

Abstract
NeuroD, a bHLH transcription factor, is implicated in differentiation of neurons and pancreatic β cells. NeuroD-null mice die shortly after birth due to severe neonatal diabetes. To examine if there is postnatal neuronal phenotype in these mice, we rescued them from neonatal lethality by introducing a transgene encoding the mouse neuroD gene under the insulin promoter. These mice survive to adulthood but display severe neurological phenotype due to neuronal deficit in the granule layers of the cerebellum and hippocampus. We show here that NeuroD is required for these postnatally generated microneurons to undergo proper differentiation, the absence of which results in cell death.
Keywords: NeuroD, neuronal differentiation, microneuron, postnatal neurogenesis, cerebellum, hippocampus
The basic helix–loop–helix (bHLH) transcription factors are involved in cell fate determination and differentiation during development of many organisms. In both flies and mice, the requirement of bHLH proteins in differentiation of many cell types has been demonstrated. Most prominent examples are the proneural bHLH proteins in fly neurogenesis and the myogenic bHLH proteins in mouse skeletal myogenesis (Weintraub et al. 1991; Jan and Jan 1993). Hematopoietic cells are also subject to regulation by bHLH proteins during differentiation (Robb et al. 1996; Zhuang et al. 1994). In the last several years, a number of mouse genes encoding neuronal bHLH proteins have been identified (for review, see Lee 1997). One such factor is NeuroD, which is highly expressed in differentiating neurons in mice and frogs (Lee et al. 1995). When ectopically expressed in Xenopus embryos, NeuroD can convert non-neuronal ectodermal cells into fully differentiated neurons, indicating that it can be a potent neuronal differentiation factor (Lee et al. 1995). In addition, neuroD is expressed abundantly in mature neurons in adult brain structures, including olfactory bulbs, hippocampus, and cerebellum, suggesting a secondary role of NeuroD in fully differentiated neurons (Lee et al. 1995). neuroD is also expressed in pancreatic β cells, and NeuroD-null mice become severely diabetic and die shortly after birth due to defects in β cell differentiation (Naya et al. 1995, 1997). To investigate whether NeuroD is involved in postnatal brain development, we have rescued NeuroD-null mice from neonatal lethality by re-expressing NeuroD in the pancreas using the transgenic constructs shown in Figure 1. The transgene (Tg) contains a short rat insulin promoter (RIP-1; Hanahan 1985) and a coding region that encodes the mouse NeuroD protein with its amino terminus fused to a Myc tag. We report here that NeuroD-null mice carrying the transgene, which we refer to as ND−/−Tg, survive to adulthood. However, these mice display severe neurological symptoms and morphological defects in the brain, indicating that NeuroD has a critical role in postnatal brain development.
Figure 1.
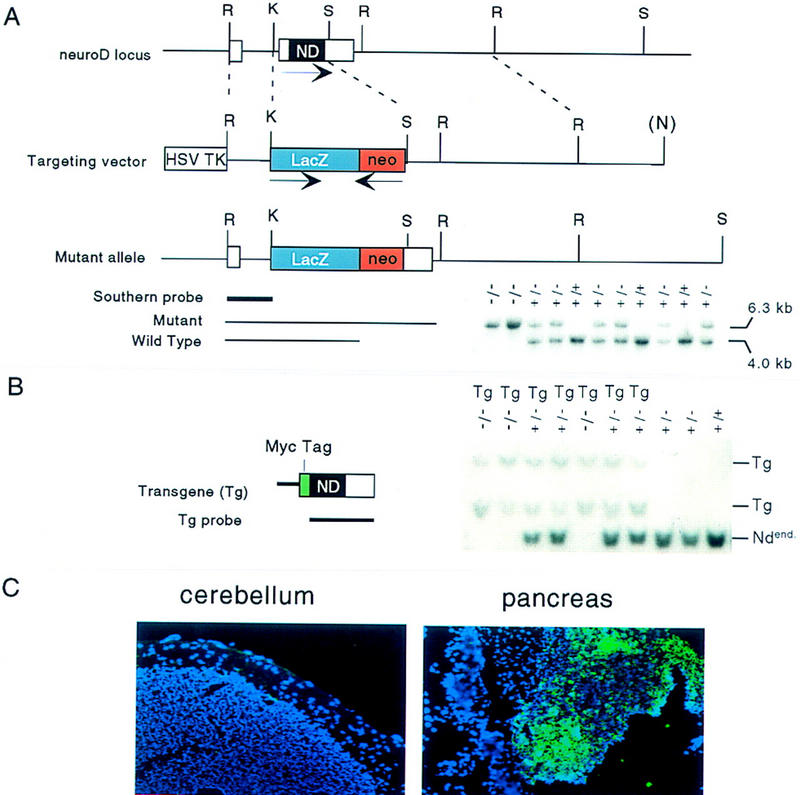
Gene targeting at the neuroD/beta-2 locus and introduction of transgene. (A). Schematic representation of the targeting vector, the wild-type neuroD locus, and the disrupted allele generated by homologous recombination. The orientations of the neuroD (noncoding regions in white; entire coding region in black), lacZ (in blue), and neoR gene (red) are indicated by arrows above below the genes. Insertion of the lacZ–neoR cassette deleted the second exon including the entire coding region. (R) EcoRI; (K) KpnI; (S) SpeI; (N) from the vector NotI. A sample of mouse genotyping by Southern blot analysis is shown below. Total mouse genomic DNA was digested with EcoRI; the blot was then hybridized with the Southern probe indicated; the presence of the mutant allele is confirmed by a 6.3-kb diagnotic band and the wild type by a 4.0-kb band. (B) Schematic representation of the transgene containing ∼700-bp RIP-1 and the 1.8-kb mouse neuroD coding region with its amino terminus fused to the Myc tag (green). A Southern blot analysis using the neuroD coding region as a probe is shown. Both the endogenous neuroD (Ndend) and Tg are detected. (C) Expression of transgene in ND−/− Tg mice. Transgene expression, as detected by immunostaining using anti-Myc tag antibody (fluorescent color), is detected in the pancreatic islets but not in the brains of P30 mice.
