

Abstract
Transcription control at the melting step is not yet understood. Here, band shift, cross-linking, and transcription experiments on diverse DNA probes were used with two bacterial RNA polymerase holoenzymes that differ in how they regulate melting. Data indicated that both σ54 and σ70 holoenzymes assume a default closed form that cannot establish single-strand binding. Upon activation the enzymes are converted to an open form that can bind simultaneously to the upstream fork junction and to the melted transcription start site. The key difference is that σ54 imposes tighter regulation by creating a complex molecular switch at −12/−11; the current data show that this switch can be thrown by activator. In this case an ATP-bound enhancer protein causes σ54 to alter its cross-linking pattern near −11 and also causes a reorganization of holoenzyme: DNA interactions, detected by electrophoretic mobility-shift assay. At a temperature-dependent σ70 promoter, elevated temperature alone can assist in triggering conformational changes that enhance the engagement of single-strand DNA. Thus, the two σ factors modify the same intrinsic opening pathway to create quite different mechanisms of transcriptional regulation.
Keywords: σ factors, transcription, promoter opening, NtrC, DNA fork junction
Bacterial RNA polymerases function primarily in two forms, in preinitiation complexes containing σ factors and in elongation complexes devoid of σ. Recent structural advances (Zhang et al. 1999) have been merged with genetics and biochemistry (for review, see Nudler 1999) to give a view of the elongation complex. A comparable level of detail is not yet available for preinitiation complexes. However, the structure of the core enzyme and recent advances from in vitro studies allow new models to be proposed. These are badly needed to understand the process of opening the DNA, which is a key point in initiating transcription.
Both open preinitiation complexes and elongation complexes contain a transcription bubble with melted DNA. A structural channel exists in the core polymerase that is believed to accommodate the melted DNA (Zhang et al. 1999). The upstream entrance to this channel is of particular interest. It maps to approximately 10 bases upstream of the point of RNA synthesis and appears to have the potential to be occluded. This occlusion could conceivably be related to controlling the filling of the channel with the transcription bubble in preinitiation complexes.
Related models are also suggested from quite different recent experiments on bubble formation in preinitiation complexes using σ70 holoenzyme. Two determinants that may help form the transcription bubble are near this same upstream location (Guo and Gralla 1998). One of these is the unique double-strand–single-strand junction that forms when base pair −11 melts. The other binding determinant is the adjacent downstream nontemplate DNA strand. Moreover, the connection between these two determinants has been shown to be the location of an inhibitory “gate” at position −10; when base pair −10 is melted, the nontemplate nucleotide at this position inhibits binding to the upstream fork. This connector position appears to coincide with the entrance to the channel, which will be filled with the transcription bubble.
Experiments with an alternate σ factor indicate that a related, but more complex, mechanism controls bubble formation. σ54 has no sequence similarity to σ70 and differs also in that its promoters are strictly regulated at the DNA-melting step (for review, see Merrick 1993). This regulation occurs using a molecular switch involving σ54 bound to a fork junction at the same location (Guo et al. 1999). In this case the complex is nonproductive in the absence of activator. The complex uses −12 consensus element and a template strand nucleotide and masks the ability to bind the nontemplate single strand. Mutations in the −12 region of the DNA or in the protein that destroy the interaction with the −12/−11 fork junction unmask the nontemplate strand binding and yield unregulated transcription (Guo et al. 1999; Wang et al. 1999). Thus, in the wild-type system the switch stays off without activator. In contrast, σ70 forms of holoenzyme have less strict opening requirements (see Helmann and deHaseth 1999). In this case the impediments to melting are weaker and can sometimes be overcome at higher temperatures without necessarily requiring the assistance of activators.
A potential common thread in these opening mechanisms is a requirement to overcome inhibition near a common position to allow the establishment of a continuous interaction between the polymerase and the DNA. This would involve the upstream fork junction and the adjacent nontemplate strand. In principle this would allow the channel to be filled and allow the active site of the polymerase to engage the melted start site region. In this paper we provide evidence to suggest homologous mechanisms of melting directed by these two σ factors, which have few sequence or regulatory features in common. The data show that σ54 activators can help establish the missing interaction with the adjacent nontemplate strand. In addition, studies with σ70 holoenzyme show a temperature-induced series of conformational changes; these overcome the much weaker inhibition at the same location and additionally assist in engagement of the transcription start site. We discuss the relevance of common mechanistic features to other transcription systems.
Results
Premelted templates can be transcribed by σ54 holoenzyme alone when the −12/−11 fork junction is not used
Prior experiments have shown that even though the function of a σ54 activator is to create a transcription bubble, certain heteroduplex templates containing preformed bubbles could not be transcribed without activator (Wedel and Kustu 1995). Subsequent binding studies using fork junction mimics showed that the exact location of the upstream junction within the bubble was very important in determining how tightly σ54 holoenzyme binds (Guo et al. 1999). The tightest binding was to the −12/−11 location, which was proposed to be important for transcriptional silencing in the absence of activator. The prior heteroduplex experiments (Wedel and Kustu 1995) had the −11 position as a terminal A:T base pair, which would be interconvertible with the −12/−11 junction by the fraying of the base pair (favored at 37°C; see Marky et al. 1981). The lack of unregulated transcription could have resulted from the use of a template with a junction that maintained proper regulation.
We tested unregulated transcription of a series of closely related templates that differ in whether the −12/−11 junction can be used. The templates were similar except that the junction position was varied systematically with positions −10 through −13 constituting the last base pair (Het-10–Het-13 in Fig. 1A). Het-10 would require the unlikely fraying of 2 base pairs to expose the −12/−11 junction (see Marky et al. 1981). Het-11 is analogous to that used previously. Het-12 is the optimally bound junction. Het-13 cannot form the optimal binding junction, as base pair −12 is mismatched, although the interpretation is complicated by the lowered binding associated with the loss of this consensus base pair. In this set Het-10 and Het-13 give the least access to the optimal binding junction.
Figure 1.

The effects of upstream fork junction location within heteroduplex template on σ54-dependent transcription. (A) The structures of DNA templates. The wild-type duplex template is a 600-bp PCR product, which includes upstream NtrC binding sites, a downstream termination site near position +360, and glnAp2 core promoter sequences (consensus GG and GC doublets at −24 and −12; the C is designated as −12 even though the start site can vary somewhat at different promoters). The top (nontemplate) strand sequences of the four heteroduplex templates (Het-10 through Het-13) are shown with mutated sequences in lower case. The −12 consensus doublet is underlined, and the last base pair at the upstream junction is indicated with a filled triangle. The bottom (template) strands of all the templates are the wild-type. (B) Transcription of wild-type σ54 RNAP on different templates using three protocols: (I) No activator, with heparin added; (II) no activator, but ATP, CTP, and GTP are added for 20 min before heparin and UTP; (III) activator is preincubated and then the 4 nucleotides and heparin are added. The autoradiographs for transcription from the five templates using each of these three conditions are shown. (C) Ratios of transcripts for each template in B. (Light bar) I/III; (solid bar) II/III. (D) Transcription on Het-12 templates with different −11 nontemplate nucleotides. (E) Transcription of amino-terminally deleted σ54 RNAP (ΔN) using protocol II on the indicated templates. (F) Single-strand binding activity detected by EMSA. The sequence of the parent probe based on Rhizobium meliloti nifH promoter is shown on the top with −12 and −24 consensus elements in bold. The most downstream C:G pair in bold is denoted as −12. A mismatched adenine is introduced into the bottom strand at −11. For wild-type (wt) lane T−12, about 5% probe is bound, and about 50% probe is bound in ΔN, lane T+1. Each probe designation refers to the position of the terminal base on the top strand.
Two protocols are used to detect transcription in the absence of activator. Protocol I is the most stringent, as the inhibitor heparin and nucleotides (but no activator) are simply added together to transcription complexes under closed complex conditions. If the complexes are not properly engaged, they are inactivated by heparin and will not transcribe. Protocol II is somewhat less stringent in that 3 nucleotides are preincubated under closed complex conditions before adding heparin and the missing nucleotide. This is less stringent because if the start site can be engaged transiently, it may direct formation of stabilizing short RNA before heparin is added. Control experiments show that neither of these protocols leads to detectable RNA synthesis on a true duplex template in the absence of activator (Fig. 1B; see right panel duplex “ds” lanes I and II compared with lane III performed in the presence of activator). All templates transcribe in control protocol III, which contains phosphorylated NtrC and ATP. The results with Het-11 are consistent with the prior heteroduplex experiment that used a similar template and protocol I (Wedel and Kustu 1995) and also showed little unregulated transcription.
