

Abstract
Hydrogen peroxide (H2O2), a reactive oxygen species (ROS), is known to induce oxidative stress and apoptosis. U937 cells treated with H2O2 were shown to produce high molecular weight (HMW) DNA fragments ∼50 to 100 kb in size in <1 min. The formation of these HMW DNA fragments is reversible and shown to be mediated by DNA topoisomerase II (TOP2). Following this initial event, formation of irreversible HMW DNA fragments and nucleosomal ladders occurs. Our results thus demonstrate a potential role of TOP2 in oxidative damage of DNA and apoptotic cell death.
Keywords: TOP2, H2O2, VM-26, oxidative stress, apoptosis
Reactive oxygen species (ROS; e.g., H2O2, hydroxyl radical, and superoxide) generated during oxidative stress are known to cause a variety of cellular injuries including DNA and protein damage, lipid peroxidation, and damages to other biomolecules (for review, see Berlett and Stadtman 1997; Croteau and Bohr 1997). Oxidative stress is also widely demonstrated to occur during apoptotic cell death (Bredesen 1995; Quillet-Mary et al. 1995; Um et al. 1996; Kroemer et al. 1997; Ha et al. 1997). In fact, H2O2, a ROS generated during oxidative stress, is known to induce apoptosis (Stangel et al. 1996). It has been reported that Bcl-2 may exert its apoptosis-suppressing effect through an antioxidant mechanism (Hockenery et al. 1993; Ellerby et al. 1996; Hedley and McCulloch 1996). One of the hallmarks of apoptotic cell death is the fragmentation of nuclear DNA into nucleosomal DNA ladders (Liu et al. 1997; Sakahira et al. 1998). However, recent studies have demonstrated that cleavage of DNA into high molecular weight (HMW) DNA fragments (∼50- to 100-kb loop-sized DNA fragments) is the early event in chromatin degradation and may be the initial committed step during apoptosis (Oberhammer et al. 1993; Cohen et al. 1994; Sun and Cohen 1994; Lagarkova et al. 1995). A number of nucleases have been implicated in HMW DNA fragmentation (Sun and Cohen 1994; Krieser and Eatman 1998). Interestingly, cellular DNA cleavage into HMW DNA fragments during apoptosis is highly reminiscent of topoisomerase II (TOP2)-mediated HMW DNA fragmentation in cells (Lagarkova et al. 1995; Stanulla et al. 1997). In fact, the pattern of HMW DNA fragmentation induced by TOP2 poisons, and that produced in apoptotic cells induced by other stimuli, are found to be similar (Lagarkova et al. 1995; Stanulla et al. 1997). However, the relationship between TOP2, oxidative stress, and excision of chromosomal loops during apoptotic cell death is still unclear.
TOP2 is known to be critically involved in both cell proliferation and cell death. Its major function is to regulate the topological state of DNA during DNA replication, and chromosome condensation and segregation through its delicate act of breaking/rejoining DNA strands (for review, see Liu 1989; Poljak and Kas 1995; Wang 1996). Many xenobiotics (e.g., antibiotics and antitumor drugs) and peptide toxins (e.g., CcdB and microcin B17) are known to poison TOP2 (i.e., to induce TOP2-mediated DNA cleavage) resulting in cell death (for review, see Liu 1989; Wang 1996). More recently, several new mechanisms including DNA structural modifications (e.g., abasic sites), enzyme modifications (e.g., thiol alkylation), and acidic pH environment (pH 5 to 6) have also been shown to activate TOP2-mediated DNA cleavage (Liu et al. 1983; Zechiedrich 1989; Frydman et al. 1997; Kingma and Osheroff 1997a,b; Kwok and Hurley 1998). It appears that TOP2 is highly vulnerable to various perturbations during its delicate cleavage/religation reactions.
To test whether ROS can activate TOP2-mediated DNA cleavage, we have examined the effect of H2O2 in the excision of loop-sized DNA fragments in U937 myeloid leukemia cells. Surprisingly, H2O2 can induce rapid and reversible cleavage of chromosomal DNA into 50- to 100-kb HMW fragments. The results presented in our current study suggest that H2O2 activates TOP2-mediated excision of chromosomal DNA loops, which may represent an early step in apoptotic cell death.
Results
H2O2 and VM-26 induce rapid excision of loop-sized DNA fragments from chromosomal DNA
U937 cells were encapsulated into agarose plugs and treated with either H2O2 or VM-26. The time course of the induction of HMW DNA fragments in U937 cells treated with 30 mm VM-26 and 30 μm H2O2 are shown in Figure 1A. Both drugs exhibited dose-dependent induction of HMW DNA fragments as revealed by pulsed-field gel electrophoresis (PFGE) and were about equicytotoxic at these concentrations (data not shown). Within 15 sec, both treatments resulted in extensive degradation of chromosomal DNA into HMW DNA fragments with a limiting size ∼50 kb (Fig. 1A). The extent of DNA fragmentation was about the same for both drugs at their respective concentrations.
Figure 1.