Results
We have previously generated NeuroD-null mice by replacing the coding region of the neuroD gene with a reporter construct containing promoterless cytoplasmic lacZ (Fig. 1A). The lacZ staining pattern reflects the endogenous neuroD gene expression pattern, which enables easy detection of neuroD promoter activity and assessment of the morphology of cells expressing lacZ. NeuroD-null mice showed all of the previously reported phenotypes including neonatal diabetes and lethality (Naya et al. 1997). The transgene that we introduced into NeuroD-null mice to rescue the pancreatic phenotype encodes mouse NeuroD whose amino terminus is fused to a Myc tag (Fig.1B). Immunostaining against the Myc tag allows us to follow the expression of the transgene. Throughout the experiment, we used sibling mice carrying Tg in either the ND+/− or wild-type background as controls. Because the insulin promoter we used is known to be transiently expressed in embryonic brain (Alpert et al. 1988), it was important to ascertain the dominant effects of the mice carrying the transgene. These control mice do not display any behavioral or morphological abnormalities, demonstrating that the transgene does not cause any dominant effects on mouse development. We have also confirmed that the transgene, as detected by an antibody against Myc tag, is expressed in the pancreatic islets, but not in the postnatal brain (Fig. 1C) of the rescued mice, thus explaining its ability to selectively rescue the pancreatic phenotype in NeuroD-null mice.
ND−/− mice and ND−/−Tg mice exhibit no overt phenotype at birth. Whereas ND−/− mice soon become severely diabetic and die within 5 days after birth, ND−/− Tg mice can be found among the surviving pups. The rescued ND−/−Tg mice display a slower growth rate compared to the control mice (Fig. 2A,C). Strikingly, the surviving ND−/−Tg mice start to show an ataxic gait as early as postnatal day 13 (P13). They fall down as often as 8–10 times per minute while moving actively (Fig. 2B). They walk around incessantly while they are awake, whereas the control mice usually have pauses and rests in their movement. When put on a narrow platform (3 cm wide), the ND−/−Tg mice fall immediately, whereas the control mice remain balanced for >5 min, thus indicating that the ND−/−Tg mice have abnormal motor functions and a failure to balance themselves. These neurological symptoms suggest defects in the sensory, motor, or CNS functions.
Figure 2.
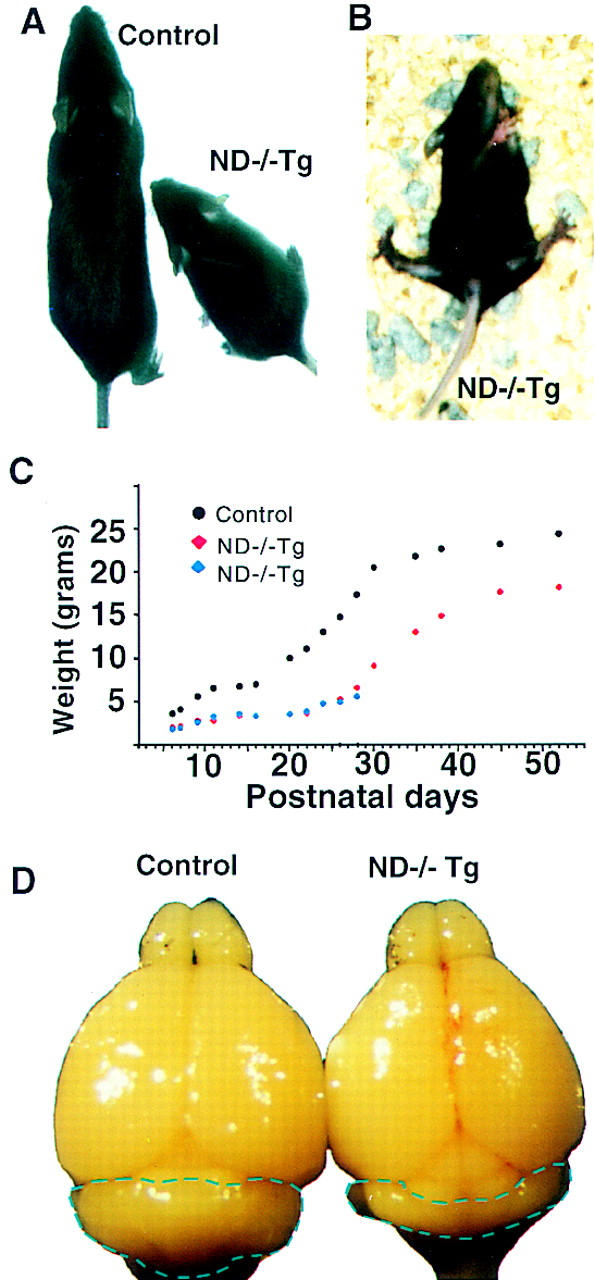
ND−/−Tg mice show retarded growth, an ataxic gait, and smaller cerebellar size. (A) Body size difference between ND+/−Tg and ND−/−Tg mice; (B) ND−/−Tg mice lose balance and fall over frequently; (C) a typical weight curve of littermates that include ND−/−Tg; (D) brains from ND+/−Tg and ND−/−Tg mice at P30.
