

Abstract
Nanoporous gold (NPG), made by dealloying low carat gold alloys, is a relatively new nanomaterial finding application in catalysis, sensing, and as a support for biomolecules. NPG has attracted considerable interest due to its open bicontinuous structure, high surface-to-volume ratio, tunable porosity, chemical stability and biocompatibility. NPG also has the attractive feature of being able to be modified by self-assembled monolayers. Here we use scanning electron microscopy (SEM) and atomic force microscopy (AFM) to characterize a highly efficient approach for protein immobilization on NPG using N-hydroxysuccinimide (NHS) ester functionalized self-assembled monolayers on NPG with pore sizes in the range of tens of nanometres. Comparison of coupling under static versus flow conditions suggests that BSA (Bovine Serum Albumin) and IgG (Immunoglobulin G) can only be immobilized onto the interior surfaces of free standing NPG monoliths with good coverage under flow conditions. AFM is used to examine protein coverage on both the exterior and interior of protein modified NPG. Access to the interior surface of NPG for AFM imaging is achieved using a special procedure for cleaving NPG. AFM is also used to examine BSA immobilized on rough gold surfaces as a comparative study. In principle, the general approach described should be applicable to many enzymes, proteins and protein complexes since both pore sizes and functional groups present on the NPG surfaces are controllable.
Introduction
Immobilization of proteins from solution onto functionalized surfaces has attracted tremendous amounts of attention, primarily due to the great interest in the development of biosensor devices, such as those for point-of-care diagnosis, and also for applications such as in bioreactors or biofuel cells.1-5 Self-assembled monolayers (SAMs) of organosulfur compounds on gold surfaces have been widely used to link proteins and other large biomolecules to gold surfaces, including both those of flat gold and of gold nanoparticles, through a variety of conjugation strategies.6-9 Nanoporous gold (NPG) presents new opportunities as a support for biomolecular immobilization for a range of applications, and its modification can also be achieved by conjugation to SAM modified NPG surfaces.10 A number of important questions concerning the efficiency of coupling of proteins to NPG and the distribution of immobilized proteins achieved on NPG under different conditions remain to be answered in order to place the possible applications of biomolecules supported on NPG on a stronger foundation. Studies are clearly needed that can examine the distribution of protein on the internal surfaces of NPG.
The influence of surface morphology is an important aspect in the study of protein adsorption or immobilization. However, the number of analytical techniques available to date that have the capability to visualize individual immobilized proteins are few, and some are beyond the reach of many academic labs. In this study, protein immobilized on NPG is characterized using atomic force microscopy (AFM). The atomic force microscope is ideally suited for characterizing nanometre scale features. It offers the capability of 3D visualization and provides both qualitative and quantitative information on many physical properties including size, morphology, surface texture and roughness. Statistical information, including size, surface area, and volume distributions, can be determined as well. Further, a wide range of specimen sizes can be characterized in the same scan, from 1 nanometre to 100s of nanometres, in air, vacuum or under experimental media, such as, a buffer solution for biological specimens.
Microscope images, such as those provided by AFM, are essential in research and development projects and can be critical when troubleshooting the packing quality of SAMs used as platforms for protein immobilization and for understanding the surface density and presentation of the proteins. AFM has several advantages for characterizing biological specimens such as proteins on surfaces. AFM allows one to study native protein without the need for any labeling or staining, in a variety of possible environments, and at nanometre scale resolution. With AFM, one can also follow dynamic changes of proteins on surfaces in response to introduction of other molecules or changes in the aqueous environment using AFM flow cell techniques.11-13
In recent decades, flat gold surfaces have most commonly been used for thiol-gold SAM preparation.8 However, recently there has been a surge of interest in functionalizing nanoporous materials for applications in catalysis, separations, and analysis.14,15 This interest has includes the emerging focus on NPG, mainly due to its large surface area and high degree of porosity, in addition to its possible role as an active catalyst itself.16 A number of studies have reported on the stability, high in-plane conductivity, and biocompatibility of NPG.17-22
Nanoporous gold is, as the name implies, gold which contains randomly directional and interconnected pores spanning the material, with these pores having lateral dimensions on the order of several nanometres to a few hundreds of nanometres depending on preparation and post-preparation annealing steps.22 In general, NPG is prepared from low carat gold alloys containing 20% to at most 50% gold via a de-alloying process in which the less noble elements are oxidized and diffuse out of the material. De-alloying is achieved either by exposure of the precursor alloy to a strong acid or to a sufficiently positive electrochemical potential.10,18 Due to its nanometre scale pore size and ligament framework, by which we mean its interconnected structure of Au ligaments each on the order of 10 to a few 100 nm in size (depending on the processing parameters),10 NPG possesses many potential applications, such as direct use as an oxidation catalyst when the average ligament size is sufficiently narrow,19,23 use as a catalyst support, as a platform for enhanced chemical sensing,24 as a gas storage medium,25 support for iterative synthesis,26 support for surface-enhanced Raman spectroscopy,17,27 support for localized or propagating surface plasmons,28 and other emerging applications. By convention, the spaces between the interconnected ligaments are referred to as pores, although they are not necessarily cylindrical in shape. When the pore and ligament framework reaches hundreds of nanometres, it becomes useful in microfluidic flow control, because the increased surface area to volume ratio greatly enhances the chemical reactivity and efficiency.29
In recent years, the use of NPG to construct electrochemical sensors and biosensors has attracted the interest of analytical chemists.10,21,30 NPG appears to be an attractive high surface area support for biomolecules due to its chemical stability, ease of modification, and apparently adequate pore sizes for accommodating proteins. A number of successful uses of NPG as a support for biosensor development or for assay development have been reported. In 2010, Chen et al., took advantage of the unique physical and chemical properties of nanoporous gold, and adapted NPG as a supporting matrix for immobilizing a redox protein hemoglobin (Hb) to develop a high-performance hydrogen peroxide biosensor. Using optimized cyclic voltammetry (CV) and chronoamperometry parameters, the detection of H2O2 was achieved over a range from 5 × 10−8 to 2 × 10−4 M with a detection limit of 2 × 10−8 M (based on S/N = 3).31
The unique architecture of NPG provides many possibilities for those who envision manufacturing nanobio-devices. However, in order to take advantage of the high surface to volume ratio of NPG to improve immobilization efficiency or electrocatalytic activity, it is important to understand the presentation and the distribution of proteins on the surfaces present throughout functionalized NPG materials. The porosity and curvature of the NPG structure introduces additional complexities beyond those of a flat Au surface. While other methods such as those based on solution depletion and supernatant analysis can provide estimations of the net amount of protein included in an NPG sample, they do not provide information on the distribution of immobilized proteins. In this paper, we report that monolithic NPG samples can be mechanically cleaved and thus their internal surfaces exposed for AFM examination. In this paper, the immobilization of two common proteins, bovine serum albumin (BSA) and immunoglobulin G (IgG) on NPG samples using AFM is reported. The ligament dimensions of NPG used here are typically in the range of 50–100 nm diameter and are larger than the dimensions of these proteins (4–15 nm), and thus individual protein features can be seen by AFM even in light of tip-surface convolution effects. In our study, we examine covalent protein immobilization under both static and flow-through conditions and image both external and internal NPG surfaces. To our knowledge, AFM visualization of proteins on NPG has thus far not been reported.
Experimental section
Compounds for SAM formation and activation
Lipoic acid (thioctic acid) and 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (referred to as EDC, purity > 99.0%) were purchased from Sigma Aldrich (St. Louis, Missouri, USA). N-hydroxysuccinimide (referred to as NHS) with a purity of more than 97.0% was purchase from Fluka (Milwaukee, Wisconsin, USA). HPLC grade ethanol solvent was purchased from Sigma Aldrich (St. Louis, Missouri, USA). Lipoic acid was dissolved in HPLC grade ethanol at a concentration of 0.5 mM. Solutions of EDC and NHS were each prepared separately in acetonitrile (99.9% purity, Fisher Scientific, Philadelphia, USA) at final concentrations of 5.0 mM. All chemicals and reagents were used as received.
Protein solutions
Bovine serum albumin (referred to as BSA, 98.0% purity) and rabbit immunoglobulin G (referred to as IgG, from rabbit serum, greater than 95.0% purity) were purchased from Sigma Aldrich (St. Louis, Missouri, USA) and used as received. The protein solutions, such as BSA (1.0, 5.0, 10.0 and 20.0 μg ml−1) and rabbit IgG (5.0 μg ml−1), were prepared in sodium phosphate buffer (pH 7.0, final ionic strength of 10.0 mM) following the protocol from New England Biolabs (Ipswich, Massachusetts, USA).
