Brewster AL, Frere SG, Pinault D, Baram TZ, Luthi A. Functional stabilization of weakened thalamic pacemaker channel regulation in rat absence epilepsy. Journal of Physiology. 2006;575:83–100. doi: 10.1113/jphysiol.2006.110486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyi M, Lopaticki S, O’Brien TJ, Reid C, Foote SJ. Genetic sequence and mRNA expression of two Ca2+ channel genes (Cav3.2 & CACNG2) in Genetic Absence Epilepsy Rats from Strasbourg. Australian Neuroscience Conference; 2006. ORAL-11-03. [Google Scholar]
- Lagae L, Pauwels J, Monte C, Verhelle B, Vervisch I. Frontal absences in children. European Journal of Paediatric Neurology. 2001;5:243–251. doi: 10.1053/ejpn.2001.0524. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Connors BW. Long-term modulation of electrical synapses in the mammalian thalamus. Science. 2005;310:1809–1813. doi: 10.1126/science.1114655. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW. Electrical synapses in the thalamic reticular nucleus. Journal of Neuroscience. 2002;22:1002–1009. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannes B, Micheletti G, Vergnes M, Marescaux C, Depaulis A, Warter JM. Relationship between spike-wave discharges and vigilance levels in rats with spontaneous petit mal-like epilepsy. Neuroscience Letters. 1988;94:187–191. doi: 10.1016/0304-3940(88)90293-5. [DOI] [PubMed] [Google Scholar]
- Lehmann J, Pryce CR, Bettschen D, Feldon J. The maternal separation paradigm and adult emotionality and cognition in male and female wistar rats. Pharmacology Biochemistry and Behavior. 1999;64:705–715. doi: 10.1016/s0091-3057(99)00150-1. [DOI] [PubMed] [Google Scholar]
- Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, et al. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nature Genetics. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- Liu L, Zheng T, Wallengren C, Morris MJ, Reid C, Clarke A, et al. The Mechanism of Carbamazepine aggravation of absence seizures. Journal of Pharmacology and Experimental Therapeutics. 2006;319:790–798. doi: 10.1124/jpet.106.104968. [DOI] [PubMed] [Google Scholar]
- Loiseau P. Human absence epilepsies. Journal of Neural Transmission. 1992;35(Suppl):1–6. doi: 10.1007/978-3-7091-9206-1_1. [DOI] [PubMed] [Google Scholar]
- Loring DW, Sheer DE, Largen JW. Forty Hertz EEG activity in dementia of the Alzheimer type and multi-infarct dementia. Psychophysiology. 1985;22:116–121. doi: 10.1111/j.1469-8986.1985.tb01570.x. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Mittmann T, van Luijtelaar G, Heinemann U. Impairment of intracortical GABAergic inhibition in a rat model of absence epilepsy. Epilepsy Research. 1995;22:43–51. doi: 10.1016/0920-1211(95)00032-6. [DOI] [PubMed] [Google Scholar]
- Luthi A, McCormick DA. Periodicity of thalamic synchronized oscillations: the role of Ca2+-mediated upregulation of Ih. Neuron. 1998;20:553–563. doi: 10.1016/s0896-6273(00)80994-0. [DOI] [PubMed] [Google Scholar]
- Manning J, Richards D, Leresche N, Crunelli V, Bowery N. Cortical-area specific block of genetically determined absence seizures by ethosuximide. Neuroscience. 2004;123:5–9. doi: 10.1016/j.neuroscience.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Marczynski TJ, Burns LL, Livezey GT, Vimal RL, Chen E. Sleep and purposive behavior: inverse deviations from randomness of neuronal firing patterns in the feline thalamus. A new form of homeostasis? Brain Research. 1984;298:75–90. doi: 10.1016/0006-8993(84)91148-x. [DOI] [PubMed] [Google Scholar]
- Marescaux C, Micheletti G, Vergnes M, Depaulis A, Rumbach L, Warter JM. A model of chronic spontaneous petit mal-like seizures in the rat: comparison with pentylenetetrazol-induced seizures. Epilepsia. 1984a;25:326–331. doi: 10.1111/j.1528-1157.1984.tb04196.x. [DOI] [PubMed] [Google Scholar]
- Marescaux C, Vergnes M, Micheletti G, Depaulis A, Reis J, Rumbach L, et al. A genetic form of petit mal absence in Wistar rats. Revue neurologique. 1984b;140:63–66. [PubMed] [Google Scholar]
- McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annual Review Physiology. 2001;63:815–846. doi: 10.1146/annurev.physiol.63.1.815. [DOI] [PubMed] [Google Scholar]
- McLean KJ, O’Brien TJ, Cook MJ, Vajda FJ. The influence of gender on the aggravation of absence seizures by in the low-dose pentylenetetrazol rat model. Seizure. 2004;13:208–216. doi: 10.1016/S1059-1311(03)00144-4. [DOI] [PubMed] [Google Scholar]