Macroscopic inspection of most brain regions at P30 shows comparable brain sizes in the regions, including the cerebrum and olfactory bulbs, to those of the sibling control transgenic mice despite the significant difference in their body weights (Fig. 2C). However, a pronounced reduction in the size of the cerebellum can be seen in the brain of ND−/−Tg mice (Fig. 2D). Further histological examination of the brain of P30 ND−/−Tg mice reveals severe depletion of granule cells in both the cerebellum and hippocampus. We discuss each of these phenotypes separately below.
At P30, any given section through the ND−/−Tg cerebellum shows a size reduction to 30%–40% of the control cerebellum (Fig. 3A,B). All of the major cerebellar lobules are recognizable with a normal, albeit shallow, foliation pattern. We performed X-gal or anti-β-galactosidase staining on the sections of these cerebella to assess the fate of the cells that no longer express NeuroD. X-Gal-staining in the granular layer shows a severe granule cell depletion in all the lobules but to different extents between anterior and posterior lobules (Fig. 3A,B). In the posterior lobules (VI–X), a near total loss of granule cells and disarrangement of Purkinje cells are observed, whereas the anterior lobules still contain some granule cells in the internal granule layer (IGL; Fig. 3). Therefore, it appears that the degree of granule cell depletion is far more substantial posterior to lobule VI.
Figure 3.
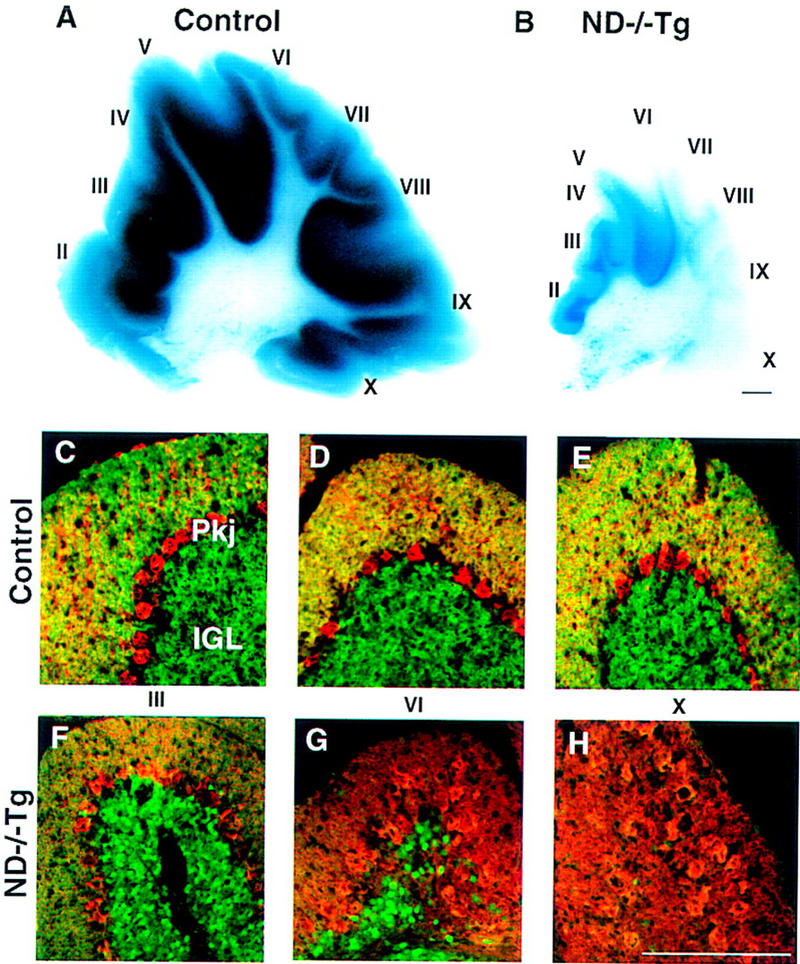
Cerebellar abnormalities in P30 ND−/−Tg mice. (A,B) X-gal-stained parasagittal sections showing a massive reduction in granule cells in ND−/−Tg mice. (C–H) β-Gal (green) and calbindin (red) double immunostaining showing the A–P difference in the degree of granule cell loss and the arrangement of Purkinje cells, respectively. (I–X) Individual cerebellar lobules. Bars, 100 μm.