Using protocol I, Het-10 shows an order of magnitude more transcription than Het-11 (Fig. 1B,C). Het-12 with the optimally bound junction shows no signal. The signal from Het-13 is weak, but this is complicated by the weak activated signal (owing to the loss of the consensus −12 base pair). A similar result is seen using protocol II, in which the Het-10 signal is nearly 40% of that seen in the presence of activator. The Het-13 signal now becomes the second strongest, reaching 20% of the activated level. The Het-12 and Het-11 templates, with the highest accessibility of the optimally bound junction, show very weak unregulated transcription in both protocols. We infer that use of the optimally bound −12/−11 junction is correlated with the silencing of unregulated transcription; when the junction is not used, as in Het-10, premelting can lead to significant deregulation. These effects on transcription mirror prior results showing that only the Het-11 and Het-12 junctions could properly restrict the single-strand binding activity of the polymerase (Guo et al. 1999).
Prior work has studied the role of nucleotide sequences at the optimal junction. −12 is the critical position; when the C:G base pair is changed, fork junction binding is weakened and deregulated transcription can occur under certain experimental conditions (Wang et al. 1999). The strength of fork junction binding is also influenced by the identity of the unpaired nucleotide at the nontemplate position −11. Binding is weakest with purines at this position and strongest with cytosine (Guo et al. 1999). The effects of these changes on transcription are not known and are explored in Figure 1D. The results show that these secondary effects on binding do not lead to detectable differences in either regulated or unregulated transcription in the context of the heteroduplex containing the optimal fork junction.
The silencing of unregulated transcription from the optimal binding fork junction has been related to forming a structure that masks the determinants needed for binding the nontemplate single strand. The amino terminus of the protein plays a central role (Wang et al. 1995, 1997; Syed and Gralla 1997; Cannon et al. 1999; Guo et al. 1999). Figure 1F shows that the identity of the unpaired −11 nucleotide is not centrally important in this regulation. When the base is removed to form an abasic position, the presence of the downstream nontemplate strand still leads to lesser binding, showing that the strand binding determinants remain fully masked (Fig. 1F, left; no lanes stronger than T−12). The overall inhibition is much weaker than that observed previously (see also Fig. 2B; cf. lanes T−12 and T−10 with the same lanes in Fig. 1F, left). This shows that any unpaired nontemplate nucleotide at −11 leads to inhibition (also see Guo et al. 1999). But the presence of a strongly inhibitory adenine at −11 does not prevent the unmasking of the nontemplate single-strand binding determinants when the amino terminus of the protein is deleted (strong band in T+1 lane in Fig. 1F, right).
Figure 2.

The effect of NtrC on σ54 RNAP binding to fork junction probes. The parent probe shown is identical to that used in Fig. 1F. (A) Effects of unphosphorylated NtrC on σ54 holoenzyme binding to probes with different junction position. The bound σ54 RNAP band is indicated with a bracket (the upper conformer varies with the preparation of core used), and a new complex of interest is marked by a filled oval in the lower panel with added NtrC. For lane B−11, about 40% of the probe is bound in the activator-independent band, and about 3% of the probe is in the band indicated with the oval. (B) The effects of top single strand sequences on the formation of the new complex of interest (oval). Without heparin, about 5% probe is bound for lane T−12 in the activator-independent band and for lane T−7 in the activator-dependent band. The difference between panels A and B for the activator-independent band is from different labeling position. (C) The effects of different protein factors on complex formation. The probe used has its junction at −12/−11 and the last 3′ top strand base at −7. (D) Effects of nontemplate-stranded nucleotide identity on NtrC-induced complex formation. A dash indicates an abasic site. Other probes are based on the parent sequence.
Transcription data show that this σ54 mutant directs transcription of all heteroduplexes, even without activator (Fig. 1E). This includes Het-11 and Het-12, which could not be transcribed in the absence of activator when using intact σ54 (see above). Taken together with prior data, it appears that unregulated transcription is silenced via the amino terminus of σ54 interacting with a fork junction at the precise junction location to enforce the masking of single-strand binding activity.
Activator NtrC establishes a connection through the −11 junction position
As just discussed, σ54 holoenzyme lacks a detectable interaction with the nontemplate strand adjacent to the −12/−11 fork junction (Guo et al. 1999). This interaction is a critical feature of open complex formation for σ70 holoenzyme (Guo and Gralla 1998; Fenton et al. 2000). It is possible that the σ54 activators help establish the missing interaction with the adjacent nontemplate strand, although this has not been shown. To learn if activator could cause a dependence on downstream single-stranded DNA sequences, we tested binding to a battery of probes (Fig. 2A), with and without NtrC. One of these (probe B-11; Fig. 2A, bottom left) showed the presence of a new band in the presence of unphosphorylated NtrC (Fig. 2A, see oval at right in bottom panel but not top panel). Probe B-11 has the nontemplate strand exposed to position −11 and, as discussed previously (above and Markey et al. 1981), should include a population with the terminal A:T frayed, creating an optimal binding fork junction. This suggested that the NtrC-dependent band might form best on a probe with the last base pair at −12 and a mismatch at −11. This turned out to be true, and the probe shown in Figure 1F was used in the next set of experiments.
Figure 2B shows that the new binding of σ54 holoenzyme induced by NtrC (see ovals in panels) requires the presence of the nontemplate-strand segment that corresponds to the previously undetectable σ70-like interaction. −11 mismatched probe T−7 is bound very strongly, but when the nontemplate strand is truncated, binding is severely reduced (compare T−7 with probes T−8 and others in the upper two panels of Fig. 2B). The strong binding is partially resistant to heparin (middle panel) and reproducibly requires NtrC (lower panel). The data confirm that this binding requires the nontemplate strand to position −7 and also is optimized by both unpaired bases at −11 (lane T+7 in Fig. 2B differs from lane B-12 in Fig. 2A only by the presence of an added mismatched base on the template strand). The lower panel confirms that NtrC is required for the new pattern of strong binding.
Figure 2C confirms these factor requirements using the optimal probe. Efficient binding requires all components: NtrC, σ54, and core polymerase (Fig. 2C, lane 5). The low level of complex formation seen in the absence of core and σ54 (Fig. 2C, lane 1) likely results from low-level contamination of NtrC with holoenzyme; because this experiment is performed in the absence of NtrC binding sites, a very high concentration of this activator is required to act from solution (North and Kustu 1997).
We tested whether the identity of the nucleotides present on the nontemplate strand was important. With the exception of a preference for an A:T or T:A at −11, these nucleotides are not conserved among σ54 promoters (Wang and Gralla 1998). The identity of the nucleotides from −11 to −7 is very important in the case of σ70, in which they are very highly conserved and bound as the nontemplate single strand (Marr and Roberts 1997; Guo and Gralla 1998). Figure 2D shows that two different substitutions of the sequence of this strand have no discernable effect on the formation of the NtrC-induced band (right panel, cf. lanes 8 and 9 with the wild-type sequence result in lane 7). Figure 2D, lane 9 uses a completely unrelated sequence, and lane 8 uses the wild-type sequence with an abasic site at position −11. We note also that the abasic site fork junction probe binds to the holoenzyme in the absence of NtrC much better than when a base is present (Fig. 2D, left, cf. lanes 4 and 2). This confirms the prior inference (above) that the presence of a base in this position enhances inhibition of fork junction binding. However, the results also show that this base is not required to respond to NtrC to form a new complex involving the nontemplate strand segment from −11 to −7; this single-strand interaction appears to be largely sequence-nonspecific for σ54.