- Meeren H, Pijn J, Van Luijtelaar E, Coenen A, Lopes da Silva F. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. Journal of Neuroscience. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren H, Van Luijtelaar G, Lopes da Silva F, Coenen A. Evolving concepts on the pathophysiology of absence seizures. Archives of Neurology. 2005;62:371–376. doi: 10.1001/archneur.62.3.371. [DOI] [PubMed] [Google Scholar]
- Melo TM, Sonnewald U, Touret M, Nehlig A. Cortical glutamate metabolism is enhanced in a genetic model of absence epilepsy. Journal of Cerebral Blood Flow and Metabolism. 2006;26:1496–1506. doi: 10.1038/sj.jcbfm.9600300. [DOI] [PubMed] [Google Scholar]
- Mercer A, West DC, Morris OT, Kirchhecker S, Kerkhoff JE, Thomson AM. Excitatory connections made by presynaptic cortico-cortical pyramidal cells in layer 6 of the neocortex. Cerebral Cortex. 2005;15:1485–1496. doi: 10.1093/cercor/bhi027. [DOI] [PubMed] [Google Scholar]
- Micheletti G, Vergnes M, Marescaux C, Reis J, Depaulis A, Rumbach L, et al. Antiepileptic drug evaluation in a new animal model: spontaneous petit mal epilepsy in the rat. Arzneimittelforschung. 1985;35:483–485. [PubMed] [Google Scholar]
- Mirsky AF, Duncan CC, Myslobodsky MS. Petit mal epilepsy: a review and integration of recent information. Journal of Clinical Neurophysiology. 1986;3:179–208. [PubMed] [Google Scholar]
- Mulle C, Madariaga A, Deschênes M. Morphology and electrophysiological properties of reticularis thalami neurons in cat: in vivo study of a thalamic pacemaker. Journal of Neuroscience. 1986;6:2134–2145. doi: 10.1523/JNEUROSCI.06-08-02134.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolelis MA, Fanselow EE. Thalamocortical [correction of Thalamcortical] optimization of tactile processing according to behavioral state. Nature Neuroscience. 2002;5:517–23. doi: 10.1038/nn0602-517. [DOI] [PubMed] [Google Scholar]
- Niedermeyer E. Sleep electroencephalograms in Petit Mal. Archives of Neurology. 1965;12:625–630. doi: 10.1001/archneur.1965.00460300073010. [DOI] [PubMed] [Google Scholar]
- Niedermeyer E. Petit mal primary generalized epilepsy and sleep. In: Se, et al., editors. Sleep and Epilepsy. Academic press; 1982. pp. 191–207. [Google Scholar]
- Niedermeyer E. Primary (idiopathic) generalised epilepsy and underlying mechanisms. Clinical Electroencephalography. 1996;27:1–21. doi: 10.1177/155005949602700103. [DOI] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: The hyperpolarization-activated cation current in neurons. Annual Review of Physiology. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Pape HC, Kanyshkova T, Broicher T, Budde T. Developmental and functional profile of the thalamic hyperpolarization-activated cation current, Ih, in absence epilepsy. Thalamus & Related Systems; 2007. (this issue) [Google Scholar]
- Pedley T. Epilepsy and the Human EEG. In: Schwartzkroin PA, Wheal HV, editors. Electrophysiology of Epilepsy. Academic Press; 1984. pp. 3–30. [Google Scholar]
- Penfield WG, Jasper HH. Epilepsy and the functional anatomy of the human brain. Little Brown & Co; 1954. [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or Neurobiotin. Journal of Neuroscience Methods. 1996;65:113–36. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- Pinault D. Cellular interactions in the rat somatosensory thalamocortical system during normal and epileptic 5–9Hz oscillations. Journal of Physiology. 2003;552:881–905. doi: 10.1113/jphysiol.2003.046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus, function and concept. Brain Research Reviews. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, et al. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. Journal of Physiology (London) 1998;509(Pt 2):449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Slezia A, Acsady L. Corticothalamic 5–9Hz oscillations are more pro-epileptogenic than sleep spindles in rats. Journal of Physiology. 2006;574:209–227. doi: 10.1113/jphysiol.2006.108498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Vergnes M, Marescaux C. Medium-voltage 5–9-Hz oscillations give rise to spike-and-wave discharges in a genetic model of absence epilepsy: in vivo dual extracellular recording of thalamic relay and reticular neurons. Neuroscience. 2001;105:181–201. doi: 10.1016/s0306-4522(01)00182-8. [DOI] [PubMed] [Google Scholar]