Rapid and reversible fragmentation of chromosomal DNA into HMW DNA fragments in U937 cells treated with VM-26 and H2O2. (A) PFGE was carried out to analyze the HMW DNA fragments generated in U937 cells treated with VM-26 (30 μm) and H2O2 (30 mm). Reversal of DNA cleavage by brief heating (55°C for 10 min) was performed as described in Materials and Methods. (B) Encapsulated U937 cells in agarose plugs were treated with H2O2 (30 mm) for 30 min (lane 1). Subsequently, the agarose plugs were incubated for another 30 min under the following reversal conditions. (Lane 2) The medium was replaced with fresh medium (removal of H2O2) and the incubation continued at 37°C; (lane 3) the agarose plug in the H2O2-containing medium was placed at 4°C; (lane 4) EDTA (final 10 mm) was added to the H2O2-containing medium and incubation continued at 37°C.
H2O2- and VM-26-induced excision of loop-sized DNA fragments is reversible
It is known that induction of HMW DNA fragments by VM-26 was due to the formation of reversible TOP2 cleavable complexes (Liu et al. 1983; Hsiang and Liu 1989). To test whether induction of HMW DNA fragments by H2O2 was also due to the formation of reversible TOP2 cleavable complexes, we examined the reversibility of the HMW DNA fragments. Heat-induced reversal (i.e., 55°C for 10 min) is one of the unique properties of TOP2 cleavable complexes (Hsiang and Liu 1989). When drug-treated cells were heated to 55°C for 10 min prior to SDS lysis, HMW DNA fragmentation (in the 50- to 100-kb range) generated by either VM-26 or H2O2 treatment completely disappeared (Fig. 1A). The reversibility of the HMW DNA fragments generated by H2O2 has also been demonstrated by three other cleavable complex reversal conditions; removal of H2O2 from the medium (H2O2-free medium), 4°C incubation, and addition of EDTA (Fig. 1B; Liu et al. 1983). These results sugget that induction of HMW DNA fragments by both VM-26 and H2O2 may be due to formation of TOP2 cleavable complexes. It was noted that whereas nearly all of the HMW DNA fragments induced by VM-26 were reversed to larger chromosomal DNA that did not migrate out of the well, about half of the HMW DNA fragments induced by H2O2 migrated to the compression zone (Fig. 1). It appeared that either the reversal was incomplete or H2O2 also damaged chromosomal DNA via other ROS generated in cells (Imlay et al. 1988; Croteau and Bohr 1997).
H2O2- and VM-26-induced excision of loop-sized DNA fragments is reduced in a TOP2 mutant cell line
The potential role of TOP2 in mediating the effect of H2O2 and VM-26 was further examined with the mutant HL-60/MX-2 cell line that was selected for resistance to mitoxantrone (a TOP2-targeting drug) and known to have reduced levels of both TOP2 isozymes (Harker et al. 1995a,b). Consequently, HL-60/MX-2 is known to exhibit cross-resistance to all known TOP2 poisons such as epipodophyllotoxins (e.g., VM-26 and VP-16), anthracyclines (e.g., doxorubicin and daunorubicin), and aminoacridines (e.g., m-AMSA) but not other anticancer drugs (Harker et al. 1995a,b). As shown in Figure 2A, the extent of excision of the loop-sized DNA fragments in HL-60/MX-2 cells treated with either VM-26 or H2O2 was greatly reduced. This result supports the notion that TOP2 is primarily responsible for the effect of H2O2 and VM-26 in the excision of loop-sized DNA fragments. We have also examined the effect of H2O2 and VM-26 in activating the nucleases involved in fragmentation of chromosomal DNA into nucleosomal ladders. Again, formation of nucleosomal DNA ladders, a hallmark of apoptotic cell death, was lacking in HL-60/MX-2 cells treated with either VM-26 or H2O2 (Fig. 2B). Furthermore, HL-60/MX-2 cells were shown to be cross-resistant to both VM-26 and H2O2 in a cytotoxicity assay (Fig. 2C). These results together suggest that the major cytotoxic target of VM-26 and H2O2 is TOP2 in HL-60 cells, and that both drugs can activate TOP2-mediated DNA damage.
Figure 2.