Core polymerase and ATP are involved in establishing the interaction with the nontemplate strand
The above experiments were performed with σ54 holoenzyme. As NtrC apparently acts only on the σ54 form of holoenzyme, it is conceivable that its effects could be felt on σ54 alone. This is testable, as σ54 alone can be bound tightly to probes that contain fork junctions near position −12 (Guo et al. 1999). Figure 3A, middle panel (marked by arrow at right), shows the binding of σ54 alone to various probes. When a high concentration of NtrC is added (Fig. 3A, right panel), the binding of σ54 is strengthened without change in the complex mobility (Fig. 3, see arrow and cf. right with middle panel). We note that this effect of NtrC does not involve the nontemplate strand sequence near position −7. This can be seen by comparing binding of probe T−7 with truncated probes such as T−8 or T−9; there is no significant difference (Fig. 3A, arrow marking the right panel).
Figure 3.

The effect of NtrC on σ54 binding to fork junction probes. (A) Effects of unphosphorylated NtrC on σ54 binding to the set of probes with differing length of top strand and the last base pair at −12. The bound σ54 band is indicated with an arrow. The upper bands of bound σ54 holoenzyme are indicated with a bracket and a filled oval. In lane Holo, 15 nm σ54 RNAP is the only protein present with probe T−7. Proteins are present as indicated below the figure. (B) Effects of ATP on the σ54 binding to junction probe in the presence of unphosphorylated NtrC.
Thus, the experiment establishes two important points: (1) NtrC can interact with σ54 directly to enhance binding to the fork junction, apparently by acting catalytically, and (2) this effect does not require the nontemplate stand. Point 2 differs from what is seen with holoenzyme, as confirmed by an internal control created by the low amount of core present in the experiment of Figure 3A (right panel). The upper band (oval) that was shown (in Fig. 2) to require holoenzyme and NtrC is clearly much stronger using probe T−7 compared with T−8, T−9, and T−10 in the very same lanes in which σ54 alone binding (arrow) is equal for this set of four probes.
The data show one other difference between the effects of NtrC on σ54 alone and on holoenzyme. Figure 3B shows that σ54 alone binding is not stimulated by ATP (arrow), whereas the NtrC-induced holoenzyme binding pattern is strongly stimulated (oval). Overall, the data imply a dual effect of NtrC: (1) It strengthens σ54 binding to the fork junction region, and (2) it uses ATP to extend strong binding of holoenzyme to the segment that includes the adjacent nontemplate-strand sequences.
Complete activation of transcription by NtrC requires the hydrolysis of ATP (Weiss et al. 1991). But how ATP binding and hydrolysis drive activation of promoter-bound σ54 RNAP is still not known. We explored how ATP functions in the establishment of the new NtrC-induced holoenzyme complex with the nontemplate strand. Figure 4A shows the effects of three analogs of ATP on formation of this complex. The most striking result is that ADP can stimulate formation of this complex by slightly more than twofold. This is quite significant, even though it is approximately half the stimulation seen by ATP. As ADP cannot be hydrolyzed, this stimulation must be a consequence of its binding.
Figure 4.

Effects of adenine nucleotides. (A) Effects of different nucleotides on unphosphorylated NtrC-induced complex formation (oval) using probe T−7 (see top). The bar graph shows the relative binding compared with the absence of nucleotide. The strength of the NtrC-independent complex (bracket) is not changed with different nucleotides. (B) Use of the complete system with ATP and NtrC phosphorylated with carbamyl phosphate.
Other ATP analogs either have a very slight stimulatory effect (AMPPNP) or are strongly inhibitory (ATPγS). This ATPγS inhibition does not simply remove the effect of ATP, but induces an actual lowering of complex formation below the level in the absence of ATP. The inhibition is unexpected in light of the ADP stimulation, because ATPγS can substitute for ATP in many reactions that do not require hydrolysis. In this system it appears that the binding of ATPγS itself in the absence of ATP causes the inhibition. It may be that the γ-phosphate binding site has an important role in this new single-strand binding activity, as indicated by the higher stimulation of ATP compared with ADP and the inhibition by ATPγS. The interaction could be a prelude to a subsequent hydrolysis step, which would be cleavage at the γ-phosphate site to produce ADP and phosphate.
Examples of ATPγS inhibition via binding rather than hydrolysis are uncommon but not unique (Palleros et al. 1993). This interpretation is supported further because NtrC is not phosphorylated and therefore should be lacking in hydrolysis activity (Weiss et al. 1991; Austin and Dixon 1992), and ATP binding by NtrC is not affected by phosphorylation (Rombel et al. 1999). Thus, the data indicate that establishing the connection with the nontemplate strand uses ATP binding rather than hydrolysis.
This leaves unexplained where ATP hydrolysis comes into play, but the data of Figures 2B and 3A present a possibility. In both experiments the NtrC-induced complex forms less well with probe T+1 than with probe T−7 (marked by ovals in both figures). Thus, the data suggest that even in the presence of NtrC the segment downstream from −7 that includes the start site is difficult to fully engage. Even when the complete system is used, that is when NtrC is phosphorylated by carbamyl phosphate and ATP is present, the T+1 probe is still less well bound than T−7 (Fig. 4B). Thus, it seems that we have not yet replicated the full effect of NtrC activation on these probes. As discussed above, one possibility is that phosphorylated NtrC may trigger polymerase to engage the nontemplate strand using ATP binding and in a subsequent, as yet undetected, step hydrolyze the ATP to trigger engagement of the start site. New types of experiments using physiological phosphorylation procedures and templates with physiological NtrC binding sites may be required to replicate this effect.
We note that in both parts of Figure 4 and in all prior experiments the NtrC-induced band does not behave as expected for a direct supershift of adding NtrC to a fork junction complex. That is, even though the strength of the new band varies greatly, the increased intensity does not occur at the expense of the holoenzyme band (see bracketed region in all lanes in Fig. 4A,B). The role of NtrC appears to be to induce formation of a new stable form of holoenzyme bound to the fork junction probe. Two subsequent experiments, involving a different activator and protein–DNA cross-linking, support this hypothesis.
A different σ54-specific activator also helps to establish the interaction with the nontemplate strand
We tested whether a second activator of σ54 promoters can induce a new binding pattern in conjunction with the nontemplate strand sequence. For this purpose, PspFΔHTH is used, which has approximately two-thirds the molecular weight of NtrC (Jovanovic et al. 1999). All features of the results were similar for activator PspFΔHTH except that the new pattern of binding was much weaker. Figure 5A shows that a new complex forms in the presence of PspFΔHTH on probe T−7 (right panel, marked by an oval). Extending the sequences to include the start site reduces binding (Fig. 5A, cf. lanes T−7 and T+1). As was true for NtrC, the complex does not form in the absence of activator (Fig. 5A, left panel) or when the nontemplate strand is truncated (Fig. 5A, right panel; cf. T−7 with T−8).
Figure 5.

Effect of PspFΔHTH on the formation of the new complex. (A) Probe requirements using PspFΔHTH (36 kD). (B) Mobility comparisons in which the left lane contains NtrC (55 kD) on probe T−7 and the right lane uses σ70 holoenzyme and an analogous λ PR′ probe with the last base pair at −12 and the top strand to +1.
The mobility of this PspFΔHTH-induced complex is identical to that formed with NtrC (cf. oval marker position in Fig. 5A,B). It is also identical to the mobility of a complex formed with σ70 holoenzyme, which can open DNA without activator (Fig. 5B); we showed previously that the interactions with the nontemplate segment are present in this σ70 complex even though no activator has been added (Guo and Gralla 1998). These comparisons suggest, but do not prove, that when the polymerase assumes the ability to interact with the nontemplate strand, it changes conformation and migrates differently in an electrophoretic mobility-shift assay (EMSA) experiment. As discussed above, the lack of classical supershift behavior also supports this interpretation. We have not been able to directly rule out the presence of activator in these complexes because even the most abundant shifted complex would contain less than 10 pg of protein (in that case, NtrC). Further evidence favoring a conformational change comes from the chemical cross-linking and temperature induction experiments presented below.
Chemical cross-linking shows that NtrC throws the fork junction switch
To obtain independent evidence of the effect of NtrC on interactions with the nontemplate −11 position, we initiated chemical cross-linking experiments. In the first experiment a probe was constructed that contained a UV cross-linkable aryl azide group (Mayer and Barany 1995; Lagrange et al. 1996) on the phosphate just upstream of the −11 nucleotide on the nontemplate strand (probe I in Fig. 6A). This probe was capable of forming the novel EMSA complex in the presence of NtrC (a modified form of probe T+1 used in prior figures). The presence of the modified phosphate did not prevent formation of this complex (data not shown). The chemical group is known to react with protein upon UV irradiation. We detected these complexes by radioactively labeling the DNA and assaying for labeled DNA–protein complexes on SDS-PAGE gels.
