

Abstract
To elucidate the cellular role of the heterotrimeric G protein Go, we have taken a molecular genetic approach in Caenorhabditis elegans. We screened for suppressors of activated GOA-1 (Goα) that do not simply decrease its expression and found mutations in only two genes, sag-1 and eat-16. Animals defective in either gene display a hyperactive phenotype similar to that of goa-1 loss-of-function mutants. Double-mutant analysis indicates that both sag-1 and eat-16 act downstream of, or parallel to, Goα and negatively regulate EGL-30 (Gqα) signaling. eat-16 encodes a regulator of G protein signaling (RGS) most similar to the mammalian RGS7 and RGS9 proteins and can inhibit endogenous mammalian Gq/G11 in COS-7 cells. Animals defective in both sag-1 and eat-16 are inviable, but reducing function in egl-30 restores viability, indicating that the lethality of the eat-16; sag-1 double mutant is due to excessive Gqα activity. Analysis of these mutations indicates that the Go and Gq pathways function antagonistically in C. elegans, and that Goα negatively regulates the Gq pathway, possibly via EAT-16 or SAG-1. We propose that a major cellular role of Go is to antagonize signaling by Gq.
Keywords: C. elegans, Go protein, Gq protein, RGS protein, signaling, regulation
Goα, a member of the Gi subfamily, is the major heterotrimeric G protein α-subunit of the brain and exists only in species with a nervous system. Although Goα homologs have been isolated biochemically from several species, including cow (Sternweis and Robishaw 1984; Van Meurs et al. 1987), Drosophila (Yoon et al. 1989), Xenopus (Olate et al. 1989), hamster (Hsu et al. 1990), and man (Lavu et al. 1988), little is known about the mechanisms through which Goα functions. To elucidate these mechanisms, we are studying Caenorhabditis elegans Goα (GOA-1), which is 81%–82% identical to mammalian homologs (Lochrie et al. 1991) and is expressed throughout the entire C. elegans nervous system (M.R. Koelle, unpubl.; Mendel et al. 1995; Ségalat et al. 1995) and apparently also in some muscles (Mendel et al. 1995; Ségalat et al. 1995). GOA-1 modulates many behaviors, including locomotion and egg laying: mutants defective in goa-1 function display hyperactive egg-laying and locomotion behaviors, whereas transgenic animals overexpressing wild-type or constitutively activated GOA-1 are lethargic and egg-laying defective (Mendel et al. 1995; Ségalat et al. 1995). Heat shock-induced expression of activated GOA-1 results in lethargy at any developmental stage, indicating that GOA-1 can function throughout the life span of the worm (Mendel et al. 1995).
G protein subunits can function as switches in signal transduction (Simon et al. 1991; Hepler and Gilman 1992). When inactive, the Gα-subunit is bound to GDP and associated with the Gβγ-subunits. Upon activation of an associated transmembrane receptor by a ligand, the α-subunit exchanges GDP for GTP and dissociates from βγ. In this state, the α-subunit is free to interact with effector molecules. GTP hydrolysis inactivates the α-subunit, returning it to Gβγ. Free βγ can also interact with effectors (Birnbaumer 1992). Substitution of leucine for a glutamine in a residue required for GTPase activity (Q205L for GOA-1 and EGL-30) renders the Gα-subunit constitutively activated (Graziano and Gilman 1989).
GOA-1 activity is thought to be regulated by EGL-10 (Trent et al. 1983), which along with the yeast SST2 gene and GAIP first defined the RGS family of proteins (de Vries et al. 1995; Dohlman et al. 1996; Koelle and Horvitz 1996). RGS proteins negatively regulate G protein activity (Arshavsky and Pugh 1998; Berman and Gilman 1998) by acting as GTPase activating proteins (GAPs) for Gα-subunits, stabilizing the transition state during hydrolysis, and facilitating a rapid return to the inactive state (Berman et al. 1996a; Hunt et al. 1996; Watson et al. 1996; Faurobert and Hurley 1997). egl-10 loss-of-function mutant animals have the opposite phenotype as goa-1 loss-of-function mutants. Eliminating EGL-10 function in a mutant lacking GOA-1 has no additional phenotypic effect, suggesting that EGL-10 may act as a GAP specific for Goα in C. elegans (Koelle and Horvitz 1996). So far, 19 mammalian RGSs have been found (Berman and Gilman 1998), and the C. elegans genome project has identified 12 genes containing the RGS core domain (Sulston et al. 1992; the C. elegans Sequencing Consortium 1998). Whereas EGL-10 may be an RGS for C. elegans Goα, an RGS that regulates C. elegans Gqα has not yet been identified.
To identify components in Goα-mediated signaling, we performed random mutagenesis looking for suppressors of constitutively activated Goα in C. elegans, and we isolated mutations in two loci that appear to act downstream of, or parallel to, Goα based on epistasis analysis: sag-1, a new locus, and eat-16 (Avery 1993). Here, we present an analysis of the function of eat-16. We positionally cloned eat-16 and found that it encodes an RGS homolog with an expression pattern similar to that of GOA-1. Based on double- and triple-mutant analysis involving Goα, Gqα, sag-1, and eat-16, we believe that EAT-16 functions as an RGS for Gqα and that Goα may negatively regulate Gqα-mediated signaling in egg laying and locomotion. Consistent with our in vivo genetic data, EAT-16 can down-regulate the endogenous mammalian Gq/G11 when transfected into COS-7 cells. SAG-1 strongly inhibits Gqα-mediated signaling and may function downstream of, or parallel to, Gqα.
Results
Isolation of sag-1 and eat-16 mutations as suppressors of activated GOA-1
We performed a genetic screen for extragenic suppressors of syIs17, an integrated transgene expressing the constitutively activated goa-1[Q205L] mutant gene under the control of a heat shock promoter (hs-GoQL). Upon heat shock, syIs17 animals progressively cease locomotion, foraging, feeding, and production and laying of eggs (Mendel et al. 1995). Animals were mutagenized with either ethylmethanesulfonate (EMS; 21,000 haploid genomes) or trimethylpsoralen and UV irradiation (11,000 haploid genomes). The grandprogeny of mutagenized animals were heat-shocked as adults, and moving or foraging mutants were selected. In this manner, 15 independent suppressor strains were isolated that displayed a hyperactive phenotype (see below) in addition to suppressing syIs17[hs-GoQL] (Fig. 1). Fourteen of these mutations mapped to the same region and failed to complement one another, defining a new locus, sag-1 (suppressor of activated G protein). The other mutation, sy438, was allelic to eat-16(ad702), which was isolated previously in a screen for defects in pharyngeal pumping (Avery 1993). ad702, the original mutation defining eat-16, could also suppress the heat shock-induced lethargy of syIs17[hs-GoQL], as could sy438/ad702 trans-heterozygotes (data not shown).
Figure 1.
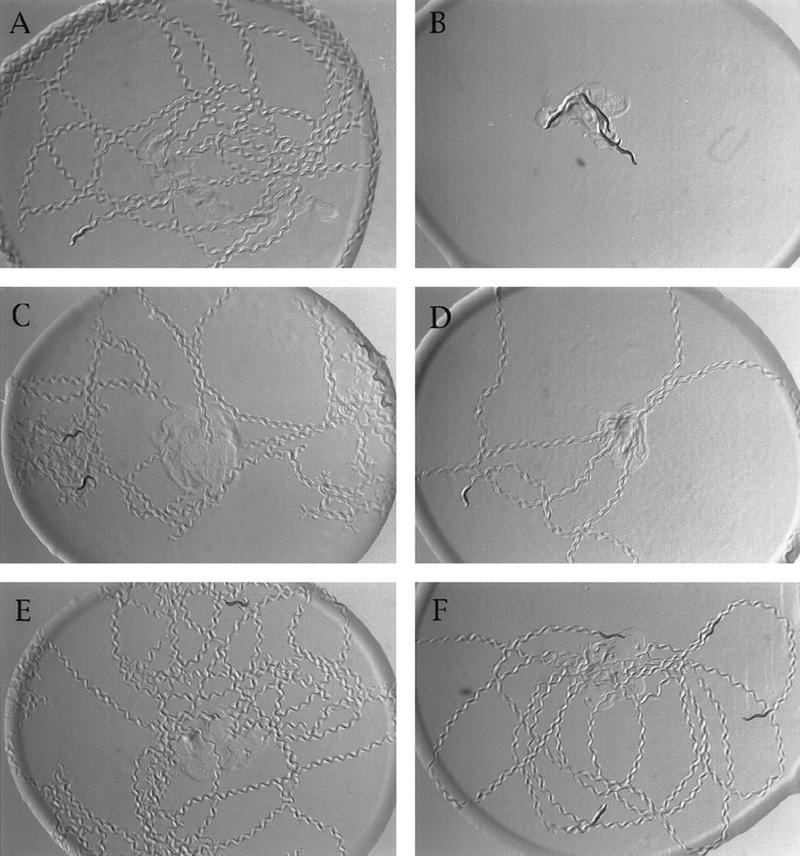
sag-1 and eat-16 mutations suppress the lethargy caused by heat shock of syIs17[hsp::goa-1(QL)]. Five adult worms were placed in the center of a bacterial lawn, allowed to crawl for 5 min, and photographed. Animals were then heat shocked and photographed 3 hr later. (A) dpy-20 syIs17 animals without heat shock. (B) dpy-20 syIs17 animals after heat shock treatment. (C) dpy-20 syIs17; sag-1(sy428). (D) The same dpy-20 syIs17; sag-1(sy428) animals after heat-shock treatment. (E) eat-16(sy438); dpy-20 syIs17. (F) The same eat-16(sy438); dpy-20 syIs17 animals after heat shock treatment.
Linkage tests eliminated the possibility that the suppression of hs-GoQL could have been due to a deletion of the syIs17 locus. All 14 sag-1 mutations were X-linked and resided close to unc-1. Three-factor mapping placed sag-1(sy428) between unc-1 and egl-17, whereas eat-16(sy438) mapped to linkage group (LG) I between unc-29 and lin-11 (see Fig. 2; Materials and Methods). In contrast, syIs17 maps to LG IV (J. Mendel, pers. comm.).
