

Abstract
The 1,310 Haloarcula marismortui proteins identified from mid-log and late-log phase soluble and membrane proteomes were analyzed in metabolic and cellular process networks to predict the available systems and systems fluctuations upon environmental stresses. When the connected metabolic reactions with identified proteins were examined, the availability of a number of metabolic pathways and a highly connected amino acid metabolic network were revealed. Quantitative spectral count analyses suggested 300 or more proteins might have expression changes in late-log phase. Among these, integrative network analyses indicated approximately 106 were metabolic proteins which might have growth-phase dependent changes. Interestingly, a large proportion of proteins in affected biomodules had same trend of changes in spectral counts. Disregard the magnitude of changes, we had successfully predicted and validated the expression changes of nine genes including the rimK, gltCP, rrnAC, and argC in lysine biosynthesis pathway which were downregulated in late-log phase. This study had not only revealed the expressed proteins but also the availability of biological systems in two growth phases, systems level changes in response to the stresses in late-log phase, cellular locations of identified proteins, and the likely regulated genes to facilitate further analyses in the postgenomic era.
Introduction
Halophilic archaea are prokaryotes living in high salt artificial or natural environments such as solar salterns, the Great Salt Lake, and the Dead Sea 1-4. Of the culturable groups of archaea, which included the methanogens, halophiles, and thermophiles, the availability of several genome sequences and genetic manipulation tools and their ease of culture had made halophiles attractive models to understand the biology of the domain archaea. Since the completion of the Halobacterium salinarum strain NRC-1 genome 5-8, at least ten other halophiles including Haloarcula marismortui 9, Natronomonas pharaonis 10, Haloquadratum walsbyi 11, Haloferax volcanii 12, Halalkalicoccus jeotgali 13, Halomicrobium mukohataei, Halorhabdus utahensis, Halorubrum lacusprofundi, Haloterrigena turkmenica, and Natrialba magadii (unpublished, Joint Genome Institute, DOE, USA; www.ncbi.nlm.nih.gov/sites/genome) had also been sequenced. Among these, H. salinarum NRC-1 had the smallest genome consisted of a large chromosome (2,014,239 bp) and two smaller replicons (191,346-bp pNRC100 and 365,425-bp pNRC200) encoding a total of approximately 2,400 unique proteins 6. In contrast, H. marismortui had the second largest (4,274,642 bp) and the most complex genome architecture consisted of nine replicons ranging from 33,303 to 3,131,724 bp 9.
Analyses of the completed halophile genome sequences had shed light on how these organisms survive in extreme environments 6, 14. For instance, examination of the H. salinarum NRC-1 genome revealed an acidic proteome with an average pI of 4.5 which is important for maintaining the solubility and functionality of proteins in the hypersaline cytoplasm 14, 15. Another discovery was the possession of an array of sensors, signal transducers, and transcriptional regulators, including multiple general transcription factors, which enable it to sense and tailor its physiology to perturbations in a diverse set of environmental factors. Comparison of the H. salinarum and H. marismortui genomes suggested both halophilic archaea use the same strategy of high surface negative charge of folded proteins as a mechanism to circumvent the salting-out phenomenon in a hypersaline cytoplasm 9. The analyses of the 4,240 predicted genes of H. marismortui revealed the presence of six opsins 16, 19 MCP and/or HAMP domain signal transducers, and an unusually large number of environmental response regulators, nearly five times as many as those encoded in H. salinarum NRC-1. This suggested H. marismortui is significantly more physiologically capable of exploiting a variety of diverse environments.
The rapid expansion of genomic data has provided a large amount of information for comprehensive prediction of most, if not all, possible biological activities present in the sequenced organisms. However, the information predicted from genome analyses are static and thus dynamic gene expression data are required to elucidate the in vivo status of the predicted biological activities. This gap can be filled either by high-throughput expression profiling of mRNAs or proteins prepared from one or more growth conditions 17-23. For instance, our previous analyses of the H. salinarum proteome by LC-MS/MS had shed light on the status of a number of biomodules in standard culture condition 22, 23.
Stresses from decreases of nutrients and oxygen concentration and increases of cell density and metabolic waste concentration in medium in the late growth phases could be easily reproduced to reveal global cellular responses. The possession of a relatively large coding capacity of the H. marismortui genome among the sequenced halophiles made it an interesting model to learn how halophiles respond to the environmental/metabolic stresses prevalent in late growth phase. In addition to cataloging the expressed proteins, defining their subcellular locations, and analyzing their expression profile changes, this study also tried to shed light on the differences in available biochemical systems and their fluctuations between the mid-log and late-log phases via integrative proteomics and network analyses.
Materials and Methods
Strain, Medium, and Culture Conditions
Haloarcula marismortui (ATCC43049) starter culture was prepared by inoculating a single colony into 4 ml of Halobacterium medium containing trace metals (CM+) 24, 25 and incubated with shaking at 225 rpm and 37°C for one week. One ml of starter culture was transferred to 1 liter of CM+ medium and incubated as above until OD600 0.7 (mid-log phase) and 1.2 (late-log-phase) for preparing the proteome samples.
Proteome Preparations
H. marismortui cells were harvested by centrifugation at 5,500×g for 15 minutes at 4°C. The cell pellets were resuspended in 25 ml of basal salt solution 24 containing 1 mM of phenylmethylsulfonyl fluoride (PMSF) and 0.5 mg each of DNase I and RNase A. The cells were lyzed via osmotic shock inside dialysis tubing (Spectra/por® membrane MWCO: 3,500; Spectrum, Rancho Dominguez, CA) and the membrane and soluble proteins were fractionated as previously described 22.
Protein Digestion and Peptide Purification
An aliquot of 100 μg of proteins was digested with 2 μg of sequencing grade modified trypsin (Promega, Madison, WI) in a final volume of 500 μl of solution containing 50 mM ammonium bicarbonate (pH 8.3) and 0.05% of SDS at 37°C for 12 hours. The resulting peptides were purified using a C18 spin column (Pierce, Rockford, IL) according to the manufacturer's procedure. The samples were then lyophilized and stored at -20°C until LC-MS/MS analyses.
Tandem Mass Spectrometry
Peptides were analyzed using a nano-HPLC (nanoAcquity UPLC systems; Waters, Milford, MA) coupled to a hybrid linear ion trap Orbitrap (LTQ-Orbitrap XL; Software: XCalibur 2.0.7, FT programs 2.0.7, and LTQ-Orbitrap XL MS2.4 SP1) mass spectrometer (Thermo Scientific, San Jose, CA) similar to previously described 26-28. Each protein sample was analyzed by five MS/MS analyses in which the MS survey scans were acquired over a full m/z range of 400∼2000 and by gas-phase fractionation within four smaller windows of 400-520, 516∼690, 685∼968, and 963∼2000 m/z. The six most intense precursor ions were sequentially isolated using data-dependent mode (isolation width: 1 m/z) and subjected to collision induced dissociation (CID) in the linear ion trap with a dynamic exclusion duration of 45 seconds and exclusion list size of 150.
Computational Analysis of MS/MS Spectra
MS raw data in Thermo XCalibur (version 2.0.7) binary format were converted to the mzXML open data format using a modified version of the ReAdW program 29 and then searched with the SEQUEST (version 27) 30 algorithm against the forward and reversed (decoy) sequences of 4,240 H. marismortui proteins from the EMBL-EBI Integr8 proteome database (www.ebi.ac.uk/integr8/EBI-Integr8-HomePage.do) 31. The search parameters included a peptide mass tolerance of ±2.1 Da 32, possible oxidation of methionine (+16.0 Da) residue, and default settings of other parameters as suggested by the software developers. Cleaving agent was not specified for non-constraint database searches. SEQUEST search results were further processed using the Trans Proteomic Pipeline 33-35 as previously described 36.
Subcellular Locations
The total numbers of peptides of each protein identified from the soluble and membrane proteomes were counted. Proteins with peptides identified exclusively (100%) or predominantly (≥75%) from the membrane and soluble proteomes were considered as potential membrane and soluble, respectively, proteins. All identified proteins were analyzed with the TMHMM program for the presence of transmembrane helices 37.
Systems Analysis
Both genomics and proteomics data were analyzed to evaluate the available systems or biomodules in H. marismortui. The number of proteins, number of reactions, and number of connected reactions (or interacting proteins) in each KEGG (Kyoto Encyclopedia of Genes and Genomes; www.genome.jp/kegg/pathway.html) metabolic pathways or cellular processes were counted to predict the available systems. To shed light on systems fluctuations, protein expression changes were estimated by spectral counting of all peptides of each protein identified in mid- and late-log phases. Integrative proteomics and metabolic network analyses were facilitated with the Cytoscape software (www.cytoscape.org).
Validation of Differential Expressions
Differential expressed candidates with at least 3-fold differences in spectral counts between the two growth phases and two immediate protein neighbors in connected reactions with the same trend of change in spectral counts were chosen as the “primary candidates” for RT-PCR analyses. Then the connected neighbors of the validated genes, namely the “extended candidates”, disregard the magnitude of spectral count changes, were chosen for another round of RT-PCR analyses. The isolation of total RNA and RT-PCR analyses with the gene specific primers (Supplementary Table 1, Supporting Information) were performed as previously described 16.