In order to achieve protein immobilization, the relatively high concentrations of BSA or IgG (as listed above) were used so as to provide an excess of protein in solution available to react with the activated functional groups on the gold surfaces. Further, NHS-ester functionalized rough gold and nanoporous gold was characterized using AFM without protein as a control experiment. A minimal amount of adsorption was observed in this blank test, confirming that the spots of greater intensity in the AFM topographs are due to protein attachment. All samples were thoroughly rinsed with 1% Tween 20, and Milli-Q water, before images were acquired using AFM.
Protein structures
The structure of BSA was obtained from Protein Data Bank (ID code 1E7I) and is shown in Fig. 1A. To highlight the lysine groups in BSA, the molecular graphics images were produced using the University of California, San Francisco (UCSF) Chimera package from the Resource for Biocomputing, Visualization, and Informatics at UCSF (supported by NIH P41 RR-01081).32 According to Ortiz et al., BSA (MW = 66382 Da) is a prolate ellipsoid (E-form conformation) with typical dimensions of 4.0 nm × 4.0 nm × 14.0 nm (a = b < c).33-35 The serum albumin protein undergoes reversible conformational isomerization with changes in pH. At pH around 8, BSA adopts an N-form conformation, with typical dimensions of 3.0 nm × 8.0 nm × 8.0 nm.35,36 For our study, the most important features are the lysine (Lys) residues (highlighted in green in Fig. 1) for covalent immobilization onto activated N-hydroxysuccinimide ester terminated SAMs on gold.37 Since Lys is the most abundant on the periphery of protein, for simplicity, we will focus only on the Lys residues on the protein surface for conjugation. Of the 607 residues in BSA, there are 58 lysines at various locations on the protein surface. Similarly, by using the (UCSF) Chimera image rendering software, the 3D space filling model of IgG (protein data bank ID code: 1IgG) is presented in Fig. 1B. The Y-shaped IgG is composed of two antigen-binding fragments, Fab, and a crystallizable fragment, Fc. Both fragments are heavily decorated with lysine groups (highlighted in green in Fig. 1B), available for covalent immobilization onto activated NHS ester termini of SAMs. The typical dimensions of IgG are approximately 4.0 nm × 8.5 nm × 14.5 nm.38 There are 83 lysine groups per IgG.
Fig. 1.
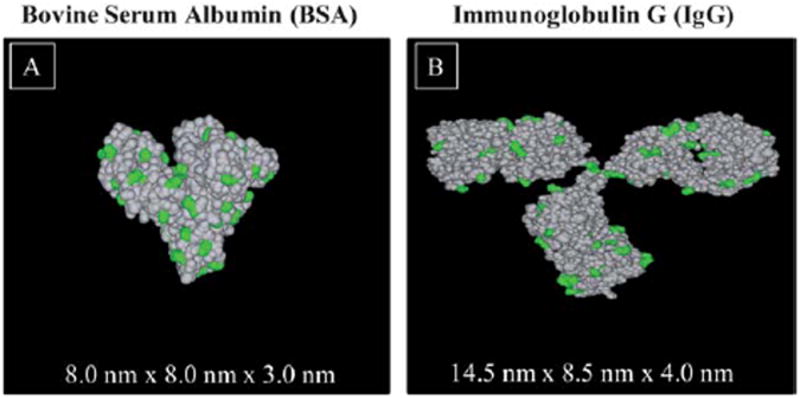
3D space filling models of protein structures revealing lysine residues. (A) N-form conformation of bovine serum albumin (BSA), and (B) immunoglobulin G (IgG). The lysine (Lys) residues are highlighted in green.
Atomic force microscopy imaging and analysis
The AFM imaging was performed in Tapping Mode™ with a Multimode™ AFM (Veeco, Santa Barbara, California, USA) operating with a Nanoscope IIIa™ controller. We used TAP300G (Budget Sensors, Sofia, Bulgaria) silicon cantilevers with a resonance frequency of about 300 kHz, and a spring constant of 40 N/m. The scan frequency was typically 1.5 Hz per line and the modulation amplitude was a few nanometres. We used a first or second order polynomial function to remove the background slope. The proteins (either BSA or IgG) were measured quantitatively from more than 30 cursor profiles per image to obtain sufficient statistics.
Preparation of free-standing nanoporous gold plates
Preparation of free-standing nanoporous gold plates was performed according to literature procedures, with a few modifications. 10,18 In brief, 10 carat white gold sheets (4.0 inch × 2.0 inch × 0.0098 inch, L × W × H, respectively) were purchased from Hoover and Strong (Richmond, Virginia, USA). The stated atomic composition of this commercial alloy is 41.8% Au, 5.0% Ag, 30–35% Cu, 8–9% Zn and 15–20% Ni. The 10 carat sheet was cut to a size of 2.0 mm × 2.0 mm × 0.25 mm, L × W × H, respectively, and placed in a concentrated nitric acid bath for 48 h (the acid solution was refreshed at the 24 h time point). The sample size was specifically chosen for convenience of handling during protein incubation and for fitting into a standard AFM sample holder. After the nitric acid de-alloying treatment, the sample was rinsed thoroughly with Milli-Q water (18.2 MΩ, Millipore Corporation, Boston, USA) to neutral pH, follow by rinsing with HPLC grade ethanol (Sigma Aldrich, St. Louis, Missouri, USA). The microstructure of NPG was characterized with AFM and also by scanning electron microscopy using a JEOL JSM-6320F field emission SEM.
Preparation of rough gold surfaces
The rough gold films used in this study were prepared by thermal evaporation of high-purity ~99.99% gold wire (Electron Microscopy Sciences, Hatfield, Pennsylvania, USA) wrapped around a small tungsten wire basket in a conventional Denton vacuum thermal-evaporator (Model DV502A, Denton Vacuum, Moorestown, New Jersey, USA) onto the surfaces of small cut pieces of Si-wafer (Nova Electronic Materials, Flower Mound, Texas, USA) which were first coated with a nichrome adhesion layer by thermal evaporation of nichrome wire (Alfa Aesar, Ward Hill, Massachusetts, USA). The chamber pressure during evaporation was less than ~5 × 10−6 torr and was achieved using a diffusion pump with a liquid nitrogen trap.
Preparation of self-assembled monolayers
The SAM preparation used in this study follows established procedures.10,18,39 The nanoporous gold template used in static immobilization experiments was transferred to lipoic acid solution (0.5 mM, in ethanol) for 24 h. The EDC (5.0 mM) and NHS (5.0 mM) solutions were prepared separately in acetonitrile. The freshly prepared acid-terminated alkyl surface was allowed to react with EDC and NHS, sequentially for 1 h at room temperature. The resulting NHS ester-terminated surface was copiously rinsed with water and PBS buffer before coupling with proteins. In the case of experiments using NPG in a flow cell, as explained in detail below, the lipoic acid in ethanol solution was flowed through the NPG plate for one hour to achieve SAM formation. For the studies on rough gold surfaces, one set of experiments used only the EDC activation step followed directly by rinsing and exposure to the protein solution. The second set of experiments on the rough gold surfaces used both the EDC activation and substitution by NHS prior to rinsing and exposure to the protein solution.
Energy dispersive X-ray analysis
Energy dispersive X-ray analysis was introduced to confirm the presence of protein. Proteins mainly consist of hydrogen, carbon, nitrogen and oxygen. Carbon compounds likely to be present as laboratory contaminants can be detected using EDS by examining the spectrum for a peak near 0.28 KeV. In contrast, observing a nitrogen peak, near 0.39 KeV, in a typical EDS spectrum due to random laboratory contamination is much less likely, and observation of such a peak can be attributed to the presence of protein in these experiments. We have captured and analyzed over 300 EDS spectra from different regions of the NPG with and without immobilized BSA. The EDS spectra (representative EDS spectrum) for NPG activated NHS-ester with and without proteins were rendered in an Excel spread sheet for comparison. Original EDS spectra (captured using Link Pentafet 6886 Model, Oxford Microanalysis Group, UK) are available in the supplementary information, Figure S1.†
Results and discussion
Covalent immobilization of bovine serum albumin on rough gold surfaces
The structure of BSA (3.0 nm × 8.0 nm × 8.0 nm), as obtained from the Protein Data Bank, is shown in Fig. 1A.35-37 Since Lys is the most abundant on the periphery of protein, for simplicity, we will focus only on the Lys residues on the protein surface for conjugation. Of the 607 residues in BSA, there are 58 lysines at various locations on the protein surface. Similarly, the 3D space filling model of IgG (4.0 nm × 8.5 nm × 14.5 nm) is presented in Fig. 1B.38 There are 83 lysine groups per IgG, available for covalent immobilization onto activated NHS ester termini of SAMs.
