

Abstract
In analogy to proteins, the function of RNA depends on its structure and dynamics, which are encoded in the linear sequence. While there are numerous methods for computational prediction of protein 3D structure from sequence, there have been very few such methods for RNA. This review discusses template-based and template-free approaches for macromolecular structure prediction, with special emphasis on comparison between the already tried-and-tested methods for protein structure modeling and the very recently developed “protein-like” modeling methods for RNA. We highlight analogies between many successful methods for modeling of these two types of biological macromolecules and argue that RNA 3D structure can be modeled using “protein-like” methodology. We also highlight the areas where the differences between RNA and proteins require the development of RNA-specific solutions.
Figure.
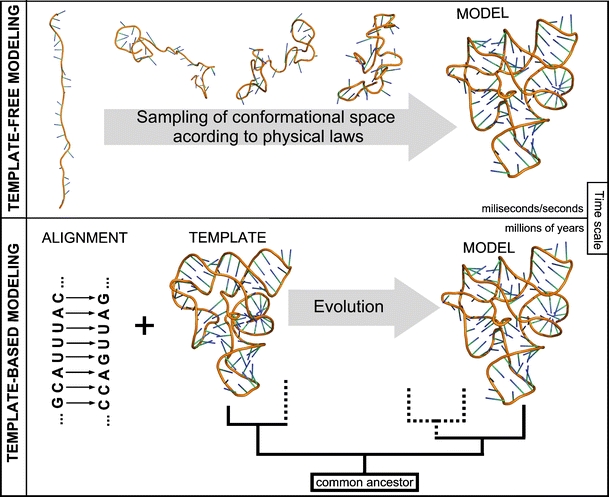
Approaches for predicting RNA structure. Top: Template-free modeling. Bottom: Template-based modeling
Keywords: Assessment, Prediction, RNA, Structure, Tertiary
Introduction
RNAs and proteins are linear polymers composed of a limited set of building blocks (ribonucleotide and amino acid residues, respectively). Despite the fundamental chemical differences of these building blocks, the higher order structure of RNA and protein molecules can be described with similar terms (Fig. 1). Each residue comprises two parts: one is common to the given type of a macromolecule and is used to form a continuous “backbone”, the other is variable and forms a “sidechain”. The order of building blocks held together by covalent bonds is called the primary structure, the local conformation of the chain stabilized mostly by hydrogen bonds is the secondary structure, while the path of the chain in three dimensions resulting from various long-range interactions is the tertiary structure.
Fig. 1.
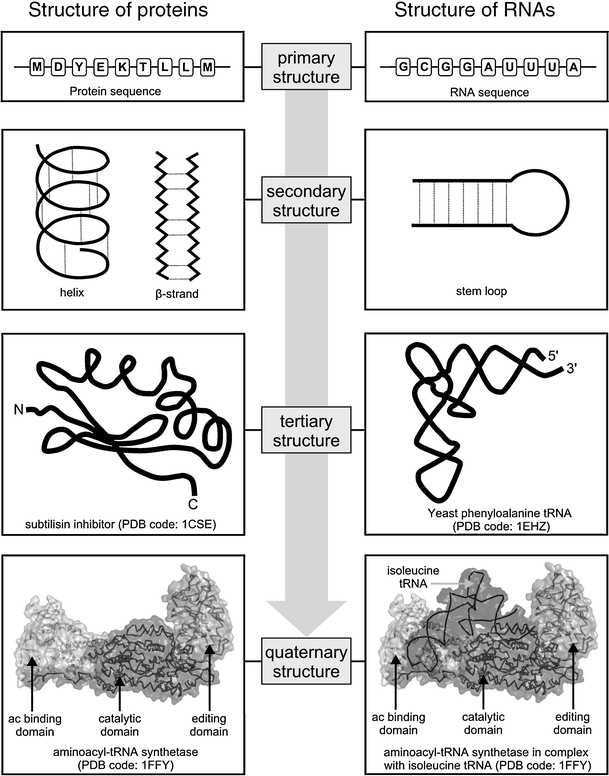
Hierarchical structure of proteins and RNAs
Most protein and RNA molecules, or at least their parts/domains, fold spontaneously into complex three-dimensional shapes [1, 2]. From a global perspective, there are a number of common principles that govern the folding of proteins and RNA molecules, but there are also important differences. The initial events leading to compaction of an RNA chain are driven by neutralization of the negative charge on the phosphate groups by counterions, whereas the compaction of a protein polypeptide chain is driven by burial of hydrophobic side-chains [3]. Besides, secondary structure in proteins is formed owing to hydrogen bonding of the main chains, while in RNA it involves hydrogen bonding between the side-chains.
The structures of biological macromolecules provide a framework for their biological functions [4]. These functions typically involve interactions with various molecules in the cell, including other proteins and RNAs. The importance of structure for the function of protein non-coding RNAs (e.g., tRNAs, ribozymes or riboswitches) has been widely accepted [5]. Recently, it has been shown that protein-coding regions of mRNAs are also highly structured, suggesting an additional role in the regulation of translation [6, 7]. However, it is also known that many proteins and RNAs undergo conformational transitions or exhibit functionally relevant structural disorder [8, 9]. Thus, the function of both proteins and RNAs depends on the three-dimensional structure and dynamics, which in turn is encoded in the linear sequence of individual molecules [10].
It should be also mentioned that mature, functional RNA and protein sequences can be modified/edited compared to the “raw” sequence information encoded in the DNA. Apart from removal or addition of sequence fragments, individual residues can be chemically altered by dedicated enzymes. Posttranscriptional modifications in RNAs and posttranslational modifications in proteins extend the basic alphabets of four nucleotides and 20 amino acids with many additional ‘letters’ that influence the structure and function of molecules that contain them [11, 12].
The knowledge of structure is very important for the understanding of RNA and protein function. However, experimental sequence determination of genes and entire genomes, from which the sequences of RNAs and proteins can be reliably inferred, is much cheaper and simpler than experimental determination of structures. As a consequence, the rate of macromolecular structure determination lags behind the rate of determination of new sequences and the gap between the number of known structures and known sequences continues to widen. It is unlikely that structures will be solved experimentally for all protein and RNA molecules. Understanding of the “1D-3D code” provides an opportunity for theoretical prediction of protein and RNA structures from their sequences. This has proven to be a very difficult task, however a few successful strategies have been identified, which now allow for reasonably accurate (practically useful) predictions of 3D structures. Most methods have been developed initially for proteins only. However, recent developments in the RNA structural bioinformatics field suggests that essentially the same principles may be applicable for modeling of those RNAs that exhibit relatively stable 3D structures.