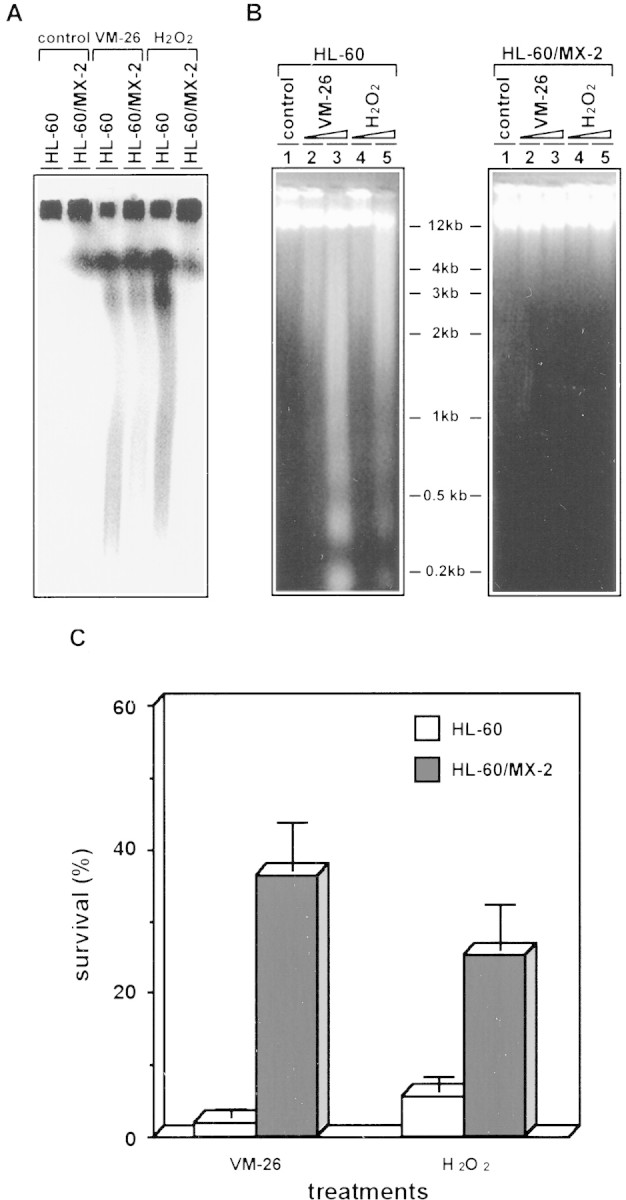
A TOP2 mutant cell line is resistant to both H2O2 and VM-26. HL-60/MX-2 is known to be cross-resistant to all TOP2 poisons primarily due to the reduced TOP2 level in the nucleus (Harker et al. 1995a,b). (A) HL-60 and HL-60/MX-2 cells were encapsulated into agarose plugs and treated with VM-26 (30 μm) and H2O2 (30 mm). PFGE was carried out as described in Materials and Methods. (B) HL-60 and HL-60/MX-2 cells were treated with VM-26 and H2O2 for 5 hr at 37°C. Subsequently, cells were lysed by SDS, cell lysates were treated with PK, and DNA was isolated by the published procedure (Shih and Shaman 1996). Gel electrophoresis was performed in 1.8% agarose gel in 1/2× TPE buffer. (Lane 1) Control; (lane 2) 0.1 μm VM-26; (lane 3) 1 μm VM-26; (lane 4) 0.1 mm H2O2; (lane 5) 1 mm H2O2. (C) (□) HL-60 and (█) HL-60/MX-2 cells were treated with 10 μm VM-26 or 10 mm H2O2 for 1 hr at 37°C. Subsequently, cells were washed once and incubated in fresh (drug-free) medium. MTT assay was performed at the end of the fourth day.
Reversible excision of loop-sized DNA fragments precedes extensive degradation of chromosomal DNA
The fate of the HMW DNA fragments in U937 cells treated with either H2O2 or VM-26 was monitored by extending the time of drug treatment up to 1000 min. Two events were noticed. First, after 100 min of treatment with either drug, the HMW DNA fragments were no longer reversible by heat treatment (Fig. 3). Second, further degradation of the HMW DNA fragments started at 400 min in cells treated with either drug (see the bracketed regions in Fig. 3). Nucleosomal ladders appeared at about the same time (data not shown). These results suggest that reversible excision of loop-sized DNA fragments precedes both irreversible excision of HMW DNA fragments and extensive degradation of chromosomal DNA by nuclease(s).
Figure 3.
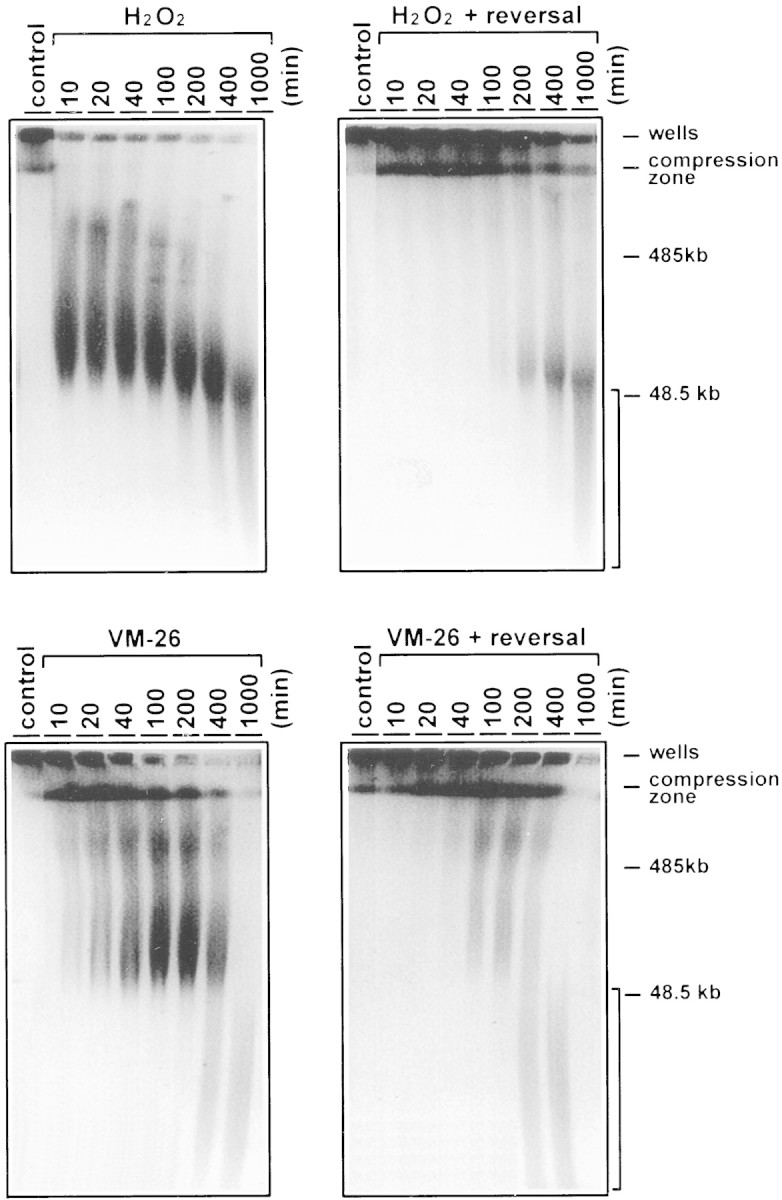
Time-dependent processing of the HMW DNA in U937 cells treated with H2O2 and VM-26. PFGE analysis of the HMW DNA was performed as described in the legend to Fig. 1, except that the time course was extended to 1000 min. The bracketed area represents DNA fragments <50 kb in size.
H2O2 induces DNA fragmentation in the presence of purified DNA TOP2
To ascertain that TOP2 is responsible for H2O2-induced excision of chromosomal DNA loops, purified topoisomerases were used for in vitro experiments. As shown in Figure 4A, H2O2, like VM-26, induced DNA fragmentation in the presence of calf thymus TOP2. The DNA cleavage activity of H2O2 was not restricted to calf thymus TOP2. Both human TOP2α- and TOP2β-mediated DNA cleavages were similarly induced by H2O2 (Fig. 4B). However, in the same concentration range, H2O2 had no effect on calf thymus TOP1-mediated DNA cleavage (Fig. 4C). Interestingly, ATP (1 mm) had a 50- to 100-fold stimulatory effect on VM-26-induced DNA cleavage. However, H2O2-induced DNA cleavage was only minimally (severalfold) affected by ATP (Fig. 4A).
Figure 4.