In order to prove that proteins immobilized on rough gold surfaces could be successfully imaged using tapping mode AFM (TM-AFM), BSA was covalently linked to EDC-activated lipoic acid SAMs on specially prepared rough gold films supported on silicon.
We observed that the irregular corrugations (grooves and ridges) of rough gold films (9.7 ± 0.2 nm, root-mean-square measurement of surface roughness determined from AFM images) on silicon are a close mimic to the rough surface of nanoporous gold (13.8 ± 4.8 nm, root-mean-square measurement of surface roughness determined from AFM images) formed by ligaments and pores. As illustrated in Fig. 2, the widely utilized covalent protein immobilization method through amide bond formation by reaction of amines on exposed lysines with NHS activated esters on the gold surface was employed.11,40-42 In brief, a carboxylic acid terminated self-assembled monolayer (SAM) was prepared by dipping the rough gold substrate in lipoic acid (0.5 mM/ethanol) for a duration of 1 h. The SAM modified gold surface was then reacted with 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC, 5.0 mM in acetonitrile). EDC reacts with a carboxyl group to form an active ester, an O-acylisourea. In general, covalent attachment of protein to the EDC reactive surface is possible.10,18 However, the EDC active surface is unstable to hydrolysis. To overcome this limitation, the use of NHS allows the conversion of the O-acylisourea intermediate into a relatively stable amine reactive NHS-ester. This NHS ester can then cross-link with amine group on the protein by forming a covalent amide bond.42
Fig. 2.
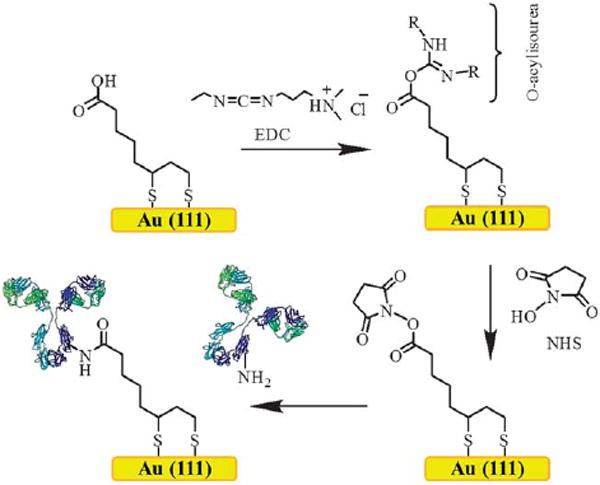
General strategy for protein immobilization on gold surfaces via the EDC/NHS coupling mechanism using a lipoic acid SAM. To tether a protein, the surface of the gold is first functionalized using a self-assembled monolayer of lipoic acid. The EDC will react with a carboxyl group to form an amine-reactive intermediate, an O-acylisourea. The use of NHS allows the conversion of the O-acylisourea intermediate into a relatively stable but reactive NHS-ester to cross-link with an amine group on the protein by forming a covalent amide bond.
Fig. 3 shows EDC/lipoic acid functionalized rough gold surfaces before and after exposure to the protein BSA, captured using TM-AFM. The EDC functionalized rough gold surfaces, Fig. 3A, before exposure to protein were imaged as a control experiment. Negligible adsorption was observed in this blank test image and serves to confirm that the numerous brighter spots in the AFM topographs shown in Fig. 3B, 3C and 3D, are due to protein attachment.
Fig. 3.

Tapping mode-AFM topographs of the EDC functionalized rough gold surface after adsorption of bovine serum albumin. (A) 2000 nm × 2000 nm, EDC treated rough gold surface without exposure to BSA. Similarly, 2000 nm × 2000 nm, AFM topographic images of EDC functionalized rough gold surface after exposure to 1.0 μg ml−1 BSA, for (B) 1 min, (C) 10 min, and (D) 15 min. (E–F) Higher resolution, AFM topographic images, 500 nm × 500 nm, of the areas indicated by the boxes in (A–D), respectively. The sample was thoroughly rinsed with 1% Tween 20, and Milli-Q water, before images were acquired using AFM.
The protein coverage on the EDC functionalized rough gold surfaces appeared to depend on the exposure time when subjected to similar immobilization conditions, specifically 1.0 ml of 1.0 μg ml−1 BSA in 1x PBS buffer (pH 7.0 at 25.0 °C) for 1 min (Fig. 3B), 10 min (Fig. 3C) and 15 min (Fig. 3D). From Fig. 3B, 3C and 3D, distinctive brighter spots decorated on the surface of the rough gold were apparent when compared to the control experiment in Fig. 3A.
Based on the observed protein coverage on the surface of rough gold, the extent of protein BSA adsorption is clearly impacted by the incubation time. While there is no obvious protein adsorbed on the control surface, coverage of approximately 1228 (8 uncertain) protein/μm2 (meaning that an additional ~8 surface features per μm2 possibly due to proteins present in aggregates or resulting from tip convolution are not included, see supplementary information for a discussion of our protein counting strategies†) was observed on surfaces as shown in Fig. 3F. For the rough gold surface after exposure to BSA for 10 and 15 min, surface coverages of 1476 (20) and 1644 (28) protein/μm2 were obtained, as shown in Fig. 3G and 3H, respectively. This study clearly indicated that protein BSA coverage is enhanced by longer incubation times by at least 25%, based on protein coverage, comparing results obtained using 15 min versus 1 min adsorption time. Based on extensive prior literature, when working with a non-flat surface, a correction (roughness) factor should be applied to correct for the surface area gained as a result of roughness that is equal to the ratio of the actual surface area to the projected surface area. The direct usage of AFM to determine this correction factor was developed by Harris et al. in 2001, and is applied here by using the total distance the tip moved over the rough gold surface compared to the lateral distance of the AFM scan, and averaging over many line scans.43 The roughness factor obtained for our rough gold is 2.5 based on multiple analyses. Upon taking the roughness factor into account, we can normalize the protein coverage on the rough gold surface by dividing the protein coverage by 2.5. Thus, protein coverages of approximately 491 (3), 590 (8) and 658 (11) protein/μm2 were obtained respectively for 1, 10 and 15 min incubation.
We also explored BSA immobilization on rough gold surfaces through conjugation of the NHS ester activated SAM with the amine groups on the proteins. Fig. 4, shows the NHS active ester functionalized rough gold surface before and after exposure to the protein BSA, captured using TM-AFM.
Fig. 4.

Tapping mode-AFM topographs of the NHS functionalized rough gold surface after adsorption of bovine serum albumin. (A) 2000 nm × 2000 nm, NHS treated rough gold surface without exposure to BSA. Similarly, 2000 nm × 2000 nm, AFM topographic images of NHS active esters rough gold surface after exposure to 1.0 μg ml−1 BSA, for (B) 1 min, (C) 10 min, and (D) 15 min. (E–H) Higher resolution, AFM topographic images, 500 nm × 500 nm, of the areas indicated by the boxes in (A–D), respectively. The sample was thoroughly rinsed with 1% Tween 20, and Milli-Q water, before images were acquired using AFM.
The topograph of the lipoic acid SAM modified rough gold treated with EDC/NHS before exposure to BSA was captured to serve as a reference to the rough gold surface after exposure to protein, Fig. 4A. The protein immobilization conditions used were similar to those used for conjugating BSA to EDC functionalized rough gold surfaces as shown in Fig. 3. From Fig. 4B (1 min exposure), 4C (10 min exposure), and 4D (15 min exposure), distinctive brighter spots decorated on the surface of the rough gold are apparent when compared to the control experiment in Fig. 4A.
The protein coverage on the NHS active esters rough gold surfaces was also found to be dependent on the protein exposure time when subjected to similar immobilization conditions. Surface coverages of 820 (12), 888 (24), 996 (32) protein/μm2 were obtained from Fig. 4F, 4G, and 4H, respectively. Accounting for the roughness factor of 2.5, coverages of 328 (5), 355 (10), 398 (13) protein/μm2 are obtained. The numbers of proteins were counted using the following dimension as the threshold: 35.0 ± 5.0 nm lateral width dimension based upon individual bright spots imaged under AFM (see supplementary information for discussion of protein coverage analysis†). This threshold is determined from bright spot capture in the low coverage region and is consistent with multiple repeated experiments. Due to the rough corrugation of the gold substrate, this threshold is used as a qualitative guide for counting proteins, as the irregular morphology of the underlying gold substrate or sharpness of the AFM probe tip may impact the lateral widths of the bright spots.