- Polack PO, Charpier S. Intracellular activity of cortical and thalamic neurones during high-voltage rhythmic spike discharge in Long-Evans rats in vivo. Journal of Physiology. 2006;571:461–476. doi: 10.1113/jphysiol.2005.100925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Farrell D. “Centrencephalic” spike and wave discharges following parenteral penicillin injection in the cat. Neurology. 1969;19:309–310. [Google Scholar]
- Proulx E, Leshchenko Y, Kokarovtseva L, Khokhotva V, El-Beheiry M, Snead OC, 3rd, et al. Functional contribution of specific brain areas to absence seizures: role of thalamic gap-junctional coupling. European Journal of Neuroscience. 2006;23:489–496. doi: 10.1111/j.1460-9568.2005.04558.x. [DOI] [PubMed] [Google Scholar]
- Pulvermuller F, Birbaumer N, Lutzenberger W, Mohr B. High-frequency brain activity: its possible role in attention, perception and language processing. Progress in Neurobiology. 1997;52:427–445. doi: 10.1016/s0301-0082(97)00023-3. [DOI] [PubMed] [Google Scholar]
- Pumain R, Louvel J, Gastard M, Kurcewicz I, Vergnes M. Responses to N-methyl-D-aspartate are enhanced in rats with petit mal-like seizures. Journal of Neural Transmission (Suppl) 1992;35:97–108. doi: 10.1007/978-3-7091-9206-1_7. [DOI] [PubMed] [Google Scholar]
- Ribary U, Ioannides AA, Singh KD, Hasson R, Bolton JP, Lado F, et al. Magnetic field tomography of coherent thalamocortical 40-Hz oscillations in humans. Proceedings of the National Academy of Sciences of the USA. 1991;88:11037–11041. doi: 10.1073/pnas.88.24.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annual Review of Physiology. 2003;65:453–80. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Rudolf G, Therese Bihoreau MF, Godfrey RP, Wilder SD, Cox R, Lathrop M, et al. Polygenic control of idiopathic generalized epilepsy phenotypes in the genetic absence rats from Strasbourg (GAERS) Epilepsia. 2004;45:301–308. doi: 10.1111/j.0013-9580.2004.50303.x. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, et al. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. Journal of Neuroscience. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. The genetics of human epilepsy. Trends in Pharmacological Sciences. 2003;24:428–433. doi: 10.1016/S0165-6147(03)00194-9. [DOI] [PubMed] [Google Scholar]
- Seidenbecher T, Pape H. Contribution of intralaminar thalamic nuclei to spike-and-wave-discharges during spontaneous seizures in a genetic rat model of absence epilepsy. European Journal of Neuroscience. 2001;13:1537–1546. doi: 10.1046/j.0953-816x.2001.01537.x. [DOI] [PubMed] [Google Scholar]
- Semba K, Szechtman H, Komisaruk BR. Synchrony among rhythmical facial tremor, neocortical ‘alpha’ waves, and thalamic non-sensory neuronal bursts in intact awake rats. Brain Research. 1980;195:281–298. doi: 10.1016/0006-8993(80)90065-7. [DOI] [PubMed] [Google Scholar]
- Shaw FZ, Liao YF. Relation between activities of the cortex and vibrissae muscles during high-voltage rhythmic spike discharges in rats. Journal of Neurophysiology. 2005;93:2435–2448. doi: 10.1152/jn.00999.2004. [DOI] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251:432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Sitnikova E, van Luijtelaar G. Cortical control of generalized absence seizures: effect of lidocaine applied to the somatosensory cortex in WAG/Rij rats. Brain Research. 2004;1012:127–137. doi: 10.1016/j.brainres.2004.03.041. [DOI] [PubMed] [Google Scholar]
- Slaght SJ, Leresche N, Deniau JM, Crunelli V, Charpier S. Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. Journal of Neuroscience. 2002;22:2323–2334. doi: 10.1523/JNEUROSCI.22-06-02323.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead OC, 3rd, Depaulis A, Vergnes M, Marescaux C. Absence epilepsy: advances in experimental animal models. Advances in Neurology. 1999;79:253–278. [PubMed] [Google Scholar]
- Snead OC., III Ganaxolone, a selective, high-affinity steroid modulator of the gamma-aminobutyric acid-A receptor, exacerbates seizures in animal models of absence. Annals of Neurology. 1998;44:688–691. doi: 10.1002/ana.410440417. [DOI] [PubMed] [Google Scholar]
- Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. Journal of Neuroscience. 2004;24:5249–5257. doi: 10.1523/JNEUROSCI.5546-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico R, de Curtis M, Frassoni C, Avanzini G. Electrophysiological characteristics of morphologically identified reticular thalamic neurons from rat slices. Neuroscience. 1988;27:629–638. doi: 10.1016/0306-4522(88)90294-1. [DOI] [PubMed] [Google Scholar]