Figure 2.

Positional cloning of eat-16. (A)Physical map of eat-16. Eat-16 was mapped to the left half of the interval between unc-29 and lin-11 and to the right of hP6 (see Materials and Methods). Two YACs covering this region were injected into syIs17 dpy-20; eat-16(sy438); lin-15(n765). The eat-16 phenotype was rescued by Y20E10 but not Y54D2. Four cosmids between hP6 and the right end of Y20E10 were tested, and C16C2 rescued eat-16(sy438). (B)Subclones of cosmid C16C2; cDNA map of eat-16 and GFP constructs. Cosmid C16C2 has four open reading frames (C. elegans Sequencing Consortium 1998). The rescuing plasmid pYH5 contains the full sequence of C16C2.2 cloned into pBluescript; the first two exons of C16C2.3 are not included. The plasmid pWJC5 is an XbaI–AccI fragment from pYH5 cloned into pBluescript; it includes the same promoter region as pYH5, but the coding sequence of EAT-16 is terminated in the middle of RGS domain at amino acid 352. Full-length cDNA sequence of eat-16 was obtained from clone yk356b3. eat-16 has 10 exons; the RGS domain is contained within Exon 8, the largest exon. Reporter construct pGP16 includes 4.7 kb of the upstream promoter region and contains most of the eat-16 coding region, including the RGS domain. Reporter construct pGR02 contains the upstream promoter region but only the first coding exon of eat-16.
sag-1 and eat-16 mutants resemble goa-1(lf) mutants
sag-1 and eat-16 mutations not only suppressed the lethargy of hs-GoQL (Fig. 1) but in a wild-type background conferred a phenotype similar to that of goa-1 loss-of-function mutants (Mendel et al. 1995; Ségalat et al. 1995). Mutants laid eggs hyperactively, that is, soon after fertilization, resulting in eggs laid as early as uncleaved (Table 1). In addition, eggs were produced more slowly than the wild type (data not shown), resulting in uteri devoid of eggs (Table 1). Forward locomotion of sag-1 and eat-16 mutants was more rapid than wild type (Table 1). Conversely, pharyngeal pumping rates were impaired (Table 1); therefore, sag-1 and eat-16 mutants were somewhat starved and had a pale, scrawny appearance. Thus, the phenotypes of sag-1 and eat-16 mutants indicated that these genes might function in a Go-mediated signaling pathway.
Table 1.
Genetic characterization of sag-1 and eat-16 mutants
| Strain
|
Egg-laying phenotype
|
Forward locomotion (sine waves/min)
|
Animals (no.)
|
Feeding (pumps/min)
|
Animals (no.)
|
|||
|---|---|---|---|---|---|---|---|---|
| cells per egg
|
eggs (no.)
|
eggs in uterus
|
animals (no.)
|
|||||
| Wild type (N2) | >10 | 50 | 12 ± 1.8 | 10 | 25.3 ± 4.9 | 10 | 218 ± 21 | 10 |
| sag-1(sy428) | 3.0 ± 1.4 | 50 | 1.5 ± 0.9 | 10 | 37.4 ± 8.7 | 10 | 161 ± 43 | 10 |
| eat-16(sy438) | 2.2 ± 1.1 | 50 | 1.3 ± 0.7 | 10 | 41.8 ± 6.8 | 12 | 156 ± 37 | 10 |
| sy438/ + | >10 | 32 | 13 ± 3 | 6 | N.D. | N.D. | ||
| eat-16(ad702) | 2.2 ± 1.0 | 50 | 2.8 ± 2.0 | 10 | 51.3 ± 8.9 | 11 | 110 ± 30 | 10 |
| ad702/ + | >10 | 30 | 12 ± 3 | 8 | N.D. | N.D. | ||
| syIs9 [GoQ205L] | 100% latea | 25 | 16 ± 4.4 | 30 | 12.1 ± 5.1 | 9 | N.D. | |
| eat-16(sy438); syIs9 | 0% latea | 10 | 7.4 ± 2.9 | 5 | 35.9 ± 11.5 | 10 | N.D. | |
| syIs9; sag-1(sy428) | 85% latea | 27 | 6.6 ± 2.2 | 30 | 40.0 ± 8.1 | 9 | ||
Animals were assayed as described in Materials and Methods.
(N.D.) Not determined.
Late-stage eggs are defined as having at least 50 cells (see Materials and Methods).
goa-1(n363) null mutants crawl backwards with deeply exaggerated flexions compared with their forward locomotion (J. Mendel, unpubl.); this behavior was not observed in other goa-1 mutants (J. Mendel, unpubl.) or in sag-1(sy428) or eat-16 mutants. Either the n363 deletion, which removes more than the entire goa-1 coding region (Ségalat et al. 1995), also removes a neighboring gene responsible for this phenotype, or the behavior is mediated via a different mechanism than that involving SAG-1 or EAT-16.
Of the 14 sag-1 mutations, sy428 was used as the reference allele for all experiments: it displays a strong hyperactive phenotype and appears to be a null or strong reduction-of-function mutation. sy428 is recessive: One hundred percent of heterozygotes were wild type in appearance and had at least seven eggs in their uteri (n = 90), and sy428/Df heterozygous animals display a similar phenotype to that of sy428 homozygotes (see Materials and Methods). eat-16(ad702) and eat-16(sy438) have similar phenotypes (Table 1) and are reduction-of-function mutations (see below).
SAG-1 and EAT-16 do not affect GOA-1 expression
syIs17[hs-GoQL] was selected as the parent strain for mutagenesis because of its stability and ease of culture; however, mutations might suppress hs-GoQL by affecting heat shock-induced protein expression in general. Two experiments addressed this possibility. First, Western blot analysis indicated that mutations in sag-1 or eat-16 do not lower heat shock-induced GOA-1 expression (data not shown). Second, we examined the ability of sag-1(sy428) and eat-16(sy438) mutations to suppress activated GOA-1 under control of its normal regulatory sequences rather than a heat shock promoter. Both sag-1(sy428) and eat-16(sy438) suppressed the lethargy of syIs9, an integrated transgene of Goα [Q205L] under control of the goa-1 promoter (GoQL; Mendel et al. 1995; Table 1). These experiments suggested that SAG-1 and EAT-16 function in GOA-1 signaling rather than in GOA-1 expression.
SAG-1 and EAT-16 function downstream of, or parallel to, GOA-1
eat-16(sy438) suppressed the lethargy and egg-laying defect of syIs9[GoQL] (see above; Table 1), indicating that EAT-16 functions downstream of, or parallel to, GOA-1 and is required for GOA-1 signaling in both behaviors. Suppression of the GoQL locomotory defect by sag-1(sy428) was similarly robust (Table 1), indicating that SAG-1 likely functions downstream of GOA-1 at least with respect to locomotion. syIs9[GoQL]; sag-1(sy428) also laid fewer late-stage eggs than did syIs9[GoQL], but the egg-laying defect was only partially suppressed (Table 1).
In addition to suppressing activated Goα, sag-1(sy428) also significantly suppressed the egg-laying and locomotory defects of reduction-of-function mutations in egl-30, a C. elegans Gqα homolog that acts antagonistically to Goα, either in parallel or downstream (see below). In all cases, suppression of egl-30 by sag-1(sy428) was stronger than that by eat-16(sy438) (Table 2; see below). We infer that SAG-1 and EGL-30 act on a common process and that SAG-1 functions downstream of, or parallel to, EGL-30.
Table 2.
Suppression of egl-30 mutant phenotypes by sag-1 and eat-16
| Strain
|
Egg-laying defects
|
Forward locomotion (sine waves/min)
|
Animals (no.)
|
|||||
|---|---|---|---|---|---|---|---|---|
| late-stage eggs (%)a
|
eggs (no.)
|
eggs about to hatch (%)
|
eggs (no.)
|
eggs retained in uterus
|
animals (no.)
|
|||
| egl-30(ad805) | 100 | 6b | 83 | 6b | 26.1 ± 5.8 | 18 | N.A.c | |
| ad805 eat-16 | 75 | 8b | 0 | 8b | 27.1 ± 6.6 | 14 | N.A.c | |
|
ad805; sag-1
|
4
|
25
|
0
|
25
|
17.6 ± 4.0
|
19
|
17.8 ± 5.1
|
5
|
| egl-30(ad809) | 63 | 35 | 0 | 35 | 24.2 ± 2.9 | 21 | N.A.c | |
| ad809 eat-16 | 37 | 38 | 0 | 38 | 21.6 ± 3.7 | 22 | N.A.c | |
| ad809; sag-1 | 0 | 40 | 0 | 40 | 7.2 ± 1.6 | 21 | 26.2 ± 5.8 | 12 |
| egl-30(md186) | 77 | 13 | 31 | 13 | 22.9 ± 4.8 | 21 | 10.1 ± 3.7 | 9 |
| md186 eat-16 | 20 | 35 | 0 | 35 | 22.0 ± 3.6 | 20 | 14.0 ± 4.7 | 11 |
| md186; sag-1 | 3 | 35 | 0 | 35 | 13.9 ± 3.4 | 20 | 19.8 ± 4.1 | 6 |
| md186 eat-16; sag-1 | 0 | 50 | 0 | 50 | 9.4 ± 2.4 | 20 | 32.3 ± 8.7 | 10 |
| egl-30(n686) | 90 | 30 | 67 | 30 | 20.2 ± 4.9 | 20 | 16.7 ± 4.4 | 18 |
| n686 eat-16 | 53 | 49 | 12 | 49 | 15.2 ± 3.3 | 20 | 29.4 ± 9.7 | 9 |
| n686; sag-1 | 4 | 24 | 0 | 24 | 6.8 ± 2.0 | 18 | 35.4 ± 6.7 | 10 |
Strains were assayed as described in Materials and Methods. Double mutants were constructed with sag-1(sy428) and eat-16(sy438).
Late stage was defined as after the comma stage (see Materials and Methods).