H2O2 stimulates TOP2- but not TOP1-mediated DNA cleavage. (A) Stimulation of calf thymus TOP2-mediated DNA cleavage by H2O2. The reaction mixture for each of the last eight lanes contained 1 mm ATP (see separately bracketed lanes). (B) Stimulation of hTOP2α- and hTOP2β-mediated DNA cleavage by H2O2; 3 mm H2O2 was used in these reactions. (C) H2O2 does not stimulate TOP1-mediated DNA cleavage. (D) DNA double-strand breaks stimulated by H2O2 require active TOP2. The reactions were performed in three sets, one set without added TOP2, one set with added TOP2, and the other with heat-inactivated TOP2 (65°C for 15 min) (see three separately bracketed lanes). (E) DNA fragments induced by H2O2 are protein linked. After incubation of cleavage reactions, one set of reactions was terminated by treatment with 1% SDS in the absence of PK (another 60 min at 37°C) (see bracketed lanes under −PK). A duplicate set of reactions was terminated with 1% SDS plus 400 μg/ml PK and then incubation continued at 37°C for another 60 min (see bracketed lanes under +PK). The last half of the panel (last 13 lanes) were identical to the first half of panel (first 13 lanes), except that labeled DNA was first reacted with VM-26 or H2O2 in the absence of TOP2. Drug-treated DNA was then extracted with phenol and precipitated with ethanol, and resuspended in reaction mixtures containing calf thymus TOP2. All topoisomerase DNA cleavage assays were performed as described in Materials and Methods. Except for B, in which recombinant hTOP2α and hTOP2β were used, calf thymus topoisomerases were used in all cleavage assays. The concentrations of VM-26 (VM), camptothecin (CPT), and hydrogen peroxide (H2O2) were as indicated at the top of each lane.
Two additional experiments were performed to show that DNA cleavage induced by H2O2 required active TOP2 and resulted in TOP2-linked DNA breaks. First, H2O2 was shown to have no effect on DNA cleavage in the absence of TOP2 (Fig. 4D, lanes 1–7) or in the presence of heat-inactivated TOP2 (Fig. 4D, lanes 15–21). Stimulation of DNA cleavage by H2O2 was observed only in the presence of active TOP2 (Fig. 4D, lanes 8–14). In the presence of active TOP2, DNA cleavage was progressively reduced with increasing concentrations of H2O2 >8 mm (Fig. 4D, lanes 8–14), possibly due to nonspecific oxidation and, hence, inactivation of TOP2. Second, all DNA fragments induced by H2O2 were shown to be protein linked as evidenced by their disappearance from the aqueous phase after phenol extraction (Fig. 4E, lanes 1–7). On the other hand, if the reaction mixtures were digested with proteinase K (PK) prior to phenol extraction, all DNA fragments were recovered in the aqueous phase (Fig. 4E, cf. +PK with −PK in lanes 2–13), suggesting that all DNA fragments were protein linked. If DNA was preincubated with VM-26 or H2O2 for 30 min at 37°C and reisolated for subsequent incubation with TOP2 (no VM-26 or H2O2 in the second stage incubation), no significant cleavage was observed (Fig. 4E, lanes 14–25).