(H. marismortui is a nonpathogenic microorganism. All laboratory procedures were conducted in accordance with the safety requirements of the Institutional Biosafety Committee in National Yang Ming University).
Results
Protein Identifications
Upon a total of forty LC-MS/MS analyses of duplicate membrane and soluble proteomes from H. marismortui grown to mid-log and late-log phases, 60,062 peptides were identified with a PeptideProphet probability (p) above 0.9 and a false discovery rate (FDR) of less than 0.4% in each dataset. A total of 1,310 proteins were detected with a ProteinProphet (P) probability above 0.9 and a minimum of two different unique peptides (Table 1; Supplementary Tables 2 to 6, Supporting Information). Among these, 692 and 913 proteins were identified from the membrane and soluble proteomes, respectively.
Table 1.
Summary of H. marismortui peptides identified by MS/MS.
| Samples | Membrane | Soluble | ||
|---|---|---|---|---|
| OD600 | 0.7 | 1.2 | 0.7 | 1.2 |
| Total # matches a | 16,758 | 15,836 | 13,334 | 14,134 |
| Total # Hma matches a | 16,746 | 15,826 | 13,317 | 14,109 |
| # decoy matches a | 12 | 10 | 17 | 25 |
| FDR b | 0.07% | 0.06% | 0.13% | 0.18% |
| # proteins identified | 596 | 626 | 842 | 862 |
| 692 | 913 | |||
| 1,310 | ||||
Only the peptide and decoy hits with a PeptideProphet probability above 0.9 were counted
FDR = # decoy matches/(total # Hma matches) × 100%
Membrane and Cytosolic Proteins
Of the 1,310 identified proteins, 441 had peptides exclusively (100% membrane: 397 proteins) or predominantly (≥ 75% membrane: 44 proteins) found in the membrane samples and were likely membrane proteins (Supplementary Table 7, Supporting Information), and 804 had peptides exclusively (100% soluble: 618 proteins) or predominantly (≥ 75% soluble: 186 proteins) found in the soluble proteomes were likely cytosolic proteins. Further TMHMM analysis indicated 63.2% and 15.9% proteins exclusively and predominantly, respectively, identified in membrane contained one or more transmembrane domain (TD), but only one (0.1%) of the 804 putative cytosolic proteins contains such a domain (Supplementary Table 8, Supporting Information). Among the detected proteins, the cell surface glycoprotein (Csg1) encoded by the 132,678 bp replicon pNG500 topped the list with 4,176 peptide identifications.
Expression Changes
Given similar numbers of peptides, 30,092 and 29,970, were identified from the mid- and late-log phases, respectively, the spectral counts were directly used to estimate expression changes (Supplementary Table 9, Supporting Information). Of the 1,310 identified proteins, 61 (each had 2 to 12 peptide identifications) were only found in mid-log phase and 69 proteins (each had 2 to 59 peptides) only in late-log phase. Among these, at least seven proteins, each had more than 10 peptide identifications, might be differentially expressed in one of the growth phases. These included the two hypothetical proteins rrnB0067 (12 peptides) and rrnAC3015 (11 peptides) identified in mid-log phase only, and thiamine biosynthesis protein ThiC (59 peptides), phosphate import ATP-binding protein PstB2 (37 peptides), sugar ABC transporter ATP-binding protein MsmX-3 (24 peptides), Sn-glycerol-3-phosphate transport system permease UgpE (23 peptides), and universal stress protein Usp25 (10 peptides) identified in late-log phase only. In addition, the expression levels of 52 and 58 proteins, each of which had at least five detected peptides and more than 3-fold higher number of peptides found in OD 0.7 and OD 1.2, respectively, might have altered due to the depletion of nutrients or other changes in the medium during late-log phase.
Systems Availability and Fluctuations
Of the 4,240 proteins predicted from the H. marismortui genome 9, 1,033 were assigned to one or more cellular processes or metabolic pathways in the KEGG database. Examination of the predicted proteins in metabolic pathways indicated at least 25 pathways had proteins catalyzing at least one cluster of five connected reactions. A total of 582 proteins with at least two different unique peptide identifications (Supplementary Table 10, Supporting Information) and 29 proteins with single unique peptide identification (Supplementary Figure 1, Supporting Information), representing 59.1% proteins with a KEGG assignment, were detected by MS/MS. Further analyses of these proteins indicated 20 pathways in mid-log phase and 21 pathways in late-log phase had enzymes catalyzed at least one cluster of five or more “connected reactions” (Table 2). These represented the likely complete or partial pathways present in an active or standby mode in these cultures. Since there were no dramatic differences between the identified proteins in these metabolic pathways or biological processes, the cells were likely possessing very similar biomodules in both growth phases. However, at least 13 pathways, in which three or more proteins had differential spectral counts, might have systems level changes in nutrient deficit late-log phase. Details of the representative available systems and systems fluctuations were summarized below.
Table 2.
Summary of the numbers of proteins, reactions, and connected reactions in KEGG metabolic pathways predicted from the genome sequence and MS/MS identified proteins (Pathways with many predicted proteins but low connectivity are shaded).
| Metabolism | Metabolic pathway/ cellular process |
Genomics Prediction | All proteomic data | OD 0.7 | OD 1.2 | OD 0.7 versus OD 1.2 b | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # predicted proteins |
# Reactions | # Connected reactions a |
#MS identified proteins (# single hit proteins) |
% identified proteins |
# Connected reactions with identified proteins a |
#MS identified proteins (# single hit proteins) |
% identified proteins |
# Connected reactions with identified proteins a |
#MS identified proteins (# single hit proteins) |
% identified proteins |
# Connected reactions with identified proteins a |
# proteins in OD 0.7 only | # proteins with ≥ 3X Peptides identified in OD 0.7 |
# proteins with ≥ 3X Peptides identified in OD 1.2 |
# proteins in OD 1.2 only | ||
| Carbohydrates | Glycolysis/ Gluconeogenesis * |
65 | 27 | 27 | 44(6) | 67.7 | 23 | 42(5) | 64.6 | 21 | 44(6) | 67.7 | 23 | 0 | 2 | 0 | 1 |
| Citrate cycle (TCA cycle) * |
29 | 17 | 16,1 | 24(1) | 82.8 | 15,1 | 24(1) | 82.8 | 15,1 | 23(1) | 79.3 | 15,1 | 1 | 1 | 0 | 0 | |
| Pentose phosphate pathway * |
24 | 17 | 8,9 | 20(4) | 83.3 | 8,6,2 | 20(4) | 83.3 | 8,6,2 | 18(3) | 75.0 | 7,6,1 | 1 | 0 | 1 | 0 | |
| Fructose and mannose metabolism * |
22 | 14 | 8,4,1,1 | 13(2) | 59.1 | 6,4 | 13(2) | 59.1 | 6,4 | 12(1) | 54.5 | 5,4 | 0 | 0 | 0 | 0 | |