Eggs were laid infrequently; therefore they were difficult to harvest.
(N.A.) Not available; mutants made few or no sinusoidal waves.
eat-16 encodes an RGS homolog
To understand the function of eat-16 in Goα-mediated signaling, we positionally cloned it by transformation rescue. We mapped eat-16 to the left half of the unc-29 lin-11 interval and to the right of mec-8 and hP6 (Fig. 2A; see Materials and Methods). Rescue was obtained with Y20E10, one of two YACs tested that reside in the left part of the interval between hP6 (Starr et al. 1989; Lundquist and Herman 1994) and lin-11 (Freyd et al. 1990), and with C16C2, one of four cosmids tested. C16C2 contains four predicted open reading frames, including C16C2.2, an RGS homolog. C16C2.2 resides entirely within one large intron of its oppositely oriented neighbor C16C2.3 (Fig. 2B). Injection of pYH5, a 7-kb Asp718–XbaI subclone containing the promoter region and all of C16C2.2 (Fig. 2B) rescued the eat-16(sy438) mutant phenotype.
All members of the RGS family have a designated 120-amino-acid RGS core domain (Tesmer et al. 1997). Some of them (RGS7, RGS9, EGL-10, and RGS11) are also highly conserved throughout the amino terminal region, which includes the DEP domain and the GGL domain. The function of the DEP domain is unknown, but the GGL domain is ∼34% identical to Gγ and can bind with Gβ in vitro (Snow et al. 1998). Full-length cDNA sequence of C16C2.2 was obtained from the clone yk356b3, a gift from Yuji Kohara (National Institute of Genetics, Mishima Japan. Sequence analysis indicates that C16C2.2 contains all three domains, making it most similar to RGS7, RGS9, and EGL-10 (Fig. 3).
Figure 3.

Sequence alignment of EAT-16 with RGS7, RGS9, and EGL-10. Regions of sequence similarity are highlighted. EAT-16 is 42% identical to mouse RGS7 in the RGS domain, 39% identical in the DEP domain, and 45% identical in the GGL domain; 41% identical to mouse RGS9 in the RGS domain, 37% identical in the DEP domain, and 34% identical in the GGL domain; and 30% identical to C. elegans EGL-10 in the RGS domain, 40% identical in the DEP domain, and 39% identical in the GGL domain. Arrowheads indicate the sites of eat-16 mutations. ad702 is AG → AA in the splice acceptor site before exon 4, sa735 is AG → AA in the splice acceptor site before exon 8, sa609 is Arg-396–Cys, and sy438 is Ser-400–Phe.
To determine whether the RGS domain of C16C2.2 is necessary to rescue the eat-16 mutant phenotype, we constructed pWJC5, a truncated genomic clone of eat-16 that lacks amino acids 353–474. The carboxy-terminal part of the RGS domain (where sa609 and sy438 are located; see below) is deleted in this construct (Fig. 2B). Injection of this plasmid failed to rescue the eat-16 phenotype, indicating that the RGS domain of C16C2.2 is required to suppress the phenotype of activated Goα, and the loss of it causes hyperactive egg laying and locomotion.
sy438 and ad702 are reduction-of-function alleles of eat-16
To verify that eat-16 encodes the C16C2.2 RGS protein, we amplified and sequenced C16C2.2 genomic DNA from each eat-16 mutant (see Materials and Methods). Each strain contained a single point mutation that was then confirmed by sequencing the opposite strand of DNA in the region of the mutation (Fig. 3). sy438 is a missense mutation that changes a conserved serine at position 400 in the RGS domain to a phenylalanine. ad702 is an AG → AA mutation in the splice acceptor site before the fourth exon, which is predicted to result in early termination before the RGS domain, although some properly spliced message is likely produced (see Aroian et al. 1993). We also sequenced two other alleles of eat-16, sa609 and sa735, kindly provided by M. Robatzek and J. Thomas (University of Washington, Seattle). sa735 is an AG → AA mutation in the splice acceptor site before the eighth exon (which contains the RGS domain), and sa609 is another missense mutation within the RGS domain that changes a conserved arginine at position 396 to a cysteine. Both sa609 and sa735 confer a phenotype similar to sy438 and ad702 (data not shown).
Because the missense mutations confer a phenotype similar to the splice acceptor site mutations, they likely reduce EAT-16 function. Although sy438 is essentially recessive (Table 1), we noticed that 6.5% of sy438/+ heterozygotes looked like sy438 homozygotes (n = 92). To test whether this effect was due to semidominance or haploinsufficiency, we examined by Nomarski optics animals heterozygous for the deficiency chromosome ces-1(n703d) qDf9 (Ellis and Kimble 1995), which deletes eat-16 and found that 58% of these animals had fewer than six eggs in their uteri and these eggs contained eight or fewer cells (n = 60 animals), indicating that the animals were laying eggs hyperactively. When placed in trans to qDf9, both sy438 and ad702 were viable and similar in phenotype to sy438 and ad702 homozygotes (data not shown). In contrast, animals bearing multiple copies of wild-type EAT-16 in the transgene syEx256 are somewhat egg-laying defective (data not shown). That presumed overexpression has a phenotype opposite that of sy438 and ad702 supports the conclusion that sy438 and ad702 reduce EAT-16 function.
The expression pattern of EAT-16 is similar to that of GOA-1
To examine expression, we made GFP translational fusions linking GFP either to the amino terminus or carboxyl terminus of eat-16. Reporter construct pGP16 (Fig. 2) contains a 7.4-kb ApaI–BamHI genomic fragment (including the eat-16 promoter region) and fuses to GFP-coding sequences in the ninth coding exon predicted by the cDNA sequence; this construct contains the entire amino-terminal region and the RGS domain. Examination of transgenic animals carrying pGP16 using confocal microscopy showed that EAT-16 is expressed in most or all neurons, including the hermaphrodite specific neuron (HSN) required for egg laying, as well as in the vulval and pharyngeal muscles (Fig. 4). Expression was also occasionally seen in the spermatheca and body wall muscles (data not shown). Similar expression patterns were seen when the GFP-coding sequences were fused to the first coding exon of eat-16. The expression pattern is consistent with the mutant phenotypes observed and is similar to the GOA-1 expression pattern (Mendel et al. 1995; Ségalat et al. 1995); therefore, eat-16 might act in many of the same cells as Goα.
Figure 4.
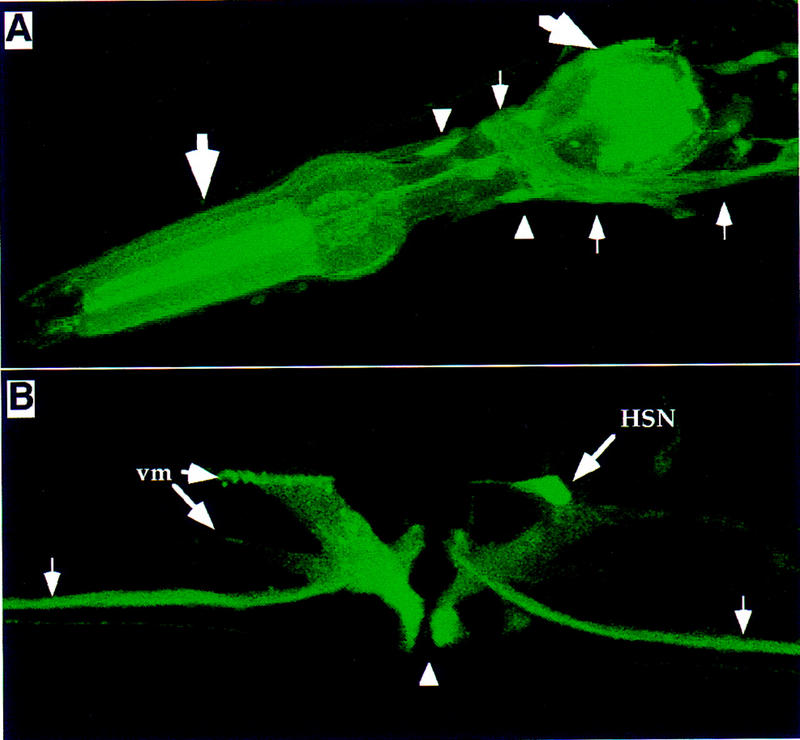
Expression of eat-16 in C. elegans. Transgenic animals carrying the eat-16::GFP reporter construct pGP16 were examined by confocal fluorescence microscopy. Similar expression patterns were seen using the reporter construct pGR02. (A) Adult head region, showing fluorescence in the cell bodies (arrowheads) and processes (small arrows) of many neurons, as well as in pharyngeal muscles (large arrows). (B) Adult vulva region. Vulval opening is indicated by the arrowhead. Fluorescence is detected in the HSN neuron, vulva muscles (vm), and ventral cord neurons (small arrows).
EAT-16 does not regulate GOA-1
RGS proteins have been shown to facilitate the inactivation of Gα-subunits by GTP hydrolysis (Berman et al. 1996a; Hunt et al. 1996; Watson et al. 1996; Faurobert and Hurley 1997); therefore, EAT-16 likely regulates one or more Gα-subunits in C. elegans. We thought it unlikely that Goα would be the target for EAT-16 based on the following arguments. First, the syIs17 parent strain expresses multiple copies of constitutively activated GOA-1(Q205L) upon heat shock, which would likely be insensitive to a wild-type RGS for Go (Berman et al. 1996b). Reducing the function of this RGS would render the excess activated subunits even more immune to down-regulation and therefore would not suppress the hs-GoQL phenotype. Second, if EAT-16 negatively regulates GOA-1, we would expect the eat-16 reduction-of-function phenotype to resemble that of activated Goα; instead, eat-16 mutants resemble goa-1 hypomorphs. Finally, eat-16 appears to act downstream of, or parallel to, goa-1 based on double-mutant analysis with syIs9[GoQL].