Figure 6.


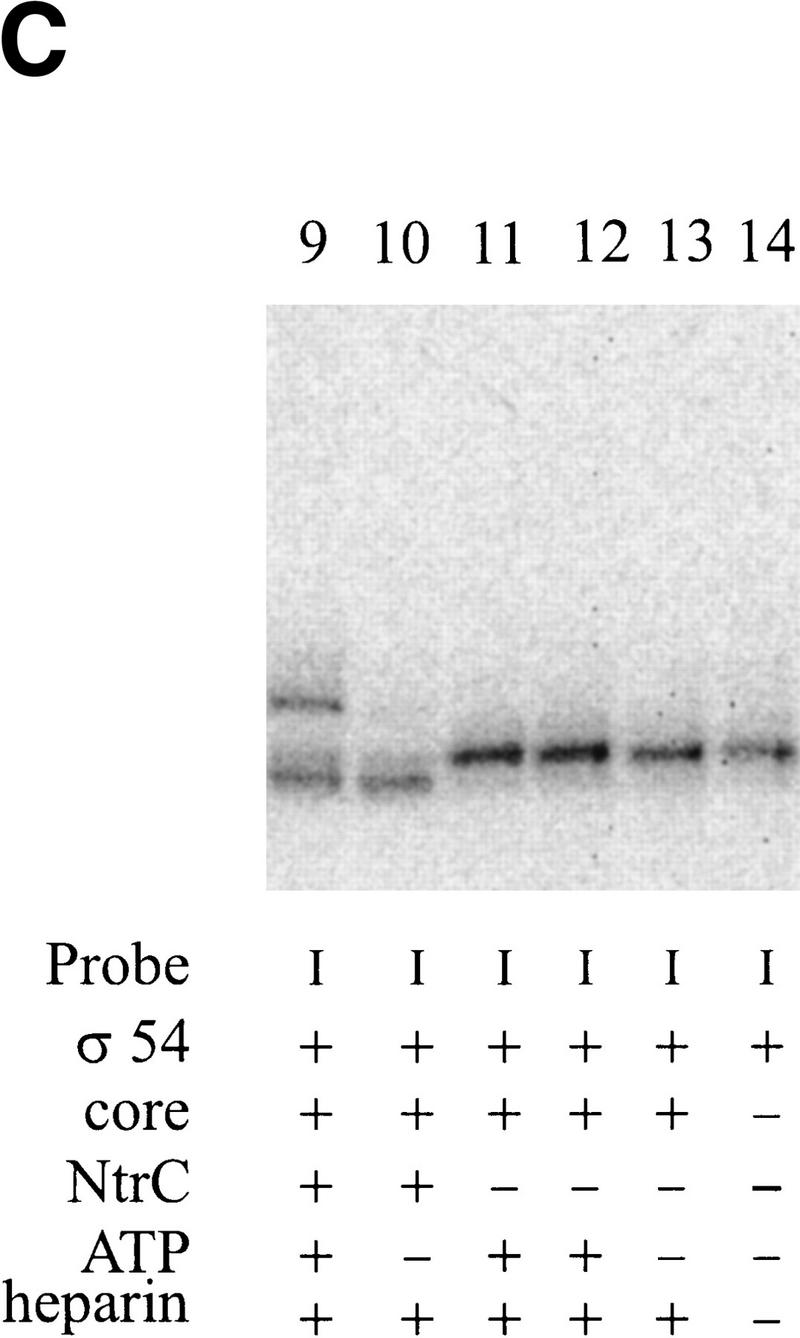
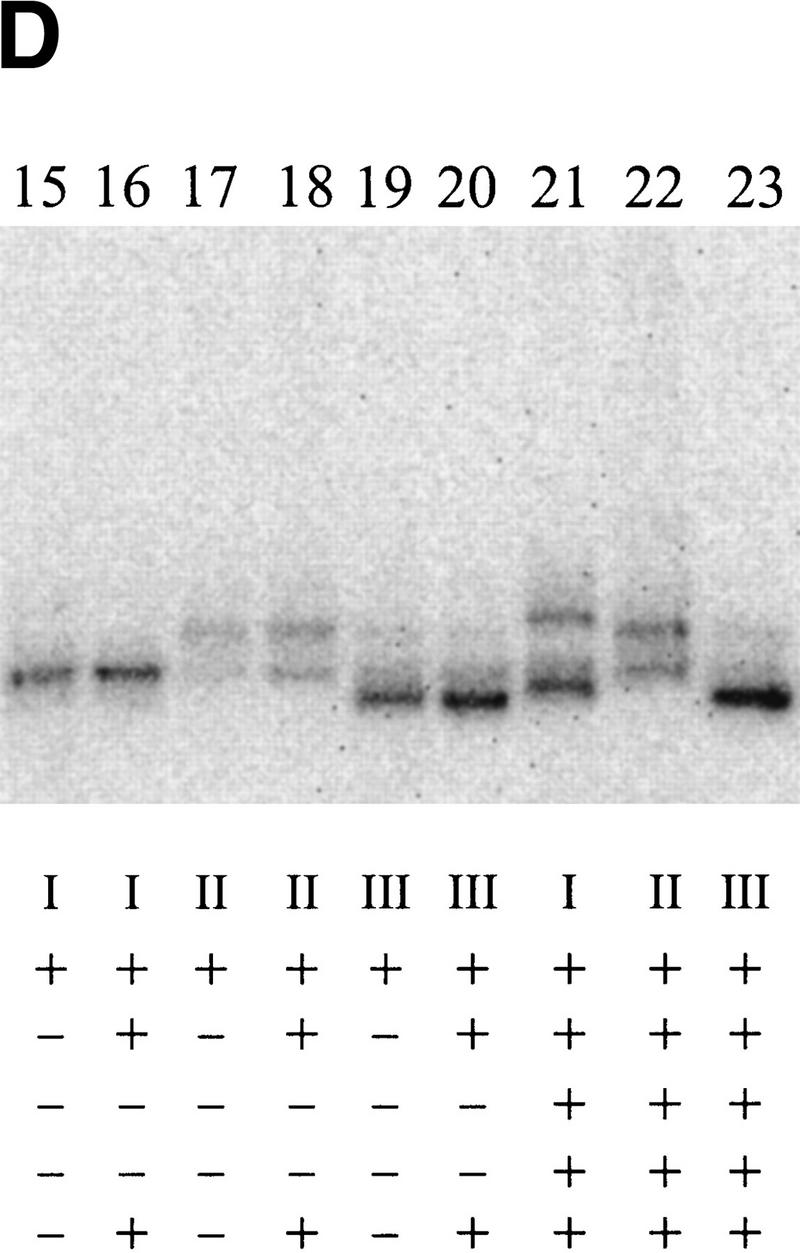
Cross-linking to the fork junction probes. (A) Schematic of the probes used, with the X indicating the position of the UV cross-linkable group, phenyl-azide. The bottom strand is identical in all probes and is nonradioactively phosphorylated. The top strands are 5′ 32P-labeled. The sequence of the probe is shown in Fig. 1F. (B–D) Results of cross-linking. The components of each reaction are listed below each lane. Where indicated, 20μg/ml of heparin is used to eliminate the top (β/β′) band. PspF reactions used 10% of the holoenzyme amount and showed no β/β′. Experiment B was not run as long as C and D, and its three parts are from separate experiments.
In this experiment σ54 alone can be labeled by the DNA in the autoradiograph of the SDS-PAGE experiment (Fig. 6B, lane 1). Controls without protein or UV irradiation did not show this band (data not shown). Repeated experiments show that the band is a closely spaced doublet, with the intensity of the lower band being somewhat variable. When holoenzyme was used, the same major σ54 band appeared, as well as an additional band with much slower mobility (Fig. 6B, lane 2). When the complex was challenged with heparin before cross-linking, only the σ54 band remained (Fig. 6B, lane 3), indicating that it is part of a specific interaction. The upper band may represent the cross-linking of the large core subunits (β/β′) in nonspecific, heparin-sensitive complexes. We conclude that when holoenzyme binds this probe, σ54 can become cross-linked to near −11 on the top strand. This is consistent with the ability of σ54 to interact at this junction sequence as detected by prior EMSA experiments (Guo et al. 1999).