Classification of methods for macromolecular 3D structure prediction
Methods for 3D structure prediction can be divided into those based on “first principles”, i.e., the fundamental laws of physics that govern the process of folding, and those based on information about other structures, available in databases. In particular, knowledge-based methods can be used to predict macromolecular structures by modeling the process of evolution (Fig. 2).
Fig. 2.
Template-dependent and template-free approaches to prediction of macromolecular structures, exemplified by the modeling of evolution and folding, respectively
Physics-based 3D structure prediction
One approach to 3D structure prediction, sometimes termed ab initio prediction, is based on the thermodynamic hypothesis formulated by Anfinsen, according to which the native structure of a protein corresponds to the global minimum of the free energy of the system comprising the macromolecule [13]. Accordingly, physics-based methods model the process of folding by simulating the conformational changes of a macromolecule while it searches for the state of minimal free energy (review: [14]). The “score” of each conformation is calculated as the true physical energy based on the interactions within the macromolecule and between the macromolecule and the solvent [15]. While in physics the term ab initio is often used to refer to find a set of wave functions and energies by solving the Schrödinger equation without external parameters, the physics-based methods described here offer a simplified approach to calculate the energy. The functional form and parameter sets used to describe the potential energy of a system is called a force field. There exist a number of software packages for simulation of protein folding in atomic detail, they typically implement various versions of molecular dynamics (MD) and Monte Carlo (MC) protocols for searching the conformational space, and force fields such as AMBER [16], CHARMM [17] or GROMOS [18] to calculate the energy.
In order to facilitate the identification of the native state as the one of the lowest energy, the energy landscape that describes the relationship between the distance from the native-like conformation and the energy should have a funnel-like shape (review: [19]). More explicitly, when plotting the energy of models versus a structural difference between the models and the native structure (e.g., expressed as the root-mean-square deviation (RMSD) between pairs of equivalent atoms in optimally superimposed structures), there should be a funnel-shaped tip at the bottom left corner of the plot. In particular, the native structure should exhibit the lowest energy, and the farther a given conformation is from the native structure, the higher its energy should be. The prediction of the native structure is easier if this relationship between the value and variability of energies and deviation from the native structure holds across the entire range of possible conformations.
Figure 3 presents diagrams comparing an ‘ideal’ (from a practical point of view of macromolecular structure prediction) relationship between the energy of models and their distance from the native structure and a ‘real life’ example of such a distribution obtained from a folding simulation of an immunoglobulin light chain-binding domain of protein L (2ptl in the Protein Data Bank), carried out using the REFINER method [20]. One conceptual difference between the energy function in a folding simulation and the real physical energy becomes apparent in this and similar plots: In reality, the energy differences between the folded and unfolded state are very small, while in practice the effective discrimination of native-like models from non-native-like ones requires maximization of the energy difference.
Fig. 3.
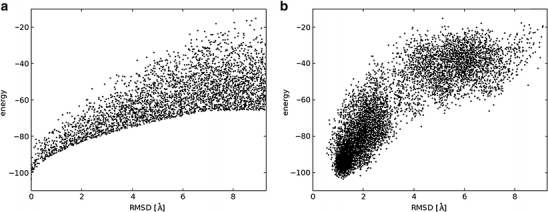
A funnel-like relationship between the value of a function for scoring of structural models and their deviation from the native structure (expressed e.g., in root mean square deviation of superimposable atoms or in some other similarity measure): (a) a hypothetical “ideal” function that maximizes the discrimination between native, native-like and non-native conformations. The minimal value of energy as well as the spread of energy values for conformations at a particular distance from the native structure (corresponding to the global energy minimum) increase monotonically with the increasing distance, so conformations closer to the native structure on the average exhibit lower energies than those farther away. Here a random sample of points that fulfill this relationship is shown. (b) results of folding simulations of an immunoglobulin light chain-binding domain of protein L (2ptl in the Protein Data Bank), carried out using the REFINER method [20], which uses a Monte Carlo sampling scheme and a statistical potential
The ab initio approach is plagued by serious problems. In particular, a full-atom structural model of a macromolecule has a large number of degrees of freedom (3*Natoms-5), which makes the search space enormous, and the function with which to calculate the energy of the system is very complex. As a result, both the sampling and energy calculations are very costly in terms of computational power required. Typically, the free energy landscape is extremely rugged, i.e., it possesses multiple local minima, and it is essentially impossible to perform an exhaustive evaluation of all these minima to identify the one with the globally lowest value. Further, some of the components of the free energy function (e.g., the entropy) are very difficult to calculate, and may not be inferred accurately for large molecules. For these reasons the use of ab initio methods is limited to very small molecules. Thus far, most of the reported successful all-atom folding simulations have been for very small proteins, such as the 20-residue “Trp-cage” miniprotein [21], and they rarely exceed the threshold of one microsecond. Further, even extended simulations, such as a ten microsecond simulation performed for a fast-folding WW domain [22] may not sample a native-like conformation. Hence, folding simulations have not yet matured to the state of a reliable method for protein 3D structure prediction.
One important problem for algorithms that deal with macromolecular structures is the representation of coordinates. Cartesian coordinates (three numbers representing distances for each atom) are used to represent the final model in a common PDB file format, but they may be impractical at some stages of modeling, as they utilize 3*Natoms degrees of freedom. To increase the efficiency of computations, the system may be transformed into internal coordinate systems and/or bond lengths and angles may be restricted to idealized values. For instance in torsion angle dynamics (e.g., as implemented in the program DYANA [23]), torsion angles are used instead of Cartesian coordinates as degrees of freedom, and the only degrees of freedom are rotations about single bonds. A biopolymer can be represented as a tree structure consisting of n + 1 atoms connected by n rotatable bonds of fixed length. The tree structure starts from a base, typically at the N-terminus of the polypeptide chain, and terminates with “leaves” at the ends of the side-chains and at the C-terminus. The conformation of the molecule is uniquely specified by the values of all torsion angles and torsion angles may be allowed to assume only discrete values. The conversion of a model from internal coordinates to a Cartesian representation can be achieved, e.g., with the Nerf algorithm [24], which requires three coordinates per atom: a bond length, a flat angle, and a torsion angle.