The observations of Fig. 3 and 4 confirm that both direct EDC and EDC/NHS treatment of the lipoic acid SAMs on rough gold can successfully result in protein immobilization. In the case studied here, the protein coverage obtained was somewhat higher for the direct EDC treatment than for the sequential treatment with EDC followed by NHS. Successful coupling of proteins to lipoic acid SAMs directly after treatment with EDC in acetonitrile has been reported previously.18,44-49 As depicted in Fig. 2, EDC reacts with carboxylic acid groups to yield an O-acylisourea, which is highly susceptible to the nucleophilic attack by lysine required for formation of an amide bond.50 In aqueous media, the O-acylisourea will be prone to rapid hydrolysis; however, it will be much more stable in acetonitrile. The subsequent treatment with NHS gives the NHS-ester intermediate, which is more stable in aqueous media, but is less reactive than the O-acylisourea intermediate and also somewhat larger in size.42,51 The hydrolysis rates will be different for these intermediates present in solution versus immobilized on a surface, and will also depend on pH and,52 if immobilized, on the structure of the surface monolayer as it impacts their reactivity/ accessibility.53 The half-life for hydrolysis of EDC in the presence of acetate has been reported to be pH dependent, with the rate 8x faster at pH 4 than at pH 7, and with the hydrolysis half-life being 806 s (3 mM EDC, 50 mM acetate, pH 7).52 It is known that the NHS-ester intermediate is prone to side reactions to form byproducts such as an anhydride or an N-acylurea.42,51 A study of the hydrolysis of NHS-ester terminated SAMs found that the hydrolysis rate can be as much as 1000 times slower for the monolayer than for analogous compounds in solution.54 The hydrolysis of the NHS-ester of mercaptohexadecanoic acid SAMs was found to have a half-life of 1700 s upon exposure to 0.01 M NaOH (pH 12), the half-life being 125 s for the analogous mercaptoacetic acid derivative.53 These half-lives would undoubtedly be much longer near pH 7. The half-life for hydrolysis of the O-acylisourea intermediate should also be expected to be much longer in the SAM environment than it would be free in solution, although a detailed study does not seem available in the literature. The exposure times used here for direct reaction of the EDC-activated rough gold surface with protein are apparently short enough that good protein coverage is obtained as a consequence of high EDC reactivity promoting amide bond formation prior to loss of EDC from the surface by hydrolysis. The addition of the second step of NHS treatment may reduce the conjugation yield to protein if the NHS-activated surface is more prone to side reactions or is simply less reactive during our selected times of protein exposure. The EDC mediated approach has also been used extensively in diverse applications to form amide bonds in peptide synthesis, in the labeling of nucleic acids through 5′ phosphate groups, and in creating amine-reactive NHS-esters of biomolecules, amongst other applications.55,56
In both approaches, the activated ester groups are accessible for reaction with lysine residues on the protein. This could be aided by the roughness of the surface contributing additional disorder into the SAM packing and making the activated ester groups more accessible. The blocking capacity towards heterogeneous electron transfer from cytochrome c across carboxylic acid terminated SAMs (14-mercaptotetradecanoic acid and 11-mercaptoundecanoic acid) on gold surfaces of varying roughness factor (r) was compared by Bowden for evaporated Au on glass (r = 2.1), Au foil (r = 1.5), single crystal Au (r = 1.1), and Au epitaxially deposited on mica (r = 1.0).57 In this study, it was found that blocking capacity and hence SAM integrity lessened as roughness increased. It has been reported that lipoic acid SAMs on flat Au(111) can have fairly disordered structures, and thus are possibly even more disordered on rough gold.58-60 The footprint of the protein on the surface will clearly cover many individual molecules of the SAM. Increases in both the surface density of the activated ester groups and their accessibility/reactivity should contribute to enhanced protein coverage. In theory, a more disordered and open presentation of O-acylisourea (or NHS ester) groups will facilitate reaction with amine groups on the peripheral lysines of the BSA protein. We hypothesize that the packing of the O-acylisourea (or NHS ester) groups on the surface of the rough gold could adopt many configurations. For example, (i) when the local radius of curvature of the gold surface is high, the chain density and spacing between O-acylisourea (or NHS ester) groups will increase with distance away from the surface. In other words, on higher curvature gold surfaces, the spacing between ω-functionalized end-groups increases when compared to the spacing between these groups if the SAM was on a flat gold surface, and the surface is able to accommodate more end-groups per unit area.61-63 For example, examination of PEG-lipoic acid conjugates on Au nanoparticles surfaces found that on smaller diameter nanoparticles, the PEG coverage density was increased due to curvature effects; that is, the smaller the AuNP, the denser its PEG layer.64 The curvature should also serve to decrease van der Waal’s interactions between neighboring molecules and disrupt the molecular packing, serving to make the reactive end-groups more mobile and accessible.65 (ii) On the other hand, if the radius of curvature of the rough gold substrate is moderate to low, the effect of the curvature on SAM structure will likely be diminished or negligible. The packing of the thiolated molecules probably shows no major differences between planar SAMs and SAMs on regions of rough substrates where the curvature is low, such as the faces of larger sized gold grains. (iii) The rough gold surfaces studied herein will present a distribution of regions of different degrees of curvature. While some of these regions will be of low curvature and present SAMs more similar to those of flat gold, other regions will present less-ordered SAMs and hence more accessible reactive end-groups (in this case, either O-acylisourea or NHS ester) thus facilitating the immobilization of a protein such as BSA through reaction with peripheral lysines and resulting in higher coverage. Our roughness factor of 2.5 for gold evaporated onto Si is close to that of 2.1 reported by Bowden for Au evaporated onto glass slides, for which the ability of SAMs of 14-mercaptotetradecanoic acid to block electron transfer from cytochrome c was severely compromised. Since EDC (or NHS) are zero length crosslinking agents, they are ideal for studying the direct conjugation of surface carboxyl termini in the SAM to the primary amines on the protein surfaces. Use of EDC or NHS removes factors accompanying the use of longer length linkers (such as polyethyleneglycol chains, or extended bifunctional linkers) including their influence on protein coverage and non-specific binding. The EDC/NHS conjugation strategy, in which both reagents are applied in a buffer at a carefully selected ratio and overall concentration of reagents, is a more commonly used strategy than that of using EDC alone. Since diffusion and reaction times will be longer for NPG monoliths exposed to protein solution and given that the NHS activated esters are more stable over longer time periods, we focus on usage of EDC/NHS for the remainder of this study concerning protein immobilization onto NPG.
Covalent immobilization of bovine serum albumin on nanoporous gold
The utilization of the rough gold substrates for protein immobilization and characterization served to set a foundation, to provide a proof of concept, and enable optimization of experimental conditions prior to the exploration of protein immobilization on nanoporous gold. The procedures and reaction conditions were then transferred to the studies on nanoporous gold. In consideration of the internal complexity of nanoporous gold and the need for much longer protein incubation times, only NHS active ester surfaces were used. The activation of lipoic acid SAMs on nanoporous gold with NHS active esters was performed following established procedures reported elsewhere.6,7,66
The nanoporous gold was prepared by de-alloying cut pieces (2 mm × 2 mm × 0.25 mm, L × W × H, respectively) of commercially available 10 K white gold plates. After de-alloying and thorough rinsing/washing, these NPG plates were first functionalized with lipoic acid, then with EDC, and finally with NHS. The nanoporous gold topographic features were captured using AFM as shown in Fig. 5A. The small size of the ligands (lipoic acid, EDC, and NHS) does not affect the observed topography. As previously noted by our lab, NPG surface morphology appears different under AFM observation than that observed using SEM observation (see Figure S3 in supplementary information for SEM image of NPG†), and this is not unexpected due to the fundamentally different imaging contrast mechanisms.10 Due to the convolution effects arising from tip-surface interactions, the pores appear more as creases or dark crevices in the images. The bumpy nature of the ligaments and the overall surface roughness is also sensed in a manner that is not revealed by the SEM image of the NPG surface.
Fig. 5.

Tapping mode-AFM topographs and amplitude images showing the exterior of the NHS functionalized nanoporous gold surfaces after conjugation of BSA. Top panels represent the amplitude images; lower panels represent the topographic images of the same areas as in top panels. (A) 500 nm × 500 nm, NHS ester functionalized gold surface without exposure to BSA. Similarly, 500 nm × 500 nm, AFM topographic images of NHS ester functionalized gold surface after exposure to BSA (4 ml of BSA, PBS buffer pH 7.0, 24 h incubation at 4 °C), at (B) 1 μg ml−1, (C) 5 μg ml−1, (D) 10 μg ml−1 and (E) 20 μg ml−1.