In the next set of experiments NtrC and ATP were included. The aim was to see whether the activator would change the ability of σ54 to cross-link with the probe near −11. In Figure 6B, a comparison of lanes 3 and 4 (and lanes 16 and 21, Fig. 6D, in an experiment with a longer electrophoresis time) shows the effect of NtrC/ATP on cross-linking to the probe. The inclusion of NtrC/ATP (Fig. 6B–D, lanes 4, 9, or 21 compared with the respective controls in lanes 3, 13, and 16) leads to the appearance of a new band higher on the gel. A band of this intensity is not seen when NtrC/ATP alone is used (Fig. 6B, cf. lane 5 with lane 4). Using a different probe (IV), a shifted band can be induced by NtrC/ATP as well as by the PspFΔHTH activator (Fig. 6B, cf. lane 6 with lane 7). The molecular masses of NtrC and PspFΔHTH differ by 20 kD, and the identical mobility shift indicates that neither is likely to be cross-linked. Thus, NtrC/ATP (and PspFΔHTH) induces formation of a new cross-linked species with the phosphate adjacent to −11 on the nontemplate strand, almost certainly to the σ54 subunit.
When the very same DNA sequence probe is used, but with the cross-linker moved from near −11 to near −7 (Fig. 6A, see probe II), a contrasting result is obtained. Now NtrC/ATP does not change the cross-linking pattern (Fig. 6D, cf. lanes 18 and 22). We infer that NtrC/ATP can induce a change in σ cross-linking near the −11 position but not near the −7 position on the very same DNA structure.
To learn if this NtrC-induced change at −11 requires the nontemplate strand segment, as did the above EMSA experiments, we assayed a truncated probe (see probe III in Fig. 6A). This probe was identical to the parent (chemical group near −11) but was truncated beyond position −10. This truncation eliminated the effect of NtrC on holoenzyme in the EMSA experiments shown above. The data show that the truncation also eliminates the effect of NtrC/ATP in these cross-linking experiments (Fig. 6D, cf. lanes 20 and 23). Thus, not only does NtrC/ATP induce a change in how σ54 interacts near −11, but this change depends on the presence of the adjacent nontemplate strand segment.
The unphosphorylated NtrC-induced presence of the new (upper) band (see above discussion of Fig. 6D, lane 21) is strongly stimulated by ATP in the cross-linking experiments (Fig. 6C, cf. lanes 9 and 10). Thus, the EMSA and cross-linking experiments are in excellent agreement. We conclude that NtrC and ATP induce a conformational change near position −11 that allows a new complex to form that relies on the adjacent nontemplate strand segment.
Inhibition in σ70 holoenzyme can be overcome by temperature-induced conformational changes
In contrast with σ54 holoenzyme, melting by σ70 holoenzyme is not as tightly regulated. Some σ70 promoters do not need activators to form open complexes. The formation of open promoter complexes by σ70 holoenzyme has long been known to be a temperature-dependent process. One likely contribution to this has been thought to be the direct use of thermal energy in melting apart the base pairs of DNA. However, conformational changes of RNAP–DNA complex are well documented (see Craig et al. 1998). The requirement for an activator-induced conformational change in the σ54 system and the existence of a potentially blocked channel in the polymerase structure raise an additional possibility for σ70. Thus, we tested the idea that the opening mechanisms by the two different holoenzymes might involve a homologous unblocking via a polymerase conformational change. This could be induced by temperature for σ70 and by activator for σ54.
As discussed above there are two types of interactions that are thought to lead to melting in the σ54 system. These are: (1) creating a connected interaction between the upstream fork junction and the nontemplate strand and (2) allowing the active site to engage the melted start site DNA. We assessed these interactions for σ70 holoenzyme using DNA probes designed to measure each process.
We consider first the connector effect, which was shown on the λ PR′ promoter to involve the −10 nontemplate strand nucleotide for σ70 holoenzyme (Guo and Gralla 1998). Because the melting behavior of the phage T7 A2 promoter is better characterized (Nierman and Chamberlin 1980; Severinov and Darst 1997), we repeated the experiment on this promoter. The result is the same, in that probe T−10 is bound less well than the probe T−11 (Fig. 7A, right panel at low temperature), using the same low-temperature (0°C) EMSA conditions applied previously. These probes differ only in that T−10 contains the unpaired connector nucleotide, and the ratio of bound T−10 to T−11 is a measure of the extent of inhibition attributable to the connecting −10 nucleotide. This inhibition is dependent on the −10 location, not the identity of the −10 nucleotide (data not shown). In preliminary experiments we found that this ratio depended somewhat on experimental conditions; higher concentrations of heparin led to less binding but gave a greater degree of inhibition.
Figure 7.

Effect of temperature on inhibition of σ70 RNAP binding to the nontemplate strand. The T7 A2 promoter is shown at the top. The 4 nucleotides shown in bold correspond to the dots and represent the last 3′ base of each probe. The unchanged radiolabeled strand is indicated with *. The upstream T in the −10 recognition element is denoted as −12. (A) The effects of temperature on probe binding with and without the −10 nucleotide (represented by probes T−10 and T−11, respectively). Autoradiographs at low and high temperature with 20 μg/ml heparin are shown at the right. The ratio was taken at various temperatures and is shown at the left. The data are the average of three experiments. (█) Results with 20 μg/ml heparin; (●) results with 100 μg/ml heparin. (B) The effects of temperature on probe binding with and without the −7 to +1 nontemplate strand segment in the presence of 20 μg/ml heparin. Autoradiographs at low and high temperature are shown at the right. The ratio was taken at various temperatures and is shown at the left. The data are the average of three experiments. (C) Raw data for parts A and B, showing percent binding with 20 μg/ml heparin (average of three experiments). (□) Probe T+i; (□) T−7; (●) T−10; (○) T−11. (D) The effects of heparin (20 μg/ml) and top (“T”) single-stranded nucleotides on σ70 RNAP binding to the probes with −12/−11 junction.
The experiment was repeated at various temperatures to assess whether a temperature-induced change could lessen this inhibition. Both high and low concentrations of heparin were used. An example of the effect of temperature is shown in Figure 7A (right panels), where at 25°C the ratio of bound T−10 to T−11 is significantly higher than at low temperature. Figure 7A (left) and Figure 7C show the data for the range of temperatures assayed.
The data show that the extent of inhibition by the −10 connector decreases as the temperature is increased. The relief of inhibition was never complete, and in the two experiments the T−10/T−11 ratio never was higher than 0.7. The interesting point, however, is that a discrete transition occurs near 10°C, in which the −10 position inhibition is significantly relieved. This transition should not reflect the melting of DNA, as the only difference between the two probes is the presence of an additional unpaired nucleotide. Thus the temperature-induced change should reflect a conformational change in the polymerase. This change leads to partial relief of the −10 position inhibition. It is possible that this is analogous to the change induced by activator in the more tightly regulated σ54 system described above.
Next, we consider the engagement of the start site. In the σ54 system we were unable to fully mimic the effect of activator; recall that the requirement for the hydrolysis of ATP and the engagement of the start site were not reproduced by EMSA. Because the σ70 system does not involve tight regulation by activator we speculated that the engagement of the start site might be triggered by increased temperature alone. Thus, we used two probes, only one of which contained the start site region (probe T+i). Figure 7B (right panels) shows that both probes are bound by σ70 holoenzyme as closely spaced doublets, with the T+i probe favoring the lower band and the T−7 probe favoring the upper band. We are not certain of the origin of the doublet nature of the binding; both are properly heparin-resistant and both require σ70 and core. However, it seems that the upper band simply represents the same complex, but with heparin bound to it. That is, Figure 7D shows that heparin induces formation of the upper band, although not equally well on all probes or at all conditions. We counted the two bands together in assessing the effect of temperature.