Another approach to reduce the number of degrees of freedom is to use coarse-grained models, which treat groups of atoms as single interaction centers, so that a smaller number of elements and interactions need to be considered (review: [25]). Actually, the first simulation of protein folding reported in the literature used a simplified chain and time-averaged forces to fold bovine pancreatic trypsin inhibitor from an open-chain conformation into a folded conformation close to that of the native molecule [26]. Another advantage of coarse-grained modeling is that the force field derived for the united interaction centers yields a much smoother energy surface than that for the all-atom energy function. As a result, many local energy minima are removed, in which the system could become trapped during the simulation. However, it must be emphasized that simplification of the model and the energy function usually leads to reduced accuracy. As of today, it is not practical to expect that a folding simulation for a macromolecule comprising more than 100 residues would confidently predict a native-like structure with a correctly estimated energy. Contemporary methods for coarse-grained protein structure prediction can be exemplified by UNRES [27], which represents side chains by ellipsoids, and the peptide bonds by united atoms located in the middle of two consecutive Cα atoms. The only degrees of freedom in the continuous space are the bond and torsion pseudoangles defined between the Cα atoms. The free energy function includes terms for interactions between the side chain centers, steric repulsion between side chains and peptide group centers, and electrostatic interactions between peptide groups. Local conformational propensities of a polypeptide are described by torsional and angle-bending potentials. Multibody interactions, which are the most important for reproducing regular secondary structure elements, are described by higher order terms.
Since the same basic laws of physics apply to all types of molecules, one can postulate that analogous methods should work for RNA as well. As mentioned earlier, RNA folding relies on the modulation of electrostatic repulsion by counterions, while protein folding relies on the formation of a hydrophobic core, and the secondary structure formation requires hydrogen-bonding either via protein side chain or RNA main chain functional groups, respectively. Nonetheless, computational methods developed to study protein folding have been successfully used to simulate RNA folding (review: [28]). Examples of all-atom simulations with general-purpose software packages such as AMBER or CHARMM include the folding of small RNA hairpins [29, 30], the analysis of H-bond stability in the anticodon loop of tRNA(Asp) [31] or modeling the interaction of “kissing loops” in the dimerization initiation site (DIS) of HIV [32]. Molecular dynamics simulations restrained by experimental data have also been used to model the conformational transitions of large macromolecular complexes involving both RNAs and proteins, such as the ribosome (review: [33]).
The modeling of nucleic acid structures can also take advantage of the use of local coordinate systems and/or coarse-graining to reduce the number of variables in the system. For instance the 3DNA program [34] constructs reference coordinate frames around bases and base pairs, while using idealized values for bond lengths and bond angles. The treatment of nucleobase moieties as rigid bodies allows one to drastically reduce the number of degrees of freedom. Further, a total of three angle and three translation variables are enough to describe the relative orientation of two bases (with parameters called propeller, buckle, opening, shear, stretch, and stagger) or two base pairs (twist, tilt, roll, shift, slide, and rise). Because these parameters are independent of the Cartesian system, they allow to directly compare two structures without the spatial superposition of coordinates. The miniCarlo program for energy minimizations and Monte Carlo simulations of nucleic acid structures applies a very similar scheme. It uses helical parameters that determine the relative position of bases in a pair and relative position of base pairs, pseudorotation parameters of sugars that determine internal geometry of sugar moieties, glycosidic angles that determine orientation of sugars relative to the bases, and torsion angles that determine the orientation of methyl groups in thymines and hydroxyl groups in riboses [35]. The commercially distributed program junction minimization of nucleic acids (JUMNA) uses a reduced coordinate approach to gain roughly an order of magnitude in the number of variables necessary to model a nucleic acid fragment [36].
One of the first applications of the coarse-grained approach for RNA 3D structure modeling involved the refinement of low-resolution structures of ribosomal RNAs with restraints from experimental data and a representation with pseudoatoms at different levels of detail - from a single pseudoatom per helix to a single pseudoatom for each nucleotide [37]. More recently, a number of new methods have been developed that allow for coarse-grained folding simulations with or without experimental data. YUP [38] and NAST [39] represent RNA by just one pseudoatom per nucleotide residue: phosphate and C3′, respectively. Vfold [40] and DMD [41] represent RNA by three pseudoatoms per residue, while HiRE-RNA [42] uses six or seven pseudoatoms for purine or pyrimidine residues, respectively. For bonded interactions (bonds, angles, and dihedrals) all these methods use parameters derived from a database of known RNA structures. For non-bonded interactions, the energy terms differ. NAST actually does not use a full energy function, it generates plausible 3D structures based on restraints supplied by the user (e.g., on secondary structure or tertiary contacts). Vfold and DMD use experimentally tabulated energy values [43] to parameterize (in different ways) base-pairing and base-stacking, as well as to estimate loop entropy. DMD also uses an explicit representation of hydrogen bonding to enforce base pairs formation and an additional term for phosphate-phosphate repulsion. With the simplifications introduced, both methods are capable of folding RNAs up to 100 residues long. Vfold has been recently used to successfully model pseudoknotted RNA structures and to estimate the conformational entropy for stem-loop tertiary contacts [44].
HiRE-RNA is an excellent example of a protein-like coarse-grained method for RNA modeling. It uses an implicit solvent force field similar to the protein OPEP force field [42]. It is expressed as a sum of local (bonded), nonbonded, and hydrogen-bond terms. All bonded interactions are described by harmonic terms. For non-bonded interactions, a Lennard-Jones potential is used, modified to mimic some of the excluded volume and screening effect that gets lost in coarse-graining. In particular, it implements a repulsive power law at short distances, an exponential tail at large distances to account for an extra screening given by the atoms that are missing from the coarse-grained representation, and a narrower well varying with the equilibrium distance. Hi-RNA energy model does not take into account electrostatic interactions explicitly, except for the repulsion between the phosphates. The base pairing is modeled by hydrogen bonding interactions consisting of 2-, 3-, and 4-body terms. The interactions taken into account include canonical A-U and G-C Watson-Crick pairs, A-U Hoogsteen pairs, G-U wobble pairs, as well as the relatively rare A-C, A-G, and U-C pairs. All the HiRE-RNA parameters were derived from a statistical study of 220 structures in the Nucleic Acids Database (NDB) and subsequently refined through the analysis of long molecular dynamics simulations for a poly-A molecule of 15 nucleotides. Tests of HiRE-RNA on two structures (22 and 36 nucleotide long) solved by NMR, have demonstrated that the method is capable of sampling the native state, however the selection of the most native-like conformation remains a challenge.