To understand the adsorption behavior of the BSA protein on NPG, we subjected the NHS-ester functionalized NPG to a concentration dependence study: incubation in 4.0 ml of BSA solution for 24 h, in pH 7.0 PBS buffer solution at 4 °C, at 1.0 μg ml−1 (Fig. 5B), 5.0 μg ml−1 (Fig. 5C), 10.0 μg ml−1 (Fig. 5D) and 20.0 μg ml−1 (Fig. 5E). The sample was thoroughly rinsed with 1% Tween 20, and Milli-Q water, before images were acquired using AFM. From Fig. 5A, the corrugation of the NPG surface appears to have rounded and interconnected ligament features with a typical lateral size range from 50 to 100 nm for the rounded features and the lengths of the ligament segments can reach above 200 nm, based on the apparent dimension obtained from AFM measurement. Fig. 5B to E, represent NPG after exposure to BSA, the topographic images appear to have many small irregularly shaped features with an average dimension of 29 ± 8 nm, observed on the surface of the NPG. These features are smaller than those of the NPG ligaments of 35–200 nm and thus can generally be distinguished as topographic features. The smooth appearance of the NPG is no longer apparent; the pores and interconnected ligament features are not as clearly prominent when compared to the NPG not exposed to protein. To assist visualization of the immobilized proteins on the nanoporous gold surface, both amplitude and height (topography) images are presented. The top panels (from Fig. 5A to E) represent the amplitude images; the lower panels (Fig. 5A1 to E1) represent the topographic images of the same areas as in the top panels. Small rounded features are vividly seen and recognizable in the amplitude images panels. There are more secondary and additional rounded features seen on the NPG surfaces incubated in the protein solutions of concentration 10.0 μg ml−1 and 20.0 μg ml−1 when compared to those incubated in the protein solutions of 1.0 μg ml−1 and 5.0 μg ml−1 BSA concentration.
In addition to protein adsorption onto the exterior NPG surface, we were interested in knowing the diffusion depth of the protein into the interior of the NPG. Thus the same samples used for concentration dependence studies, as shown in Fig. 5, were physically removed from the AFM scanner. Examination of the interior is made possible by the fact that we have discovered that macroscopic NPG samples can be cleaved. The top layer (at various depths, for repeated data acquisition on a same sample) of the sample is then physically peeled off from the NPG substrate with the assistance of a conventional double sided tape. To ensure the experimental results are consistent, multiple regions of the same sample were characterized, and similar results were observed. Fig. 6A, showing a region of 7 μm × 7 μm, represents the exterior of the NHS-ester functionalized NPG captured using AFM. Although the NPG morphology can be seen in Fig. 6A, it is not highly pronounced and the sample is relatively smooth when compared to the interior of the NPG, seen in Fig. 6B. The irregular corrugations or terrain like features are noticeable and expected as a consequence of the mechanical cleaving procedure. It is noteworthy to mention that when characterizing protein adsorbed onto the inner NPG surfaces, at higher resolution, such as 500 nm × 500 nm, the effect of additional corrugation of the interior surface of the NPG samples does not hinder AFM data acquisition. Flat plateaus ranging from hundreds of nanometres to a few microns in size can be located by systematically zooming in from a large scanned area. To determine the depth of a typical peeled NPG substrate, SEM was utilized to acquire the cross section thickness of blank and peeled NPG (peeled only once). Fig. 6C and 6D represents the top view of typical blank and peeled NPG, respectively. The morphology of the peeled NPG appeared to be rougher, and resembles the terrain of a dry cracked mud parch pattern/texture, when compared to the exterior. Fig. 6E and 6F, are the cross-section views of the NPG. Based on SEM measurement, the thickness of a typical NPG is 333 μm and 288 μm for peeled NPG. Thus, the interior surfaces that are imaged on one-time peeled NPG are approximately 50 μm from the exterior. This distance is typical of what is obtained by our peeling procedure, at present we do not have a strategy for precisely controlling the depth at which cleavage occurs.
Fig. 6.
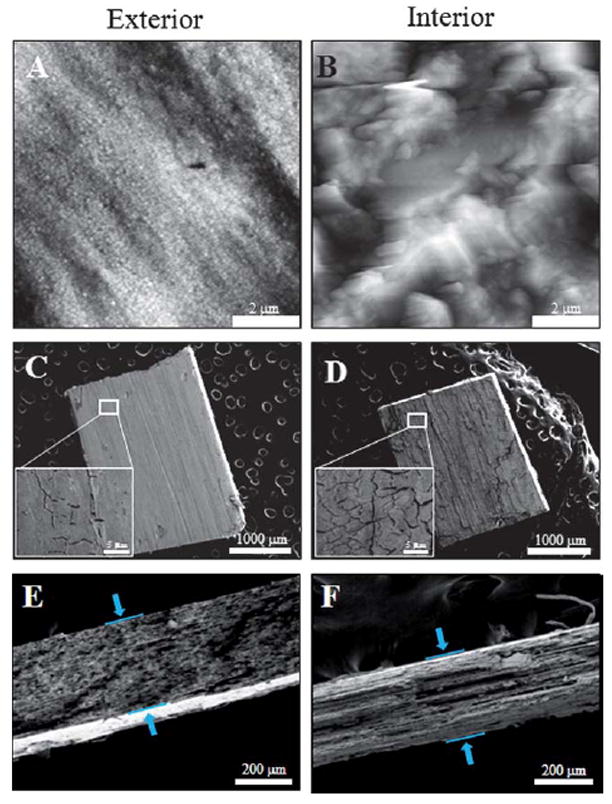
Morphology of nanoporous gold. (A) 7 μm × 7 μm, nanoporous gold before peeling, (B) after peeling and captured using AFM. (C) and (D) SEM images of the porous gold before and after peeling. The inset in (C) and (D) are 23 μm × 17 μm zoomed in images of the NPG. (E) and (F) are the side view of the porous gold before and after peeling, respectively, revealing the thickness of the gold plates.
The newly exposed inner surface of the NPG was then characterized using the same AFM probes without any modification to the sample or probes. From Fig. 7A, the image of the interior of a typical blank NPG is seen to resemble that of the topographic images of the exterior blank NPG, Fig. 5A. Similarly no protein was observed on the interior of NPG samples that were exposed to 1.0 μg ml−1, 5.0 μg ml−1, and 10.0 μg ml−1 of BSA. However, minute amounts of BSA were observed on the interior of NPG samples that were incubated in the 20.0 μg ml−1 BSA solution. This indicated that a high concentration of protein is needed to allow protein to diffuse from higher concentration regions in the external solution to the low concentration interior of NPG during the 24 h period used in these preparations.
Fig. 7.

Tapping mode-AFM topographs and amplitude images showing the interior of the NHS functionalized nanoporous gold surfaces after adsorption of BSA. Top panels represent the amplitude images; lower panels represent the topographic images of the same area as in top panels. (A) 500 nm × 500 nm, NHS ester functionalized gold surface without exposure to BSA. Similarly, 500 nm × 500 nm, AFM topographic images of NHS ester functionalized gold surface after exposure to BSA (4 ml of BSA, PBS buffer pH 7.0, 24 h incubation at 4 °C), at (B) 1 μg ml−1, (C) 5 μg ml−1, (D) 10 μg ml−1 and (E) 20 μg ml−1.
Bovine serum albumin and immunoglobulin G coupling under flow-through conditions
Based on the above reproducible experimental results, BSA immobilization onto the surface of the NHS-functionalized NPG is evident. However, the protein adsorption studies thus far are based on a static incubation method. Given that the pores of NPG range from tens to hundreds of nanometres in size and form a deep network of tortuous paths, the nano-environment may severely hinder protein diffusion throughout the interior of the NPG substrate. The possibility for hindered diffusion of the protein solution or low diffusion depths could explain why minute amount of proteins were observed at the interior of NPG under the conditions used. To overcome the apparent low penetration of protein throughout the nanoporous gold sample, a homemade flow cell system was designed and used in conjunction with a commercially available peristaltic pump (Model 77390–00, Cole-Parmer Instrument Company, Illinois, USA) equipped with polytetrafluoroethylene (PTFE) tubing. A schematic representation of the flow cell system is available in supplementary information, Figure S3.† The design allows a typical de-alloyed NPG plate, 8 mm × 8 mm × 0.25 mm, to be inserted into a sealed chamber. A typical SEM image of the dealloyed NPG is shown in Figure S3, and a typical pore size range from 50 to 100 nm is observed. To minimize damaging the brittle NPG plate or contaminating the NPG, the setup allows in situ functionalization of NPG with lipoic acid (0.5 mM / ethanol, 1 h at 1.0 ml min−1), activation using EDC (5.0 mM / acetonitrile, 1 h) and NHS (5.0 mM / acetonitrile, 1 h), followed by protein injection at 1.0 ml min −1. We anticipate that the flow cell system permits the protein solution to be forced to flow through the porous environment and hence protein can then be tethered more efficiently onto the surfaces throughout the interior of the sample.