| Starch and sucrose metabolism * |
19 | 18 | 4,3,3, 3,2,1, 1,1 |
12(4) | 63.2 | 3,3,3, 1,1 |
10(3) | 52.6 | 2,2,1, 1,1 |
12(4) | 63.2 | 3,3,3, 1,1 |
0 | 0 | 1 | 1 | |
| Amino sugar and nucleotide sugar metabolism * |
18 | 15 | 7,4,2,2 | 13(3) | 72.2 | 6,2,2,2 | 9(0) | 50.0 | 2,2,2, 2,1 |
13(3) | 72.2 | 2,4,2, 2,2 |
0 | 0 | 0 | 0 | |
| Pyruvate metabolism * |
44 | 19 | 19 | 34(1) | 77.3 | 18 | 34(1) | 77.3 | 17 | 34(1) | 77.3 | 17 | 0 | 3 | 1 | 0 | |
| Glyoxylate and dicarboxylate metabolism |
15 | 11 | 9,1,1 | 10(0) | 66.7 | 9,1 | 8(0) | 53.3 | 4,4,1 | 10(0) | 66.7 | 6.4 | 0 | 0 | 2 | 2 | |
| Propanoate metabolism |
38 | 11 | 7,2,1,1 | 32(0) | 84.2 | 7,2,1,1 | 31(0) | 81.6 | 5,2,1,1 | 32(0) | 84.2 | 5,2,1,1 | 0 | 2 | 1 | 1 | |
| Butanoate metabolism | 46 | 14 | 4,3,2, 1,1,1, 1,1 |
36(1) | 78.3 | 4,3,2, 1,1 |
36(1) | 78.3 | 4,3,2, 1,1 |
34(1) | 73.9 | 4,3,2,1 | 2 | 4 | 2 | 0 | |
|
| |||||||||||||||||
| Energy | Nitrogen metabolism * |
30 | 15 | 12,2,1 | 14(1) | 46.7 | 9,1 | 13(1) | 43.3 | 9,1 | 13(0) | 43.3 | 8,1 | 0 | 0 | 5 | 1 |
|
| |||||||||||||||||
| Lipids | Fatty acid metabolism * | 48 | 27 | 4,4,3,3(×5),1 | 36(1) | 75.0 | 4,4,3,3(×5),1 | 35(1) | 72.9 | 4,4,3,3(×5),1 | 36(1) | 75.0 | 4,4,3, 3(5),1 |
0 | 2 | 3 | 1 |
| Glycerolipid metabolism |
12 | 4 | 4 | 7(1) | 58.3 | 2,1 | 6(0) | 50.0 | 1,1 | 7(1) | 58.3 | 2,1 | 0 | 0 | 1 | 0 | |
|
| |||||||||||||||||
| Nucleotides | Purine metabolism * | 64 | 54 | 49,1,1, 1,1,1,1 |
43(2) | 67.2 | 32,5,1, 1,1 |
42(1) | 65.6 | 32,5,1, 1,1 |
39(1) | 60.9 | 30,5,1, 1,1,1 |
3 | 4 | 1 | 0 |
| Pyrimidine metabolism * |
51 | 45 | 40,1,1, 1,1,1 |
38(4) | 74.5 | 39,1,1 | 37(2) | 72.5 | 39 | 38(4) | 74.5 | 39,1,1 | 1 | 3 | 2 | 0 | |
|
| |||||||||||||||||
| Amino Acids | Alanine, aspartate and glutamate metabolism * | 25 | 17 | 17 | 19(2) | 76.0 | 13 | 19(2) | 76.0 | 13 | 17(0) | 68.0 | 11 | 0 | 3 | 1 | 0 |
| Glycine, serine and threonine metabolism * |
35 | 17 | 6,9,1,1 | 24(1) | 68.6 | 9,6,1 | 23(1) | 65.7 | 9,6,1 | 23(0) | 65.7 | 8,6,1 | 0 | 2 | 4 | 1 | |
| Cysteine and methionine metabolism * |
19 | 19 | 12,4,1, 1,1 |
13(2) | 68.4 | 8,3,4, 1,1 |
13(2) | 68.4 | 8,3,4, 1,1 |
11(0) | 57.9 | 6,3,2, 1,1,1 |
0 | 1 | 1 | 0 | |
| Valine, leucine and isoleucine degradation * |
40 | 24 | 4(×3),2(×3), 2,2,2 |
34(0) | 85.0 | 4(×3),4,2(×3),2 | 33(0) | 82.5 | 4(×3),4,2(×3),2 | 34(0) | 85.0 | 4(3),4,2(3),2 | 0 | 1 | 3 | 1 | |
| Valine, leucine and isoleucine biosynthesis * |
25 | 24 | 24 | 22(2) | 88.0 | 24 | 22(2) | 88.0 | 24 | 20(1) | 80.0 | 23 | 1 | 1 | 2 | 0 | |
| Lysine biosynthesis * |
16 | 15 | 8,4,3 | 13(2) | 81.3 | 7,4,3 | 12(1) | 75.0 | 7,3,3 | 13(2) | 81.3 | 7,4,3 | 0 | 2 | 0 | 0 | |
| Lysine degradation | 27 | 6 | 2,2,1,1 | 23(0) | 85.2 | 2,2,1,1 | 23(0) | 85.2 | 2,2,1,1 | 23(0) | 85.2 | 2,2,1,1 | 0 | 1 | 1 | 0 | |
| Arginine and proline metabolism * |
44 | 24 | 15,3,2, 1,1,1,1 |
28(4) | 63.6 | 7,3,3, 2,1,1, 1,1 |
26(3) | 59.1 | 6,3,3, 2,1,1, 1,1 |
26(2) | 59.1 | 4,3,3, 3,1,1, 1,1 |
0 | 2 | 1 | 1 | |
| Histidine metabolism * |
25 | 15 | 7,7,1 | 15(0) | 60.0 | 6,2,2, 1,1 |
14(0) | 56.0 | 6,2,1, 1,1 |
15(0) | 60.0 | 6,2,2, 1,1 |
0 | 1 | 1 | 1 | |
| Tyrosine metabolism |
24 | 6 | 2,1,1, 1,1 |
12(1) | 50.0 | 1,1,1 | 11(1) | 45.8 | 1,1,1 | 12(1) | 50.0 | 1,1,1 | 0 | 2 | 1 | 1 | |
| Tryptophan metabolism |
33 | 10 | 2,2,2, 1,1,1,1 |
27(0) | 81.8 | 2,2,1, 1,1,1,1 |
27(0) | 81.8 | 2,2,1, 1,1,1,1 |
27(0) | 81.8 | 2,2,1, 1,1,1,1 |
0 | 1 | 3 | 0 | |
| Phenylalanine, tyrosine and tryptophan biosynthesis * |
26 | 19 | 19 | 15(2) | 57.7 | 12,2,1 | 15(2) | 57.7 | 12,2,1 | 15(2) | 57.7 | 12,2,1 | 0 | 3 | 1 | 0 | |
|
| |||||||||||||||||
| Other Amino Acids | Beta-Alanine metabolism * | 22 | 7 | 3,2,1,1 | 16(1) | 72.7 | 3,2 | 15(0) | 68.2 | 2,2 | 16(1) | 72.7 | 3,2 | 0 | 0 | 1 | 0 |
| Selenoamino acid metabolism * |
10 | 8 | 4,4 | 6(0) | 60.0 | 3,3 | 6(0) | 60.0 | 3,3 | 6(0) | 60.0 | 3,3 | 0 | 0 | 0 | 0 | |
|
| |||||||||||||||||
| Cofactors and Vitamins | Ubiquinone and other terpenoid-quinone biosynthesis | 8 | 10 | 4,2,2, 1,1 |
3(1) | 37.5 | 4,1,1 | 3(1) | 37.5 | 4,1,1 | 3(1) | 37.5 | 4,1,1 | 0 | 0 | 0 | 0 |
| One carbon pool by folate * |
17 | 15 | 15 | 13(1) | 76.5 | 12 | 10(1) | 58.8 | 11 | 12(0) | 70.6 | 11 | 0 | 0 | 4 | 3 | |
| Thiamine metabolism * |
9 | 8 | 6,2 | 5(0) | 55.6 | 4,1 | 3(0) | 33.3 | 3,1 | 5(0) | 55.6 | 4,1 | 0 | 1 | 1 | 2 | |
| Riboflavin metabolism * |
5 | 5 | 4,1 | 3(1) | 60.0 | 2,1 | 3(1) | 60.0 | 2,1 | 3(1) | 60.0 | 2,1 | 0 | 0 | 1 | 0 | |
| Pantothenate and CoA biosynthesis |
15 | 11 | 6,3,1,1 | 12(3) | 80.0 | 5,3,1 | 10(1) | 66.7 | 5,1 | 10(2) | 66.7 | 5,3,1 | 1 | 0 | 0 | 0 | |
| Folate biosynthesis |
9 | 12 | 5,3,2, 1,1 |
3(1) | 33.3 | 2,1,1 | 2(0) | 22.2 | 1,1 | 3(1) | 33.3 | 1,1,1 | 0 | 0 | 1 | 0 | |
| Nicotinate and nicotinamide metabolism * |
12 | 11 | 11 | 5(1) | 41.7 | 7 | 4(0) | 33.3 | 2,2,1 | 5(1) | 41.7 | 7 | 0 | 1 | 0 | 0 | |
| Porphyrin and chlorophyll metabolism * |
25 | 32 | 15,7,6, 2,1,1 |
20(4) | 80.0 | 8,7,4, 4,2,1 |
18(2) | 72.0 | 8,7,3, 2,1,1, 1,1 |
17(3) | 68.0 | 7,4,3, 1,1,1,1 |
2 | 0 | 0 | 0 | |
|
| |||||||||||||||||
| Biosynthesis of Polyketides and Terpenoids | Terpenoid backbone biosynthesis * | 14 | 11 | 4,3,3,1 | 11(0) | 78.6 | 4,2,1 | 11(0) | 78.6 | 4,2,1 | 11(0) | 78.6 | 4,2,1 | 0 | 1 | 1 | 0 |
| Carotenoid biosynthesis |
2 | 5 | 5 | 2(0) | 100.0 | 5 | 2(0) | 100.0 | 5 | 2(0) | 100.0 | 5 | 0 | 0 | 0 | 0 | |
Number of reactions within each “cluster of linked reaction(s)” separated by commas. Number in parenthesis indicates the number reaction cluster catabolized by the same enzyme(s).
The numbers included only the proteins with at least 2 unique peptide identifications.
Indicate the biomodules seemed to contain sufficient number of connected reactions (predicted proteins) to drive the functions.
Genetic Information Processing
A total of 178 (73.6%) of the 242 predicted proteins involved in genetic information processing including those for DNA replication and repair, transcription, translation, and protein folding, sorting, and degradation, were identified by MS/MS. Of these, a significant fraction of the RNA polymerase and ribosome subunits, basal transcription factors, and DNA replication proteins had relatively fewer peptides detected in the late-log phase suggesting the cells might be less active in DNA replication and gene expression (Table 3 and Supplementary Table 11, Supporting Information). These included 33 of the 44 ribosomal proteins, 9 of the 11 RNA polymerase subunits (including all the five subunits encoded by the putative rpoN-rpoB′-rpoB″-rpoA-rpoC operon), and the three basal transcription factors TATA-box binding protein E (TbpE), transcription initiation factors IIB (TfbA), and IIE alpha subunit (TfeA).
Table 3.