Evolution-based structure prediction
At the other end of the methodological spectrum there are approaches based on the principles of evolution. After experimental determination of the first handful of protein structures it became clear that evolutionarily related (homologous) proteins usually retain the same three-dimensional fold (i.e., the 3D arrangement and connectivity of secondary structure elements) despite the accumulation of divergent mutations [45]. It was also found that structural divergence is much slower than sequence divergence, although these two features are strongly correlated. Thus, methods have been developed to align the sequence of one protein (a target) to the structure of another protein (a template), model the overall fold of the target based on that of the template and infer how the target structure will change due to substitutions, insertions and deletions (indels), as compared with the template (reviews: [46, 47]). The process of identification of a structurally related template has been termed “fold recognition”, while the transformation of atomic coordinates of the template structure into the target has been typically referred to as “homology modeling” or “comparative modeling” (the latter takes into account a possibility that the template is not homologous, as long as it is structurally similar to the target). This entire approach has been termed “template-based modeling”.
Comparative analyses of evolutionarily related RNAs (see e.g., [48]), revealed patterns of conservation that are analogous to those observed in proteins: the secondary and tertiary structure is usually more conserved than sequence, and core regions important to stability and function tend to be more conserved at all levels. In general, it can be stated that in families of homologous RNAs, the 3D fold is often conserved and alignment of sequences and secondary structure patterns can be used to recognize such structural conservation, enabling template-based modeling.
Template-based modeling has two main limitations. First, the modeling of the “target” structure starts with another known structure of a structurally similar molecule to be used as a ”template”, hence if such a structure does not exist or cannot be identified reliably, then the model cannot be built or almost certainly will be completely wrong. Further, each element of the target sequence must be aligned to the structurally equivalent element in the template sequence/structure. In particular, homologous residues should be aligned to each other. High sequence similarity is not a prerequisite for template-based modeling. In fact, it is possible to create good homology models even if the sequence identity between the target and the template is zero [49]. However, on the average, molecules with higher sequence similarity tend to exhibit more similar structures [45]. Besides, for highly similar sequences it is generally easier to generate a correct alignment (to find homologous residues between the target and the template). Therefore, using templates with higher sequence similarity is recommended. Apart from sequence divergence, structures may also change because of environmental factors, e.g., the binding of other molecules or the composition of the solution (salt, pH) [50]. This is particularly true for RNA, where the binding of metal ions is often a key factor enabling a stable tertiary structure [51]. It is generally the responsibility of the user of the homology modeling software to choose a template, whose biological state corresponds best to the desired biological state of the target to be modeled. With an incorrectly chosen template and/or wrong alignment, the model will always be very far from the native structure. These limitations concern all homology modeling tools, as templates and alignments are always necessary in this approach [52].
Finally, it must be noted that like proteins, homologous RNAs need not retain the same structure in all details. Topological variability (e.g., preserving the overall 3D structure while changing the pattern of secondary structure elements) has been observed in many protein families [53], as well as in RNA families, with one prominent example being the RNA subunit of RNase P from Escherichia coli (type A) and Bacillus subtilis (type B) [54]. However, methods for automated template-based modeling of macromolecules assume that the overall fold is conserved between the template and the target, and special intervention of the user is usually required to model topological variations.
Two major approaches have been developed for template-based modeling of proteins. One is to model the structure by copying the coordinates of the template (both the backbone and the side-chains) in the aligned core regions, which can also include “averaging” over coordinates of multiple templates. The variable regions are modeled by taking fragments with similar sequence from a database of previously observed loops, followed by replacing the mutated side-chains with rotamers that satisfy the stereochemical criteria, and (optionally) limited energy optimization, as implemented in SWISS-MODEL [55]. The other possibility is to use the distance and torsion angles and interatomic distances from the aligned regions of the template(s) as modeling restraints, which permits the use of information from multiple structures. This approach also requires the idealization of geometry and packing of the entire chain by satisfying stereochemical constraints derived from the database of protein structures, as implemented in MODELLER [56].
The same two types of methods have been recently proposed for RNA modeling. The Altman group has implemented a MODELLER-like strategy in RNABuilder, an extension to the SimTK molecular modeling toolkit [57]. The force field consists of forces and torques which act to fold the RNA molecule according to the restraints specified by the user. No forces act between nucleotide residues unless specified by the user, except stacking forces. A coarse-grained simulation is carried out to fold the model into a conformation that minimizes the violation of restraints. RNABuilder starts with an extended representation of the target sequence and threads it onto the template structure(s), guided by restraints derived from the target-template sequence alignment, optionally using additional user-specified restraints on base pairing, stacking and tertiary interactions, rigidifying portions of the molecule, and many more. It detects runs of three or more consecutive Watson-Crick base pairs and automatically enforces helical geometry. Given a complete description of the structural interactions, RNABuilder is able to construct an RNA model that satisfies all restraints. The structure may however get caught in local minima, especially for longer RNA, where the method cannot satisfy all constraints without further action from the user. RNABuilder has been recently used to construct a homology model of the Azoarcus group I intron, using structural information from two template structures.
Our group has recently developed a protein-like RNA comparative modeling method ModeRNA (http://iimcb.genesilico.pl/moderna/ [58]), inspired by the SWISS-MODEL method for protein structure modeling. ModeRNA interprets a pairwise sequence alignment as a set of instructions that are used to create a model by copying the conserved core from a template structure, and introducing the variable parts by taking fragments from a database of experimentally determined structures. A highlight of ModeRNA is that it can automatically add and remove nucleotide modifications. ModeRNA also offers a scripting interface that allows the users to perform more complex manipulations, such as recombination of fragments taken from unrelated structures.