Although the granular neuronal population is severely affected, Purkinje cells appear to have differentiated normally (Fig. 3C–H). However, their average total number on a 10-μm sagittal section is slightly less (337, n = 5) than that of corresponding sections in control cerebellum (398, n = 4) at P30. Their spatial arrangement is disturbed significantly in the posterior lobules and they no longer form a monolayer (Fig. 3H), possibly as a secondary result of lacking granule cells. Despite the severely disturbed Purkinje cell distribution in the posterior region, the number of Purkinje cells varies little along the anteroposterior (A–P) axis. The Toluidine blue staining of the deep nuclear neurons indicates that these neurons are not grossly affected (data not shown).
To elucidate the pathological mechanisms of this cerebellar phenotype, we performed histological analyses on younger tissues of both control and ND−/−Tg mice. During mouse development, neuroD is highly expressed in the inner layer of cerebellar external granular layer (EGL), where the postmitotic premigratory neurons are situated. A lower level of neuroD expression is also detected in postmigratory cells in the IGL (Fig. 4A,B). However, neuroD expression is not found in the outer EGL, which consists of dividing granule precursor cells, or in Purkinje cells (Fig. 4A,B). The pattern of neuroD expression also differs from those of radial glial cells or Bergmann glial cells stained with anti-tenescin or GFAP. Differential expression patterns from embryonic to early postnatal cerebellar development indicate that neuroD is expressed exclusively in the granule layers and not in the layers containing glial cells.
Figure 4.
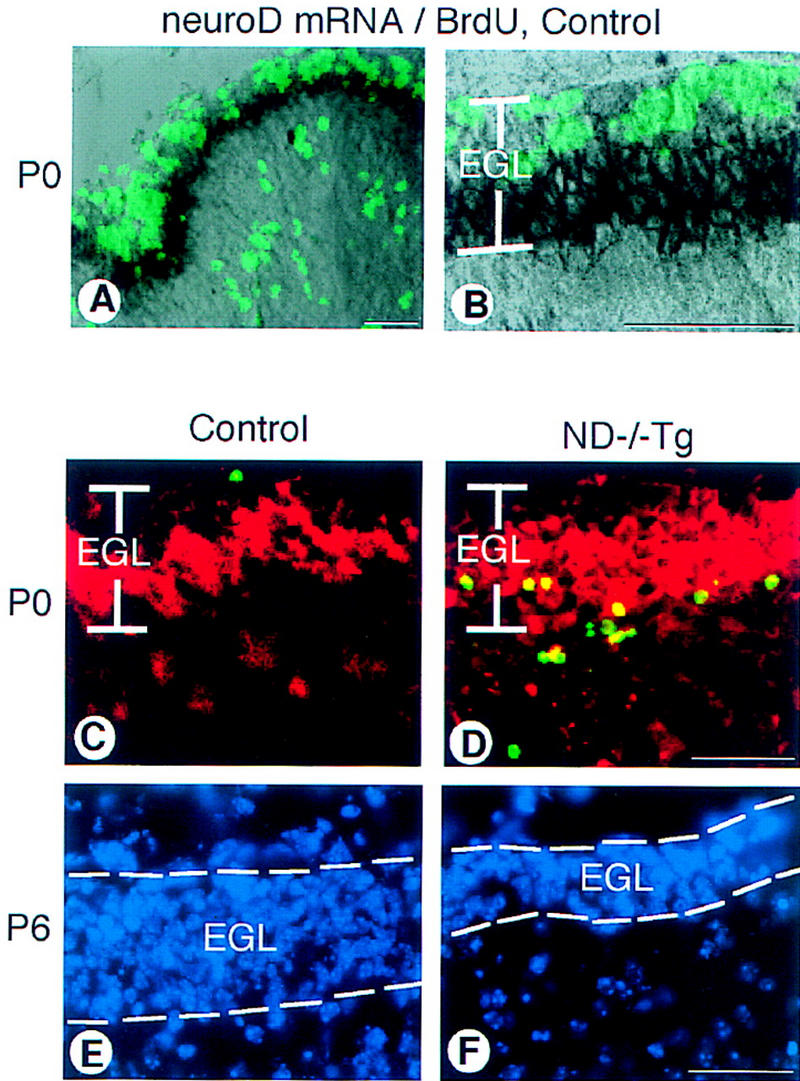
Cerebellar abnormalities in the ND−/−Tg mice. (A) neuroD in situ hybridization with BrdU labeling on control mice at P0. neuroD mRNA (black) is expressed in the inner zone of the EGL; the outer zone is cells incorporating BrdU (green). (B) A higher magnification of the EGL shown in A. (C,D) Apoptotic cells in the P0 cerebellar cortex. In the posterior lobules of ND−/−Tg mice, many TUNEL+ cells (green) are found in the inner EGL where immunostaining with antibody to β-galactosidase (red) is also found. (E,F) DAPI staining shows the reduced EGL thickness in the posterior lobules of ND−/−Tg cerebellum at P6. Bars, 20 μm.