Immobilization of BSA and IgG on NHS-ester activated NPG surfaces
Fig. 8 represent the topographic images of functionalized NPG surfaces before and after exposure to bovine serum albumin and rabbit immunoglobulin G captured using tapping mode AFM. By using the flow cell, approximately 15.0 ml of the protein solution (5.0 μg ml−1 of BSA or IgG) in 1x PBS buffer at pH 7.0 was introduced to the NHS-ester functionalized NPG and continued to circulate for a duration of 2 h at 1.0 ml min −1. Upon washing with 1% Tween 20 and Milli-Q water (18.2 MΩ) by passage through the flow cell, the surface was then imaged by AFM and is shown Fig. 8.
Fig. 8.
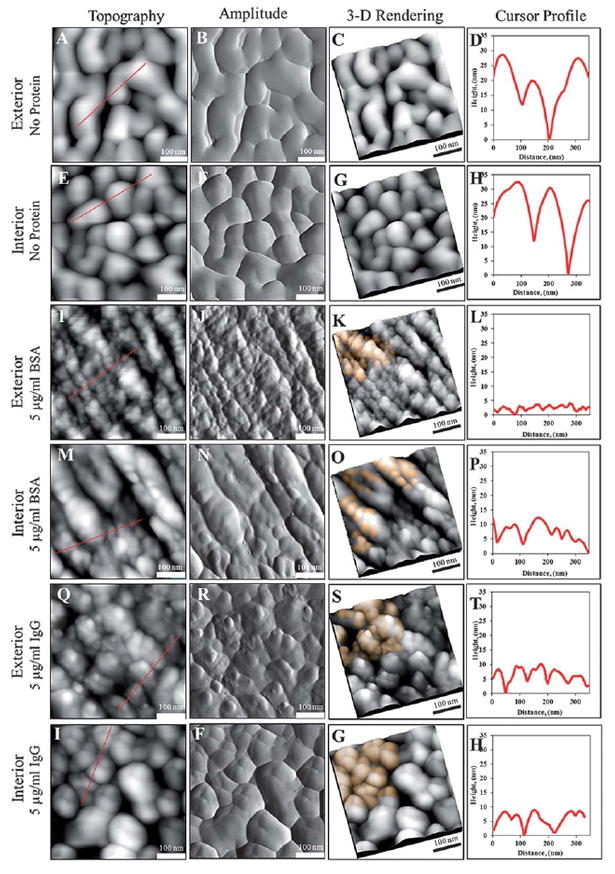
Tapping mode-AFM topographs and amplitude images showing BSA and IgG coupling under flow conditions. Column 1, 2, 3, and 4, represent topographic, amplitude, 3D rendered image of column 1, and AFM cursor profile, respectively. The flow system was utilized to assist protein passage into the nanoporous gold plate for immobilization. Showing in panel 1 and 2, are side-by-side comparison of the exterior and interior of, 500 nm × 500 nm images, NHS ester functionalized gold surface without exposure to protein. In panel 3 and 4, protein BSA immobilized on the exterior and interior is distinctive from panel 1 and 2, respectively. TheNHS ester functionalized gold surface was exposed to BSA under 15.0 ml of 5.0 μg ml−1 BSA, PBS buffer pH 7.0, 2 h incubation at 25 °C. For robustness test, protein IgG (15.0 ml of 5.0 μg ml−1 BSA, PBS buffer pH 7.0, 2 h incubation at 25 °C), was passed into the nanoporous gold plate for immobilization. Similar results were obtained; IgG was successfully immobilized on the exterior (panel 5) and interior (panel 6) of nanoporous gold surfaces using the flow system.
The influence of surface morphology is an important aspect in the study of protein adsorption/immobilization. The interior surfaces of the cleaved NPG have a different macroscopic roughness than the exterior surface. The cleaved surface presents many regions which on average are less uniformly horizontal than the exterior surface, and this slope is seen to affect the tip-surface interactions and increase the apparent dimensions of the protein features. Figure S4, in the supplementary information,† is a schematic drawing demonstrating the dependence of the feature shape on the finite tip radius and the local topography of the substrate. From panel 1 in Fig. 8, the topographic image of the external surface of NHS-ester activated NPG appears to have many irregular shape ligaments and nanopores with typical lateral dimension range from 50 to 500 nm. A similar morphology is observed for the interior of the NPG, as show in panel 2. It is also noteworthy to mention, because both metallic ligaments and nanopore channels are irregular in morphology and random in orientation, it is difficult to visually determine the sizes of nanopores and ligaments by direct measurements. Nevertheless, upon exposure of NHS-ester activated NPG to protein solution, the morphology of the NPG drastically changes as shown in panel 3, 4, 5, and 6. Panel 3 is the exterior of NPG with immobilized BSA. Many small rounded features decorated on the irregular shape ligaments are vivid and recognizable when compare directly to the NPG without protein in panel 1. These results for BSA average dimension are consistent with those previously based on Fig. 5. After peeling off approximately 50 μm of the NPG layer, the interior of the NPG also presented small rough features decorated homogeneously on the surface. The corrugation can be seen in the amplitude channel images. The AFM measurement indicates that the protein related features on the exterior NPG surface are approximately 15 ± 10 nm in lateral dimension versus 30 ± 10 nm for protein associated features on the exposed interior surface. The dimensions of 15 ± 10 nm were used as thresholds in protein counting for determining the BSA coverage on the exterior NPG surface. Considering the dimensions of the BSA molecules, 3.0 nm × 8.0 nm × 8.0 nm, coupled with lateral convolution and the curvature of the substrate, the lateral dimensions obtained from AFM measurement indicated BSA protein was successfully immobilized onto the NPG surface. The 3D rendering images in Fig. 8 K and 8O, are manually colorized to assist visualization of the immobilized protein on the NPG surfaces. Fig. 8L and 8P represent the cursor profiles of the immobilized protein BSA on the exterior and interior NPG surfaces, respectively. In theory, a wide array of proteins of a few tens of nanometre in dimension (less than the smaller nanopore dimensions) can be introduced and immobilized on NPG via the flow cell system.
To test the applicability of the flow system for covalent protein immobilization, a larger size protein IgG (in term of lateral dimension, 4.0 nm × 8.5 nm × 14.5 nm) was introduced for coupling with NHS-ester activated NPG under flow-through conditions. Panel 5 and 6 represent the exterior and interior, respectively, of the NPG after passage of IgG under similar conditions as used for BSA. Based on Fig. 8Q and 8I, the protein related features on the exterior NPG surface are approximately 25 ± 10 nm in lateral dimension versus 35 ± 10 nm for protein associated features on the exposed interior surface (see supplementary information for protein coverage analysis, Figure S2†). The dimensions of 25 ± 10 nm were used as thresholds in protein counting for determining the IgG coverage on the exterior NPG surface. The larger features observed for IgG versus BSA are consistent with the known larger dimensions of IgG and provide additional confidence that the topographic features examined are indeed immobilized proteins. The lateral dimensions obtained from AFM cursor profile measurement, appear to be reasonable when accounting for the lateral convolution and the curvature of the substrate, indicating that protein IgG was successfully immobilized onto the NPG surface. To confirm the passage of the protein solution through the NPG nano-environment, the front and back sides of the NPG sample were peeled and examined at different locations on the same sample. The IgG protein coverage, on average 368 (14) protein/μm2, was found to be similar on each peeled side after peeling, signifying that the protein immobilization is homogeneous in nature; no non-specific protein adsorption or immobilization gradient was formed inside NPG by using the flow cell system. In this direct counting determination of surface coverage, the area used is that of the scan size. It must be kept in mind that a significant portion of this surface represents pore space above which the AFM probe will not be able access interactions with proteins on ligaments deeper below the surface or on the lower portions of surface ligaments curved away from access to the tip. An estimation of the total surface area of the outer most surface layer of NPG is included in the supplementary information section.† It must also be kept in mind that the NPG surface with which the tip interacts is itself rough. This also demonstrated that the flow through method successfully forced the protein solution into the porous environment of NPG and promoted the protein conjugation reaction and formation of amide bonds with the NHS-functionalized NPG surfaces. These results are significant for applications requiring biomolecule immobilization throughout NPG samples of thicknesses in the microns range and greater. They are also of significance for applications requiring diffusion and reaction inside larger macroscopic or monolithic NPG samples. The quantitative information on surface coverage obtained by AFM for the two proteins (BSA or IgG) immobilized on the rough and porous gold surfaces is summarized in Table 1.
Table 1.