Hybrid methods
In the protein structure prediction field the most successful approach combines the features of physics-based folding with the use of previously solved structures. The known structures that may be used explicitly as templates or implicitly, as the source of information to calculate a scoring function that may complement or replace the ’purely physical‘ energy. This type of structure prediction is often termed ’de novo modeling‘, and should not be confused with the ab initio modeling, as it heavily relies on information from databases. De novo methods for structure prediction share many problems with the ab initio approach, including a high computational cost of the conformational sampling and uncertainty as to which of the large number of alternative conformations generated is the most native-like structure. Nonetheless, so far in blind tests such as the CASP benchmark, they have outperformed methods based on either ‘pure physics’ or ‘pure evolution’ [59, 60]. Methods such as ROSETTA [61] improve the efficiency of the conformational search by restricting local conformations to those taken from known structures, which should correspond to locally energy-minimized structures. Hence, the main type of conformational transition in ROSETTA involves a replacement of conformational parameters for a short fragment in the modeled protein by parameters taken from a randomly selected fragment in a previously solved structure of another protein. Additional conformational changes are required to refine the local structure. The ROSETTA energy function combines parameters that are based on physics and statistics. Other methods such as CABS [62] restrict the conformational space by projecting all possible conformations onto a discrete three-dimensional lattice. In CABS the scoring function is entirely based on a statistical potential. REFINER is an off-lattice variant of CABS [20], which makes the method slower, but potentially more accurate. TASSER [63] goes even further into hybrid modeling by combining the fragment assembly (if any starting models are available from template-based modeling) with lattice-based modeling (in particular for fragments that lack any template). All these methods initially use a simplified (coarse-grained) model and the final refinement is usually carried out after rebuilding a full-atom model and with an energy function that is enriched into high-resolution physics-based terms.
A number of methods based on the principle of fragment assembly have been recently proposed also for RNA 3D modeling. In particular, FARNA/FARFAR [64, 65] is essentially ‘ROSETTA for RNA’. The FARNA procedure assembles an RNA 3D structure from short linear fragments, using a knowledge-based energy function, which takes into account preferences of the backbone and side-chains conformations, and of base-pairing and base-stacking interactions, derived from experimentally determined RNA structures. Fragments for the assembly of RNA structure were taken from the large ribosomal subunit of Haloarcula marismortui (PDB code: 1ffk). FARFAR is an extension of FARNA, which uses a full-atom refinement in order to optimize the RNA structures generated by FARNA. The full-atom energy function is supplemented with harmonic constraints placed between Watson-Crick edge atoms in the two residues that are assumed to form each bounding canonical base pair and a term to approximately describe the screened electrostatic interactions between phosphates. It also includes terms derived from the earlier work on proteins: a potential for weak carbon hydrogen bonds, an alternative orientation-dependent model for desolvation based on occlusion of protein moieties. In the authors’ own tests of folding 32 RNA targets, 14 cases gave at least one of five FARFAR models with better than 2.0 Å all-heavy-atom RMSD to the experimentally observed structure.
MC-Fold|MC-Sym [66] is based on a principle related to FARNA, as it assembles RNA structures from a library of ‘nucleotide cyclic motifs’, i.e., fragments in which all nucleotides are circularly connected by covalent, pairing or stacking interactions. MC-Fold|MC-Sym implements two energy functions, one based on non-bonded terms (van der Waals and stacking interactions) from the AMBER package and another one based on statistics of the experimentally determined structures, but neither of them can discriminate native-like models from misfolded ones. Recently, inspired by the CABS and REFINER methods for protein structure modeling, we developed SimRNA for RNA structure modeling (M.B., Konrad Tomala, Pawel Łukasz, T.P., K.R., J.M.B., in preparation). SimRNA represents the nucleotide chain by three pseudoatoms per nucleotide residue, similarly to Vfold and DMD, but instead of a physics-based potential, both bonded and non-bonded terms in its energy function are based entirely on database statistics. A conceptually related CG model represents RNA with five pseudoatoms per residue and uses a statistical potential to describe all the nonbonded interactions, including the excluded volume repulsive, the attractive force, and the electrostatic force between nonbonded particles, as well as the solvation forces due to the environment [67].
There exist methods for interactive (user-guided) modeling of macromolecular structures based on assembly of fragments derived from various structures that are predicted to be similar to different parts of the target. Computational tools and the graphics front-end facilitate the choice, the manipulation, and the visualization of fragments, and often provide specialized algorithms for local optimization of geometry to seal breaks in the chain or relieve steric clashes. The approach that allows the expert user to rearrange and recombine multiple template structures has been particularly widely used in the RNA modeling field, with methods such as S2S/Assemble [68, 69], ERNA-3D [70], or RNA2D3D [71]. However, similar methods including the ‘Frankenstein’s Monster approach’ [72] and the “protein lego” approach [73] have also been applied to model protein structures (review: [74]).
Critical assessment and benchmarking of protein and RNA structure prediction
For a very long time, the field of RNA 3D structure modeling has been dominated by methods based on interactive graphical interfaces that allow human experts to manipulate sequences and structures in 3D. Only recently have a number of automated methods been developed, many of which are based on concepts previously used with success in the protein 3D structure modeling field (Table 1). Thus, we conclude that protein and RNA modeling present more similarities than differences, and that it may be worthwhile for these two fields of research to inspire and ‘bootstrap’ each other to overcome some of the existing bottlenecks.
Table 1.
Automated methods for protein and RNA modeling reviewed in this article, arranged according to the analogous principles used
| Prediction method class | Protein | RNA | |
|---|---|---|---|
| Template-based, comparative modeling | Restraints-based | MODELLER | RNABuilder |
| Fragments-based | SWISS-MODEL | ModeRNA | |
| Template-free, physics-based | All-atom | AMBER, CHARMM | |
| Coarse-grained | UNRES | Vfold, DMD, HiRE-RNA | |
| Automated hybrid (statistics + physics) | All-atom, fragment-based | ROSETTA | MC-Fold |
| Coarse-grained | CABS, TASSER, REFINER | SimRNA, CG | |
The development of useful methods for protein structure prediction has been driven by the benchmarking experiments, in which blind predictions are objectively compared to the experimentally solved structures. In the protein structure prediction community there are periodic evaluation experiments that rigorously test the accuracy of prediction methods, e.g., CASP (biannually; http://www.predictioncenter.org/casp8/) and Livebench (continuously; http://meta.bioinfo.pl/livebench.pl). The ability to objectively assess the structure prediction methods, their relative performance as well as the typical accuracy of predictions using an established set of measures [75] has proven indispensable for progress in this field of research.