Although cerebella of ND−/−Tg and control mice are indistinguishable in size and morphology at P0, the overall size reduction in ND−/−Tg mice becomes apparent as early as P2, indicating that cell depletion takes place early during postnatal development. To assess whether cell depletion is due to cell death among neuroD-expressing postmitotic granule cells, we performed the TUNEL assay (Gavrieli et al. 1992) at P0. Normally, a few TUNEL+ cells are expected to be found in the outer half of the EGL, which contains dividing cells at P0 (Fig. 4C; Wood et al. 1993). In both ND−/− or ND−/−Tg mice at P0, in addition to the small number of TUNEL+ cells in this outer zone, many are found in deeper areas, most prominently in the inner half of the EGL (Fig. 4D). However, there is an A-P variation of TUNEL staining at P0 when the morphological phenotypic difference is not yet apparent in the A–P axis: Many TUNEL+ cells are detected in the posterior half, whereas an almost normal level is observed in the anterior half. Granule cell loss and accompanying morphological change become apparent in the ND−/−Tg cerebellum at P6, most notably in the posterior region. Whereas the anterior lobules still contain the normal 8- to 10-cell thick layer of EGL, the posterior lobules have a thinner EGL of only 4- to 5-cell thickness (Fig. 4E,F). At P30, extensive TUNEL labeling of the remaining anterior IGL cells is observed (data not shown), indicating that a significant number of the surviving granule cells is also degenerating. However, at P88, we still detect ∼5%–10% of granule cells remaining in the anterior cerebellum. These granule cells do not label with TUNEL but can be stained with antibodies against the neurofilament or β-galactosidase, which stains granular axons in the molecular layer (data not shown). Although differentiation occurs to some extent among a small population of anterior granule cells in ND−/−Tg mice, the majority of granule cells die in the inner EGLs at the time of differentiation or after migration to the IGL. From these results, we conclude that NeuroD regulates the expression of differentiation genes, including those that are essential to the survival of postmitotic granule cells in the cerebellum.
The granule cells of the cerebellum share many developmental, morphological, and cellular characteristics with the granule cells found in the hippocampus (Altman 1967; Vicario-Abejon et al. 1995; Yang et al. 1966). Therefore, we examined the granule cell population of the hippocampus of ND−/−Tg mice and found that these cells exhibited similar defects in differentiation. During the development of the hippocampus in wild-type mice, neuroD is expressed in the Ammon’s horn (AH), which includes a CA1–CA3 pyramidal layer, as well as both the proliferating precursor and mature population of granule cells that make up the dentate gyrus (DG) (Fig. 5A,B). We find that mature ND−/−Tg brains have no structure corresponding to the DG, whereas AH formation is normal (Fig. 5G–J). The defect in DG formation is evident as early as E18.5, even in ND−/− mice that do not carry the RIP1–ND transgene, as confirmed by Toluidine blue and X-gal staining (data not shown). By P6, no X-gal staining is detected in the region corresponding to the DG, whereas the staining of AH neurons is normal. The lack of calretinin immunoreactivity, which is normally found in DG granule cells, also confirms the absence of these granule cells in ND−/−Tg mice at P30 (Fig. 5I,J).
Figure 5.
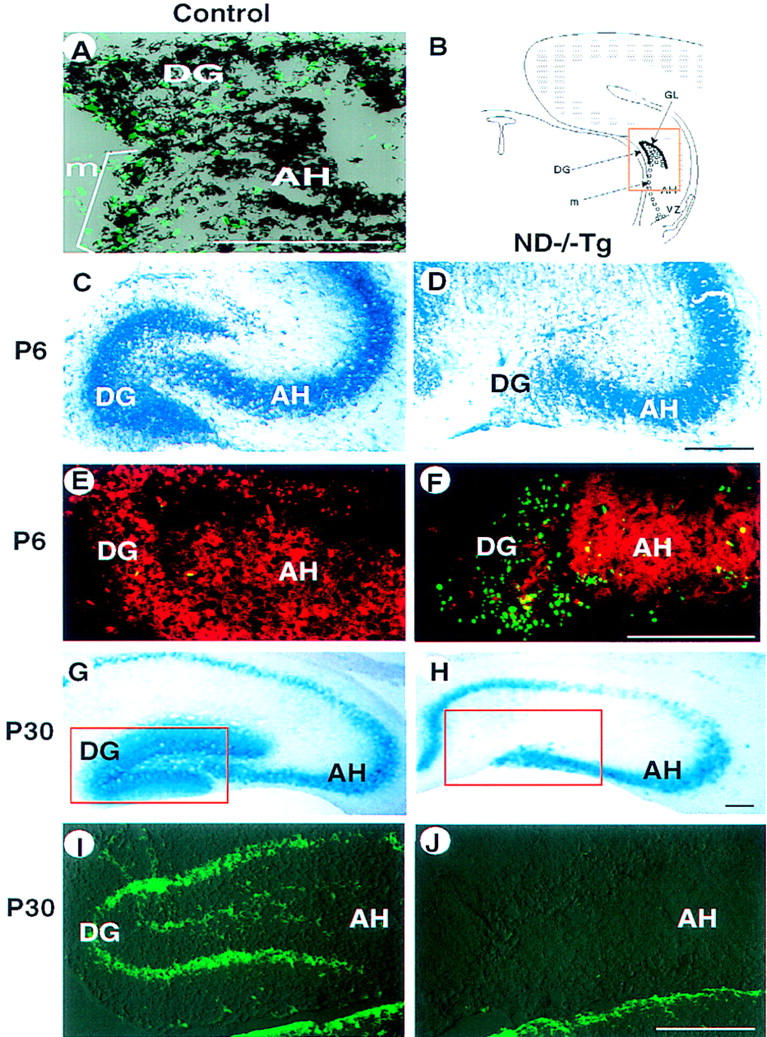
Hippocampal abnormalities in ND−/−Tg mice. (A) In situ hybridization at P0 showing that neuroD mRNA is expressed (black) in DG-forming granule cells and progenitor cells that are labeled with BrdU (green). (B) Schematic presentation of development of the DG in normal mice. The DG is formed by the granule neurons (●) generated by progenitor cells (○) after migration (m) from the VZ along the AH. (C–J) Degeneration of DG in ND−/−Tg mice as shown by staining with toluidine blue (C,D), anti-β-gal immunostaining (red in E,F), X-gal staining (G,H), or anti-calretinin immunostaining (I and J, corresponding to the squared areas in G and H, respectively; staining in the thalamic area below is also shown) at P6 (C–F) or P30 (G–J). TUNEL staining at P6 (E,F) shows many apoptotic cells (green) in the poorly formed DG in ND−/−Tg mice. (B–F) The squared region in B. Bars, 100 μm.