Comparison of protein surface coverages achieved on different gold substrates using different immobilization conditions
| Protein | Substrate | Activating molecule | Immobilization method | Incubation time (min) | Protein coverage (protein/μm2) |
|---|---|---|---|---|---|
| aBSA | rough gold | EDC | static | 1 | 491 (3) |
| 10 | 590 (8) | ||||
| 15 | 658 (11) | ||||
| aBSA | rough gold | NHS-ester | static | 1 | 328 (5) |
| 10 | 355 (10) | ||||
| 15 | 398 (13) | ||||
| BSA | NPG | NHS-ester | flow | 120 | 627 (36) (exterior coverage) |
| IgG | NPG | NHS-ester | flow | 120 | 368 (14) (exterior coverage) |
Protein coverages reported here on rough gold represent values accounting for the roughness factor of 2.5 found for rough gold. The values in parenthesis are the plausible numbers of additional proteins identified by image analysis as present in aggregates or possibly subject to tip convolution.
Energy-dispersive X-ray spectrometry analysis confirmation of the presence of immobilized protein on NPG
An alternate analytical approach, energy dispersive X-ray analysis, was introduced to confirm the presence of protein on NPG. This approach is simple and promising, because (1) the structures of most proteins are known from the Protein Data Bank and their stoichiometric composition can be extracted, and (2) energy dispersive X-ray analysis, also known as EDS, has detection limits of approximately 0.1% in bulk materials.67,68 Fig. 9 shows an EDS spectrum obtained using NPG (modified with a NHS-ester activated SAM) and such NPG then covalently modified with BSA. The peak in green (interior) and red (exterior) is from a control (NPG with NHS-ester SAM only), in which a nitrogen peak is clearly not observed in the region near 0.39 KeV. For simplicity, only NPG previously conjugated to BSA under flow through conditions was used for EDS analysis. From Fig. 9, a carbon peak is clearly seen at 0.28 KeV and a well-defined nitrogen peak at 0.39 KeV is evident. The increased carbon peak intensity is reasonable because proteins contain high amounts of carbon. The previously unseen nitrogen peak is seen clearly both on the exterior and interior of the NPG (blue and black lines near 0.39 KeV) after conjugation to BSA. Based on the EDS spectra, the plausibility of protein presence on both the interior and exterior NPG surfaces is confirmed, and the similarity in peak intensities for BSA on the exterior and interior regions signifies that the protein immobilization is homogeneous in nature; no specific protein immobilization gradient was formed inside NPG by using the flow cell system.
Fig. 9.
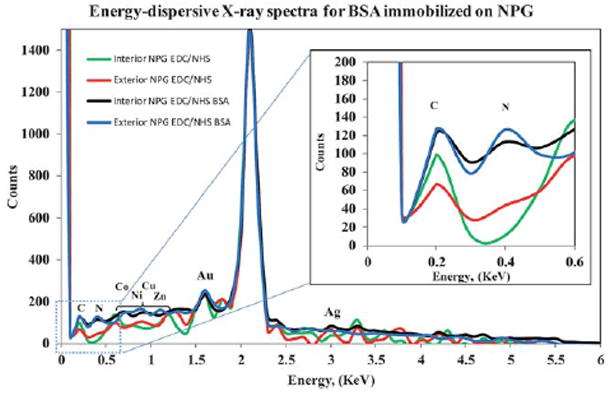
Energy-dispersive X-ray spectra for nanoporous gold with activated NHS-ester in the presence and absence of BSA. The previously unseen nitrogen peak (from NPG with NHS-ester SAM only, red and green lines) is seen clearly both on the exterior (blue line) and interior (black line) of the NPG. The K emission lines for C (0.28 keV), N (0.39 keV) and Au (1.65 and 2.12 keV) are clearly visible, as well as the K emission line for various metal indicated in spectra in the range from 0.6 to 1.5 KeV.
Conclusions
The purpose of this study was to use AFM to investigate the formation of BSA and IgG adlayers conjugated to SAM modified NPG, with a typical pore size ranging from fifty to a few hundreds of nanometres. Covalent immobilization under static versus flow-through conditions was compared. As a proof of concept and also as a comparison with NPG, the proteins were imaged immobilized on rough gold films. The AFM results reveal that immobilization of BSA onto rough gold surfaces is favored by longer incubation times. It was also observed that immobilization after treatment with EDC (in acetonitrile) alone will give a surface coverage that is actually greater than that for EDC followed by NHS. The coverage obtained for BSA on the exterior surface of NPG was comparable to that found on the rough gold surface. IgG coverage on the exterior surface of NPG is lower than that of BSA, as expected given that IgG is larger than BSA. AFM observation of protein on the exterior and interior of the NPG shows that use of a flow cell system greatly enhances the protein solution passage through NPG and hence protein can be tethered more efficiently onto the surfaces throughout the interior of the sample. Many variables with respect to NPG structure, SAM structure and formation time, surface activation and protein incubation, as well as comparisons of static versus flow conditions remain to be explored and we are actively investigating the influence of these parameters on protein coverage inside NPG. The present study demonstrates that use of the flow through conditions presented here can greatly enhance protein coverage and distribution inside NPG. The existence of the carbon and nitrogen peaks near 0.28 KeV and 0.39 KeV, respectively, in energy dispersive X-ray analysis, also qualitatively confirm the presence of protein both on and in NPG.
AFM is a uniquely suited technique for examining protein immobilization on NPG, especially on internal surfaces produced by cleaving NPG monoliths. For the alternate case of NPG formed as a film on a substrate, exposure of internal surfaces could likely also be achieved by adhesion and pealing onto mica or glass slides. In principle, it should be possible to cleave different NPG samples at roughly the same depth into the interior but after different times of exposure to a protein solution. AFM imaging of protein on these internal surfaces exposed after different times could reveal information about the rate at which protein diffuses into and becomes immobilized within NPG. Sampling effects would need to be carefully accounted for, but such experiments could provide unique insights into protein diffusion inside NPG and how coverage of the interior of NPG progresses with time and depth and possibly with flow related parameters. If the cleavage of the NPG could be achieved at a series of depths, then protein coverages determined as a function of depth at a single time point could also provide information on protein diffusion inside NPG. It would be of interest to study protein diffusion inside the torturous pathways within NPG and to examine different pore sizes and protein sizes to look for evidence of, for example, bottleneck effects or pore clogging. Such study provides important information with implications for understanding the distribution of immobilized protein on and within NPG and should be beneficial in the engineering of protein sensing platforms and other biological applications based on protein modified nanoporous gold.