Comparison of the number of peptides of the major genetic information processing proteins identified in OD600 0.7 and 1.2.
| Locus name | Gene name | Protein description | Total # peptides | OD 0.7/1.2 | |
|---|---|---|---|---|---|
| OD 0.7 | OD 1.2 | ||||
| RNA polymerase | |||||
|
| |||||
| rrnAC0067 | rpoN | DNA-directed RNA polymerase subunit N | 2 | 0 | ∞ |
| rrnAC2829 | rpoE | DNA-directed RNA polymerase subunit E′ | 22 | 7 | 3.1 |
| rrnAC2432 | rpoH | DNA-directed RNA polymerase subunit H | 9 | 3 | 3.0 |
| rrnAC2428 | rpoA | DNA-directed RNA polymerase | 70 | 41 | 1.7 |
| rrnAC2430 | rpoB | DNA-directed RNA polymerase subunit B | 54 | 34 | 1.6 |
| rrnAC2830 | rpoE | DNA-directed RNA polymerase subunit E | 3 | 2 | 1.5 |
| rrnAC2429 | rpoB | DNA-directed RNA polymerase β subunit | 56 | 40 | 1.4 |
| rrnAC2427 | rpoC | RNA polymerase Rpb1 domain 5 | 22 | 16 | 1.4 |
| rrnAC0259 | rpb4 | RNA polymerase Rpb4 | 20 | 17 | 1.2 |
| rrnAC1396 | rpoL | DNA-directed RNA polymerase subunit L | 4 | 4 | 1.0 |
| rrnAC0062 | rpoD | DNA-directed RNA polymerase subunit D | 4 | 7 | 0.6 |
|
| |||||
| DNA replication proteins | |||||
|
| |||||
| rrnAC0455 | gyrA | DNA gyrase subunit A | 13 | 4 | 3.2 |
| rrnAC2691 | polC | DNA polymerase II large subunit | 3 | 1 | 3.0 |
| rrnAC1645 | topA | DNA topoisomerase I | 4 | 2 | 2.0 |
| rrnAC2565 | rfcS | Replication factor C small subunit | 12 | 7 | 1.7 |
| rrnAC0457 | top6B | Type II DNA topoisomerase VI subunit B | 37 | 24 | 1.5 |
| rrnAC0456 | gyrB | DNA gyrase subunit B | 7 | 5 | 1.4 |
| rrnAC2487 | rfcC2 | Replication factor C small subunit | 5 | 4 | 1.3 |
| rrnAC1831 | polB1 | DNA polymerase B elongation subunit | 1 | 2 | 0.5 |
| rrnAC0459 | top6A | Type II DNA topoisomerase VI subunit A | 2 | 5 | 0.4 |
|
| |||||
| DNA repair and recombination proteins | |||||
|
| |||||
| rrnAC2996 | uvrB | UvrABC system protein B | 2 | 0 | ∞ |
| rrnB0041 | xseA2 | Probable exodeoxyribonuclease VII large subunit | 3 | 0 | ∞ |
| rrnAC0455 | gyrA | DNA gyrase subunit A | 13 | 4 | 3.3 |
| rrnAC2267 | uvrD | Repair helicase | 27 | 9 | 3.0 |
| rrnAC0832 | phrB1 | Deoxyribodipyrimidine photolyase | 2 | 1 | 2.0 |
| rrnAC2159 | dcd3 | Probable deoxyuridine 5′-triphosphate nucleotidohydrolase | 2 | 1 | 2.0 |
| rrnAC2532 | mutS1 | DNA mismatch repair protein mutS 1 | 2 | 1 | 2.0 |
| rrnAC1840 | smc1 | Structural maintenance of chromosomes | 2 | 1 | 2.0 |
| rrnAC2847 | hjr | Holliday junction resolvase | 4 | 2 | 2.0 |
| rrnAC1645 | topA | DNA topoisomerase I | 4 | 2 | 2.0 |
| rrnAC2116 | uvrA | Excinuclease ABC subunit A | 14 | 8 | 1.8 |
| rrnAC2744 | rrnAC2744 | Hypothetical protein | 5 | 3 | 1.7 |
| pNG2007 | pNG2007 | Hypothetical protein | 18 | 11 | 1.6 |
| rrnAC2015 | nfo | Probable endonuclease 4 | 3 | 2 | 1.5 |
| rrnAC0456 | gyrB | DNA gyrase subunit B | 7 | 5 | 1.4 |
| rrnAC1622 | nrdA | Ribonucleoside-diphosphate reductase α chain | 108 | 91 | 1.2 |
| rrnAC0669 | polX | DNA polymerase IV | 9 | 8 | 1.1 |
| pNG7116 | nthC | Endonuclease III | 4 | 4 | 1.0 |
| rrnAC2910 | radA | DNA repair and recombination protein RadA | 25 | 27 | 0.9 |
| rrnAC2851 | pcn | DNA polymerase sliding clamp | 24 | 33 | 0.7 |
| rrnAC1133 | rpaA | Replication protein A | 12 | 17 | 0.7 |
| rrnAC2550 | mutL | DNA mismatch repair protein mutL | 2 | 3 | 0.7 |
| rrnAC0265 | apl | AP-endonuclease/AP-lyase | 4 | 7 | 0.6 |
| pNG6094 | xseA1 | Probable exodeoxyribonuclease VII large subunit | 0 | 2 | 0 |
|
| |||||
| Basal transcription factors | |||||
|
| |||||
| rrnAC0016 | tfbA | Transcription initiation factor IIB | 4 | 0 | ∞ |
| rrnAC0861 | tfeA | Transcription initiation factor IIE α subunit | 9 | 3 | 3.0 |
| rrnAC0681 | tbpE | TATA-box binding protein E | 22 | 12 | 1.8 |
|
| |||||
| Ribosome | |||||
|
| |||||
| rrnAC1596 | rpl6p | 50S ribosomal protein L6P | 5 | 0 | ∞ |
| rrnAC1418 | rpl12p | 50S ribosomal protein L12P | 3 | 0 | ∞ |
| rrnAC1607 | rps19p | 30S ribosomal protein S19P | 12 | 2 | 6.0 |
| rrnAC1604 | rpl29p | 50S ribosomal protein L29P | 5 | 1 | 5.0 |
| rrnAC0059 | rps4p | 30S ribosomal protein S4P | 27 | 7 | 3.9 |
| rrnAC0061 | rps11p | 30S ribosomal protein S11P | 23 | 6 | 3.8 |
| rrnAC1598 | rpl5p | 50S ribosomal protein L5P | 13 | 4 | 3.3 |
| rrnAC0058 | rps13p | 30S ribosomal protein S13P | 21 | 8 | 2.6 |
| rrnAC2488 | rps24e | 30S ribosomal protein S24e | 16 | 6 | 2.7 |
| rrnAC1605 | rps3p | 30S ribosomal protein S3P | 53 | 21 | 2.5 |
| rrnAC1417 | rplP0 | 50S ribosomal protein L10E | 17 | 8 | 2.1 |
| rrnAC1429 | rps3Ae | 30S ribosomal protein S3Ae | 80 | 39 | 2.1 |
| rrnAC1600 | rps4e | 30S ribosomal protein S4E | 34 | 17 | 2.0 |
| rrnAC2212 | rps6e | 30S ribosomal protein S6e | 14 | 7 | 2.0 |
| rrnAC1610 | rpl4p | 50S ribosomal protein L4P | 2 | 1 | 2.0 |
| rrnAC2357 | rpl10e | 50S ribosomal protein L10e | 14 | 7 | 2.0 |
| rrnAC1592 | rps5p | 30S ribosomal protein S5P | 63 | 35 | 1.8 |
| rrnAC1611 | rpl3p | 50S ribosomal protein L3P | 91 | 57 | 1.6 |
| rrnAC0064 | rpl18e | 50S ribosomal protein L18e | 41 | 26 | 1.6 |
| rrnAC1595 | rpl32e | 50S ribosomal protein L32e | 8 | 5 | 1.6 |
| rrnAC1426 | rps15p | 30S ribosomal protein S15P/S13e | 18 | 12 | 1.5 |
| rrnAC3179 | rps19e | 30S ribosomal protein S19e | 22 | 15 | 1.5 |
| rrnAC0070 | rps2p | 30S ribosomal protein S2P | 26 | 18 | 1.4 |
| rrnAC2065 | rpl15e | 50S ribosomal protein L15e | 16 | 11 | 1.5 |
| rrnAC1608 | rpl2p | 50S ribosomal protein L2P | 24 | 17 | 1.4 |
| rrnAC3112 | rpl39e | 50S ribosomal protein L39e | 7 | 5 | 1.4 |
| rrnAC1511 | rps8e | 30S ribosomal protein S8e | 12 | 9 | 1.3 |
| rrnAC1591 | rpl30p | 50S ribosomal protein L30P | 9 | 7 | 1.3 |
| rrnAC1603 | rps17p | 30S ribosomal protein S17P | 10 | 8 | 1.3 |
| rrnAC0260 | rpl21e | 50S ribosomal protein L21e | 5 | 4 | 1.3 |
| rrnAC0055 | rps17e | 30S ribosomal protein S17e | 6 | 5 | 1.2 |
| rrnAC1606 | rpl22p | 50S ribosomal protein L22P | 12 | 10 | 1.2 |
| rrnAC0065 | rpl13p | 50S ribosomal protein L13P | 30 | 27 | 1.1 |
| rrnAC1597 | rps8p | 30S ribosomal protein S8P | 7 | 7 | 1.0 |
| rrnAC2424 | rps12p | 30S ribosomal protein S12P | 15 | 15 | 1.0 |
| rrnAC0103 | rpl7ae | 50S ribosomal protein L7Ae | 16 | 16 | 1.0 |
| rrnAC2423 | rps7p | 30S ribosomal protein S7P | 35 | 40 | 0.9 |
| rrnAC1602 | rpl14p | 50S ribosomal protein L14P | 21 | 29 | 0.7 |
| rrnAC3113 | rpl31e | 50S ribosomal protein L31e | 4 | 6 | 0.7 |
| rrnAC1415 | rpl1p5 | 50S ribosomal protein L1P | 1 | 2 | 0.5 |
| rrnAC1594 | rpl19e | 50S ribosomal protein L19e | 2 | 5 | 0.4 |
| rrnAC1590 | rpl15p | 50S ribosomal protein L15P | 10 | 28 | 0.4 |
| rrnAC2405 | rps10p | 30S ribosomal protein S10P | 1 | 3 | 0.3 |
| rrnAC3513 | rps27E | 30S ribosomal protein S27e | 1 | 3 | 0.3 |
Ratios of proteins with lower spectral counts in OD600 1.2 are shaded.