However, the similar expression patterns of EAT-16 and GOA-1 encouraged us to further test the possibility that EAT-16 regulates GOA-1. We overexpressed wild-type EAT-16 in a goa-1 null mutant background and found that the transgene syEx256[eat-16(+)] suppressed the hyperactive egg-laying behavior of goa-1(n363): Ten percent of eggs laid by goa-1; syEx256 animals were premature (n = 48), compared with 97% of eggs laid by their nontransgenic siblings (n = 34) (see Materials and Methods). If EAT-16 preferentially regulates GOA-1, we would have seen no suppression of goa-1(n363), because the n363 lesion deletes the entire goa-1 coding region (Ségalat et al. 1995). Therefore, we conclude that the major function of EAT-16 is not to regulate GOA-1 activity.
Genetic evidence that EAT-16 regulates EGL-30 Gqα
Because GOA-1 was an unlikely target for EAT-16, we considered other C. elegans Gα-subunits. Two of many Gα-subunits identified in C. elegans have been shown to affect the same sets of behaviors as Goα: EGL-30, the Gqα homolog (Brundage et al. 1996), and GSA-1, the Gsα homolog (Korswagen et al. 1997). Because the crystal structure of Gsα as well as biochemical evidence suggests that Gsα is not regulated by an RGS (Berman et al. 1996b; Tesmer et al. 1997; Natochin and Artemyev 1998), we focused our attention on Gqα. Whereas the putative null mutation, ad810, is lethal (Brundage et al. 1996), reduction-of-function mutations in egl-30 result in a lethargic and egg laying-defective phenotype roughly opposite to that of goa-1 reduction-of-function or null mutations. ad809 is a splice-donor site mutation, and ad805 and n686 are splice-acceptor site mutations; all result in reduced copies of full-length EGL-30 (Brundage et al. 1996). md186 is a missense mutation that reduces EGL-30 activity (Miller et al. 1996; L. Brundage, pers. comm.). The phenotypes of these reduction-of-function mutants vary in severity with ad805 having the strongest phenotype (Brundage et al. 1996). Because egl-30 and eat-16 reduction-of-function mutations have essentially opposite phenotypes, we asked whether EAT-16 might regulate EGL-30 activity.
We reasoned that if EAT-16 accelerates Gqα GTPase activity, reducing EAT-16 function should allow more Gqα-subunits to remain active, thereby alleviating the phenotype of a Gqα hypomorph, whereas reducing EAT-16 function in a null Gqα background should have no phenotypic effect. To test this hypothesis, we built double mutants between eat-16(sy438) and several egl-30 mutations. Although sy438 did not suppress the lethality of the putative null allele ad810 (see Materials and Methods), we found that sy438 partially suppressed the egg-laying defect of all hypomorphs tested and partially suppressed the locomotory defect of n686 (Table 2). These results support the hypothesis that EAT-16 inhibits EGL-30 activity.
To examine whether multiple copies of EAT-16 could compensate for EGL-30 overexpression, we overexpressed EAT-16 (using the transgene syEx256) in two different egl-30 transgenic strains (see Materials and Methods). Animals bearing syIs36[egl-30(+)], an integrated transgene overexpressing wild-type EGL-30 (L. Brundage, pers. comm.), move and lay eggs hyperactively (see Brundage et al. 1996). syIs36/+; syEx256 transgenic animals displayed various phenotypes (probably due to mosaicism of the syEx256 transgene), ranging from hyperactive (similar to syIs36) to slightly egg-laying defective (similar to syEx256); however, suppression of the pale, scrawny phenotype of syIs36[egl-30(+)] was observed in 50% of animals (n = 189). In contrast, overexpression of EAT-16 did not suppress the phenotype of overexpression of activated EGL-30(Q205L) under control of a heat shock promoter (L. Brundage, C. Bastiani, P.W. Sternberg, and M.I. Simon, unpubl.; data not shown). The Q205L mutation renders α-subunits insensitive to regulation by an RGS protein (Berman et al. 1996b). That we see suppression of wild-type, but not constitutively activated, EGL-30 by EAT-16 is consistent with a model in which EAT-16 inactivates EGL-30.
EAT-16 reduces endogenous Gq/G11 activity in COS-7 cells
The M1 receptor is coupled specifically to Gq/G11 in mammalian cells, and the activity of Gq/G11 can be measured by a PLCβ-IP3 assay (Berstein et al. 1992; Wu et al. 1992; Offermanns et al. 1994). We cotransfected COS-7 cells with expression constructs of M1 receptor and eat-16 (Fig. 5A) and observed that the addition of EAT-16 to the system significantly reduces the PLCβ activity caused by endogenous Gq/G11. A similar result was obtained when we cotransfected EGL-30 and EAT-16 (Fig. 5B; no M1 receptor was added), but because the stimulation of PLCβ activity by EGL-30 is not much greater than the background of endogenous Gq/G11, we cannot infer from this experiment that EAT-16 down-regulates EGL-30.
Figure 5.

EAT-16 down-regulates the PLCβ activity of endogenous Gq/G11 in COS-7 cells. (A) M1 receptor cotransfection. COS-7 cells were transfected with either control vector pCIS (left lane), 0.25 μg of M1 receptor (middle lane), or 0.25 μg of M1 receptor + 0.25 μg of EAT-16 (right lane). The total concentration of DNA was normalized to 1.0 μg per well using pCIS vector. (The M1 receptor activates endogenous Gq/G11.) Shown is the measured [3H]Inositol phosphate level 48 hr after transfection. Higher concentrations of EAT-16 (up to 1.5 μg per well) gave similar results (data not shown). (B) EGL-30 cotransfection. EAT-16 was cotransfected at various concentrations with EGL-30 into COS-7 cells. Total DNA concentration was normalized to 2.0 μg per well with pCIS. (No M1 receptor was added in this experiment.) PLCβ activity was caused by both endogenous Gq/G11 and EGL-30. (Top) COS-7 cells were transfected with control vector pCIS (left), 0.5 μg of EGL-30 (second from left), or 0.5 μg of EGL-30 with various concentrations of EAT-16 (right lanes). (Bottom) Same conditions as at top except that 1.0 μg of EGL-30 was transfected. (Shaded bars) s.d.; (solid bars) average.
Reducing EGL-30 function restores viability to eat-16; sag-1 mutants
We constructed a strain that segregates eat-16; sag-1 double mutants and found that >99% of the double mutants die (see Materials and Methods), arresting during larval development (Fig. 6A). Because each suppressor mutation results in a starved phenotype, one might argue that the lethality is caused by an additive starvation effect. However, goa-1(n363) has a more severe phenotype than either suppressor, and goa-1(n363); sag-1(sy428) double mutants are viable. Therefore SAG-1 and EAT-16 appear to act synergistically and are functionally redundant for survival.
Figure 6.
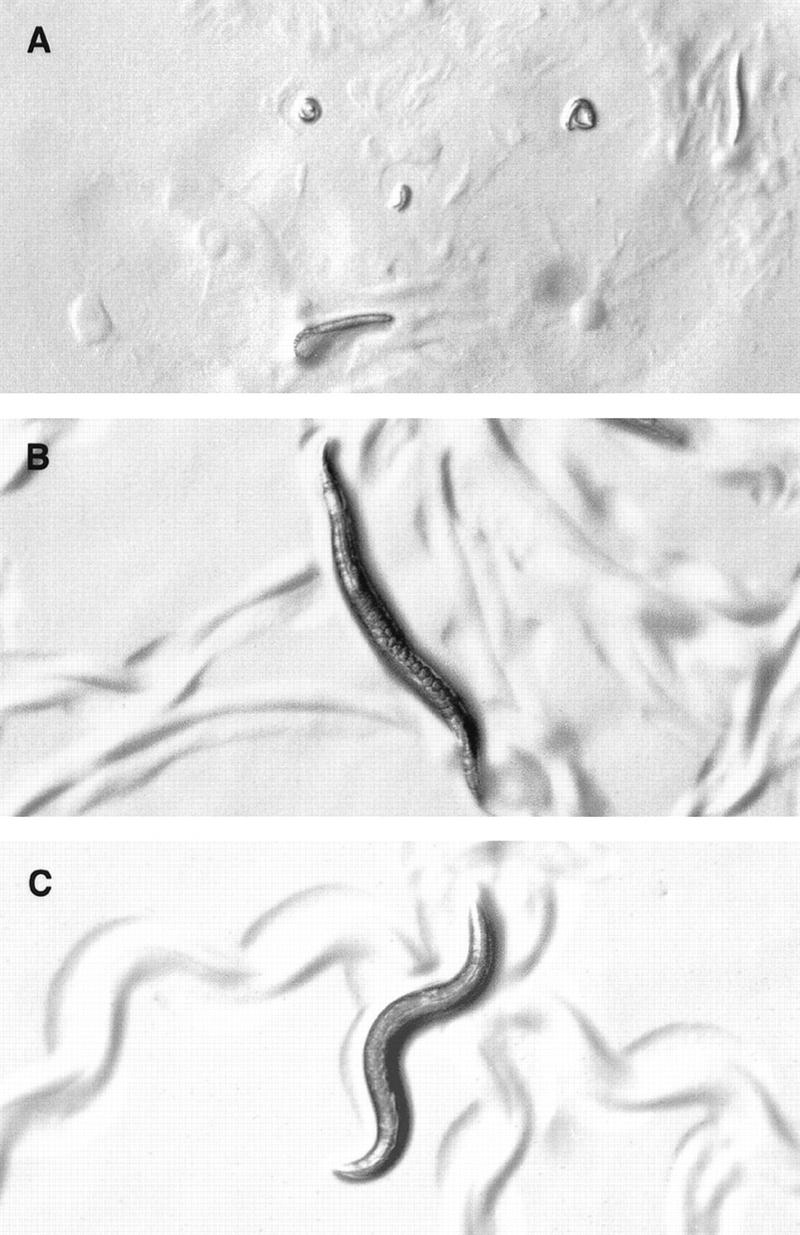
Reducing EGL-30 function restores viability to eat-16; sag-1 double mutants. All animals were photographed at the same magnification. (A) eat-16(sy438); sag-1(sy428) double mutants arrest as young larvae. (B) egl-30(md186) mutants are lethargic and egg-laying defective and leave flattened tracks (Miller et al. 1996). (C) egl-30(md186) eat-16(sy438); sag-1(sy428) triple mutants are viable and active, leaving sinusoidal tracks and laying eggs.