Supplementary Material
Acknowledgments
The authors thank Professor Fraundorf, Dr David Osborn and Dr Dan Zhou of the UM-St. Louis Center for Nanoscience for usage and discussion of the AFM and SEM. This work was supported by UM-St. Louis and by the NIGMS award R01-GM090254.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c1nr10427f
References
- 1.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 2.Yaropolov A, Skorobogat’ko O, Vartanov S, Varfolomeyev S. Appl Biochem Biotechnol. 1994;49:257–280. [Google Scholar]
- 3.Gupta G, Rajendran V, Atanassov P. Electroanalysis. 2003;15:1577–1583. [Google Scholar]
- 4.Barton SC, Kim H-H, Binyamin G, Zhang Y, Heller A. J Am Chem Soc. 2001;123:5802–5803. doi: 10.1021/ja010408b. [DOI] [PubMed] [Google Scholar]
- 5.Heller A. Phys Chem Chem Phys. 2004;6:209–216. [Google Scholar]
- 6.Frasconi M, Mazzei F, Ferri T. Anal Bioanal Chem. 2010;398:1545–1564. doi: 10.1007/s00216-010-3708-6. [DOI] [PubMed] [Google Scholar]
- 7.Rusmini F, Zhong Z, Feijen J. Biomacromolecules. 2007;8:1775–1789. doi: 10.1021/bm061197b. [DOI] [PubMed] [Google Scholar]
- 8.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem Rev. 2005;105:1103–1170. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 9.Tan YH, Stine KJ. In: Langmuir Monolayers in Thin Film Technology. Sherwin JA, editor. Nova Science Publishers; 2011. [Google Scholar]
- 10.Shulga OV, Jefferson K, Khan AR, D’Souza VT, Liu J, Demchenko AV, Stine KJ. Chem Mater. 2007;19:3902–3911. doi: 10.1021/cm070238n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan YH, Liu M, Nolting B, Go JG, Gervay-Hague J, Liu G-y. ACS Nano. 2008;2:2374–2384. doi: 10.1021/nn800508f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu J-J, Nolting B, Tan Y, Grvay-Hague XLJ, Lu G-y. Nano Biotechnology. 2005;1:201–210. [Google Scholar]
- 13.Haensch C, Herzer N, Hoeppener S, Schubert US. Nanotechnology. Wiley-VCH Verlag GmbH & Co. KGaA; 2010. [Google Scholar]
- 14.Zhang XY, Zhang LD, Lei Y, Zhao LX, Mao YQ. J Mater Chem. 2001;11:1732–1734. [Google Scholar]
- 15.Sander MS, Tan LS. Adv Funct Mater. 2003;13:393–397. [Google Scholar]
- 16.Wittstock A, Zielasek V, Biener J, Friend CM, Bäumer M. Science. 2010;327:319–322. doi: 10.1126/science.1183591. [DOI] [PubMed] [Google Scholar]
- 17.Qian LH, Yan XQ, Fujita T, Inoue A, Chen MW. Appl Phys Lett. 2007;90:153120–153123. [Google Scholar]
- 18.Shulga OV, Zhou D, Demchenko AV, Stine KJ. Analyst. 2008;133:319–322. doi: 10.1039/b712760j. [DOI] [PubMed] [Google Scholar]
- 19.Hartmann M, Jung D. J Mater Chem. 2010;20:844–857. [Google Scholar]
- 20.Seker E, Reed M, Begley M. Materials. 2009;2:2188–2215. [Google Scholar]
- 21.Erlebacher J, Aziz MJ, Karma A, Dimitrov N, Sieradzki K. Nature. 2001;410:450–453. doi: 10.1038/35068529. [DOI] [PubMed] [Google Scholar]
- 22.Ding Y, Erlebacher J. J Am Chem Soc. 2003;125:7772–7773. doi: 10.1021/ja035318g. [DOI] [PubMed] [Google Scholar]
- 23.He X, Antonelli D. Angew Chem Int Ed. 2002;41:214–229. doi: 10.1002/1521-3773(20020118)41:2<214::aid-anie214>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 24.Tierney MJ, Kim HOL. Anal Chem. 1993;65:3435–3440. [Google Scholar]
- 25.Ding Y, Chen M. Mat Res Soc. 2009;34:569–576. [Google Scholar]
- 26.Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem Commun. 2009:1834–1836. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- 27.Kucheyev SO, Hayes JR, Biener J, Huser T, Talley CE, Hamza AV. Appl Phys Lett. 2006;89:053102–053103. [Google Scholar]
- 28.Maaroof AI, et al. J Phys D: Appl Phys. 2007;40:5675. [Google Scholar]
- 29.Moreau F, Bond GC. Catal Today. 2007;122:215–221. [Google Scholar]
- 30.Manso J, Mena ML, Yáñez-Sedeño P, Pingarrón JM. Electrochim Acta. 2008;53:4007–4012. [Google Scholar]
- 31.Kafi AKM, Ahmadalinezhad A, Wang J, Thomas DF, Chen A. Biosens Bioelectron. 2010;25:2458–2463. doi: 10.1016/j.bios.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Rixman MA, Dean D, Macias CE, Ortiz C. Langmuir. 2003;19:6202–6218. [Google Scholar]
- 34.Wright AK, Thompson MR. Biophys J. 1975;15:137–141. doi: 10.1016/s0006-3495(75)85797-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carter DC, Ho JX. In: Advances in Protein Chemistry. Anfinsen CB, Edsall JT, Richards FM, Eisenberg DS, editors. Vol. 45. Academic Press; 1994. pp. 153–203. [DOI] [PubMed] [Google Scholar]
- 36.Tsai D-H, DelRio FW, Keene AM, Tyner KM, MacCuspie RI, Cho TJ, Zachariah MR, Hackley VA. Langmuir. 2011;27:2464–2477. doi: 10.1021/la104124d. [DOI] [PubMed] [Google Scholar]
- 37.Renner C, Piehler J, Schrader T. J Am Chem Soc. 2005;128:620–628. doi: 10.1021/ja0560229. [DOI] [PubMed] [Google Scholar]
- 38.Bagçi H, Kohen F, Kusçuoglu U, Bayer EA, Wilchek M. FEBS Lett. 1993;322:47–50. doi: 10.1016/0014-5793(93)81108-c. [DOI] [PubMed] [Google Scholar]
- 39.Qiu H, Xu C, Huang X, Ding Y, Qu Y, Gao P. J Phys Chem C. 2009;113:2521–2525. [Google Scholar]
- 40.Ducker RE, Montague MT, Leggett GJ. Biointerphases. 2008;3:59–65. doi: 10.1116/1.2976451. [DOI] [PubMed] [Google Scholar]
- 41.Droumaguet BL, Nicolas J. Polym Chem. 2010;1:563–598. [Google Scholar]
- 42.Tan YH, Pandey B, Sharma A, Bhattarai J, Stine K. Glob J Biochem. 2011 accepted for advance online publication. [Google Scholar]
- 43.Olson LG, LoBeebe Y-S, Harris JM. Anal Chem. 2001;73:4268–4276. doi: 10.1021/ac000873b. [DOI] [PubMed] [Google Scholar]
- 44.Dong Y, Shannon C. Anal Chem. 2000;72:2371–2376. doi: 10.1021/ac991450g. [DOI] [PubMed] [Google Scholar]
- 45.Fowler JM, Stuart MC, Wong DKY. Anal Chem. 2006;79:350–354. doi: 10.1021/ac061175f. [DOI] [PubMed] [Google Scholar]
- 46.Labib M, Hedström M, Amin M, Mattiasson B. Biotechnol Bioeng. 2009;104:312–320. doi: 10.1002/bit.22395. [DOI] [PubMed] [Google Scholar]
- 47.Ducey JMW, Meyerhoff ME. Electroanalysis. 1998;10:157–162. [Google Scholar]
- 48.Duan C, Meyerhoff ME. Microchim Acta. 1995;117:195–206. [Google Scholar]
- 49.Duan C, Meyerhoff ME. Anal Chem. 1994;66:1369–1377. doi: 10.1021/ac00081a003. [DOI] [PubMed] [Google Scholar]
- 50.Hermanson GT. Bioconjugate Techniques. Second. Academic Press; New York: 2008. pp. 391–395. [Google Scholar]
- 51.Gooding JJ, Ciampi S. Chem Soc Rev. 2011;40:2704–2718. doi: 10.1039/c0cs00139b. [DOI] [PubMed] [Google Scholar]
- 52.Wrobel N, Schinkinger M, Mirsky VM. Anal Biochem. 2002;305:135–138. doi: 10.1006/abio.2002.5646. [DOI] [PubMed] [Google Scholar]
- 53.Dordi B, Schönherr H, Vancso GJ. Langmuir. 2003;19:5780–5786. [Google Scholar]
- 54.Wang J, Kenseth JR, Jones VW, Green J-BD, McDermott MT, Porter MD. J Am Chem Soc. 1997;119:12796–12799. [Google Scholar]
- 55.Staros JV, Wright RW, Swingle DM. Anal Biochem. 1986;156:220–222. doi: 10.1016/0003-2697(86)90176-4. [DOI] [PubMed] [Google Scholar]
- 56.Grabarek Z, Gergely J. Anal Biochem. 1990;185:131–135. doi: 10.1016/0003-2697(90)90267-d. [DOI] [PubMed] [Google Scholar]
- 57.Leopold MC, Bowden EF. Langmuir. 2002;18:2239–2245. [Google Scholar]
- 58.Willey TM, Vance AL, Bostedt C, van Buuren T, Meulenberg RW, Terminello LJ, Fadley CS. Langmuir. 2004;20:4939–4944. doi: 10.1021/la049868j. [DOI] [PubMed] [Google Scholar]
- 59.Volkert AA, Subramaniam V, Ivanov MR, Goodman AM, Haes AJ. ACS Nano. 2011;5:4570–4580. doi: 10.1021/nn200276a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong Y, Abaci S, Shannon C, Bozack MJ. Langmuir. 2003;19:8922–8926. [Google Scholar]
- 61.Braun PV. Nat Mater. 2004;3:281–282. doi: 10.1038/nmat1125. [DOI] [PubMed] [Google Scholar]
- 62.Jackson AM, Myerson JW, Stellacci F. Nat Mater. 2004;3:330–336. doi: 10.1038/nmat1116. [DOI] [PubMed] [Google Scholar]
- 63.Daniel M-C, Astruc D. Chem Rev. 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 64.Zhang G, Yang Z, Lu W, Zhang R, Huang Q, Tian M, Li L, Liang D, Li C. Biomaterials. 2009;30:1928–1936. doi: 10.1016/j.biomaterials.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chu Y, Seo B, Kim J. Bull Korean Chem Soc. 2010;31:3407–3410. [Google Scholar]
- 66.Wong LS, Khan F, Micklefield J. Chem Rev. 2009;109:4025–4053. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 67.Wollman DA, Irwin KD, Hilton GC, Dulcie LL, Newbury DE, Martinis JM. J Microsc. 1997;188:196–223. [Google Scholar]
- 68.Dunham AC, Wilkinson FCF. X-Ray Spectrom. 1978;7:50–56. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.