Amino Acid Metabolism
The identification of a number of amino, dipeptide, and oligopeptide transporters, peptidases, and amino acid metabolic proteins suggested the amino acids and peptides in medium were actively transported and metabolized in the cells (Supplementary Tables 10, 12, and 13, Supporting Information). Among the 158 identified proteins (including 17 with single unique peptide identification) in 13 amino acid metabolic networks, acetolactate synthase small subunit (IlvN) was only found in mid-log phase and sacrosine dehydrogenase/glycine cleavage T-protein (GcvT1), methylmalonyl-CoA mutase subunit α (McmA2), imidazole glycerol phosphate synthase subunit (HisF), zinc-binding dehydrogenase (Adh10), and arginase (Arg1) were only detected in late-log phase. The examination connectivity of reactions with identified proteins indicated there were no differences in the availability of amino acid metabolic systems between the two growth phases (Table 2).
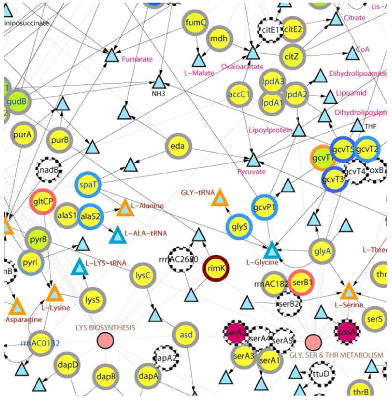
Despite only a limited number of proteins had more than 3-fold changes in spectral counts, a trend toward an increase or decrease of a high proportion of proteins catalyzing clusters of connected reactions were noticed in some of the pathways. For example, histidine metabolism might have upregulated in late-log phase. In addition to the HisF (found only in late-log phase), 1-(5-phosphoribosyl)-5-[(5-phosphoribosylamino)methylideneamino]imidazole-4-carboxamide isomerase (HisA) (>3 fold), and histidine ammonia-lyase (HutH), imidazolonepropionase (HutI), and urocanate hydratase (HutU) (2 to 3 fold), and four other proteins linked to the major connected reaction cluster also had slightly more peptides identified in the late culture (Figure 1A). Similarly, the lysine biosynthesis might have reduced because 9 of the 11 proteins (including two > 3 fold and one 2 to 3-fold) had fewer peptides detected in late-log phase (Figure 1B).
Figure 1. Comparative protein-metabolite networks analysis of the proteins identified in the mid-log and late-log phases for histidine metabolism (A), lysine biosynthesis (B), alanine, aspartate, glutamate metabolism (C), and phenylanaline, tyrosine, and tryptophan metabolism (D).

Circles and triangles indicate the enzymes and metabolites, respectively. Color scales shown on the bottom right corner indicate the log2 ratio of the number of peptides identified in OD600 0.7 and 1.2 (perimeters of the circles; top color scale) and the percentage of peptides identified in the soluble fraction (color of the circles; bottom color scale). Triangles with orange and black colored perimeters depict the amino acids and the other metabolic intermediates, respectively. Purple circles with dotted perimeter denote proteins with single unique peptide identification. Pink circles indicate the metabolic pathways and they are linked to the participating enzymes with thin lines. Single and double headed arrows indicate unidirectional and reversible, respectively, reactions. White circles with dotted circumference indicate those proteins not detected by MS/MS. The expression of tyrA (ellipse in D), which had no peptide identification in mid- and late-log phases, was confirmed by RT-PCR (data not shown). Gene names in red and blue colors indicate relatively more peptides were observed in mid-log and late-log, respectively.
Spectral counts also suggested some other amino acid metabolism pathways might have subsystem level changes or were only partially available in the cell. For instance, the two clusters of aspartate and glutamate subnetworks in the alanine, aspartate, and glutamate metabolism pathway seemed to have opposite changes in protein expressions (Figure 1C). Possibly due to sufficient supply of tryptophan in the medium, the tryptophan biosynthesis subnetwork in the phenylanaline, tyrosine, and tryptophan biosynthesis pathway might have little or no activity during the culture. Of the 12 predicted Trp proteins in this pathway, only the tryptophan synthase α chain (TrpA), anthranilate phosphoribosyltransferase (TrpD), and anthranilate synthase component I 1 (TrpE1) were identified raised the question as to whether most of these proteins were not expressed or expressed at undetectable low levels (Figure 1D). Although 23 (85.2%) of the 27 predicted proteins in lysine degradation, 27 (81.8%) of the 33 predicted proteins in the tryptophan metabolism, and 13 (54.2%) of the 24 predicted proteins in tyrosine metabolism were identified by MS/MS, the fact that they had low connectivity in reactions (≤ 2) suggested none of these pathways had enough connected reactions to drive the metabolisms.
Nucleotide Metabolism
The purine and pyrimidine metabolism pathways consisted of 64 and 51, respectively, predicted proteins were two of the most highly connected metabolic pathways in KEGG database (Table 2). Most of the proteins in purine metabolism pathway catalyzed a large cluster of 49 connected reactions. Proteomic and network analyses indicated approximately 32 and 30 of the 49-linked biochemical reactions had proteins identified in mid-log and late-log phases, respectively. Similarly, most of the predicted proteins in pyrimidine metabolism pathway also catalyzed a big cluster of 40 linked reactions. Proteomics and network analyses indicated approximately 39 of the 40-connected pyrimidine reactions had proteins identified in each growth phase. Other than the decrease of RNA polymerase level as suggested by lower spectral counts of nine of the subunit proteins in late-log phase, there were no obvious patterns of shifts in spectral counts to conclude other subsystems level changes in these pathways.
Cofactors and Vitamins Metabolism
The fact that Haloarcula marismortui could grow well in CM+ medium 24, 25 suggested it could synthesize all essential cofactors and vitamins. Among the eleven cofactor and vitamin metabolism pathways, only five had predicted proteins catalyzing more than five connected reactions (Table 2). The one carbon pool by folate metabolism pathway had a relative large number of connected reactions and a high percentage of the predicted proteins identified by MS/MS. Network analysis indicated the 17 predicted proteins in the pathway formed a single cluster of 15 connected reactions. The identified proteins catalyzed a cluster of 11 connected reactions in both cultures. Although the riboflavin metabolism network had only five predicted proteins, the presence of the genes of RibB, H, and C for converting ribulose 5-phosphate from the pentose phosphate pathway to 3,4-dihydroxy 2-butanone 4-phosphate,6,7-dimethyl-8-ribityllumazine, and the final product riboflavin suggested the presence of this pathway. However, the fact that none of these enzymes were identified by MS/MS implied these proteins might not have detectable levels in the samples. The nicotinate and nicotinamide metabolism pathway had 12 predicted proteins catalyzing a single cluster of reactions, among which five were identified in this study. The linkage between nicotinamide and the metabolic intermediates derived from aspartate in the culture medium or from the alanine, aspartate, and glutamate metabolism or quinolinate from tryptophan metabolism suggested this might be a functional pathway.
Analyses of the thiamine, vitamin B6, biotin, and lipoic acid metabolisms, and pantothenate and CoA, folate, and ubiquinone and other terpenoid-quinone biosynthesis pathways in KEGG database indicated these pathways might be partially functional or absent in H. marismortui. The thiamine metabolism pathway had three of the nine predicted proteins identified in both growth phases and another two proteins, thiamine biosynthesis protein (ThiC) (59 peptide identifications) and putative transcriptional regulator (TenA-2) (9 peptides), were only identified in late-log phase. An examination of the predicted pathway suggested H. marismortui might be able to utilize, but not synthesize thiamine. Although 12 of the 15 proteins in the Pantothenate and CoA biosynthesis pathway were identified by MS/MS, the absence of the genes of at least three key enzymes, pantothenate synthetase (PanC), pantothenate kinase (CoaA), and dephospho-CoA kinase (CoaE), suggested the pathway is incomplete. The folate and the ubiquinone and other terpenoid-quinone biosynthesis networks had eight and nine, respectively, predicted proteins but both lacked sufficient connectivity for carrying out the functions. Both vitamin B6 and biotin metabolism pathways did not seem to have enough enzymes for synthesizing the vitamins.