H2O2-induced TOP2-mediated DNA cleavage in vitro is reversible
To test whether the TOP2-mediated DNA cleavage stimulated by H2O2 was due to the increased formation of reversible cleavable complexes, the cleavage reaction was subjected to two standard reversal tests (Liu et al. 1983; Hsiang and Liu 1989). First, a brief heating (65°C for 15 min) of the reaction mixture just prior to termination (with SDS) of the cleavage reaction was shown to reverse DNA cleavage (heat-induced reversal) (Fig. 5A). Second, addition of excess EDTA (10 mm) to chelate Mg (II) ion was shown to completely reverse DNA cleavage induced by H2O2 and VM-26 (Fig. 5B).
Figure 5.

TOP2-mediated DNA cleavage stimulated by H2O2 is reversible. TOP2-mediated DNA cleavage assays were performed as described in Materials and Methods. Drug concentrations were indicated at the top of each lane. After incubation, duplicate reactions were subjected to different reversal treatments (see bracketed lanes under reversal) for 15 min prior to termination with SDS plus PK. (A) The reversal treatment was brief heating at 65°C for 15 min. (B) The reversal treatment was performed with 10 mm EDTA at 37°C for 15 min.
Discussion
Molecular basis for H2O2-dependent activation of TOP2-mediated DNA cleavage
The molecular basis for TOP2 poisoning (i.e., activation of TOP2-mediated DNA cleavage) by H2O2 is unclear. However, recent studies have demonstrated that TOP2 can be poisoned by multiple mechanisms (Liu et al. 1983; Zechiedrich 1989; Frydman et al. 1997; Kingma and Osheroff 1997a,b; Kwok and Hurley 1998). For example, in addition to drugs that bind TOP2 or TOP2–DNA complexes, DNA lesions have been shown to act as site-specific TOP2 poisons (Kingma and Osheroff 1997a,b; Kwok and Hurley 1998). We have therefore tested whether DNA damaged by H2O2 is responsible for activation of TOP2-mediated DNA cleavage. Our results do not support this possibility. DNA preincubated with H2O2 [with or without Fe (II) and EDTA] did not lead to significant stimulation of TOP2-mediated DNA cleavage (Fig. 4E; unpublished results).
Another new mechanism for poisoning TOP2 is through thiol alkylation of TOP2 (Frydman et al. 1997). The characteristics of TOP2 cleavage stimulated by H2O2 are quite reminiscent of that by thiol-alkylating naphthoquinones and other thiol-reactive compounds. Thiol alkylators have been suggested to activate TOP2-mediated DNA cleavage by covalently modifying the enzyme–DNA complex (Frydman et al. 1997; H. Wang and L.F. Liu, unpubl.). A cysteineless mutant yeast TOP2 was shown to be completely resistant to activation by thiol-alkylating naphthoquinones, suggesting that cysteine modification may be involved in activation of TOP2-mediated DNA cleavage (H. Wang and L.F. Liu, unpubl.). It seems conceivable that H2O2 may activate TOP2-mediated DNA cleavage through oxidation of a critical thiol group(s) on TOP2. Clearly, further studies are necessary to elucidate this interesting TOP2 poisoning mechanism.
TOP2-mediated DNA damage in oxidative stress and apoptotic cell death
TOP2 poisons are known to induce apoptotic cell death (Walker et al. 1991; Clarke et al. 1993; Lowe et al. 1993). Furthermore, TOP2 poisons have also been used to excise chromosomal DNA loops in cells (Lagarkova et al. 1995; Poljak and Kas 1995). Because oxidative stress and HMW fragmentation are commonly observed during the early stages of apoptosis (Quillet-Mary et al. 1995; Ha et al. 1997), poisoning of TOP2 by H2O2 may have interesting implications in apoptotic cell death. Oxidative stress is thought to cause DNA breaks and base modifications through hydroxyl radicals. The DNA breaks are primarily in the form of single-strand breaks (Imlay et al. 1988; Croteau and Bohr 1997). Our observation that H2O2 can activate TOP2-mediated DNA damage, which is primarily in the form of double-strand breaks, raises the question of which form of DNA damage, if any, is primarily responsible for apoptotic cell death. On the basis of the known fact that TOP2-mediated DNA damage induced by TOP2 poisons can efficiently trigger apoptotic cell death (Walker et al. 1991; Clarke et al. 1993; Lowe et al. 1993), one should consider the possibility that TOP2-mediated DNA damage induced by H2O2 may be involved in triggering apoptotic cell death. In agreement with this notion, H2O2 has been suggested to be a common mediator in ceramide- and polyamine-analog-induced apoptotic cell death (Quillet-Mary et al. 1995; Ha et al. 1997). In addition, generation of H2O2, HMW DNA fragmentation and apoptosis can be inhibited by antioxidants, catalase, and polyamine oxidase (PAO) inhibitors (Quillet-Mary et al. 1995; Ha et al. 1997).