The data show that probes T+i and T−7 are bound about equally at low temperature but that T+i is favored over T−7 at high temperatures (see Fig. 7B, right panels, and Fig. 7C). The experiment was repeated over a wide temperature range, and the ratio of binding to the two probes was calculated. Figure 7B, left, shows that temperatures above 20°C induce a transition that allows increased binding to the probe (T+i) containing the start site region. As discussed above, the increased temperature has not been used to melt DNA, as the probes differ only by the addition of unpaired nucleotides. We infer that the σ70 polymerase can undergo another conformational change that allows the start site region to be better engaged. Thus, it appears that σ70 polymerase can undergo two changes: In one the fork junction is more easily connected to the nontemplate strand, and in the other the downstream start site is more easily engaged. In the following section we discuss the generality of promoter recognition and melting using such processes for both the σ70 and σ54 systems.
Discussion
Diverse mechanisms have been suggested to be capable of contributing to promoter melting in prokaryotes and eukaryotes. These include helicases (Tirode et al. 1999), single-strand binding activities (Marr and Roberts 1997), topoisomerases (Parvin and Sharp 1993), and fork junction binding activities (Guo and Gralla 1998). The current data show that for diverse Escherichia coli RNA polymerase holoenzymes, control of promoter melting occurs by simple variations on a common theme. To fully engage the transcription bubble, holoenzymes need to establish a stable connected interaction between the fork junction at the upstream edge of the bubble and the melted downstream DNA. Initially σ factors appear to organize the polymerase into default conformations that cannot support the single-stranded DNA binding; without σ, core polymerase binds both single-stranded DNA and DNA:RNA hybrids readily (Sidorenkov et al. 1998). The data suggest that σ54 and σ70 induce different requirements for changing the holoenzyme from this default conformation, leading to different mechanisms of activation.
The data show that determinants near position −11 are prime foci for σ-dependent regulation of DNA melting. Early during promoter melting a −12/−11 fork junction must be created, and this interacts with σ54 (Guo et al. 1999) and σ70 holoenzyme (Guo and Gralla 1998). Both types of holoenzyme are inhibited initially from proceeding with the required conformational changes, the inhibition being very strong when directed by σ54. For σ54, activator relieves this inhibition. For σ70, elevated temperature can promote the change, involving the −10 position. The −12 to −10 region is coincident with the entrance to the proposed melted DNA channel in the structure of core polymerase (Zhang et al. 1999). This suggests that different σ factors can direct different types of regulatory mechanisms by facilitating different methods of opening the channel to allow connection between the upstream fork junction and the remainder of the melted DNA.
Open complex formation by σ70 holoenzyme
σ70 promoters do not necessarily require activators, and when they do, the assistance can be either in promoter binding or melting (isomerization) (McClure 1985; Hochschild and Dove 1998). Open complex formation is known to involve several intermediates and to involve conformational changes in both the DNA and the holoenzyme (see Craig et al. 1998). In Figure 7 we detected two conformational changes in the holoenzyme and, most important, identified them as impacting specific segments of melted DNA. These experiments involved the T7 A2 promoter, at which open complex formation is simply induced by elevated temperature (see Severinov and Darst 1997). In this experiment temperature induced the changes; one partially relieved the −10 position inhibition and the other assisted in the engagement of the melted nontemplate strand covering the start site region. It is important to note that these changes involved premelted DNA. This suggests that the enzyme converts from closed to open form and that the latter optimally exposes single-strand binding determinants. The final open complex includes the critical melted nontemplate strand consensus sequence between −7 and −11 (Marr and Roberts 1997; Guo and Gralla 1998). Together, these conformational changes to the “open” form of the enzyme promote the connected interaction between fork junction and start site that is required for stable transcription bubble formation.
In this view σ70 organizes the free holoenzyme predominantly into a closed form in which its single-strand binding activities are not fully exposed. If DNA melting begins prematurely, the nucleotide at −10 assists in maintaining this closed conformation of the holoenzyme. For temperature-regulated promoters, elevated temperature helps convert the enzyme from the closed to the open form. For regulated promoters, the activator could convert the enzyme to the open form (isomerization) or enhance binding, allowing sufficient time for the enzyme to undergo the change while in contact with the DNA. Open complex formation occurs when the open enzyme uses its newly revealed activities to bind the single-stranded DNA.
Open complex formation by σ54 holoenzyme and other systems
σ54 promoters are enhancer-dependent and are strictly regulated at the melting step. Thus, the holoenzymes are bound tightly in closed promoter complexes, which provide convenient activation targets for the looping enhancer proteins. Before this activation event, melting is held in check by a proposed “molecular switch” that uses determinants within the amino terminus of σ54 and the region surrounding position −12 in the DNA (Guo et al. 1999; Wang et al. 1999). As discussed above, the −12 and −11 locations cooperate with σ54 to maintain a conformation in which the single-strand binding activities of the holoenzyme are unavailable.
The current data show that activator throws this switch by reorganizing how σ54 interacts with the DNA near this location, detected by EMSA and cross-linking. These data and prior evidence of conformational changes in σ54 (Casaz and Buck 1997) indicate that activator triggers a change in holoenzyme conformation to overcome the transcriptional silencing associated with the hiding of single-strand binding activities. This conformational change allows binding to the adjacent nontemplate strand sequences (Fig. 6, and supported by Figs. 2–5). That is, in contrast to the σ70 situation, interaction with this element is only detectable when activator is present (Fig. 2).
This conformational change in the enzyme, however, is not sufficient to complete the connection to the unpaired start site region. This final connection requires changes involving the amino terminus of σ54 (Wang et al. 1995; Cannon et al. 1999; Gallegos et al. 1999; Guo et al. 1999); mutation of this region allows partial engagement of the start site even without activator (Fig. 1; Wang et al. 1995, 1997; Syed and Gralla 1997). One clue here is that the activator-dependent establishment of the connected interaction between the fork junction and the adjacent nontemplate strand requires ATP binding but not hydrolysis (Fig. 4). As full open complex formation requires hydrolysis, we speculate that hydrolysis can drive the second enzyme conformational change that antagonizes the effect of the amino terminus and allows the full connection to be made to the start site region. This remains speculative because we have been unsuccessful in mimicking this hydrolysis effect in vitro by EMSA. The use of two such conformational changes to fully open the enzyme would also be closely analogous to the situation described above for σ70 holoenzyme. The main difference is that σ54 has organized the holoenzyme so that activator is strictly required to reveal the determinants that bind tightly to melted DNA.
NtrC contacts σ54 holoenzyme transiently and triggers conformational changes (Popham et al. 1989). Our data indicate that NtrC has multiple roles in the promoter opening process. Most unexpectedly, it has two ATP hydrolysis-independent activities, one strengthening σ54 binding (Fig. 3) and one allowing use of the nontemplate strand between −11 and −7 (Fig. 4). Thus, NtrC appears to catalyze multiple conformational changes to open holoenzyme, and these changes direct single-strand DNA binding from the upstream fork junction to the melted start site.
As discussed above, changes in the conformation of both holoenzymes critically involve promoter positions −12–−10. In the case of σ54 the presence of any base at the −11 position helps prevent unregulated binding to the nontemplate strand, as judged by the properties of abasic templates (Fig. 2D). This location appears to be coincident with the channel entrance in bacterial RNA polymerase and is rich with both structural and functional features. The location is proposed to act critically during control of elongation and termination when the channel is filled mostly with DNA–RNA hybrid (Mooney and Landick 1999). We note also that eukaryotic polymerase cores are quite homologous based on both sequence and structural comparisons with bacterial cores (Woychik and Young 1990; Fu et al. 1999; Zhang et al. 1999). Several general transcription factors have regions of homology with σ factors (Hoffman et al. 1990; Ohkuma et al. 1991). Among the eukaryotic polymerases, some (pol II) (Wang et al. 1992; Yan and Gralla 1999) require ATP, like σ54, and the others do not (Kassavetis et al. 1992; Lofquist et al. 1993), like σ70. The location of the upstream fork junction is similar for RNA polymerase II and both bacterial holoenzymes (see Yan and Gralla 1999). If all the cores are built similarly, as predicted, then this common mechanism in bacterial transcription may simply show further interesting variations in the more complex eukaryotic systems.
Materials and methods
Proteins
E. coli core enzyme and σ70 holoenzyme are from Epicentre (Madison, WI). Wild-type E. coli σ54 and amino-terminally deleted σ54 (Wang et al. 1997) and NtrC are purified as reported (Moore et al. 1993). His-tagged PspFΔHTH is purified by Shun Lee as described (Jovanovic et al. 1999). σ54 holoenzyme was prepared by incubating σ54 and core enzyme with molar ratio of 2.5:1 for 30 min on ice.