The DG is formed by a complicated pattern of cell migration and cytogenesis (Fig. 5B; Altman and Bayer 1990). The TUNEL assay at P0 shows significantly more (15–20) apoptotic cells in the DG primordium of ND−/− Tg mice than in control mice (<3 cells; data not shown). Many more TUNEL+ cells were found in the DG remnant in ND−/−Tg mice at P6 (Fig. 5 E,F). Proliferation of DG precursor cells is also affected. At P0, whereas the initial migratory cells from the ventricular zone (VZ) show a normal level of BrdU incorporation (30–40 cells), <50% labeling of BrdU is observed in the poorly forming DG primordium of ND−/−Tg mice (17–20 cells/10-μm section as compared to 40–50 labeled cells in the control; data not shown). However, we cannot ascertain whether such reduction of BrdU labeling in the ND−/−Tg DG is due to extensive cell death among proliferating cells or a true reduction in the number of proliferating cells. neuroD expression is detected in the mitotic population of the hippocampal granule progenitors (Fig. 5A,B), unlike elsewhere in the brain where it is expressed in the postmitotic neurons. Whether such mitotically active migrating DG precursors in the hippocampus represent cells with some aspects of differentiated characteristics is not known. From these results, we conclude that the disappearance of DG that occurs within the first postnatal week in ND−/−Tg mice can be attributed mainly to death of granule progenitor cells shortly after initiation of migration.
Although neuroD is expressed in other areas of the brain (E. Marcora and J.E. Lee, in prep.), structures other than the cerebellum and hippocampus seem to develop normally. We are currently investigating whether there are more subtle histological defects in other regions of the brain where neuroD expression is normally found.
Discussion
In summary, we have shown that the absence of NeuroD in the mouse causes severe defects in brain development. Both cerebellar granular neurons and hippocampal DG cells, which show the most obvious and severe defects, belong to a postnatally generated group of neurons called microneurons that mainly work as interneurons whose output is local (Altman 1967). Such a phenotype in the development of microneurons indicates that NeuroD is critical for postnatal neurogenesis in the mouse brain. Interestingly, we have not been able to find an equally severe phenotype in olfactory neurons, possibly due to their ability to extensively regenerate themselves. Although NeuroD is expressed in different types of neurons in the brain, it seems that there is a functional requirement or selectivity of NeuroD for granule cells.
The degree of defects in the cerebellum differs considerably between anterior and posterior to the midline of lobule VI, where the expression of several genes showing an A–P segmental pattern have been reported to be bordered (Herrup and Kuemerle 1997). Although granule cells are completely depleted in the posterior region of the cerebellum, a small population of seemingly healthy granule cells survive in the anterior portion of the cerebellum. Whether these remaining granular neurons display the normal physiology of granule cells remains to be determined. More dramatic nonuniform A–P differences have been observed in other mutations involving cerebellar granule cell depletion, such as the leaner (tgla/tgla) (Herrup and Wilczynski 1982) and meander tail (mea/mea) (Ross et al. 1989) mutations in mice. Our results also support existence of A–P compartmentalization in the cerebellum and suggest that the granule cell depletion in ND−/−Tg mice is under the influence of the genes regulating such regional differences in the cerebellum (for review, see Hatten and Heitz 1995). We also cannot rule out the possibility that the anterior cerebellum contains a population of granule cells, represented by those surviving in ND−/−Tg mice, that are functionally and physiologically distinct from the majority of cerebellar granule cells that are affected by the absence of NeuroD.
