

Abstract
Structure-based design of synthetic inhibitors of protein-protein interactions requires adept molecular design and synthesis strategies as well as knowledge of targetable complexes. To address the significant gap between the elegant design of helix mimetics and their sporadic use in biology, we analyzed the full set of helical protein interfaces in the Protein Data Bank to obtain a snapshot of how helices that are critical for complex formation interact with the partner proteins. The results of this study are expected to guide systematic design of synthetic inhibitors of protein-protein interactions. We have experimentally evaluated new classes of protein complexes that emerged from this dataset – highlighting the significance of the results described herein.
Interactions of proteins with partner proteins control essential cellular processes, and misregulation of these interactions is often implicated in disease states.1 However, despite their fundamental role, protein-protein interactions (PPIs) are generally not considered attractive targets for drug design because of their large, and often flat, contact surfaces.2–4 A promising rational design approach for the discovery of PPI inhibitors is centered on the role of protein secondary structures at protein interfaces. Analysis suggests that although protein interfaces are large, often a small subset of the residues contributes significantly to the free energy of binding.5–8 Secondary structures are common scaffolds for the organization of these “hot spots” in proteins.4,9,10 It has been demonstrated that synthetic molecules that reproduce key elements of energetically significant protein secondary structures can inhibit chosen interfaces with high affinity and specificity. 11–23
We recently analyzed the full set of helical protein interfaces in the Protein Data Bank to identify potentially suitable candidates for inhibition by small molecules or helix mimetics.24,25 We began by identifying protein complexes that feature helical segments at interfaces and computationally evaluating the energetic contribution of helices to complex formation (Figure 1). Although several examinations of protein–protein interactions have been performed, our approach is unique in its focus on interfaces involving a specific secondary structure. The key motivation behind this structure-based dissection of interfaces is to aid systematic design of synthetic inhibitors of PPIs.
Figure 1.

Evaluation of structures from the Protein Data Bank to identify and assess helical interfaces in protein-protein interactions. The helical interfaces were evaluated by computational alanine scanning mutagenesis.
In earlier reports we categorized helical protein interfaces identified with our algorithm by cellular functions24 and proposed a predictive scale for inhibition of protein-protein interactions by synthetic ligands.25 These studies focused on the disposition and energetic contributions of “hot spot” residues within interfacial helices, and provided a list of interactions that have not previously been inhibited along with candidate helices whose mimics may serve as potent inhibitors. Based on these predictions, we have designed cell-permeable synthetic α-helices that interfere with protein-protein interactions that control transcription of hypoxia inducible genes and Ras signaling.13,14 Here we examine the composition and characteristics of helical domains identified to be critical for protein complex formation. We analyzed the full set of available protein complexes in the PDB to assess amino acid propensity at helical interfaces, location and positioning of hot spot residues on helices, and contact residues on partner proteins.
Examination of entries in the PDB (version August 2009) shows that multiprotein complexes constitute roughly 15% of the data-bank.24,25 Of these 62% feature a helix at the interface, highlighting the role of α-helices in protein-protein interactions. However, presence of a helix at the interface does not imply a critical role for the particular helix in the interaction. To evaluate the energetic contribution of each helix to the complex formation, we employed computational alanine scanning mutagenesis scans within Rosetta to identify residues that contribute most strongly to complex formation.26,27 Alanine scanning mutagenesis is a standard approach for identifying hot spot residues.28 The results of this analysis have been reported along with a full list of filtered PPIs.25
Three general strategies have been used to develop helix mimetics: helix stabilization, helical foldamers, and helical surface mimetics.29,30 Helix stabilizing methods based on side chain crosslinks18,31 and hydrogen-bond surrogates32 preorganize amino acid residues and initiate helix formation. Helical foldamers,11,33 such as β-peptides34–36 and peptoids,37 are composed of amino acid analogs and are capable of adopting conformations similar to those found in natural proteins. Helical surface mimetics utilize conformationally restricted scaffolds with attached functional groups that resemble the i, i+3, i + 4, and i + 7 pattern of side chain positioning on an α-helix (Figure 2a). Surface mimetics typically impart functionality from one face of the helix,38 while stabilized peptide helices and foldamers are able to reproduce functionality present on multiple faces of the target helix. A key advantage of the helix surface mimicry is that it affords low molecular weight compounds as modulators of protein interactions.39–44
Figure 2.

Energetic contributions of residues on different faces of interfacial helices. (a) Positioning of side chain residues on a canonical α-helix, (b) percent occurrence of hot spot residues on one, two or three helical faces (total number helices in each category shown in parentheses), (c) percent occurrence of hot spot residues as a function of helix position, (d–f) examples of protein complexes with hot spot residues on one face, two faces and three faces (PDB codes: 1xl3, 1xiu, and 1or7).