Among the cofactors and vitamins metabolism pathways containing five or more connected reactions, several proteins in the one carbon pool by folate metabolism network might have a higher expression level in the late-log phase. Except for the thymidylate synthase (ThyX) and serine hydroxymethyltransferase (GlyA) catalyzing the bidirectional conversion of 5,6,7,8-tetrahydrofolate and 5,10-methylene tetrahydrofolate, all of the other proteins had a higher number of peptides identified in the late culture (Table 2). These included four proteins with at least 3-fold more peptides and another three proteins with peptides exclusively detected in late-log phase. Although the folate biosynthesis pathway did not seem to contain a highly linked cluster of reactions, all of the four proteins detected by MS/MS showed higher spectral counts in late growth phase. These included the N(5)N(10)-methenyltetrahydromethanopterin cyclohydrolase (Mch) (>3 fold), and dihydropteroate synthase (FolP2) and F420-dependent N5N10-methylene-tetrahydromethanopterin reductase (Mer1) identified only in the late culture.
Carbohydrate Metabolism
Previous studies indicated H. marismortui could metabolize several carbohydrates including glucose, fructose, arabinose, xylulose, sucrose, and lactose 38-42 and synthesize glycoproteins 43-45. Analyses of the carbohydrate metabolism pathways revealed eight pathways contained at least one cluster of five connected reactions (Table 2). The glycolysis/gluconeogenesis pathway consisted of 65 predicted proteins to catalyze a single cluster of 27 linked reactions in glycolysis, conversion of pyruvate to acetyl-CoA and ethanol, and most of the gluconeogenesis reactions. Although amino acids and citrate were the only organic compounds in the medium, 42 proteins in 21 linked reactions and 44 proteins in 23 linked reactions were identified in mid-log and late-log phases, respectively. H. marismortui might also have a semi-complete fructose and mannose metabolism pathway consisted of 22 predicted proteins (14 predicted reactions). The cluster containing eight connected reactions in the pathway might allow fructose metabolism to occur. The detections of 13 (catalyzed 10 of the 14 predicted reactions) and 12 (catalyzed 9 reactions) proteins raised the possibility of constitutive expression of this pathway.
Apparently the pentose phosphate pathway for production of 5-phospho-α-D-ribose 1-diphosphate (PRPP), an important substrate for nucleotide synthesis, lacked the critical enzyme dihydroxyacid dehydratase (IlvD) in the KEGG network. Upon the examination of the H. marismortui proteome, the annotated IlvD protein (rrnAC0302), which had 38% identity (92% sequence coverage) and 30% identity (79% coverage) with the E. coli IlvD and 6-phosphogluconate dehydratase (Edd), respectively, was likely the missed enzyme. Proteomics analysis identified 20 and 18 of the 24 predicted proteins in mid-log and late-log phases, respectively, suggesting the presence of this pathway.
The pyruvate metabolism pathway contained 44 predicted proteins that catalyzed a cluster of 19 linked reactions, among which 34 proteins catalyzing 17 linked reactions were identified in both growth phases. The citrate cycle had 24 and 23 of the 29 predicted proteins, which catalyzed a cluster of 15 linked reactions and an orphan reaction, identified in mid-log and late-log phase, respectively. The starch and sucrose metabolism pathway contained the key enzymes to metabolize sucrose to α-D-glucose and β-D-fructose which might be further degraded in the glycolysis/gluconeogenesis and/or the fructose and mannose metabolism pathways. Among these, the proteins α-glucosidase (AglA1) and sucrose-6-phosphate hydrolase (ScrB) for degrading sucrose to β-D-fructose and α-D-glucose were identified by MS/MS. On the other hand, the other carbohydrate metabolism pathways such as pentose and glucuronate interconversion, galactose metabolism, and ascorbate and aldarate metabolism, which had very few predicted proteins and very limited connected reactions, might not exist in H. marismortui.
Miscellaneous proteins
There were other proteins, which did not have well defined biomodule or network relationships as those discussed above but had similar annotations or functions, might also have expression changes in late-log phase. For instance, of the 33 predicted universal stress proteins (Usp), 10 of the 23 identified proteins had more than 3-fold difference in spectral counts between the two growth phases. These included four exclusively found in the late-log phase and five had approximately 3- to 5.5-fold higher and one had 5-fold lower spectral counts in late-log phase (Table 4).
Table 4.
Summary of the 23 universal stress proteins identified by MS/MS.
| Locus name | Gene name | Protein description | Total # peptides | OD 0.7/1.2 | |
|---|---|---|---|---|---|
| OD 0.7 | OD 1.2 | ||||
| pNG7373 | usp25 | Universal stress protein | 0 | 10 | 0.00 |
| pNG7142 | usp31 | Universal stress protein | 0 | 8 | 0.00 |
| rrnAC2359 | usp16 | Universal stress protein | 0 | 2 | 0.00 |
| rrnB0291 | usp27 | Universal stress protein | 0 | 2 | 0.00 |
| rrnAC3171 | usp19 | Universal stress protein | 2 | 11 | 0.18 |
| rrnAC3225 | usp22 | Universal stress protein | 3 | 12 | 0.25 |
| rrnAC2000 | usp9 | Universal stress protein family | 3 | 11 | 0.27 |
| rrnAC3395 | usp24 | Universal stress protein family | 10 | 30 | 0.33 |
| rrnAC3508 | usp7 | Universal stress protein | 1 | 3 | 0.33 |
| rrnAC0716 | usp6 | Universal stress protein | 3 | 8 | 0.38 |
| rrnAC0758 | usp17 | Universal stress protein | 7 | 12 | 0.58 |
| rrnAC2809 | usp21 | Universal stress protein | 3 | 5 | 0.60 |
| rrnAC0738 | usp29 | Universal stress protein | 5 | 8 | 0.63 |
| rrnAC3125 | usp33 | Universal stress protein | 31 | 45 | 0.69 |
| rrnAC3460 | usp26 | Universal stress protein | 16 | 23 | 0.70 |
| rrnAC0402 | usp15 | Universal stress protein | 8 | 9 | 0.89 |
| pNG5052 | usp8 | Universal stress protein | 12 | 13 | 0.92 |
| pNG7139 | usp13 | Universal stress protein | 8 | 7 | 1.14 |
| rrnAC1404 | usp28 | Universal stress protein | 37 | 31 | 1.19 |
| rrnAC2223 | usp2 | Universal stress protein family | 6 | 5 | 1.20 |
| pNG7083 | usp23 | Putative universal stress protein family | 4 | 3 | 1.33 |
| rrnAC2220 | usp5 | Universal stress protein | 9 | 6 | 1.50 |
| rrnB0192 | usp1 | Universal stress protein 1 | 10 | 2 | 5.00 |
Ratios of proteins with higher spectral counts in OD600 1.2 are shaded and rows with more than 3-fold difference are bolded.
System level changes among the 117 identified transporters (total 274 predicted) were also suggested by the spectral counts (Supplementary Table 13, Supporting Information). For instance, four phosphonates transporter proteins (total of seven predicted) encoded by the phnD1, phnC2, phnE, and phnD2 gene cluster were all identified with a relatively lower number (3.4 to 7.3-fold decreases) of peptides in late-log phase. Of the nine phosphate transporter proteins, the phosphate import ATP-binding proteins (PstB1 and PstB2), phosphate ABC transporter permease proteins (PstA1 and PstC2), and phosphate ABC transporter binding protein (YqgG) were predominantly or exclusively found in late-log phase. Most of the dipeptide and oligopeptide transporter proteins, except for oligopeptide ABC transporter solute-binding protein (OppA) and oligopeptide ABC transporter ATPase component (OppD2), had relatively fewer peptides identified in late-log phase. These included the proteins encoded by the “dipeptide ABC transporter ATP-binding proteins (DppF and DppD) and dipeptide ABC transporter permease (DppB1)” and “oligopeptide ABC transporter permease protein (OppC), dipeptide ABC transporter permease (DppB), and dipeptide ABC transporter dipeptide-binding protein (DppA)” gene clusters. A total of 10 dipeptide transporters were identified with 18 to 551 peptides. Interestingly all of them had relatively fewer (0.71 to 0.96 folds) peptides identified in the late-log samples. A total of 24 amino acid transporter proteins were detected by MS/MS, among which 17 had slightly higher to 3-fold more peptides in late-log phase. Despite the absence of carbohydrate in the culture medium, 16 sugar transporter proteins were identified in either one or both growth phases (Supplementary Table 13, Supporting Information). Among these, the sugar ABC transporter ATP-binding protein (MsmX-3; 24 peptides), Sn-glycerol-3-phosphate transport system permease (UgpE; 23 peptides), ABC transporter permease protein (UgpA; 9 peptides), and putative ABC transporter substrate binding protein (RbsB-3; 9 peptides) were only detected in the late-log phase.