- Staak R, Pape HC. Contribution of GABA(A) and GABA(B) receptors to thalamic neuronal activity during spontaneous absence seizures in rats. Journal of Neuroscience. 2001;21:1378–1384. doi: 10.1523/JNEUROSCI.21-04-01378.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein OK. Genes and mutations in human idiopathic epilepsy. Brain Development. 2004;26:213–218. doi: 10.1016/S0387-7604(03)00149-9. [DOI] [PubMed] [Google Scholar]
- Steriade M. Coherent oscillations and short-term plasticity in corticothalamic networks. Trends in Neurosciences. 1999;22:337–345. doi: 10.1016/s0166-2236(99)01407-1. [DOI] [PubMed] [Google Scholar]
- Steriade M. The Intact and Sliced Brain. MIT Press; 2001a. [Google Scholar]
- Steriade M. The GABAergic reticular nucleus: a preferential target of corticothalamic projections. Proceedings of the National Academy of Sciences of the USA. 2001b;98:3625–3627. doi: 10.1073/pnas.071051998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. Journal of Neuroscience. 1995;15:623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss U, Kole MH, Brauer AU, Pahnke J, Bajorat R, Rolfs A, et al. An impaired neocortical Ih is associated with enhanced excitability and absence epilepsy. European Journal of Neuroscience. 2004;19:3048–3058. doi: 10.1111/j.0953-816X.2004.03392.x. [DOI] [PubMed] [Google Scholar]
- Stroud LM, O’Brien TJ, Jupp B, Wallengren C, Morris MJ. Neuropeptide Y suppresses absence seizures in a genetic rat model. Brain Research. 2005;1033:151–156. doi: 10.1016/j.brainres.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Depaulis A, Perez-Reyes E, Bayliss DA. Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Molecular Brain Research. 2000;75:159–165. doi: 10.1016/s0169-328x(99)00307-1. [DOI] [PubMed] [Google Scholar]
- Tallon-Baudry C, Bertrand O, Delpuech C, Permier J. Oscillatory gamma-band (30–70Hz) activity induced by a visual search task in humans. Journal of Neuroscience. 1997;17:722–734. doi: 10.1523/JNEUROSCI.17-02-00722.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Bazhenov M, Sejnowski T, Steriade M. Cortical hyperpolarization-activated depolarizing current takes part in the generation of focal paroxysmal activities. Proceedings of the National Academy of Sciences of the USA. 2002;99:9533–9537. doi: 10.1073/pnas.132259899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape HC. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. Journal of Neuroscience. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C, Depaulis A. Mapping of spontaneous spike and wave discharges in Wistar rats with genetic generalized non-convulsive epilepsy. Brain Research. 1990;523:87–91. doi: 10.1016/0006-8993(90)91638-w. [DOI] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C. Cortical and thalamic lesions in rats with genetic absence epilepsy. Journal of Neural Transmission. 1992;35(Suppl):71–83. doi: 10.1007/978-3-7091-9206-1_5. [DOI] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C, Micheletti G, Reis J, Depaulis A, Rumbach L, et al. Spontaneous paroxysmal electroclinical patterns in rat: a model of generalized non-convulsive epilepsy. Neuroscience Letters. 1982;33:97–101. doi: 10.1016/0304-3940(82)90136-7. [DOI] [PubMed] [Google Scholar]
- von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nature Genetics. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- Wallengren C, Li S, Morris MJ, Jupp B, O’Brien TJ. Aggravation of absence seizures by carbamazepine in a genetic rat model does not induce neuronal c-Fos activation. Clinical Neuropharmacology. 2005;28:60–65. doi: 10.1097/01.wnf.0000159955.87511.bc. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen S, Nolan MF, Siegelbaum SA. Activity-dependent regulation of HCN pacemaker channels by cyclic AMP: signaling through dynamic allosteric coupling. Neuron. 2002;36:451–461. doi: 10.1016/s0896-6273(02)00968-6. [DOI] [PubMed] [Google Scholar]
- Williams D. A study of thalamic and cortical rhythms in petit mal. Brain. 1953;76:50–69. doi: 10.1093/brain/76.1.50. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Deschênes M. Intracortical axonal projections of lamina VI cells of the primary somatosensory cortex in the rat: a single-cell labeling study. Journal of Neuroscience. 1997;17:6365–6379. doi: 10.1523/JNEUROSCI.17-16-06365.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]