The assessment of model accuracy requires reliable and meaningful metrics for comparisons between the models and the experimentally determined structures used as a “gold standard”. One of the measures used commonly for comparison of macromolecular models is the RMSD between pairs of equivalent atoms in the optimally superimposed structures. Typically only backbone atoms are considered, e.g., Cα in protein structures or P in RNA structures, but RMSD can also be calculated for any (or all) atoms. However, RMSD is not a perfect measure. A small perturbation in just one part of the structure (e.g., a hinge movement of two domains) can create a large RMSD suggesting that the two structures are very different overall. To take into account both local and global structural similarities, several metrics have been developed. The global distance test (GDT_TS) score [76] and the template matching (TM) score [77] are examples of metrics developed for comparison of protein structures that have been generally accepted in the protein structure prediction field and used by assessors in the CASP experiment; they can also be applied to compare RNA structures and measure the accuracy of RNA models.
Many metrics of structural similarity are dependent on the molecule size: if randomly selected molecules of the same size are compared, the score deteriorates with the molecule size. To eliminate the dependence on protein size, Levitt and Gerstein converted the structure similarity score into the P-value, i.e., a statistical significance score, based on the statistics of random structure comparisons [78]. Recently, Hajdin et al. have analyzed the dependence of the structure similarity on the molecule size in small RNAs ( < 161 nt length) with relatively complex tertiary structures [79]. They found that the compactness of folded RNA molecules is slightly lower than for proteins with the same mass. Based on their analysis they defined an expression relating RMSD with the P-value that describes prediction significance.
Measures of structural similarity developed for protein models are not always ideal for RNA structures. They may capture the general 3D shape, local deviations of the structure, intradomain deformation, or interdomain deviations, but are agnostic about important features that are unique to RNA, i.e., the base-pairing and base-stacking patterns. Parisien et al. developed an RNA 3D structure comparison measure called the deformation index (DI), which evaluates the deviations between two RNA 3D structures by calculating the proportion of base interactions (stacking and pairing) that are identical in both structures [80]. They also developed another measure called a deformation profile (DP) that highlights dissimilarities between structures at the residue level for both intradomain and interdomain interactions. DP can also be used for proteins.
CASP for RNA has not fully materialized yet, hence it is difficult to objectively assess how different methods and approaches for RNA modeling compare with each other and how well they perform in the hands of different users. The number of crystal and NMR structures solved for RNA molecules that are sufficiently large for meaningful analysis is probably still too small to provide a sufficient number of targets for CASP-like intense modeling over a few months every year. In the meantime we have started a project similar to Livebench (again, an inspiration from the field of protein structural bioinformatics), which aims to become an objective benchmark of fully automated methods for RNA structure prediction. The CompaRNA web server (http://comparna.amu.edu.pl, T.P., K.R., Łukasz Kozłowski, Ewa Tkalińska, J.M.B., manuscript in preparation) provides a continuous benchmark for standalone and web server methods. Currently it addresses only fully automated methods for RNA secondary structure prediction, but we intend to extend it to include methods for RNA 3D structure prediction that will become available as public web servers and/or local installations that can be run in a fully automated mode with default parameters and do not require large computing resources. While this approach excludes expert-based modeling and methods that are not yet fully automated or require high performance computing, we hope it will contribute to the assessment of the progress in the RNA structure prediction field.
Acknowledgments
Our work on template-free modeling of RNA structures was supported by the Polish Ministry of Science (HISZPANIA/152/2006 grant to J.M.B.), and by the EU (6FP grant “EURASNET”, LSHG-CT-2005-518238). Our work on template-based modeling of RNA structures was supported by the Faculty of Biology, Adam Mickiewicz University (PBWB-03/2009 grant to M.R.) and by the Polish Ministry of Science (PBZ/MNiSW/07/2006 grant to M.B.). Software development in the Bujnicki laboratory in IIMCB has been supported by the EU structural funds ([POIG.02.03.00-00-003/09). K.R. was independently supported by the German Academic Exchange Service (grant D/09/42768).
We thank Konrad Tomala and Pawel Łukasz for their participation in the development of our RNA modeling methods. We thank present and former members of the Bujnicki laboratory, in particular Ewa Wywiał, Pawel Skiba, Piotr Byzia, Irina Tuszynska, Joanna Kasprzak, Jerzy Orlowski, Tomasz Osiński, Marcin Domagalski, Anna Czerwoniec, Stanisław Dunin-Horkawicz, Marcin Skorupski, and Marcin Feder, for their comments and constructive criticism during development of our software. We also thank Neocles Leontis, Eric Westhof, Rob Knight, Sandra Smit, Magda Jonikas, Alain Laederach, Andrzej Kolinski, and Francois Major for stimulating discussions and helpful advice on various occasions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Kristian Rother, Magdalena Rother and Michał Boniecki contributed equally to the manuscript
Note
This article is a modified version of a chapter “Template-based and template-free modeling of RNA 3D structure: inspirations from protein structure modeling”, K.R., M.R., M.B., T.P., K. Tomala, P. Lukasz, J.M.B., in a book "RNA 3D Structure Analysis and Prediction" edited by N.B. Leontis and E. Westhof, Springer 2011. Reproduced with permission
References
- 1.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 2.Ferre-D'Amare AR, Doudna JA. RNA folds: insights from recent crystal structures. Annu Rev Biophys Biomol Struct. 1999;28:57–73. doi: 10.1146/annurev.biophys.28.1.57. [DOI] [PubMed] [Google Scholar]