Unlike the cerebellum, where a small number of cells do survive, DG granule cells in the hippocampus are completely absent in ND−/−Tg mice. It is also striking that migrating DG cells are mitotically active and express neuroD at the same time while neuroD is expressed only in the postmitotic population in most other brain regions. Whether these migrating and mitotic DG cells display molecular differentiation characteristics is currently not known. The complete absence of DG cells in ND−/−Tg mice implies that neuronal precursors existing in the adult DG (Gage et al. 1998) may also be dependent on NeuroD for survival.
NeuroD function is critical for β cell differentiation. In the absence of NeuroD, β cells are born but undergo massive cell death around E18.5 (Naya et al. 1997). It is possible that the time at which the β and granule cells die represents equivalent developmental stages in these two cell lineages. They may represent the initial differentiation point that requires NeuroD, the absence of which can cause physiological damage. Cell deaths observed in these two cell types could also involve common molecular mechanisms. As we have shown in the case of the cerebellum, ND−/− cells are able to exit mitosis. Evidence also indicates that no increase in proliferation among β cells occurs in the NeuroD-null pancreas (data not shown). Therefore, it is unlikely that these cells die as a result of the commonly accepted model of incompatibility of proliferation and differentiation. Unlike enteroendocrine cells, which keep cycling in the absence of NeuroD (Mutoh et al. 1998), we propose that NeuroD-null granule and β cells die because of physiological defects resulting from failure to properly differentiate, including misregulated ion channels, which are known to be activated during differentiation. We are currently exploring this possibility. Therefore, it seems that there are at least two different responses that cells take when NeuroD is absent. Either cells die during differentiation or later as mature cells, as is the case with β and granule cells in the brain, or cells continue to proliferate, as observed with enteroendocrine cells. Thus, NeuroD is implicated in differentiation and maintenance of differentiated state, as well as cell cycle arrest during differentiation of certain neuronal cell lineages.
Another member of the bHLH protein family, MATH-1, is expressed in the proliferating other EGL of the cerebellum. The MATH-1-null mice fail to make any granule cells in the cerebellum, indicating that MATH-1 has a critical role in generation of granule precursors (Ben-Arie et al. 1997). This observation and our finding that NeuroD is involved in differentiation of postmitotic cerebellar granule cells establish a hierarchy of bHLH proteins involved in cerebellar granule cell development, with MATH-1 being upstream of NeuroD.
Although NeuroD might also have a critical role in embryonic neurogenesis, other embryonically expressed bHLH proteins, such as NeuroD2, MATH-2, and MATH-3, may compensate functionally for NeuroD. We also cannot rule out the possibility that our transgene is expressed in embryonic structures, thereby rescuing prenatal phenotypes. We are currently investigating this possibility.
We have shown that NeuroD is required for postnatal neurogenesis in the cerebellum and hippocampus. The transgenic method of rescuing neonatal lethality revealed the postnatal brain phenotype that could be confirmed in neonatal ND−/− mice. Future study on ND−/− and ND−/−Tg mice will enhance our understanding of the molecular mechanisms governing postnatal neuronal differentiation. It will also enable us to examine the basis of its selectivity for microneurons and establish the epistatic relationship of different transcription factors involved in granule cell differentiation.
Materials and methods
Targeted disruption of the neuroD gene
The pPNT vector containing a positive selection marker, PGK–neoR, and a negative selection marker, HSV–TK, was used to perform the targeting experiment. A 1.5-kb EcoRI–KpnI fragment from the 5′ upstream region fused to cytoplasmic lacZ and 6.5-kb SpeI–NotI (from the λ vector) from the 3′ region were used for generation of the targeting construct. Cytoplasmic lacZ was used to stain the entire cell body. Upon correct integration, deletion of the entire second exon containing all of the coding region is achieved. Chimeras obtained from three independent targeted ES clones were maintained in both pure 129/SV background or mixed background with C57BL/6.
Generation of transgenic animals
Transgenic animals carrying the Myc-tagged-NeuroD transgene were generated by the standard pronuclear injection. Briefly, the egg-donor mice (C57CBA) were mated with ND+/− males to generate either ND+/− or wild-type embryos. Once the transgene was injected into the nuclei, the embryos were implanted and the founder transgenic mice were obtained. ND−/−Tg mice were obtained by mating ND+/− female mice with ND+/−Tg males. ND−/− animals carrying the transgene were identified by Southern analysis, as described in the legend to Figure 1. After being anesthetized with avertin, the animals were perfused transcardially with 4% paraformaldehyde. Isolated brains were postfixed in 4% paraformaldehyde from 2 hr to overnight, immersed in 20% sucrose, and embedded in OCT freezing compound. Frozen 10-μm sections were obtained for further analyses.
Immunohistochemistry and X-gal staining
Toluidine blue or antibody staining was done as described (Miyata et al. 1996). BrdU labeling into DNA was achieved by BrdU injection of 100 μg/gram body weight, intraperitoneally, 1 hr before the mice were sacrificed. The antibodies used in the experiments include anti-β-gal (5′-3′ five prime three prime, 1:500), BrdU (Boerhinger, 1:100), calbindin (Sigma, 1:200), and calretinin (Chemicon; 1:500). X-gal staining was done as described (Spector et al. 1998).