Heteroduplex templates
These are prepared using biotin-labeled DNA methods (Ring et al. 1996). Mutated sequences are introduced into pTH8, which contains the glnAp2 promoter. A 600-base pair segment is amplified by PCR using primers flanking the NtrC binding sites and transcription terminator site. One of the two primers is 5′ biotin-labeled. The biotin-labeled primer corresponds to the sequence unwanted in the heteroduplex. PCR fragments from the wild-type and mutant templates are mixed 1:1 in 20 mm NaCl and 5 mm EDTA (pH 8.0) and heated to 100°C. The mixture is slowly cooled to 65°C and is kept for 10 min at this temperature. Then it is cooled down to room temperature, streptavidin is added to 0.3 mg/ml, and the mixture is incubated for 20 min at 37°C before it is separated by 1% agarose gel. The unshifted band is cut out and the heteroduplex DNA is recovered by the glass milk method (Bio101, Vista, CA). The purified fragment is checked by adding streptavidin, and no visible shift is detected.
EMSA
Promoter probes for EMSA assays were prepared as reported (Guo et al. 1999). Mobility assays are as follows: Holoenzyme or σ54 was added to a 10 μl reaction mixture containing 1x buffer T (50 mm Hepes-HCl at pH 7.9, 100 mm KCl, 10 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, 0.05 μg/ml BSA, 2.8% PEG-8000, 6.0 ng/μl dIdC) with annealed promoter probe. For σ70 holoenzyme, 25 ng/μl protein is incubated with 1 nm probe. After a 10-min incubation at the desired temperature, 1 μl of 1mg/ml heparin is added for 5 min before analysis by 5% PAGE (Minigel system, Bio-Rad) with 1x TBE buffer, which is adjusted to the same temperature as the reaction. The gel is run at 200 V in a water bath kept at the desired temperature. The internal temperature of the gel was monitored after the run and was slightly higher. The signals are visualized and quantitated by phosphoimager. For σ54 holoenzyme, 7.5 nm protein is incubated with 1 nm probe for 10 min at 37°C. When noted, 0.6 μm NtrC or 1 μm PspFΔHTH is added, and the ATP concentration is 4 mm. The pH of ADP (Sigma) is adjusted to near 7.0. Reaction mixture is run at 300 V at room temperature. For the σ54 alone assay, 0.2 μm protein is used.
In vitro transcription
Activated transcription (protocol III) is as follows: 50 nm σ54 RNAP is incubated with 2 nm DNA template, 0.6 μm NtrC, 10 mm carbamyl phosphate, 4 mm ATP in 10 μl 1x buffer T with 0.7% PEG for 20 min at 37°C before 1 μl NTP–heparin mixture (0.5 mm ATP, CTP, GTP each, 50 nm UTP, 0.2 μCi/μl [32P]α-UTP and 100 μg/ml heparin) is added. This is incubated for 10 min at 37°C, followed by 6% PAGE with urea. RNA is analyzed with a phosphoimager. For heparin-challenged unactivated transcription (protocol I), NtrC is left out. In the alternative unactivated transcription (protocol II), the above nucleotide mixture without UTP and heparin is mixed with σ54 holoenzyme and DNA in 10 μl 1×; buffer T with 0.7% PEG. This is incubated for 20 min at 37°C before the UTP-heparin mixture is added and is then continued for 10 min.
Cross-linking
Phosphorothioated oligodeoxyribonucleotides (Operon) are derivatized with p-azidophenacyl bromide (Sigma) as described (Mayer and Barany 1995), purified through a Sephadex G-50 column in 10 mm Tris-HCl (pH 7.5), 32P-labeled, and annealed to the complementary strand. Cross-linking assays are as follows: Holoenzyme or σ54 is added to a 10 μl reaction mixture containing 1x buffer X (50 mm Hepes-HCl at pH 7.9, 50 mm KCl, 10 mm MgCl2, 0.1 mm EDTA, 1 mm β-mercaptoethanol, 0.05 μg/ml BSA, 0.7% PEG) and derivatized annealed promoter probe. Where stated, 1.2 μm NtrC and 4 mm ATP are also added to the reaction mixture. For the PspFΔHTH reaction, 0.75 nm holoenzyme and 2 μm PspFΔHTH were used. Following a 10 min incubation at 37°C, 0.5 μl of 0.4 mg/ml heparin is added and incubated for an additional 5 min. The mixture is then UV-irradiated at 365 nm for 4.5 minutes (1.08 J/cm2) (Lagrange et al. 1996) using a CL-1000L cross-linker (UVP, Upland, CA) and loaded onto an 8% SDS-PAGE with SDS buffer and run at 200 V at room temperature. Bands are analyzed by a phosphoimager. Preliminary EMSA experiments showed that the derivatization at the stated position did not inhibit appropriate DNA binding.
Acknowledgments
We thank P. Model for the PspFΔHTH plasmid, S. Lee for purifying the protein, and members of the group for their advice. This work was supported by a National Institutes of Health grant (GM 35754).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gralla@mbi.ucla.edu; FAX (310) 267-2302.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.794800.
References
- Austin S, Dixon R. The prokaryotic enhancer binding protein NTRC has an ATPase activity which is phosphorylation and DNA dependent. EMBO J. 1992;11:2219–2228. doi: 10.1002/j.1460-2075.1992.tb05281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon W, Gallegos MT, Casaz P, Buck M. Amino-terminal sequences of σN (σ54) inhibit RNA polymerase isomerization. Genes & Dev. 1999;13:357–370. doi: 10.1101/gad.13.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaz P, Buck M. Probing the assembly of transcription initiation complexes through changes in σN protease sensitivity. Proc Natl Acad Sci. 1997;94:12145–12150. doi: 10.1073/pnas.94.22.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig ML, Tsodikov OV, McQuade KL, Schlax PEJ, Capp MW, Saecker RM, Record MTJ. DNA footprints of the two kinetically significant intermediates in formation of an RNA polymerase-promoter open complex: Evidence that interactions with start site and downstream DNA induce sequential conformational changes in polymerase and DNA. J Mol Biol. 1998;283:741–756. doi: 10.1006/jmbi.1998.2129. [DOI] [PubMed] [Google Scholar]
- Fenton M, Lee SJ, Gralla JD. E. coli promoter opening and −10 recognition: Mutational analysis of σ70. EMBO J. 2000;19:1130–1137. doi: 10.1093/emboj/19.5.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Gnatt AL, Bushnell DA, Jensen GJ, Thompson NE, Burgess RR, David PR, Kornberg RD. Yeast RNA polymerase II at 5 Å resolution. Cell. 1999;98:799–810. doi: 10.1016/s0092-8674(00)81514-7. [DOI] [PubMed] [Google Scholar]
- Gallegos MT, Cannon WV, Buck M. Functions of the σ54 region I in trans and implications for transcription activation. J Biol Chem. 1999;274:25285–25290. doi: 10.1074/jbc.274.36.25285. [DOI] [PubMed] [Google Scholar]
- Guo Y, Gralla JD. Promoter opening via a DNA fork junction binding activity. Proc Natl Acad Sci. 1998;95:11655–11660. doi: 10.1073/pnas.95.20.11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Wang L, Gralla JD. A fork junction DNA–protein switch that controls promoter melting by the bacterial enhancer-dependent sigma factor. EMBO J. 1999;18:3736–3745. doi: 10.1093/emboj/18.13.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann JD, deHaseth PL. Protein–nucleic acid interactions during open complex formation investigated by systematic alteration of the protein and DNA binding partners. Biochemistry. 1999;38:5959–5967. doi: 10.1021/bi990206g. [DOI] [PubMed] [Google Scholar]
- Hochschild A, Dove SL. Protein–protein contacts that activate and repress prokaryotic transcription. Cell. 1998;92:597–600. doi: 10.1016/s0092-8674(00)81126-5. [DOI] [PubMed] [Google Scholar]