A scheme summarizing our results is shown in Figure 6. In this scheme, hydrogen peroxide (H2O2) can covalently modify TOP2, which is located at the base of the chromosomal loops (30–100 kb in size) leading to the formation of reversible TOP2 cleavable complexes. TOP2-directed anticancer drugs act similarly except that their interaction with TOP2–DNA complexes is noncovalent. At this stage, addition of SDS to lyse cells would release the HMW DNA fragments. However, due to the reversibility of the cleavable complexes, a number of reversal conditions (e.g., drug removal, incubation at 55°C or 4°C, and addition of EDTA) can abolish the release of the HMW DNA fragments. With increasing time, irreversible excision of loop-sized DNA fragments occurs. In this case, the fragmentation process can no longer be reversed by the reversal conditions. The nature of this transition is unclear but may involve nuclease activation (Sun and Cohen 1994; Huang et al. 1997; Krieser and Eatman 1998). Following this stage, HMW DNA fragments are further digested by nucleases into smaller DNA fragments, which can be revealed as nucleosomal DNA ladders. Cytochrome c has been shown to activate a nuclease [DNA fragmentation Factor (DFF)/ caspase-activated deoxyribonuclease, (CAD)] involving caspase 3 (Liu et al. 1997; Sakahira et al. 1998; Zhang et al. 1998). It seems possible that CAD-dependent degradation of nuclear DNA could be responsible for the late stage events observed in our studies.
Figure 6.

A schematic illustrating the kinetic events of chromosomal DNA degradation due to activation of TOP2 by H2O2 or TOP2-directed anticancer drugs.
TOP2, being located at the base of the chromosomal DNA loops, has been suggested to control the loop topology that can modulate critical cellular processes such as DNA replication and RNA transcription (for review, see Wang 1996). Recent studies have suggested an interaction between yeast TOP2 and SGS1. SGS1, an Escherichia coli RecQ homolog, is known to be involved in aging of yeast (Sinclair and Guarente 1997). Interestingly, the two human RecQ homologs, BLM (the Bloom’s syndrome protein) and WRN (the Werner’s syndrome protein), are involved in genome instability and premature aging, respectively (for review, see Kirkwood 1996; Wang 1996). Oxidative stress is often proposed to be closely associated with the process of aging (for review, see Jazwinski 1996; Kirkwood 1996; Berlett and Stadtman 1997). Whether oxidative activation of TOP2 may be important in both genome instability and aging remains to be answered. Our studies have demonstrated such a possibility.
Materials and methods
Chemicals and cell lines
VM-26 was obtained from Bristol-Myers Squibb Co. H2O2, ATP, and DMSO were purchased from Sigma Chemical Co. VM-26 was dissolved in DMSO and kept frozen in aliquots at −20°C. [α-32P]dATP (3000 Ci/mmole) was purchased from DuPont. U937 and HL-60 cells were obtained from Dr. Eric H. Rubin and Dr. K.V. Chin, respectively (The Cancer Institute of New Jersey, New Brunswick, NJ). The mitoxantrone-resistant variant of HL-60, HL-60/MX-2, was obtained from ATCC (American Type Culture Collection).
Enzymes and nucleic acids
TOP2 was purified to near homogeneity from calf thymus glands with the published procedure (Liu et al. 1983). Human TOP2β (hTOP2β) cDNA was isolated by RT–PCR with mRNA isolated from human U937 cells. hTOP2β cDNA was then used to replace the TOP2α cDNA in YEpWob6 (unpubl.). The resulting plasmid, YEphTOP2β, was used to transform protease-deficient yeast BCY123. Purification of recombinant TOP2 isozymes followed the published procedure with slight modification (Wasserman et al. 1993).
Topoisomerase cleavage assay
TOP2 cleavage assay was performed as described previously (Tewey et al. 1985). Briefly, the reaction mixture (20 μl each) containing 40 mm Tris-HCl (pH 7.5), 100 mm KCl, 10 mm MgCl2, 1.0 mm ATP (unless indicated otherwise), 0.5 mm dithiothreitol, 0.5 mm EDTA, 30 μg/ml bovine serum albumin, 20 ng of 3′ end-labeled YEpG plasimd DNA, 10 ng of TOP2, and drug were incubated at 37°C for 30 min. The reactions were terminated by the addition of 5 μl of 5% SDS and 1 mg/ml PK with an additional 1 hr incubation at 37°C. Following addition of sucrose (5% final concentration) and bromophenol blue (0.05 mg/ml final concentration), DNA samples were loaded onto a 1% agarose gel in 1/2× TPE (45 mm Tris-phosphate, 1 mm EDTA at pH 8.0) buffer. After electrophoresis, gels were then dried onto Whatman 3MM chromatographic paper and autoradiographed at −80°C with Kodak XAR-5 films. TOP1 cleavage assay (Chen et al. 1993) was performed in a similar manner with the following differences; 10 ng of calf thymus TOP1 was used instead of TOP2, and ATP was omitted. After SDS/PK treatment, samples were alkali denatured by addition of NaOH, EDTA, sucrose, and bromophenol blue to final concentrations of 75 mm, 5 mm, 2.5%, and 0.05 mg/ml, respectively, prior to loading onto a neutral agarose gel described above.