A catalog of PPIs predicting energetic contributions of residues on different faces of interfacial helices should provide an invaluable starting point for design of synthetic inhibitors of protein complex formation. Such a dataset would enable design of an appropriate mimic for a particular interface of interest. Based on this hypothesis, we analyzed the occurrence of hot spot residues on different helical faces. Hot spot residues are defined as residues that upon mutation to alanine are predicted to decrease the binding energy by a threshold value ΔΔGbind ≥ 1.0 kcal mol−1, as measured in Rosetta energy units.5,7,8,26 We used a cut-off value of ΔΔGavg ≥ 2.0 kcal mol−1 to define strongly and weakly interacting interfaces.25 This average binding energy difference accounts for all hot-spot residues at an interface. Our current dataset consists of 480 “strongly interacting” interfaces, which were closely examined. The number of such complexes will grow as new entries are deposited in the PDB.
Analysis reveals that roughly 60% of helical interfaces in the dataset feature helices with hot spot residues on one face of the helix (Figure 2b,d), a third of the complexes utilize helices with hot spots on two faces (Figure 2b,e) and roughly 10% require all three faces for interaction with target protein partner (Figure 2b,f). The full list of protein-protein interactions that correspond to each category is included in the Supporting Information. Residues i, i+1, and i + 2 reside on different faces of a single helical turn; we examined models of each interfacial helix individually as the non-integer number of residues per helical turn makes it difficult to classify locations of non-contiguous residues on helical faces. Overall percent occurrence of hot spot residues at the first twelve positions in interfacial helices is depicted in Figure 2c. Our inquiry suggests that helix surface mimetics may prove to be a highly effective class of synthetic inhibitors; however, a significant fraction of protein-protein interactions will require mimetics that array protein-like functionality on multiple faces. Figure 3 shows the targeting potential of various helix mimetics. Terphenyls, the prototypical helix surface mimetics, imitate one helical face, side chain crosslinked helices can reproduce functionality of up to two faces; although the linker itself may interact with the protein pocket. Hydrogen bond surrogate (HBS) helices and β-peptide foldamers potentially afford complete replicas of functionality present on protein α-helices. We categorized the functions of protein-protein interactions featuring hot spots on different number of helical faces as defined in the PDB (Figure 4). Some interactions could fall into more than one function category. The four largest categories for each type are gene regulation, enzymatic function, cell cycle, and signaling.
Figure 3.
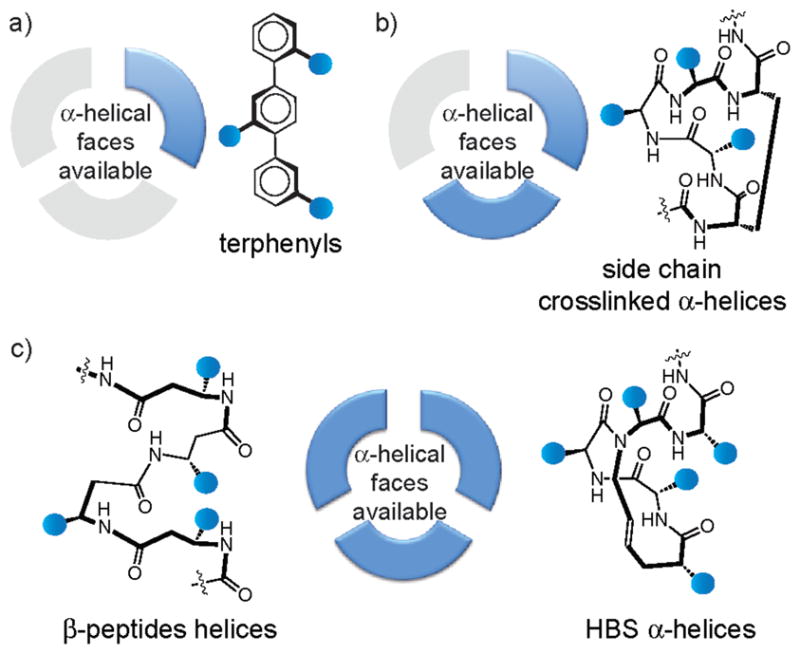
Potential of various helix mimetics to reproduce functionality of one, two or all three faces of protein α-helices.
Figure 4.

Functions associated with protein-protein interactions featuring hot spots on (a) one helical face, (b) two helical faces and (c) three helical faces.