- Hoffman A, Sinn E, Yamamoto T, Wang J, Roy A, Horikoshi M, Roeder RG. Highly conserved core domain and unique N terminus with presumptive regulatory motifs in a human TATA factor (TFIID) Nature. 1990;346:387–390. doi: 10.1038/346387a0. [DOI] [PubMed] [Google Scholar]
- Jovanovic G, Rakonjac J, Model P. In vivo and in vitro activities of the Escherichia coli σ54 transcription activator, PspF, and its DNA-binding mutant, PspFΔHTH. J Mol Biol. 1999;285:469–483. doi: 10.1006/jmbi.1998.2263. [DOI] [PubMed] [Google Scholar]
- Kassavetis GA, Blanco JA, Johnson TE, Geiduschek EP. Formation of open and elongating transcription complexes by RNA polymerase III. J Mol Biol. 1992;226:47–58. doi: 10.1016/0022-2836(92)90123-2. [DOI] [PubMed] [Google Scholar]
- Lagrange T, Kim TK, Orphanides G, Ebright YW, Ebright RH, Reinberg D. High-resolution mapping of nucleoprotein complexes by site-specific protein–DNA photocrosslinking: Organization of the human TBP–TFIIA–TFIIB–DNA quaternary complex. Proc Natl Acad Sci. 1996;93:10620–10625. doi: 10.1073/pnas.93.20.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofquist AK, Li H, Imboden MA, Paule MR. Promoter opening (melting) and transcription initiation by RNA polymerase I requires neither nucleotide β–γ hydrolysis nor protein phosphorylation. Nucleic Acids Res. 1993;21:3233–3238. doi: 10.1093/nar/21.14.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marky LA, Canuel L, Jones RA, Breslauer KJ. Calorimetric and spectroscopic investigation of the helix-to-coil transition of the self-complementary deoxyribonucleotide ATGCAT. Biophys Chem. 1981;13:141–149. doi: 10.1016/0301-4622(81)80013-0. [DOI] [PubMed] [Google Scholar]
- Marr MT, Roberts JW. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- Mayer AN, Barany F. Photoaffinity cross-linking of TaqI restriction endonuclease using an aryl azide linked to the phosphate backbone. Gene. 1995;153:1–8. doi: 10.1016/0378-1119(94)00752-e. [DOI] [PubMed] [Google Scholar]
- McClure WR. Mechanism and control of transcription initiation in prokaryotes. Annu Rev Biochem. 1985;54:171–204. doi: 10.1146/annurev.bi.54.070185.001131. [DOI] [PubMed] [Google Scholar]
- Merrick MJ. In a class of its own—the RNA polymerase sigma factor σ54 (sigma N) Mol Microbiol. 1993;10:903–909. doi: 10.1111/j.1365-2958.1993.tb00961.x. [DOI] [PubMed] [Google Scholar]
- Mooney RA, Landick R. RNA polymerase unveiled. Cell. 1999;98:687–690. doi: 10.1016/s0092-8674(00)81483-x. [DOI] [PubMed] [Google Scholar]
- Moore JB, Shiau SP, Reitzer LJ. Alterations of highly conserved residues in the regulatory domain of nitrogen regulator I (NtrC) of Escherichia coli. J Bacteriol. 1993;175:2692–2701. doi: 10.1128/jb.175.9.2692-2701.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierman WC, Chamberlin MJ. Studies of RNA chain initiation by Escherichia coli RNA polymerase bound to T7 DNA. Direct analysis of the kinetics of RNA chain initiation at T7 promoter A2. J Biol Chem. 1980;255:1819–1823. [PubMed] [Google Scholar]
- North AK, Kustu S. Mutant forms of the enhancer-binding protein NtrC can activate transcription from solution. J Mol Biol. 1997;267:17–36. doi: 10.1006/jmbi.1996.0838. [DOI] [PubMed] [Google Scholar]
- Nudler E. Transcription elongation: Structural basis and mechanisms. J Mol Biol. 1999;288:1–12. doi: 10.1006/jmbi.1999.2641. [DOI] [PubMed] [Google Scholar]
- Ohkuma Y, Sumimoto H, Hoffmann A, Shimasaki S, Horikoshi M, Roeder RG. Structural motifs and potential σ homologies in the large subunit of human general transcription factor TFIIE. Nature. 1991;354:398–401. doi: 10.1038/354398a0. [DOI] [PubMed] [Google Scholar]
- Palleros DR, Reid KL, Shi L, Welch WJ, Fink AL. ATP-induced protein-Hsp70 complex dissociation requires K+ but not ATP hydrolysis. Nature. 1993;365:664–666. doi: 10.1038/365664a0. [DOI] [PubMed] [Google Scholar]
- Parvin JD, Sharp PA. DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell. 1993;73:533–540. doi: 10.1016/0092-8674(93)90140-l. [DOI] [PubMed] [Google Scholar]
- Popham DL, Szeto D, Keener J, Kustu S. Function of a bacterial activator protein that binds to transcriptional enhancers. Science. 1989;243:629–635. doi: 10.1126/science.2563595. [DOI] [PubMed] [Google Scholar]
- Ring BZ, Yarnell WS, Roberts JW. Function of E. coli RNA polymerase sigma factor σ70 in promoter-proximal pausing. Cell. 1996;86:485–493. doi: 10.1016/s0092-8674(00)80121-x. [DOI] [PubMed] [Google Scholar]
- Rombel I, Peters-Wendisch P, Mesecar A, Thorgeirsson T, Shin YK, Kustu S. MgATP binding and hydrolysis determinants of NtrC, a bacterial enhancer-binding protein. J Bacteriol. 1999;181:4628–4638. doi: 10.1128/jb.181.15.4628-4638.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Darst SA. A mutant RNA polymerase that forms unusual open promoter complexes. Proc Natl Acad Sci. 1997;94:13481–13486. doi: 10.1073/pnas.94.25.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorenkov I, Komissarova N, Kashlev M. Crucial role of the RNA:DNA hybrid in the processivity of transcription. Mol Cell. 1998;2:55–64. doi: 10.1016/s1097-2765(00)80113-6. [DOI] [PubMed] [Google Scholar]
- Syed A, Gralla JD. Isolation and properties of enhancer-bypass mutants of σ54. Mol Microbiol. 1997;23:987–995. doi: 10.1046/j.1365-2958.1997.2851651.x. [DOI] [PubMed] [Google Scholar]
- Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: Assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3:87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- Wang L, Gralla JD. Multiple in vivo roles for the −12-region elements of σ54 promoters. J Bacteriol. 1998;180:5626–5631. doi: 10.1128/jb.180.21.5626-5631.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Carey M, Gralla JD. Polymerase II promoter activation: Closed complex formation and ATP-driven start site opening. Science. 1992;255:450–453. doi: 10.1126/science.1310361. [DOI] [PubMed] [Google Scholar]
- Wang JT, Syed A, Hsieh M, Gralla JD. Converting Escherichia coli RNA polymerase into an enhancer-responsive enzyme—role of an NH2-terminal leucine patch in σ54. Science. 1995;270:992–994. doi: 10.1126/science.270.5238.992. [DOI] [PubMed] [Google Scholar]
- Wang JT, Syed A, Gralla JD. Multiple pathways to bypass the enhancer requirement of σ54 RNA polymerase: Roles for DNA and protein determinants. Proc Natl Acad Sci. 1997;94:9538–9543. doi: 10.1073/pnas.94.18.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Guo Y, Gralla JD. Regulation of σ54 dependent transcription by core promoter sequences: Role of −12 region nucleotides. J Bacteriol. 1999;181:7558–7565. doi: 10.1128/jb.181.24.7558-7565.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel A, Kustu S. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes & Dev. 1995;9:2042–2052. doi: 10.1101/gad.9.16.2042. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Batut J, Klose KE, Keener J, Kustu S. The phosphorylated form of the enhancer-binding protein NTRC has an ATPase activity that is essential for activation of transcription. Cell. 1991;67:155–167. doi: 10.1016/0092-8674(91)90579-n. [DOI] [PubMed] [Google Scholar]
- Woychik NA, Young RA. RNA polymerase II: Subunit structure and function. Trends Biochem Sci. 1990;15:347–351. doi: 10.1016/0968-0004(90)90074-l. [DOI] [PubMed] [Google Scholar]
- Yan M, Gralla JD. The use of ATP and initiating nucleotides during postrecruitment steps at the activated adenovirus E4 promoter. J Biol Chem. 1999;274:34819–34824. doi: 10.1074/jbc.274.49.34819. [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]