Validation of expression changes
The primary differential expressed candidate genes arg2 (arginase), rimK (Ribosomal protein S6 modification protein), fmdA (formamidase), gcvT1 (sacrosine dehydrogenase/glycine cleavage T-protein), acaB4 (3-ketoacyl-CoA thiolase) and ech4 (enoyl-CoA hydratase), which had (i) more than 3-fold differences in spectral counts between the two growth phases or more than nine peptides exclusively identified in mid- or late-log phase and (ii) at least two connected neighbors in the pathway or were expressed from genes in an operon with the same trend of changes in spectral counts, were chosen for expression validation. The RT-PCR results indicated their mRNA profiles are comparable with the spectral counts (Figure 2A, B, and C). To validate the down regulation of lysine biosynthesis in late-log phase, the mRNA levels of the ribosomal protein S6 modification protein (RimK) and glutamate carboxypeptidase (GltCP), which showed a 3-fold decrease in spectral counts in late-log phase, were analyzed by RT-PCR. As shown in Figure 2D, E, and F, the mRNA expression levels of rimK and gltCP were reduced by approximately 4.5- and 4.8-fold, respectively, in late-log phase. Further RT-PCR examinations of their adjacent neighbors (i.e. the “extended candidates”), rrnAC0132 (lysine-ketoglutarate reductase/saccharopine dehydrogenase) and argC (N-acetyl-γ-glutamyl-phosphate reductase), which only had minor decreases in spectral counts, also showed an mRNA decrease in late-log phase.
Figure 2. Validation of the differential expressed protein candidates predicted by spectral count.
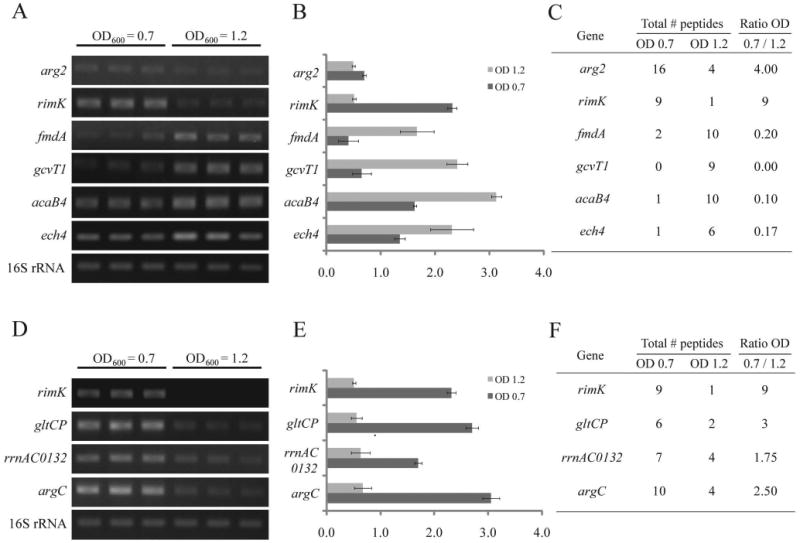
RT-PCR results of six “primary candidate” genes that had 3-folds or more changes in peptide ratio (A) and the primary candidate rimK and its “extended candidates” in lysine biosynthesis pathway that had the same trend of changes in spectral counts (D). The 16S rRNA is served as the internal control. Expression level in (A) and (D) normalized against the 16S rRNA signals are shown in (B) and (E), respectively. The spectral counts of the tested genes are shown in (C) and (F).
Discussion
H. marismortui grows well in the Halobacterium medium 25, 46 which provides the amino acids, peptides, citrate and salts for the syntheses of other simple or more complex molecules. With the completed genome sequence and recent advances in proteomic and bioinformatic technologies, it became possible to investigate at a systems level of how nucleotides, carbohydrates, lipids, amino acids, cofactors, vitamins, etc. are synthesized 9, 23.
Systems analyses indicated H. marismortui had a complex metabolic network to utilize amino acids. To shed light on the availabilities, interactions, and fluctuations of different amino acid metabolism pathways, the networks of amino acid metabolisms and TCA cycle were integratively analyzed with the proteomic data (Figure 3). Of the 244 predicted proteins in these pathways, 166 and 167 proteins were identified in the mid-log and late-log phase, respectively. The discovery of less than two protein identification differences in each pathway between the growth phases suggested very similar amino acid metabolic systems were present in both growth phases. The integrative analyses suggested ten of the amino acids might be converted to acetyl Co-A (alanine, glycine and serine), fumarate or oxaloacetate (arginine, aspartate, asparagine, and proline), or oxoglutarate (glutarate, glutamate, and histidine), and entered the TCA cycle for energy production or as the metabolic intermediates for synthesizing other metabolites. Despite the fact that similar proteins in amino acid metabolism pathways were found, the differences in spectral counts between the mid- and late-log phases suggested the expressions of at least some of the proteins might have changed. Among these, dramatic changes in spectral counts of three or more proteins were noticed in the “alanine, aspartate, and glutamate metabolism”, “glycine, serine, and threonine metabolism”, and “phenylalanine, tyrosine and tryptophan biosynthesis” pathways. Concurrent changes for a significant fraction of the proteins (some of the proteins < 3-fold) in histidine metabolism and lysine biosynthesis pathways were also noticed in our analyses (Table 2 and Figure 1). The down regulation of the lysine biosynthesis module is supported by lower transcript levels of the four genes rimK, gltCP, rrnAC0132 (lys1), and argC (Figure 2)
Figure 3. Integrative view of amino acid metabolisms and TCA cycle.

Complete network: Large circles represent the enzymes and small circles denote the pathways (A). Triangles with orange, blue, black colored perimeters indicate the amino acids, aminoacyl-tRNA, and metabolic intermediates, respectively. Color scales shown on the bottom right corner indicate the log2 ratio of the number of peptides identified in OD600 0.7 and 1.2 (perimeters of the circles; top color scale) and the percentage of peptides identified in the soluble fraction (color of the circles; bottom color scale). Large purple circles represent proteins with single unique peptide identification. The arrows represent the direction of enzymatic reactions (double arrows denote reversible reactions) and thin lines link the enzymes to their corresponding pathways. The metabolic pathways information was derived from the KEGG database and the network was displayed using the Cytoscape software. A simplified network of amino acid metabolisms and TCA cycle is shown in (B).
In addition to the proteins involved in metabolisms or cellular processes, a number of other proteins which might be important to H. marismortui were also suggested by the proteomic data. For instance, the cell surface glycoprotein (Csg1), a critical structural component for maintaining cell morphology, had the highest number of peptide (4,176) identifications (Supplementary Table 12, Supporting Information). Interestingly, the second to the ninth most identified proteins (754 to 2633 peptides), which had more than 98% of peptides detected from the membrane proteome, were also membrane proteins. These included in descending order of the number of detected peptides two hypothetical proteins RrnAC1803 and RrnAC3098, proteinase IV-like protein (Edp), H+-ATP synthase subunit C (AtpC1), dipeptide ABC transporter ATP-binding (DppD), phosphate ABC transporter binding (YqgG), dipeptide ABC transporter dipeptide-binding (DppA), and an halocyanin-like protein (Hcp9). The top ten most identified protein list ended with the cytosolic proteins malate dehydrogenase (Mdh) of the TCA cycle that had a total of 483 peptides identified from the soluble proteomes and 3 peptides from membrane proteomes. Although the two highly detected hypothetical proteins RrnAC1803 and RrnAC3098 did not have a known function, the fact that they both contained one TMHMM predicted transmembrane domain (23 aa) near their N-termini suggested they might play an important role in the cell surface.
An important candidate that might have orchestrated the expression changes of metabolic proteins was the transcription regulator TrmB. Previous studies had shown TrmB regulated diverse, but functionally linked metabolic pathways in Halobacterium salinarum strain NRC-1 47. H. marismortui had four TrmB homologues (encoded by rrnAC0914, rrnAC2354, rrnAC0924, and rrnAC0172) with sequence identities between 28 and 57% with the TrmB in NRC-1. Among these, only the regulators encoded by rrnAC0914 (TrmB1) and rrnAC2354 were identified in both growth phases. Since less than three peptides were identified in each sample, we cannot say for sure whether the TrmB-1, which had the highest homology with the NRC-1 homologue, or RrnAC2354 had expression change in response to the stimulations in late-log phase. Nonetheless, we could correlate the expression changes of some other regulators with their putative target proteins. For instance, the expressions of the phosphate regulatory protein-like Prp1 (mid-log:late-log peptides = 7:26) and the phosphate ABC transporter proteins PstA1 (14:39), PstB2 (0:37), PstC2 (1:11), and YqgG (79:729) encoded by adjacent genes might have increased to raise phosphate uptake in late-log phase. The increase of the transcription activator TenA (3:21) might be related to the higher peptide detection rates of the other proteins including cysteine desulfurase (Csd1), thiamine biosynthesis protein (ThiC), and putative transcriptional regulator (TenA-2) in thiamine metabolism pathway.
Conclusion
We had performed integrative network analyses on the genomic and proteomic data to shed light on H. marismortui metabolic capacities and systems fluctuations between the mid-log and late-log phases. By examining the connectivity of metabolic reactions and the proteins in cellular processes (Table 2), we could predict whether the available systems were likely present in a functional or standby mode. Spectral count analyses of the identified proteins (Table 2, Figure 3, and Supplementary Table 10, Supporting Information) suggested some of the biomodules might have system or subsystem level changes in response to the metabolic and other stresses in the late-log phase. Based on the presence of concurrent increases or decreases in spectral counts among the proteins in connected reactions or among proteins encoded by the same operon, the expression profiles of several differential candidates were validated by RT-PCR. Once the expression changes of the proteins with dramatic changes were validated, the mRNA levels of their juxtaposed proteins in metabolic networks, or those encoded by the same operon, were also found to be in agreement with the spectral count estimations. Thus the identification of expression changes of at least some of the proteins, disregarding the magnitude of fluctuations, could be facilitated by integrative network analyses. Despite the fact that how these genes were regulated in H. marismortui have yet to be elucidated, a list of possibly regulated genes or systems, which may be useful for understanding how this organism manage metabolic stresses and other environmental perturbations, has been obtained in this study.