- 3.Thirumalai D, Hyeon C. RNA and protein folding:common themes and variations. Biochemistry. 2005;44:4957–4970. doi: 10.1021/bi047314+. [DOI] [PubMed] [Google Scholar]
- 4.Laskowski RA, Thornton JM. Understanding the molecular machinery of genetics through 3D structures. Nat Rev Genet. 2008;9:141–151. doi: 10.1038/nrg2273. [DOI] [PubMed] [Google Scholar]
- 5.Laederach A. Informatics challenges in structured RNA. Brief Bioinform. 2007;8:294–303. doi: 10.1093/bib/bbm026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watts JM, Dang KK, Gorelick RJ, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kertesz M, Wan Y, Mazor E, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hazy E, Tompa P. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem. 2009;10:1415–1419. doi: 10.1002/cphc.200900205. [DOI] [PubMed] [Google Scholar]
- 9.Fulle S, Gohlke H. Constraint counting on RNA structures:linking flexibility and function. Methods. 2009;49:181–188. doi: 10.1016/j.ymeth.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Anfinsen CB, Scheraga HA. Experimental and theoretical aspects of protein folding. Adv Protein Chem. 1975;29:205–300. doi: 10.1016/S0065-3233(08)60413-1. [DOI] [PubMed] [Google Scholar]
- 11.Grosjean H. Fine-Tuning of RNA Functions by Modification and Editing. Berlin Heidelberg: Springer; 2009. [Google Scholar]
- 12.Walsh CT (2005) Posttranslational modification of proteins:Expanding nature's inventory. Roberts, Greenwood Village, CO
- 13.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 14.Hardin C, Pogorelov TV, Luthey-Schulten Z. Ab initio protein structure prediction. Curr Opin Struct Biol. 2002;12:176–181. doi: 10.1016/S0959-440X(02)00306-8. [DOI] [PubMed] [Google Scholar]
- 15.Scheraga HA. Recent developments in the theory of protein folding:searching for the global energy minimum. Biophys Chem. 1996;59:329–339. doi: 10.1016/0301-4622(95)00126-3. [DOI] [PubMed] [Google Scholar]
- 16.Case DA, Cheatham TE, III, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks BR, Brooks CL, III, Mackerell AD, Jr, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christen M, Hunenberger PH, Bakowies D, et al. The GROMOS software for biomolecular simulation:GROMOS05. J Comput Chem. 2005;26:1719–1751. doi: 10.1002/jcc.20303. [DOI] [PubMed] [Google Scholar]
- 19.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 20.Boniecki M, Rotkiewicz P, Skolnick J, et al. Protein fragment reconstruction using various modeling techniques. J Comput Aided Mol Des. 2003;17:725–738. doi: 10.1023/B:JCAM.0000017486.83645.a0. [DOI] [PubMed] [Google Scholar]
- 21.Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. J Am Chem Soc. 2002;124:11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 22.Freddolino PL, Liu F, Gruebele M, et al. Ten-microsecond molecular dynamics simulation of a fast-folding WW domain. Biophys J. 2008;94:L75–L77. doi: 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein EG, Rice LM, Brunger AT. Torsion-angle molecular dynamics as a new efficient tool for NMR structure calculation. J Magn Reson. 1997;124:154–164. doi: 10.1006/jmre.1996.1027. [DOI] [PubMed] [Google Scholar]
- 24.Parsons J, Holmes JB, Rojas JM, et al. Practical conversion from torsion space to Cartesian space for in silico protein synthesis. J Comput Chem. 2005;26:1063–1068. doi: 10.1002/jcc.20237. [DOI] [PubMed] [Google Scholar]
- 25.Tozzini V (2009) Multiscale modeling of proteins. Acc Chem Res [DOI] [PubMed]
- 26.Levitt M, Warshel A. Computer simulation of protein folding. Nature. 1975;253:694–698. doi: 10.1038/253694a0. [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Liwo A, Scheraga HA. Energy-based de novo protein folding by conformational space annealing and an off-lattice united-residue force field:application to the 10-55 fragment of staphylococcal protein A and to apo calbindin D9K. Proc Natl Acad Sci USA. 1999;96:2025–2030. doi: 10.1073/pnas.96.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDowell SE, Spackova N, Sponer J, et al. Molecular dynamics simulations of RNA:an in silico single molecule approach. Biopolymers. 2007;85:169–184. doi: 10.1002/bip.20620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuo G, Li W, Zhang J, et al. Folding of a small RNA hairpin based on simulation with replica exchange molecular dynamics. J Phys Chem B. 2010;114:5835–5839. doi: 10.1021/jp904573r. [DOI] [PubMed] [Google Scholar]
- 30.Deng NJ, Cieplak P. Free energy profile of RNA hairpins:a molecular dynamics simulation study. Biophys J. 2010;98:627–636. doi: 10.1016/j.bpj.2009.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auffinger P, Westhof E. H-bond stability in the tRNA(Asp) anticodon hairpin:3 ns of multiple molecular dynamics simulations. Biophys J. 1996;71:940–954. doi: 10.1016/S0006-3495(96)79298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarzynska J, Reblova K, Sponer J, et al. Conformational transitions of flanking purines in HIV-1 RNA dimerization initiation site kissing complexes studied by CHARMM explicit solvent molecular dynamics. Biopolymers. 2008;89:732–746. doi: 10.1002/bip.21001. [DOI] [PubMed] [Google Scholar]
- 33.Sanbonmatsu KY, Tung CS. High performance computing in biology:multimillion atom simulations of nanoscale systems. J Struct Biol. 2007;157:470–480. doi: 10.1016/j.jsb.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu XJ, Olson WK. 3DNA:a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulyanov NB, Gorin AA, Zhurkin VB. Conformational mechanics of the DNA double helix. A combined Monte Carlo and energy minimization approach. In: Kartashev LP, Kartashev SI, editors. Proceedings of the International Conference on Supercomputing'89: Supercomputer Applications: St. Petersburg: FL; 1989. pp. 368–370. [Google Scholar]
- 36.Lavery R, Zakrzewska K, Sklenar H. JUMNA (junction minimisation of nucleic acids) Comput Phys Commun. 1995;91:135–158. doi: 10.1016/0010-4655(95)00046-I. [DOI] [Google Scholar]
- 37.Malhotra A, Tan RK, Harvey SC. Prediction of the three-dimensional structure of Escherichia coli 30 S ribosomal subunit: a molecular mechanics approach. Proc Natl Acad Sci USA. 1990;87:1950–1954. doi: 10.1073/pnas.87.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan RKZ, Petrov AS, Harvey SC. YUP:A molecular simulation program for coarse-grained and multiscaled models. J Chem Theor Comput. 2006;2:529–540. doi: 10.1021/ct050323r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonikas MA, Radmer RJ, Laederach A, et al. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao S, Chen SJ. A new computational approach for mechanical folding kinetics of RNA hairpins. Biophys J. 2009;96:4024–4034. doi: 10.1016/j.bpj.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding F, Sharma S, Chalasani P, et al. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA. 2008;14:1164–1173. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasquali S, Derreumaux P. HiRE-RNA: a high resolution coarse-grained energy model for RNA. J Phys Chem B. 2010;114:11957–11966. doi: 10.1021/jp102497y. [DOI] [PubMed] [Google Scholar]