Because sag-1(sy428) significantly suppresses the phenotype of egl-30 hypomorphs (see above; Table 2), it seemed likely that SAG-1 and EAT-16 function synergistically to reduce EGL-30 signaling. If so, lowering EGL-30 signaling might suppress the lethality of eat-16; sag-1 double mutants. To test this hypothesis we constructed the egl-30(md186) eat-16(sy438); sag-1(sy428) triple mutant (see Materials and Methods) and found that it is viable to adulthood (Fig. 6C); this result indicates that excess EGL-30 activity is responsible for the lethality of eat-16; sag-1 double mutants. The lethargic and egg laying-defective phenotype of egl-30(md186) (Fig. 6B) was almost completely suppressed in the triple mutant (Table 2; Fig. 6C).
Goα antagonizes Gqα in C. elegans
Reduction-of-function mutations in goa-1 and egl-30 have essentially opposite phenotypes (Mendel et al. 1995; Ségalat et al. 1995; Brundage et al. 1996), suggesting that Goα and Gqα function antagonistically in C. elegans. egl-30(ad805) goa-1(n363) double mutants are lethargic and egg laying-defective like egl-30(ad805) animals (L. Brundage, P.W. Sternberg, and M.I. Simon, unpubl.), indicating that EGL-30 functions downstream of, or parallel to, GOA-1. Although on a gross level the double mutant resembled the ad805 single mutant, ad805 n363 had more active egg laying than ad805 alone: Fifteen percent of ad805 n363 eggs (n = 34) versus 100% of ad805 eggs (n = 12) were laid >5 hr after fertilization, respectively. The partial suppression of ad805 by n363 might be due to the presumably low level of wild-type EGL-30 activity expressed by the splice acceptor site mutant ad805 (especially because the egl-30 null phenotype is lethal; Brundage et al. 1996), which would be enhanced when GOA-1 activity is reduced. Nonetheless, the partial suppression of the egg-laying defect of egl-30(ad805) by goa-1(n363) is similar to that by eat-16(sy438) (see Table 2), consistent with GOA-1 and EAT-16 both negatively regulating EGL-30.
As with reducing gene function, overexpression of wild-type or activated GOA-1 and EGL-30 have opposite phenotypic effects (Mendel et al. 1995; Ségalat et al. 1995; Brundage et al. 1996). We reasoned that if the lethargy of syIs17[hs-GoQL] is due to excessive negative regulation of Gqα activity by activated Goα, then simultaneously overexpressing Gqα might suppress this lethargy. We tested this hypothesis by overexpressing both Gα-subunits in wild-type animals (see Materials and Methods) and found that syIs36[egl-30(+)], which overexpresses multiple copies of wild-type EGL-30, suppressed the heat shock-induced lethargy of syIs17[hsGoQL] (89 of 90 animals tested). Supporting these observations, our identification of genes involved in EGL-30 signaling as suppressors of activated GOA-1 suggests that Goα negatively regulates Gqα activity in C. elegans.
Discussion
To elucidate the largely unknown role of Goα in signal transduction, we screened for suppressors of activated GOA-1, the C. elegans Goα homolog. Because in C. elegans loss-of-function mutations occur at a frequency of 1 in 5000 to 1 in 2000 EMS-mutagenized gametes (Brenner 1974), our screen of 21,000 EMS-mutagenized gametes should be fairly representative of the C. elegans genome. In this EMS screen we isolated 13 mutations in sag-1 and 1 mutation in eat-16. The frequency with which we identified sag-1 mutations is consistent with their being reduction-of-function mutations. Mutations of eat-16 appear to be reduction-of-function alleles based on both genetic and molecular criteria. The low frequency with which eat-16 mutations were isolated suggests that weak mutations in EAT-16 might not suppress syIs17[hs-GoQL] well enough to be detected in our screen. In addition, Avery (1993) did not isolate any mutations in eat-16 in an F2 EMS mutagenesis of similar size; ad702 was isolated in an F1 screen designed to recover mutations at 100% efficiency (Avery 1993). These results and ours indicate that mutations in eat-16 are not easily recoverable; perhaps only mutations affecting the RGS domain (one exon) confer starvation and suppression of hs-GoQL. eat-16 and sag-1 mutants display a hyperactive phenotype similar to that of goa-1 loss-of-function mutants and are required for GOA-1 signaling. The simplest interpretation of our results is that EAT-16 and/or SAG-1 function as effectors for GOA-1; however, it is also possible that the effectors of GOA-1 either do not mutate to a viable phenotype or are numerous and functionally redundant.
EAT-16 and EGL-10 distinguish between Gq and Gi/Go subfamilies
Several lines of evidence indicate that EAT-16 does not inhibit Goα. eat-16 reduction-of-function mutations suppress the phenotype of an overexpressed, constitutively activated form of GOA-1 and phenotypically resemble goa-1 loss-of-function mutants; moreover, overexpression of EAT-16 compensates for a complete deletion of goa-1. We have presented genetic and biochemical data consistent with a model in which EAT-16 regulates the Gqα homolog EGL-30. A missense mutation in the RGS domain of eat-16 alleviates the phenotypes of several egl-30 reduction-of-function mutations but not that of the putative null allele ad810. Overexpression of EAT-16 can suppress the phenotype caused by overexpression of wild-type, but not constitutively activated, EGL-30. Cotransfection of EAT-16 and M1 receptor in COS-7 cells significantly reduces PLCβ activity resulting from endogenous mammalian Gq/G11, and a similar result was observed in cells cotransfected with EAT-16 and EGL-30. These results taken together argue that EAT-16 functions as a GAP for EGL-30.
EGL-10 and EAT-16, which are homologous to each other in both the amino-terminal region and the RGS domain, have similar GFP expression patterns but opposite phenotypic effects, indicating that they are selectively regulating different G proteins within the same cell. Eliminating EGL-10 function in a goa-1 null background has no additional phenotypic effect, suggesting that EGL-10 regulates Goα (Koelle and Horvitz 1996). Our results indicate that EAT-16 does not regulate Goα activity but instead regulates Gqα activity. Previous in vitro experiments on RGS proteins have provided some evidence that RGS proteins can act on Gq (Heximer et al. 1997; Zhang et al. 1998), but most RGS proteins examined could also act on Gi/o family members (Heximer et al. 1997; Zhang et al. 1998). Our results provide in vivo evidence that RGS7 homologs can distinguish among major families of Gα-subunits.
Negative regulation of Gq by Go
Analysis of double mutants involving goa-1 and egl-30 indicates that Goα and Gqα function antagonistically in C. elegans. Although it is possible that Gq and Go antagonize each other by positively and negatively regulating a common target, such as intracellular calcium, the identification of genes required for Goα signaling that negatively regulate Gqα signaling argues that Goα regulates behavior by modulating Gqα activity. Because we identified only one gene (eat-16) apparently upstream of egl-30 in a fairly extensive screen for downstream targets of GOA-1, the number of steps between Go and the Gq pathway might be small. In that view, Go might antagonize Gq directly or might antagonize a downstream target of Gq, perhaps via SAG-1. A third possibility is that Go antagonizes Gq signaling via EAT-16, in which case EAT-16 might be a direct effector for Goα as well as being an RGS for Gqα. Go could modulate the activity of other Gα-subunits by activating one or more RGS proteins such as EAT-16 that in turn could down-regulate other Gα-subunits.
The data presented here suggest a model for the functions of GOA-1, EGL-30, EAT-16, and SAG-1 (Fig. 7). GOA-1 negatively regulates EGL-30 activity, possibly via EAT-16 or SAG-1. EGL-10 selectively regulates GOA-1 activity, whereas EAT-16 selectively regulates EGL-30 activity. EGL-30 has been shown to activate PLCβ in COS-7 cells (Brundage et al. 1996); therefore, second messengers generated from stimulation of PLCβ by Gqα are probably produced downstream of EGL-30. Reducing EAT-16 function would result in elevated levels of active EGL-30 subunits, which would in turn result in higher levels of second messengers and increased mobilization of internal calcium stores (Berridge 1993). SAG-1 negatively regulates the EGL-30 pathway. Whereas moderate overexpression of wild-type EGL-30 has been shown to cause hyperactive egg-laying and locomotion behaviors (Brundage et al. 1996), more intense overexpression of EGL-30 results in lethality (L. Brundage, P.W. Sternberg, and M.I. Simon, unpubl.). We have shown that the eat-16(sy438); sag-1(sy428) double mutant is also inviable. The synthetic lethality of sag-1 and eat-16 mutations, as well as the lethality caused by expressing multiple copies of EGL-30, could be due to an excessive production of second messengers and/or excessive calcium release downstream of activated EGL-30. Reducing EGL-30 activity (and hence the level of downstream signaling) restores viability to eat-16; sag-1 mutants.
Figure 7.

A model for regulation of behavior by GOA-1, EAT-16, and SAG-1. Goα regulates behavior by antagonizing EGL-30-mediated signaling, either via EAT-16 and/or SAG-1 or an unknown effector. SAG-1 and EAT-16 function downstream, or parallel to, Goα and operate synergistically to negatively regulate Gqα signaling. EAT-16 regulates EGL-30 by accelerating EGL-30 GTPase activity. SAG-1 also negatively regulates EGL-30 signaling and likely functions downstream of EGL-30, based on the stronger suppression of egl-30 hypomorphs by sag-1(sy428).
Behavior is modulated through a network of G proteins
Our results are consistent with a model in which a network of G protein pathways within cells can affect behavior by both positive and negative cross talk. Although synergistic effects between Gi/o and Gq pathways have been observed (for review, see Selbie and Hill 1998), our results indicate negative regulation of Gqα or its downstream targets by Goα. That Go and Gq function antagonistically in some way was implied from the opposite phenotypes of goa-1 and egl-30 mutations (Brundage et al. 1996). The isolation and analysis of GOA-1 suppressors involved in Gqα signaling support the model that Goα functions to modulate behavior by down-regulating the Gq pathway in C. elegans and perhaps in other species as well. These results are analogous to the stimulatory and inhibitory effects of Gs and Gi on adenylyl cyclase (Hepler and Gilman 1992), raising the possibility that antagonistically acting G protein subunits are more universal than previously thought.