The helical interfaces that form this dataset allow a detailed analysis of basic interactions that underlie protein complex formation. Examination of these fundamental forces will inform design of PPI inhibitors. We calculated the percentage of each helical residue that contributes strongly to binding. (Glycine and proline residues were exempted from alanine scanning since substitutions of proline or glycine to alanine may cause a conformational change in the protein backbone.) Leucine dominates the interface region (Figure 5a), which is not surprising as leucine is also the most prevalent residue in proteins in general. When normalized for natural abundance,45 we find that aromatic residues and arginine, along with leucine, are overrepresented as hot spots at helical interfaces in comparison to polar residues (Figure 5c). These results correspond with previous studies of the types of amino acids appearing as hot spot residues in protein interfaces (Supporting Information, Figure S2);5,9,10,46,47 although our dataset is considerably larger than those previously examined. We expect these results to help guide design of helix mimetics libraries.40,43,44,48–50
Figure 5.

(a) Percent occurrence of hot spot amino acids in helix-mediated protein interfaces, (b) percent occurrence of hot spot residues classified into similar groups, (c) representation of hot spot amino acids normalized to natural abundance of amino acids in proteins, and (d) average predicted decrease in binding energy of helical interfaces upon mutation of hot spot residues to alanine. Color code: aromatic (phenylalanine, tryptophan and tyrosine), white; hydrophobic (isoleucine, leucine and valine), green; negatively charged (aspartic acid and glutamic acid), blue; polar neutral (asparagine, cysteine, glutamine, serine and threonine), gray; positively charged (arginine, histidine and lysine) red.
Hydrophobic and aromatic residues constitute a majority of hot spot residues; however, polar and charged residues are also significant contributors at interfaces (Figure 5b).51 This analysis supports the common perception that protein-protein interactions are generally hydrophobic but feature key salt-bridges and other polar interactions that appreciably influence the binding energy landscape.8 This view is further supported by the evaluation of residues on the partner protein that are within 5 Å of the helical hotspot residue (Supporting Information, Figure S3). Not surprisingly a majority of residues that are within the specified radius of a hydrophobic residue are themselves hydrophobic, which is consistent with the hypothesis that the burial of a hot spot in a hydrophobic environment is a major stabilizing influence.5 In this respect, it is interesting to note that, on average, mutations of aromatic residues to alanine are more destabilizing than substitution of other interfacial residues, with the effect being dependent on the size of the aromatic ring (Figure 5d).
Helical protein-protein interactions have so far been successfully targeted by a diverse array of mimetics.12,14,16,18,21,23 Preliminary success in this field validates helix design concepts from multiple research groups and provides an impetus for designing inhibitors of interactions previously considered to be intractable to inhibition by synthetic ligands. A key motivation for our approach is to bridge the significant chasm between the elegant design of helix mimetics and their sporadic use in biology. This study provides a list of targets to be considered for different classes of helix mimetics based on the number of contact surfaces the target helix utilizes for interactions with partner proteins. We have successfully used this information to identify two new classes of protein-protein interactions amenable to disruption by helix mimetics,13,14 supporting the basic hypotheses and results of these computational efforts.
Supplementary Material
Acknowledgments
This work was financially supported by the National Institutes of Health (GM073943) and National Science Foundation (CHE 0848410). B.N.B. thanks the New York University for a Kramer Pre-doctoral Fellowship and A.L.J. thanks NYU for the Dean’s Dissertation Fellowship.
Footnotes
Supporting Information. Lists of helical protein-protein interactions with predicted occurrences of hot spot residues on different faces of target helix, and summary of helix contact residues. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ryan DP, Matthews JM. Curr Opin Struct Biol. 2005;15:441. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 3.Hajduk PJ, Greer J. Nat Rev Drug Discov. 2007;6:211. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 4.Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 5.Bogan AA, Thorn KS. J Mol Biol. 1998;280:1. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 6.Moreira IS, Fernandes PA, Ramos MJ. Proteins. 2007;68:803. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 7.Clackson T, Wells JA. Science. 1995;267:383. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 8.Keskin Z, Gursoy A, Ma B, Nussinov R. Chem Rev. 2008;108:1225. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 9.Guharoy M, Chakrabarti P. Bioinformatics. 2007;23:1909. doi: 10.1093/bioinformatics/btm274. [DOI] [PubMed] [Google Scholar]