Encapsulation of cells into agarose plugs
Logarithmically growing cells were pelleted and resuspended to a density of 107 cells/ml in RPMI-1640 supplemented with 10% fetal calf serum, l-glutamate (2 mm), penicillin (100 U/ml), and streptomycin (100 mg/ml). Following mixing with an equal volume of 1.5 % (wt/vol) low-melting point agarose premelted in the same culture medium (37°C), the mixture (100 ml each) was loaded into agarose plug makers. After solidification (5 min at room temperature), these agarose plugs were placed into multiple-well tissue culture plates and incubated in a CO2 incubator at 37°C.
Drug treatment and heat-induced reversal
To induce excision of chromosomal DNA loops, the agarose blocks containing cells were incubated at 37°C in RPMI-1640 medium supplemented with 30 μm VM-26 or 30 mm H2O2. At each indicated time point, one plug from each well was removed and placed in an eppendorf tube containing 1 ml of the same medium (with or without drug) preheated to 55°C. Heat reversal was carried out in a 55°C heat block for 10 min. Another agarose plug was subjected to lysis immediately (see the next section).
Cell lysis and PFGE
Cell lysis in the plugs was accomplished by placing plugs into new Eppendorf tubes containing 500 ml of TEN buffer (50 mm Tris at pH 8.0, 100 mm EDTA, and 100 mm NaCl) with 1% SDS and 0.5 mg/ml of PK. Following incubation at 50°C for 24 hr, the integrity of nuclear DNA in the plugs was analyzed by PFGE. PFGE was performed in the cold room (4°C) for 45 hr with circulation with 4 V/cm and the following switching times; 60 sec (21 hr), 80 sec (7 hr), and 120 sec (17 hr). Following electrophoresis, the gel was stained with 1 mg/ml ethidium bromide and photographed.
Acknowledgments
This work was supported by the National Institutes of Health (grant nos. GM27731 and CA 39962).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lliu@umdnj.edu; FAX (732) 235-4073.
References
- Berlett BS, Stadtman ER. Protein oxidative in aging, disease and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- Chen AY, Yu C, Liu LF. DNA minor groove binding ligands: A different class of mammalian DNA topoisomerase I inhibitors. Proc Natl Acad Sci. 1993;90:8131–8137. doi: 10.1073/pnas.90.17.8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- Cohen GM, Sun XM, Fearnhea H, MacFarlane M, Brown DG, Snowden RT, Dinsdale D. Formation of large molecular weight fragments of DNA is a key committed step of apoptosis in thymocytes. J Immunol. 1994;153:507–516. [PubMed] [Google Scholar]
- Croteau DL, Bohr V. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- Frydman B, Marton LJ, Sun JS, Neder K, Liu AA, Wang HM, Mao Y, Wu HY, Sanders MM, Liu LF. Induction of DNA topoisomerase II-mediated DNA cleavage by beta-lapachone and related naphthoquinones. Cancer Res. 1997;57:620–627. [PubMed] [Google Scholar]
- Ha HA, Woster PM, Yager JD, Casero RA., Jr The role of catabolism in polyamine analog-induced programmed cell death. Proc Natl Acad Sci. 1997;94:11557–11562. doi: 10.1073/pnas.94.21.11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harker WG, Slade DL, Parr RL, Feldhoff PW, Sullivan DM, Holguin HM. Alteration in the topoisomerase IIα gene, message RNA, and subcellular protein distribution as well as reduced expression of the DNA topoisomerase IIβ enzyme in a mitoxantrone-resistant HL-60 human leukemia cell line. Cancer Res. 1995a;55:1707–1716. [PubMed] [Google Scholar]
- Harker WG, Slade DL, Parr RL, Holguin HM. Selective use of an alternative stop codon and polyadenylation signal within intron sequences leads to a truncated topoisomerase IIα message RNA and protein in human HL-60 leukemia cell line selected for resistance to mitoxantrone. Cancer Res. 1995b;55:4962–4971. [PubMed] [Google Scholar]
- Hedley DW, McCulloch EA. Generation of reactive oxygen intermediates after treatment of blasts of acute myeloblastic leukemia with cytosine arabinoside: Role of bcl-2. Leukemia. 1996;10:1143–1149. [PubMed] [Google Scholar]
- Hockenery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- Hsiang Y-H, Liu LF. Evidence for the reversibility of cellular DNA lesion induced by mammalian topoisomerase II poisons. J Biol Chem. 1989;264:9713–9715. [PubMed] [Google Scholar]
- Huang P, Ballal K, Plunkett W. Biochemical characterization of the protein activity responsible for high molecular weight DNA fragmentation during drug-induced apoptosis. Cancer Res. 1997;57:3407–3414. [PubMed] [Google Scholar]
- Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- Jazwinski SM. Longevity, genes and aging. Science. 1996;273:54–58. doi: 10.1126/science.273.5271.54. [DOI] [PubMed] [Google Scholar]