Materials and methods
Nematodes were cultured and handled according to standard procedures (Brenner 1974). All experiments were performed at 20°C except where otherwise noted. The following mutations and strains were used in this study for mapping experiments and double-mutant constructions: LGI egl-30(ad805), egl-30(ad809), DA1096 egl-30(ad810)/szT1 [lon-2(e678)] (Brundage et al. 1996), egl-30(md186), egl-30(n686), unc-55(e402), MT363 goa-1(n363) (Ségalat et al. 1995), dpy-5(e61), unc-29(e1072), SP1726 unc-29(h1) hP6 dpy-24(s71) (a gift from J.A. Powell-Coffman, Iowa State University, Ames, IA), mec-8(e398), lin-11(n566), JK1553 ces-1(n703d) qDf9/unc-29(e1702) lin-11(n566) (Ellis and Kimble 1995). LGII: unc-4(e120). LGIII: unc-32(e189). LGIV: dpy-20(e1282ts), PS1681 dpy-20 syIs17[hsp::goa-1(Q205L)] (Mendel et al. 1995), unc-31(e169). LGV: unc-42(e270), him-5(e1490). LGX: TY2137 meDf6; yDp13 (Akerib and Meyer 1994), PS1104 egl-17(e1313) sli-1(sy143) unc-1(e719), dpy-3(e27), unc-20(e112), lin-15(n765ts). Linkage unknown: syIs9[goa-1(Q205L)] (Mendel et al. 1995), syIs36[egl-30(+)] (L. Brundage, P.W. Sternberg, and M.I. Simon, unpubl.).
Genetic screen
dpy-20(e1282) syIs17[hsp::goa-1(Q205L)] animals (Mendel et al. 1995) were mutagenized with ethylmethanesulfonate (21,000 haploid genomes) or trimethylpsoralen + UV irradiation (Yandell et al. 1994; 11,000 haploid genomes). F2 progeny were heat-shocked (33°C, 30 min) as adults; moving animals were selected the next morning. All suppressors were backcrossed three times to the syIs17 parent strain, with the suppression of heat shock-induced lethargy used as the criterion for scoring; the reference alleles were then outcrossed to N2 for characterization by selecting for the empty uterus and pale, scrawny appearance. eat-16(sy438) was originally isolated with another linked mutation that was removed by recombination during mapping experiments.
Characterization of mutants and double-mutant strains
To characterize the egg laying phenotype, animals were examined 24–28 hr after selecting them as L4 larvae, except for syIs9[goa-1(Q205L)] and egl-30(n686) strains, which were selected from mixed stage plates as gravid young adults before excess egg retention. A large number of staged adults were placed on a plate with Escherichia coli OP50. Newly laid eggs were harvested every 10–20 min and examined at 125× or with Nomarski optics (for syIs9 strains). Cells in premature or wild-type eggs could be easily counted at this magnification. Later stage eggs were categorized qualitatively as follows: 20–50 cells (2–3 hr after fertilization), ∼50 cells, precomma (gastrulation is beginning, before comma stage), comma (∼5 hr after fertilization), twofold (∼7 hr after fertilization), threefold (∼9 hr after fertilization), and about to hatch. Eggs were considered premature if they contained eight or fewer cells. To count the number of eggs in the uterus, adults were examined at 125× magnification 24 hr after selecting as L4 larvae. N2, eat-16/+, syIs9, and Egl strains were bleached (as in Koelle and Horvitz 1996) to facilitate counting eggs. Hyperactive mutants were examined without bleaching.
To calculate pharyngeal pumps per minute, similarly staged adults were placed on individual plates seeded with OP50 and left undisturbed at least 15 min before counting. Pharyngeal pumps were counted for 2 min by pressing a counter once every three pumps; then, the numbers were multiplied by 1.5 to yield pumps per minute.
To calculate forward locomotion rate, staged adult animals were observed under conditions that maximize forward locomotion and minimize other behaviors (J. Mendel, pers. comm.): Two hundred microliters of a 5-ml OP50 culture was spread over the entire surface of a fresh 60-mm NGM plate preincubated at 20°C. Plates were left uncovered for the lawns to dry. After drying (which generally took ∼1 hr), the plates were stored with lids on and used within 2 hr after drying. The result was a very thin lawn that covered the entire plate. Animals were left undisturbed on the lawns at least 5 min and then observed for 2 min. Seconds elapsed per sine wave (counting anterior flexing just posterior to the pharynx) were recorded using software written for this purpose by Hou-Pu Chou and Chieh Chang. Only forward flexing was counted, and waves right before or after a reversal were not included. Entries for all animals were then converted to waves/second and averaged. Averages and standard deviations were multiplied by 60 to yield waves per minute.
Because the presence of excess eggs in the uterus might affect locomotion rate, egl-30 and syIs9 strains were not staged as above; instead, young gravid adults with a single row of eggs in the uterus were selected from mixed-stage plates. Because of syIs9 animals’ tendency to travel in a circular manner (J. Mendel, unpubl.), one side of the body would often make a more visible flexion and the other side would not flex much, if at all; counting was done using the side that made the deeper flexions. Occasional animals did not move normally and may have been harmed during transfer; these animals’ data were not included in the totals.
Characterization of sag-1/meDf6
meDf6; yDp13 males were mated to unc-4(e120); sag-1(sy428) dpy-3(e27) hermaphrodites and all nonUnc cross-progeny were selected as L4 larvae and examined 24 hr later. Cross-progeny were either non-Dpy or Dpy. meDf6 deletes sag-1 and dpy-3, and yDp13 likely covers sag-1 as well as dpy-3; therefore, Dpy progeny were assumed to be sag-1 dpy-3/meDf6, and Dp-bearing non-Dpy animals were examined in parallel as a control. All 16 meDf6/sag-1(sy428) dpy-3(e27) heterozygotes examined had empty uteri, and at least 15 of them suppressed the syIs17[hs-GoQL] lethargy (the sixteenth animal crawled off the plate after heat shock treatment and could not be scored).
Mapping experiments
sag-1(sy428) was mapped between egl-17 and unc-1 by selecting Egl non-Unc recombinant progeny from sy428/egl-17(e1313) sli-1(sy143) unc-1(e719) heterozygous animals; 14 of 16 recombinants carried the sy428 mutation. The following three-factor crosses determined the map position of eat-16(sy438). First, eat-16 was mapped between unc-29 and lin-11 by selecting recombinants from unc-29 lin-11/eat-16; dpy-20 syIs17 heterozygotes. Nine of 13 Lin non-Unc recombinants carried sy438 (scored by heat shock). Then, sy438 was mapped right of mec-8 by selecting Eat non-Dpy recombinants from dpy-5 eat-16/mec-8; dpy-20 syIs17 heterozygotes. One of seven recombinants carried mec-8. (Recombinants were scored for sy438 by the hyperactive phenotype and by heat shock.) Finally, sy438 was mapped right of hP6 by building + mec-8 + eat-16+/unc-29 + hP6 + dpy-24 heterozygotes and selecting for non-Mec recombinants with empty uteri. Unc progeny from these recombinants were homozygosed, and the presence of hP6 was determined by PCR amplification (Williams et al. 1992) using a mixture of three primers, 618 Tc1 primer (Williams et al. 1992), and two primers designed by J.A. Powell-Coffman (pers. comm.): hP6 (5′-TAGATTTTGATCGTCTTCG) and hP62 (5′-TGTCTCGCCTACGATCTGATATTGC). Two of 10 Eat non-Mec recombinants carried hP6.
Transformation rescue
Animals were microinjected according to standard protocols (Mello et al. 1991; Mello and Fire 1995). The lin-15 rescuing plasmid pbLH98 at 50 ng/μl (Huang et al. 1994) was used as the coinjection marker for all rescue experiments. pBluescript was included as carrier DNA to bring total DNA concentrations to 150–200 ng/μl. Strains bearing the temperature-sensitive lin-15(n765) mutation were cultured at 15°C before injection; afterwards, they were cultured at 22°C–23°C for 4–5 days, and non-Muv transformants were selected. Rescue was scored after at least one generation by heat-shocking non-Muv animals and looking for no suppression. Y20E10, C16C2, and three other cosmids contained within Y20E10 were each injected at ∼50 ng/μl into eat-16(sy438); dpy-20 syIs17; lin-15(n765) animals. Subclone pYH5 was injected at a concentration of 30 ng/μl, and pWJC5 was injected at a concentration of 25 ng/μl.
Sequencing of eat-16 mutations
A 3-kb genomic DNA fragment was amplified (Williams et al. 1992) from ad702 mutant animals in three independent reactions and from sy438 animals in 10 independent reactions using the Expand long-range PCR kit (Boehringer Mannheim) with the following primers (from 5′ to 3′): AGACAGCTTCGTCGTATGTCTCAC (“P1”) and GCAGTGTTGGGTGGTTCGAGATTG (“P2”); the products from each strain were gel-purified (Qiagen) and pooled. The ad702 fragment was amplified a second time with P2 and the nested primer TGTCGAGCTGATTGAGACACGCTG (‘S1’) in 10 independent reactions; the products were purified as above and pooled. For both strains, PCR fragments were cloned into pGEM vectors (Stratagene), and the entire predicted gene product was sequenced (Kretz et al. 1989) in two plasmids per strain. The point mutations were then confirmed in the second strand and in both strands of three additional plasmids for sy438 and four additional plasmids for ad702. For sa735 and sa609 mutants, products amplified as above in three independent PCR reactions were gel-purified, pooled, and sequenced directly.