- 43.Mathews DH, Sabina J, Zuker M, et al. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 44.Cao S, Giedroc DP, Chen SJ. Predicting loop-helix tertiary structural contacts in RNA pseudoknots. RNA. 2010;16:538–552. doi: 10.1261/rna.1800210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krieger E, Nabuurs SB, Vriend G. Homology modeling. Methods biochem anal. 2003;44:509–523. doi: 10.1002/0471721204.ch25. [DOI] [PubMed] [Google Scholar]
- 47.Cohen-Gonsaud M, Catherinot V, Labesse G, et al. From molecular modeling to drug design. In: Bujnicki JM, et al., editors. Practical Bioinformatics. Berlin: Springer-Verlag; 2004. pp. 35–71. [Google Scholar]
- 48.Dror O, Nussinov R, Wolfson H (2005) ARTS:alignment of RNA tertiary structures. Bioinformatics 21 Suppl 2:ii47-ii53 [DOI] [PubMed]
- 49.Chothia C, Gerstein M. Protein evolution. How far can sequences diverge? Nature. 1997;385:579–581. doi: 10.1038/385579a0. [DOI] [PubMed] [Google Scholar]
- 50.Kumar S, Ma B, Tsai CJ, et al. Folding and binding cascades:dynamic landscapes and population shifts. Protein Sci. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pyle AM. Metal ions in the structure and function of RNA. J Biol Inorg Chem. 2002;7:679–690. doi: 10.1007/s00775-002-0387-6. [DOI] [PubMed] [Google Scholar]
- 52.Fiser A, Feig M, Brooks CL, 3rd, et al. Evolution and physics in comparative protein structure modeling. Acc Chem Res. 2002;35:413–421. doi: 10.1021/ar010061h. [DOI] [PubMed] [Google Scholar]
- 53.Grishin NV. Fold change in evolution of protein structures. J Struct Biol. 2001;134:167–185. doi: 10.1006/jsbi.2001.4335. [DOI] [PubMed] [Google Scholar]
- 54.Krasilnikov AS, Xiao Y, Pan T, et al. Basis for structural diversity in homologous RNAs. Science. 2004;306:104–107. doi: 10.1126/science.1101489. [DOI] [PubMed] [Google Scholar]
- 55.Peitsch MC. Protein Modelling by E-mail. Bio/Technology. 1995;13:658–660. doi: 10.1038/nbt0795-658. [DOI] [Google Scholar]
- 56.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 57.Flores SC, Wan Y, Russell R et al. (2010) Predicting RNA structure by multiple template homology modeling. Pac Symp Biocomput 216-227 [DOI] [PMC free article] [PubMed]
- 58.Rother M, Rother K, Puton T et al. (2011) ModeRNA:A tool for comparative modeling of RNA 3D structure. Nucleic Acids Res. (in press) [DOI] [PMC free article] [PubMed]
- 59.Ben-David M, Noivirt-Brik O, Paz A, et al. Assessment of CASP8 structure predictions for template free targets. Proteins. 2009;77(Suppl 9):50–65. doi: 10.1002/prot.22591. [DOI] [PubMed] [Google Scholar]
- 60.Cozzetto D, Kryshtafovych A, Fidelis K, et al. Evaluation of template-based models in CASP8 with standard measures. Proteins. 2009;77(Suppl 9):18–28. doi: 10.1002/prot.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simons KT, Kooperberg C, Huang E, et al. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 62.Kolinski A, Bujnicki JM. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins. 2005;61(Suppl 7):84–90. doi: 10.1002/prot.20723. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc Natl Acad Sci USA. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci USA. 2007;104:14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Das R, Karanicolas J, Baker D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Meth. 2010;7:291–294. doi: 10.1038/nmeth.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 67.Xia Z, Gardner DP, Gutell RR, et al. Coarse-grained model for simulation of RNA three-dimensional structures. J Phys Chem B. 2010;114:13497–13506. doi: 10.1021/jp104926t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jossinet F, Westhof E. Sequence to Structure (S2S):display, manipulate and interconnect RNA data from sequence to structure. Bioinformatics. 2005;21:3320–3321. doi: 10.1093/bioinformatics/bti504. [DOI] [PubMed] [Google Scholar]
- 69.Jossinet F, Ludwig TE, Westhof E (2010) Assemble:an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics 26:2057–2059 [DOI] [PMC free article] [PubMed]
- 70.Zwieb C, Muller F (1997) Three-dimensional comparative modeling of RNA. Nucleic Acids Symp Ser 69-71 [PubMed]
- 71.Martinez HM, Maizel JV, Jr, Shapiro BA. RNA2D3D:a program for generating, viewing, and comparing 3-dimensional models of RNA. J Biomol Struct Dyn. 2008;25:669–683. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kosinski J, Cymerman IA, Feder M, et al. A "FRankenstein's monster" approach to comparative modeling:merging the finest fragments of Fold-Recognition models and iterative model refinement aided by 3D structure evaluation. Proteins. 2003;53(Suppl 6):369–379. doi: 10.1002/prot.10545. [DOI] [PubMed] [Google Scholar]
- 73.Venclovas C. Comparative modeling in CASP5:progress is evident, but alignment errors remain a significant hindrance. Proteins. 2003;53(Suppl 6):380–388. doi: 10.1002/prot.10591. [DOI] [PubMed] [Google Scholar]
- 74.Bujnicki JM. Protein-structure prediction by recombination of fragments. Chembiochem. 2006;7:19–27. doi: 10.1002/cbic.200500235. [DOI] [PubMed] [Google Scholar]
- 75.Moult J, Fidelis K, Kryshtafovych A, et al. Critical assessment of methods of protein structure prediction - Round VIII. Proteins. 2009;77(Suppl 9):1–4. doi: 10.1002/prot.22589. [DOI] [PubMed] [Google Scholar]
- 76.Zemla A. LGA:A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 78.Levitt M, Gerstein M. A unified statistical framework for sequence comparison and structure comparison. Proc Natl Acad Sci USA. 1998;95:5913–5920. doi: 10.1073/pnas.95.11.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hajdin CE, Ding F, Dokholyan NV, et al. On the significance of an RNA tertiary structure prediction. RNA. 2010;16:1340–1349. doi: 10.1261/rna.1837410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parisien M, Cruz JA, Westhof E, et al. New metrics for comparing and assessing discrepancies between RNA 3D structures and models. RNA. 2009;15:1875–1885. doi: 10.1261/rna.1700409. [DOI] [PMC free article] [PubMed] [Google Scholar]