- Kingma PS, Osheroff N. Apurinic sites are position-specific topoisomerase II poisons. J Biol Chem. 1997a;272:1148–1155. doi: 10.1074/jbc.272.2.1148. [DOI] [PubMed] [Google Scholar]
- ————— Spontaneous DNA damage stimulates topoisomerase II-mediated DNA cleavage. J Biol Chem. 1997b;272:7488–7493. doi: 10.1074/jbc.272.11.7488. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB. Human senescence. BioEssays. 1996;18:1009–1016. doi: 10.1002/bies.950181211. [DOI] [PubMed] [Google Scholar]
- Krieser RJ, Eatman A. The cloning and expression of human deoxyribonuclease II. A possible role in apoptosis. J Biol Chem. 1998;273:30909–30914. doi: 10.1074/jbc.273.47.30909. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:43–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Kwok Y, Hurley LH. Topoisomerase II site-directed alkylation of DNA by peorospermin and its effect on topoisomerase II-mediated DNA cleavage. J Biol Chem. 1998;273:33020–33026. doi: 10.1074/jbc.273.49.33020. [DOI] [PubMed] [Google Scholar]
- Lagarkova MA, Iarovaia OV, Razin SV. Large-scale fragmentation of mammalian DNA in the course of apoptosis proceeds via excision of chromosomal DNA loops and their oligomers. J Biol Chem. 1995;270:20239–42021. doi: 10.1074/jbc.270.35.20239. [DOI] [PubMed] [Google Scholar]
- Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- Liu LF, Rowe TC, Yang L, Tewey K, Chen GL. Cleavage of DNA by mammalian DNA topoisomerase II. J Biol Chem. 1983;258:15365–15370. [PubMed] [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that fuctions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulate the cytotoxicity of antitumor drugs. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Walker AE, Sikorska M. Apoptotic death in epithelial cells: Cleavage of DNA to 300 and/or 50 kb fragments prior to or in the presence of internucleosomal fragmentation. EMBO J. 1993;12:3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poljak L, Kas E. Resolving the role of topoisomerase II in chromatin structure and function. Trends Cell Biol. 1995;5:348–354. doi: 10.1016/s0962-8924(00)89068-6. [DOI] [PubMed] [Google Scholar]
- Quillet-Mary A, Jaffrezou J-P, Mansat V, Bordier C, Naval J, Laurent G. Implication of mitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J Biol Chem. 1995;272:21388–21395. doi: 10.1074/jbc.272.34.21388. [DOI] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Shih SC, Shaman O. Cell cycle-dependent tumor necrosis factor apoptosis. Cancer Res. 1996;57:1591–1601. [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles-a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Stangel M, Zettl UK, Mix E, Zielasek J, Toyka KV, Hartung HP, Gold R. H2O2and nitric oxide-mediated oxidative stress induce apoptosis in rat skeletal muscle myoblasts. J Neuropathol Exp Neurol. 1996;55:36–43. doi: 10.1097/00005072-199601000-00004. [DOI] [PubMed] [Google Scholar]
- Stanulla M, Wang J, Chervinsky DS, Thandla S, Aplan PD. DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher-order chromatin fragmentation during the initial stages of apoptosis. Mol Cell Biol. 1997;17:4070–4079. doi: 10.1128/mcb.17.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XM, Cohen GM. Mg(2+)-dependent cleavage of DNA into kilobase pair fragments is responsible for the initial degradation of DNA in apoptosis. J Biol Chem. 1994;269:14857–14860. [PubMed] [Google Scholar]
- Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin induced DNA damage mediated by mammalian topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- Um HD, Orenstein JM, Wahl SM. Fas mediates apoptosis in human monocytes by a reactive oxygen intermediate dependent pathway. J Immunol. 1996;156:3469–3477. [PubMed] [Google Scholar]
- Walker PR, Smith C, Youdale T, Leblanc J, Whitfield JF, Sikorska M. Topoisomerase II-reactive chemotherapeutic drugs induce apoptosis in thymocytes. Cancer Res. 1991;51:1078–1085. [PubMed] [Google Scholar]
- Wang JC. DNA topoisomerases. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- Wasserman RA, Austin CA, Fisher LM, Wang JC. Use of yeast in the study of antitumor drugs targeting DNA topoisomerases: Expression of a functional recombinant human topoisomerase IIa in yeast. Cancer Res. 1993;53:3591–3596. [PubMed] [Google Scholar]
- Zechiedrich EL, Christiansen K, Anderson AH, Westergaard O, Ocheroff N. Double-stranded cleavage/religation reaction of eukayotic topoisomerase II: Evidence for a nicked DNA intermediate. Biochemistry. 1989;28:6229–6236. doi: 10.1021/bi00441a014. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu X, Scherer DC, van Kaer L, Wang X, Xu M. Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc Natl Acad Sci. 1998;95:12480–12485. doi: 10.1073/pnas.95.21.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]