Sequencing of eat-16 cDNA
Full-length cDNA sequence of eat-16 was obtained from clone yk356b3, kindly provided by Yuji Kohara. Phage clones were excised in vitro and amplified in SOLR cells (Maniatis et al. 1982). Purified phagemids were then sequenced by the primers used for sequencing the eat-16 mutations and primers for the T3 and T7 promoters. The splicing pattern was obtained by comparing yk356b3 with wild-type eat-16 genomic sequence obtained from the GenBank database.
GFP-tagged expression of eat-16
Genomic DNA fragments including the eat-16 promoter region and some coding exons were cloned into GFP expression vectors provided by A. Fire, J. Ahnn, G. Seydoux, and S. Xu (pers. comm.). Reporter construct pGP16 contains the 7.4-kb ApaI–BamHI fragment and fuses to GFP-coding sequences in the ninth coding exon of eat-16. Reporter construct pGR02 contains the same upstream sequence but fuses to GFP-coding sequences in the first coding exon of eat-16. Both constructs were injected at 80 ng/μl into lin-15(n765) animals along with the lin-15 rescuing plasmid pL15EK at 50 ng/μl (Clark et al. 1994) as a coinjection marker.
Double-mutant constructions
egl-30 eat-16 linked double mutants were constructed as follows: dpy-5 eat-16/++ males were mated to egl-30 hermaphrodites (or egl-30/szT1 heterozygotes, in the case of ad810). Non-Egl F1 progeny were picked individually and removed the following day to synchronize the F2 progeny. Plates with Dpy F2 progeny were saved, and Eat non-Dpy animals were selected based on the empty uterus phenotype (which was transitory in these recombinants due to the semidominance of the egl-30 alleles; see Brundage et al. 1996). Egl non-Dpy F3 progeny were saved, and the presence of eat-16(sy438) was confirmed by mating with N2 males and reisolating Eat non-Egl F2 recombinants from all of several F1 cross-progeny.
In the case of ad810, two Eat non-Dpy recombinants segregated Eat, Eat Dpy and arrested larvae; no viable Egl progeny were seen. Recombinants were mated with szT1 males to balance the lethal chromosome. A parallel comparison of the lethal progeny of ad810 sy438/szT1 and ad810/szT1 was done by placing 16 worms of each strain on a plate with a thin bacterial lawn and removing them the next day. Dead larvae on both plates were observed over the course of several days and appeared similar.
Construction of double mutants between unlinked genes was straightforward; the strains were all confirmed either by complementation tests or by crossing with N2 males and reisolating both mutations. egl-30(ad805) goa-1(n363); him-5(e1490) was built by L. Brundage.
Transgenic strains
Strains containing eat-16 transgenes were constructed by following the marker lin-15(n765) for syEx256, whereas Go and Gq transgenes were followed by using dpy-20(e1282) as a rescuing marker. For example, a cross between dpy-20 males and dpy-20; lin-15; syEx256 hermaphrodites was kept for one day at 20°C to allow mating to occur and then cultured at 15°C to reduce the severity of the Dpy phenotype of the F1 progeny. The resulting dpy-20; lin-15; syEx256 males were mated to dpy-20; syIs36; lin-15 hermaphrodites at 20°C, and non-Lin progeny were saved. The goa-1(n363); syEx256 strain was constructed by mating dpy-20/+; lin-15; syEx256 males to goa-1(n363); lin-15 hermaphrodites and saving non-Dpy, non-Muv transgenic F2 animals whose Lin (i.e., nontransgenic) progeny were all homozygous for goa-1(n363). To test animals expressing both Go and Gq transgenes, dpy-20(e1282) syIs17 males were mated to dpy-20(e1282); syIs36 hermaphrodites, and the resulting male progeny were heat-shocked.
Synthetic lethality of eat-16 and sag-1
To build the eat-16; sag-1 double mutant, mec-8 lin-11/++; sag-1 males were mated to dpy-5 eat-16 hermaphrodites. Non-Dpy F1 progeny were picked to individual plates, and homozygous Sag F2s were picked from plates with Mec Lin progeny. To score penetrance of the lethality, 20 dpy-5 + eat-16+/+mec-8 + lin-11; sag-1 L4 heterozygotes were placed on individual plates and transferred daily for 4 days. The total number of progeny was counted at the L4-Adult stage, yielding 2193 heterozygous, 936 Mec, and 52 Dpy animals. Dpy animals were saved and followed for a generation to determine their genotype. Of the 50 viable Dpy animals, 48 were either dpy++/dpy mec lin or dpy++/++lin recombinants, one was a spontaneous male whose genotype could not be determined, and one escaped to adulthood and produced a few inviable progeny, a 0.1% survival rate. Arrested larvae were seen among the progeny of all 20 heterozygous mothers.
egl-30 eat-16; sag-1 triple-mutant construction
+eat-16+/mec-8 + lin-11; him-5; sag-1 males were mated to egl-30(md186) eat-16 hermaphrodites, and F1 progeny with empty uteri were picked to individual plates. From two plates that segregated Eat, Egl, and dead larvae but no Mec Lin (i.e., egl eat/+ eat; sag-1/+), 26 Egl F2 progeny were picked to individual plates. None of the Egls produced lethal progeny. Seven of 26 Egls gave only Egl-30-like progeny, but on 18 plates, about one-fourth of the progeny were active, and one plate had only active progeny. Homozygosity of sag-1 was confirmed by mating dpy-20 syIs17 males with the triple mutant; male progeny were heat-shocked, and all were active 8 hr following heat shock (n = 36). As a control, egl-30(md186) eat-16(sy438) animals were also crossed to syIs17 males; the male progeny resulting from this cross were lethargic 8 hr following heat shock (n = 20). Homozygosity of eat-16 was confirmed by sequencing across the portion of the eat-16 locus containing the sy438 mutation in both strands, using a protocol similar to that described above.
COS-7 cell transfection and IP3 assay
A 2.4-kb XhoI–XbaI fragment from yk356b3 (EAT-16 cDNA) was cloned into the pCIS vector to make pWJC4 for COS-7 cell transfection. M1 receptor (Bo Yu, pers. comm.) and EGL-30 (L. Brundage, pers. comm.) were also cloned in the same vector. Cells were transfected as described (Wu et al. 1992; Liu and Simon 1996) and seeded (1 × 105 per well) to 12-well plates the night before transfection. The pCIS vector was used as carrier DNA to normalize the total concentration of DNA in each well to 1.0 μg (for M1 receptor experiments) or 2.0 μg (for EGL-30 transfections). Lipofectin (5 μl) was added to each well. M1 receptor was activated by Carbachol (1 μm), and IP3 assays were done as described (Liu and Simon 1996). Each transfection was performed in duplicate.
Acknowledgments
We thank Jane Mendel for the locomotion assay conditions; Hou-Pu Chou and Chieh Chang for writing software used to record locomotion data; John DeModena for technical assistance; Yuji Kohara for cDNA; Andrew Fire, Joohong Ahnn, Geraldine Seydoux, and Siqun Xu for GFP vectors; Merrilee Robatzek and James Thomas for sa609 and sa735; Bo Yu, Jennifer Gu, and Valeria Mancino for assistance with COS-7 cell assays; Lorna Brundage, Carol Bastiani, Stephen Nurrish, Laurant Ségalat, Joshua Kaplan, Ron Ellis, and Jo Anne Powell-Coffman for sharing strains and communicating unpublished results; and Lorna Brundage, L. René Garcia, Jane Mendel, Carol Bastiani, Maureen Barr, Nadeem Moghal, Henry Lester, Mel Simon, and members of our laboratories for stimulating discussions and critical reading of this manuscript. Some of the strains used in this study were provided by the Caenorhabditis Genetics Center. This project is supported by HHMI, with which P.W.S. is an investigator.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pws@cco.caltech.edu; FAX (626) 568-8012.