- 10.Jones S, Thornton JM. Prog Biophys Mol Bio. 1995;63:31. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 11.Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Proc Natl Acad Sci U S A. 2009;106:14751. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. J Am Chem Soc. 2010;132:941. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patgiri A, Yadav K, Arora PS, Bar-Sagi D. Nature Chem Biol. 2011;7 doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang D, Lu M, Arora PS. Angew Chem Int Ed. 2008;47:1879. doi: 10.1002/anie.200704227. [DOI] [PubMed] [Google Scholar]
- 16.Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Nature. 2009;462:182. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison RS, Shepherd NE, Hoang HN, Ruiz-Gomez G, Hill TA, Driver RW, Desai VS, Young PR, Abbenante G, Fairlie DP. Proc Natl Acad Sci U S A. 2010;107:11686. doi: 10.1073/pnas.1002498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith BA, Daniels DS, Coplin AE, Jordan GE, McGregor LM, Schepartz A. J Am Chem Soc. 2008;130:2948. doi: 10.1021/ja800074v. [DOI] [PubMed] [Google Scholar]
- 20.Harker EA, Schepartz A. ChemBiochem. 2009;10:990. doi: 10.1002/cbic.200900049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cummings CG, Hamilton AD. Curr Opin Chem Biol. 2010;14:341. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Hammond MC, Harris BZ, Lim WA, Bartlett PA. Chem Biol. 2006;13:1247. doi: 10.1016/j.chembiol.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Ko E, Liu J, Burgess K. Chem Soc Rev. 2011;40:4411. doi: 10.1039/c0cs00218f. [DOI] [PubMed] [Google Scholar]
- 24.Jochim AL, Arora PS. Mol BioSyst. 2009;5:924. doi: 10.1039/b903202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jochim AL, Arora PS. ACS Chem Biol. 2010;5:919. doi: 10.1021/cb1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kortemme T, Kim DE, Baker D. Sci STKE. 2004;2004:l2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 27.Kortemme T, Baker D. Proc Natl Acad Sci U S A. 2002;99:14116. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cunningham BC, Wells JA. Science. 1989;244:1081. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 29.Henchey LK, Jochim AL, Arora PS. Curr Opin Chem Biol. 2008;12:692. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards TA, Wilson AJ. Amino acids. 2011;41:743. doi: 10.1007/s00726-011-0880-8. [DOI] [PubMed] [Google Scholar]
- 31.Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891. [Google Scholar]
- 32.Patgiri A, Jochim AL, Arora PS. Acc Chem Res. 2008;41:1289. doi: 10.1021/ar700264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gellman SH. Acc Chem Res. 1998;31:173. [Google Scholar]
- 34.Cheng RP, Gellman SH, DeGrado WF. Chem Rev. 2001;101:3219. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- 35.Horne WS, Gellman SH. Acc Chem Res. 2008;41:1399. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seebach D, Gardiner J. Acc Chem Res. 2008;41:1366. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- 37.Yoo B, Kirshenbaum K. Curr Opin Chem Biol. 2008;12:714. doi: 10.1016/j.cbpa.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 38.Marimganti S, Cheemala MN, Ahn JM. Org Lett. 2009;11:4418. doi: 10.1021/ol901785v. [DOI] [PubMed] [Google Scholar]
- 39.Plante JP, Burnley T, Malkova B, Webb ME, Warriner SL, Edwards TA, Wilson AJ. Chem Commun. 2009:5091. doi: 10.1039/b908207g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaginian A, Whitby LR, Hong S, Hwang I, Farooqi B, Searcey M, Chen J, Vogt PK, Boger DL. J Am Chem Soc. 2009;131:5564. doi: 10.1021/ja810025g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Restorp P, Rebek J., Jr Bioorg Med Chem Lett. 2008;18:5909. doi: 10.1016/j.bmcl.2008.06.074. [DOI] [PubMed] [Google Scholar]
- 42.Tosovska P, Arora PS. Org Lett. 2010;12:1588. doi: 10.1021/ol1003143. [DOI] [PubMed] [Google Scholar]
- 43.Lee JH, Zhang Q, Jo S, Chai SC, Oh M, Im W, Lu H, Lim HS. J Am Chem Soc. 2011;133:676. doi: 10.1021/ja108230s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. ACS Chem Biol. 2009;4:335. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson DL, Lehninger AL, Cox MM. Principles of Biochemistry. 5. W.H. Freeman; New York: 2008. [Google Scholar]
- 46.Kossiakoff AA, Koide S. Curr Opin Struct Biol. 2008;18:499. doi: 10.1016/j.sbi.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 47.Argos P. Protein Eng. 1988;2:101. doi: 10.1093/protein/2.2.101. [DOI] [PubMed] [Google Scholar]
- 48.Ko E, Liu J, Perez LM, Lu G, Schaefer A, Burgess K. J Am Chem Soc. 2010;133:462. doi: 10.1021/ja1071916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campbell F, Plante JP, Edwards TA, Warriner SL, Wilson AJ. Org Biomol Chem. 2010;8:2344. doi: 10.1039/c001164a. [DOI] [PubMed] [Google Scholar]
- 50.Murray JK, Farooqi B, Sadowsky JD, Scalf M, Freund WA, Smith LM, Chen JD, Gellman SH. J Am Chem Soc. 2005;127:13271. doi: 10.1021/ja052733v. [DOI] [PubMed] [Google Scholar]
- 51.Sheinerman FB, Norel R, Honig B. Curr Opin Struct Biol. 2000;10:153. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.