In situ hybridization and cell death assay
Nonradioactive in situ hybridization with digoxigenin-labeled probes was performed as described (Lee et al. 1995). After visualization of the signal with an alkaline phosphatase-conjugated secondary antibody, sections were processed further for BrdU immunostaining. Apoptotic cells were detected by use of a TUNEL stain kit (Boehringer).
Acknowledgments
We thank M. Moore, C. Snyder, and K. Swihart for technical support; K. Jones for the insulin promoter construct; and T. Jessell, E. Morrow, and members of the Lee laboratory for critical reading of the manuscript and helpful discussions. T.M. thanks Dr. M. Ogawa for constant support and R. Miyata and M. Miyata for encouragement. This work was supported by The Japan Society for the Promotion of Science (to T.M.), the Juvenile Diabetes Foundation International, and the National Institutes of Health (R01NS35118) (to J.E.L.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Jackie.Lee@colorado.edu; FAX (303) 492-7744.
References
- Alpert S, Hanahan D, Teitelman G. Hybrid insulin genes reveal a developmental lineage for pancreatic endocrine cells and imply a relationship with neurons. Cell. 1988;53:295–308. doi: 10.1016/0092-8674(88)90391-1. [DOI] [PubMed] [Google Scholar]
- Altman J. Postnatal growth and differentiation of the mammalian brain, with implications for a morphological theory of memory. In: Quarton GC, Melnechuk T, Schmitt FO, editors. The neurosciences. New York, NY: The Rockefeller University Press; 1967. pp. 723–743. [Google Scholar]
- Altman J, Bayer SA. Mosaic organization of the hippocampal neuroepithelium and the multiple germinal sources of dentate granule cells J. Comp Neurol. 1990;301:325–342. doi: 10.1002/cne.903010302. [DOI] [PubMed] [Google Scholar]
- Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:439–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- Herrup K, Kuemerle B. The compartmentalization of the cerebellum. Annu Rev Neurosci. 1997;20:61–90. doi: 10.1146/annurev.neuro.20.1.61. [DOI] [PubMed] [Google Scholar]
- Herrup K, Wilczynski S. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience. 1982;7:2185–2196. doi: 10.1016/0306-4522(82)90129-4. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. HLH proteins, fly neurogenesis, and vertebrate myogenesis. Cell. 1993;75:827–830. doi: 10.1016/0092-8674(93)90525-u. [DOI] [PubMed] [Google Scholar]
- Lee JE. bHLH genes in neural development. Curr Opin Neurobiol. 1997;7:13–20. doi: 10.1016/s0959-4388(97)80115-8. [DOI] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lippnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Miyata T, Nakajima K, Aruga J, Takahashi S, Ikenaka K, Mikoshiba K, Ogawa M. Distribution of a reeler gene-related antigen in the developing cerebellum: An immunohistochemical study with an allogeneic antibody CR-50 on normal and reeler mice. J Comp Neurol. 1996;372:215–228. doi: 10.1002/(SICI)1096-9861(19960819)372:2<215::AID-CNE5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Mutoh H, Naya FL, Tsai MJ, Leiter AB. The basic helix-loop-helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes & Dev. 1988;12:820–830. doi: 10.1101/gad.12.6.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Stellrecht CMM, Tsai J-J. Tissue-specific regulation of the insulin gene by a novel basic helix–loop–helix transcription factor. Genes & Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Huang H-P, Qui Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai M-J. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD-deficient mice. Genes & Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L, Elwood NJ, Elefanty AG, Kontgen F, Li R, Barnett LD, Begley CG. The scl gene product is required for the generation of all hematopoietic lineages in the adult mouse. EMBO J. 1996;15:4123–4129. [PMC free article] [PubMed] [Google Scholar]
- Ross ME, Fletcher C, Mason CA, Hatten ME, Heintz N. Meander tail reveals a developmental unit in mouse cerebellum. Proc Natl Acad Sci. 1989;87:4189–4192. doi: 10.1073/pnas.87.11.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL, Goldman RD, Leinwand LA. Cells: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Vicario-Abejon C, Cunningham MG, McKay RD. Cerebellar precursors transplanted to the neonatal dentate gyrus express features characteristic of hippocampal neurons. J Neurosci. 1995;15:6351–6363. doi: 10.1523/JNEUROSCI.15-10-06351.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell T, Turner D, Rupp R, Hollenberg S. The myoD gene family: Nodal point during specification of the muscle cell lineage. Science. 1991;251:761–766. doi: 10.1126/science.1846704. [DOI] [PubMed] [Google Scholar]
- Wood KA, Dipasquale B, Youle RJ. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- Yang XW, Zhong R, Heintz N. Granule cell specification in the developing mouse brain as defined by expression of the zinc finger transcription factor RU49. Development. 1996;122:555–566. doi: 10.1242/dev.122.2.555. [DOI] [PubMed] [Google Scholar]
- Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell. 1994;79:875–884. doi: 10.1016/0092-8674(94)90076-0. [DOI] [PubMed] [Google Scholar]