References
- Akerib CC, Meyer BJ. Identification of X chromosome regions in Caenorhabditis elegans that contain sex-determination signal elements. Genetics. 1994;138:1105–1125. doi: 10.1093/genetics/138.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian RV, Levy AD, Koga M, Ohshima Y, Kramer JM, Sternberg PW. Splicing in Caeonorhabditis elegans does not require an AG at the 3′ splice acceptor site. Mol Cell Biol. 1993;13:626–637. doi: 10.1128/mcb.13.1.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshavsky VY, Pugh EN., Jr Lifetime regulation of G protein-effector complex: Emerging importance of RGS proteins. Neuron. 1998;20:11–14. doi: 10.1016/s0896-6273(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Avery L. The genetics of feeding in Caenorhabditis elegans. Genetics. 1993;133:897–917. doi: 10.1093/genetics/133.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DM, Gilman AG. Mammalian RGS proteins: Barbarians at the gate. J Biol Chem. 1998;273:1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- Berman DM, Kozasa T, Gilman AG. The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J Biol Chem. 1996a;271:27209–27212. doi: 10.1074/jbc.271.44.27209. [DOI] [PubMed] [Google Scholar]
- Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-Activating Proteins for the Gi subfamily of G protein α subunits. Cell. 1996b;88:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berstein G, Blank JL, Jhon DY, Exton JH, Rhee SG, Ross EM. Phospholipase C-beta 1 is a GTPase-activating protein for Gq/11, its physiologic regulator. Cell. 1992;70:411–418. doi: 10.1016/0092-8674(92)90165-9. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L. Receptor-to-effector signaling through G proteins: Roles for beta gamma dimers as well as alpha subunits. Cell. 1992;71:1069–1072. doi: 10.1016/s0092-8674(05)80056-x. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, Sternberg PW, Simon MI. Mutations in a C. elegans Gqalpha gene disrupt movement, egg laying, and viability. Neuron. 1996;16:999–1009. doi: 10.1016/s0896-6273(00)80123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology [published erratum appears in Science 1999 Jan 1; 283: (5398)35] Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Clark SG, Lu X, Horvitz HR. The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics. 1994;137:987–997. doi: 10.1093/genetics/137.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson A, Sulston J, Brenner S, Karn J. Toward a physical map of the genome of the nematode Caenorhabditis elegans. Proc Natl Acad Sci. 1986;83:7821–7825. doi: 10.1073/pnas.83.20.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson A, Waterston R, Kiff J, Sulston J, Kohara Y. Genome linking with yeast artificial chromosomes. Nature. 1988;335:184–186. doi: 10.1038/335184a0. [DOI] [PubMed] [Google Scholar]
- De Vries L, Mousli M, Wurmser A, Farquhar MG. GAIP, a protein that specifically interacts with the trimeric G protein G alpha i3, is a member of a protein family with a highly conserved core domain. Proc Natl Acad Sci. 1995;92:11916–11920. doi: 10.1073/pnas.92.25.11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Song J, Ma D, Courchesne WE, Thorner J. Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: Expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein alpha subunit) Mol Cell Biol. 1996;16:5194–5209. doi: 10.1128/mcb.16.9.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Thorner J. RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem. 1997;272:3871–3874. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Kimble J. The fog-3 gene and regulation of cell fate in the germ line of Caenorhabditis elegans. Genetics. 1995;139:561–577. doi: 10.1093/genetics/139.2.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faurobert E, Hurley JB. The core domain of a new retina specific RGS protein stimulates the GTPase activity of transducin in vitro. Proc Natl Acad Sci. 1997;94:2945–2950. doi: 10.1073/pnas.94.7.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyd G, Kim SK, Horvitz HR. Novel cysteine-rich motif and homeodomain in the product of the Caenorhabditis elegans cell lineage gene lin-11. Nature. 1990;344:876–879. doi: 10.1038/344876a0. [DOI] [PubMed] [Google Scholar]
- Graziano MP, Gilman AG. Synthesis in Escherichia coli of GTPase-deficient mutants of Gs alpha. J Biol Chem. 1989;264:15475–15482. [PubMed] [Google Scholar]
- Hepler JR, Gilman AG. G proteins. Trends Biochem Sci. 1992;17:383–387. doi: 10.1016/0968-0004(92)90005-t. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gq alpha function. Proc Natl Acad Sci. 1997;94:14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin MN, Pettitt TR, Martin A, Michell RH, Pemberton AJ, Wakelam MJ. Diacylglycerols and phosphatidates: Which molecular species are intracellular messengers? Trends Biochem Sci. 1998;23:200–204. doi: 10.1016/s0968-0004(98)01200-6. [DOI] [PubMed] [Google Scholar]
- Hsu WH, Rudolph U, Sanford J, Bertrand P, Olate J, Nelson C, Moss LG, Boyd AE, Codina J, Birnbaumer L. Molecular cloning of a novel splice variant of the alpha subunit of the mammalian Go protein. J Biol Chem. 1990;265:11220–11226. [PubMed] [Google Scholar]
- Huang LS, Tzou P, Sternberg PW. The lin-15 locus encodes two negative regulators of Caenorhabditis elegans vulval development. Mol Biol Cell. 1994;5:395–411. doi: 10.1091/mbc.5.4.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt TW, Fields TA, Casey PJ, Peralta EG. RGS10 is a selective activator of Gai GTPase activity. Nature. 1996;383:175–177. doi: 10.1038/383175a0. [DOI] [PubMed] [Google Scholar]
- Koelle MR. A new family of G-protein regulators—the RGS proteins. Curr Opin Cell Biol. 1997;9:143–147. doi: 10.1016/s0955-0674(97)80055-5. [DOI] [PubMed] [Google Scholar]
- Koelle MR, Horvitz HR. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84:115–125. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- Korswagen HC, Park JH, Ohshima Y, Plasterk RH. An activating mutation in a Caenorhabditis elegans Gs protein induces neural degeneration. Genes & Dev. 1997;11:1493–1503. doi: 10.1101/gad.11.12.1493. [DOI] [PubMed] [Google Scholar]
- Kretz KA, Carson GS, O’Brien JS. Direct sequencing from low-melt agarose with Sequenase [published erratum appears in Nucleic Acids Res. 1990. 18: (2)400] Nucleic Acids Res. 1989;17:5864. doi: 10.1093/nar/17.14.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavu S, Clark J, Swarup R, Matsushima K, Paturu K, Moss J, Kung HF. Molecular cloning and DNA sequence analysis of the human guanine nucleotide-binding protein Go alpha [published erratum appears in Biochem. Biophys. Res. Commun. 1988. 153: (1)487] Biochem Biophys Res Commun. 1988;150:811–815. doi: 10.1016/0006-291x(88)90463-9. [DOI] [PubMed] [Google Scholar]
- Liu M, Simon MI. Regulation by cAMP-dependent protein kinease of a G-protein-mediated phospholipase C. Nature. 1996;382:83–87. doi: 10.1038/382083a0. [DOI] [PubMed] [Google Scholar]
- Lochrie MA, Mendel JE, Sternberg PW, Simon MI. Homologous and unique G protein alpha subunits in the nematode Caenorhabditis elegans. Cell Regul. 1991;2:135–154. doi: 10.1091/mbc.2.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist EA, Herman RK. The mec-8 gene of Caenorhabditis elegans affects muscle and sensory neuron function and interacts with three other genes: unc-52, smu-1 and smu-2. Genetics. 1994;138:83–101. doi: 10.1093/genetics/138.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: A laboratory manual. Vol. 1. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. pp. 2.60–2.63. [Google Scholar]
- Mello C, Fire A. DNA Transformation. In: Shakes DC, Epstein HF, editors. Caenorhabditis elegans: Modern biological analysis of an organism. San Diego, CA: Academic Press; 1995. pp. 451–482. [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, Plasterk RH, Sternberg PW. Participation of the protein Go in multiple aspects of behavior in C. elegans. Science. 1995;267:1652–1655. doi: 10.1126/science.7886455. [DOI] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natochin M, Artemyev NO. A single mutation Asp229 → Ser confers upon Gs alpha the ability to interact with regulators of G protein signaling. Biochemistry. 1998;37:13776–13780. doi: 10.1021/bi981155a. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Wieland T, Homann D, Sandmann J, Bombien E, Spicher K, Schultz G, Jakobs KH. Transfected muscarinic acetylcholine receptors selectively couple to Gi-type G proteins and Gq/11. Mol Pharmacol. 1994;45:890–898. [PubMed] [Google Scholar]
- Olate J, Jorquera H, Purcell P, Codina J, Birnbaumer L, Allende JE. Molecular cloning and sequence determination of a cDNA coding for the alpha-subunit of a Go-type protein of Xenopus laevis oocytes [published erratum appears in FEBS Lett. 1990 Jul 16; 267: (2)316] FEBS Lett. 1989;244:188–192. doi: 10.1016/0014-5793(89)81190-1. [DOI] [PubMed] [Google Scholar]
- Ségalat L, Elkes DA, Kaplan JM. Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science. 1995;267:1648–1651. doi: 10.1126/science.7886454. [DOI] [PubMed] [Google Scholar]
- Selbie LA, Hill SJ. G protein-coupled-receptor cross-talk: The fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol Sci. 1998;19:87–93. doi: 10.1016/s0165-6147(97)01166-8. [DOI] [PubMed] [Google Scholar]
- Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- Snow BE, Krumins AM, Brothers GM, Lee SF, Wall MA, Chung S, Mangion J, Arya S, Gilman AG, Siderovski DP. A G protein gamma subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gbeta5 subunits. Proc Natl Acad Sci. 1998;95:13307–13312. doi: 10.1073/pnas.95.22.13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondek J, Lambright DG, Noel JP, Hamm HE, Sigler PB. GTPase mechanism of G proteins from the 1.7-A crystal structure of transducin alpha-GDP-AIF-4. Nature. 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- Starr T, Howell AM, McDowall J, Peters K, Rose AM. Isolation and mapping of DNA probes within the linkage group I gene cluster of Caenorhabditis elegans. Genome. 1989;32:365–372. doi: 10.1139/g89-456. [DOI] [PubMed] [Google Scholar]
- Sternweis PC, Robishaw JD. Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J Biol Chem. 1984;259:13806–13813. [PubMed] [Google Scholar]
- Sulston J, Du Z, Thomas K, Wilson R, Hillier L, Staden R, Halloran N, Green P, Thierry-Mieg J, Qiu L, et al. The C. elegans genome sequencing project: A beginning. Nature. 1992;356:37–41. doi: 10.1038/356037a0. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AIF4–activated Gia1: Stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsung N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Voorn L, Gebbink M, Plasterk RHA, Ploegh HL. Characterization of a G-protein b-subunit gene from the Caenorhabditis elegans. J Mol Biol. 1990;213:17–26. doi: 10.1016/s0022-2836(05)80118-4. [DOI] [PubMed] [Google Scholar]
- Van Meurs KP, Angus CW, Lavu S, Kung HF, Czarnecki SK, Moss J, Vaughan M. Deduced amino acid sequence of bovine retinal Go alpha: Similarities to other guanine nucleotide-binding proteins. Proc Natl Acad Sci. 1987;84:3107–3111. doi: 10.1073/pnas.84.10.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-proteins a-subunits. Nature. 1996;383:172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- Williams BD, Schrank B, Huynh C, Shownkeen R, Waterston RH. A genetic mapping system in Caenorhabditis elegans based on polymorphic sequence-tagged sites. Genetics. 1992;131:609–624. doi: 10.1093/genetics/131.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem. 1992;267:1811–1817. [PubMed] [Google Scholar]
- Yandell MD, Edgar LG, Wood WB. Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc Natl Acad Sci. 1994;91:1381–1385. doi: 10.1073/pnas.91.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J, Shortridge RD, Bloomquist BT, Schneuwly S, Perdew MH, Pak WL. Molecular characterization of Drosophila gene encoding Go alpha subunit homolog. J Biol Chem. 1989;264:18536–18543. [PubMed] [Google Scholar]
- Zhang S, Watson N, Zahner J, Rottman JN, Blumer KJ, Muslin AJ. RGS3 and RGS4 are GTPase activating proteins in the heart. J Mol Cell Cardiol. 1998;30:269–276. doi: 10.1006/jmcc.1997.0591. [DOI] [PubMed] [Google Scholar]