Supplementary Material
Synposis.
The evaluations of in vivo available biomodules and systems level fluctuations upon perturbation could be facilitated by the analyses of proteomics data in protein-interaction networks. Despite only a portion of the proteome was identified and the protein expression changes were not always dramatic, the trends of up- or down-regulation of a number of biomodules in mid- and late-log phase cultures were revealed after overlaid the expression information in protein networks.
Acknowledgments
This work was supported in part by Grant 96-2628-B-010-003-MY3 from the National Science Council and intramural funding derived from the Aim for the Top University Grant awarded to National Yang Ming University from the Ministry of Education in Taiwan, Republic of China and by the University of Washington's Proteomics Resource (UWPR95794). The work was also supported by U.S.A. grants from the National Center for Research Resources (1S10RR023044-01) and NIEHS Center for Ecogenetics and Environmental Health (P30ES07033).
Footnotes
SUPPORTING INFROMATION AVAILABLE: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.DasSarma S, RajBhandary UL, Khorana HG. High-frequency spontaneous mutation in the bacterio-opsin gene in Halobacterium halobium is mediated by transposable elements. Proc Natl Acad Sci U S A. 1983;80(8):2201–5. doi: 10.1073/pnas.80.8.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dundas IE. Physiology of halobacteriaceae. Adv Microb Physiol. 1977;15:85–120. doi: 10.1016/s0065-2911(08)60315-x. [DOI] [PubMed] [Google Scholar]
- 3.Simsek M, DasSarma S, RajBhandary UL, Khorana HG. A transposable element from Halobacterium halobium which inactivates the bacteriorhodopsin gene. Proc Natl Acad Sci U S A. 1982;79(23):7268–72. doi: 10.1073/pnas.79.23.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DasSarma P, Klebahn G, Klebahn H. Translation of Henrich Klebahn's ‘Damaging agents of the klippfish - a contribution to the knowledge of the salt-loving organisms’. Saline Systems. 2010;6:7. doi: 10.1186/1746-1448-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeiffer F, Schuster SC, Broicher A, Falb M, Palm P, Rodewald K, Ruepp A, Soppa J, Tittor J, Oesterhelt D. Evolution in the laboratory: the genome of Halobacterium salinarum strain R1 compared to that of strain NRC-1. Genomics. 2008;91(4):335–46. doi: 10.1016/j.ygeno.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Ng WV, Kennedy SP, Mahairas GG, Berquist B, Pan M, Shukla HD, Lasky SR, Baliga NS, Thorsson V, Sbrogna J, Swartzell S, Weir D, Hall J, Dahl TA, Welti R, Goo YA, Leithauser B, Keller K, Cruz R, Danson MJ, Hough DW, Maddocks DG, Jablonski PE, Krebs MP, Angevine CM, Dale H, Isenbarger TA, Peck RF, Pohlschroder M, Spudich JL, Jung KW, Alam M, Freitas T, Hou S, Daniels CJ, Dennis PP, Omer AD, Ebhardt H, Lowe TM, Liang P, Riley M, Hood L, DasSarma S. Genome sequence of Halobacterium species NRC-1. Proc Natl Acad Sci U S A. 2000;97(22):12176–81. doi: 10.1073/pnas.190337797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng WV, Berquist BR, Coker JA, Capes M, Wu TH, DasSarma P, DasSarma S. Genome sequences of Halobacterium species. Genomics. 2008;91(6):548–52. doi: 10.1016/j.ygeno.2008.04.005. author reply 553-4. [DOI] [PubMed] [Google Scholar]
- 8.Ng WV, Ciufo SA, Smith TM, Bumgarner RE, Baskin D, Faust J, Hall B, Loretz C, Seto J, Slagel J, Hood L, DasSarma S. Snapshot of a large dynamic replicon in a halophilic archaeon: megaplasmid or minichromosome? Genome Res. 1998;8(11):1131–41. doi: 10.1101/gr.8.11.1131. [DOI] [PubMed] [Google Scholar]
- 9.Baliga NS, Bonneau R, Facciotti MT, Pan M, Glusman G, Deutsch EW, Shannon P, Chiu Y, Weng RS, Gan RR, Hung P, Date SV, Marcotte E, Hood L, Ng WV. Genome sequence of Haloarcula marismortui: a halophilic archaeon from the Dead Sea. Genome Res. 2004;14(11):2221–34. doi: 10.1101/gr.2700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falb M, Pfeiffer F, Palm P, Rodewald K, Hickmann V, Tittor J, Oesterhelt D. Living with two extremes: conclusions from the genome sequence of Natronomonas pharaonis. Genome Res. 2005;15(10):1336–43. doi: 10.1101/gr.3952905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolhuis H, Palm P, Wende A, Falb M, Rampp M, Rodriguez-Valera F, Pfeiffer F, Oesterhelt D. The genome of the square archaeon Haloquadratum walsbyi: life at the limits of water activity. BMC Genomics. 2006;7:169. doi: 10.1186/1471-2164-7-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartman AL, Norais C, Badger JH, Delmas S, Haldenby S, Madupu R, Robinson J, Khouri H, Ren Q, Lowe TM, Maupin-Furlow J, Pohlschroder M, Daniels C, Pfeiffer F, Allers T, Eisen JA. The complete genome sequence of Haloferax volcanii DS2, a model archaeon. PLoS One. 2010;5(3):e9605. doi: 10.1371/journal.pone.0009605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roh SW, Nam YD, Nam SH, Choi SH, Park HS, Bae JW. Complete genome sequence of Halalkalicoccus jeotgali B3(T), an extremely halophilic archaeon. J Bacteriol. 2010;192(17):4528–9. doi: 10.1128/JB.00663-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy SP, Ng WV, Salzberg SL, Hood L, DasSarma S. Understanding the adaptation of Halobacterium species NRC-1 to its extreme environment through computational analysis of its genome sequence. Genome Res. 2001;11(10):1641–50. doi: 10.1101/gr.190201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanyi JK. Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol Rev. 1974;38(3):272–90. doi: 10.1128/br.38.3.272-290.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu HY, Lin YC, Chang YN, Tseng H, Huang CC, Liu KC, Huang CS, Su CW, Weng RR, Lee YY, Ng WV, Yang CS. A novel six-rhodopsin system in a single archaeon. J Bacteriol. 2010 doi: 10.1128/JB.00642-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 18.Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Light-directed, spatially addressable parallel chemical synthesis. Science. 1991;251(4995):767–73. doi: 10.1126/science.1990438. [DOI] [PubMed] [Google Scholar]
- 19.Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D, Winkler J, Lockhart DJ, Morris MS, Fodor SP. Accessing genetic information with high-density DNA arrays. Science. 1996;274(5287):610–4. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 20.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 21.Choi H, Fermin D, Nesvizhskii AI. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics. 2008;7(12):2373–85. doi: 10.1074/mcp.M800203-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goo YA, Yi EC, Baliga NS, Tao WA, Pan M, Aebersold R, Goodlett DR, Hood L, Ng WV. Proteomic analysis of an extreme halophilic archaeon, Halobacterium sp. NRC-1. Mol Cell Proteomics. 2003;2(8):506–24. doi: 10.1074/mcp.M300044-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Gan RR, Yi EC, Chiu Y, Lee H, Kao YC, Wu TH, Aebersold R, Goodlett DR, Ng WV. Proteome analysis of Halobacterium sp. NRC-1 facilitated by the biomodule analysis tool BMSorter. Mol Cell Proteomics. 2006;5(6):987–97. doi: 10.1074/mcp.M500367-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.DasSarma S, Fleischmann EM. Archaea: A Laboratory Manual - Halophiles. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1995. [Google Scholar]
- 25.Oesterhelt D, Stoeckenius W. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 1974;31(Pt A):667–78. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 26.Ducret A, Van Oostveen I, Eng JK, Yates JR, 3rd, Aebersold R. High throughput protein characterization by automated reverse-phase chromatography/electrospray tandem mass spectrometry. Protein Sci. 1998;7(3):706–19. doi: 10.1002/pro.5560070320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi EC, Lee H, Aebersold R, Goodlett DR. A microcapillary trap cartridge-microcapillary high-performance liquid chromatography electrospray ionization emitter device capable of peptide tandem mass spectrometry at the attomole level on an ion trap mass spectrometer with automated routine operation. Rapid Commun Mass Spectrom. 2003;17(18):2093–8. doi: 10.1002/rcm.1150. [DOI] [PubMed] [Google Scholar]
- 28.Gao Q, Doneanu CE, Shaffer SA, Adman ET, Goodlett DR, Nelson SD. Identification of the interactions between cytochrome P450 2E1 and cytochrome b5 by mass spectrometry and site-directed mutagenesis. J Biol Chem. 2006;281(29):20404–17. doi: 10.1074/jbc.M601785200. [DOI] [PubMed] [Google Scholar]
- 29.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22(11):1459–66. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 30.Yates JR, 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67(8):1426–36. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 31.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 32.Hsieh EJ, Hoopmann MR, MacLean B, MacCoss MJ. Comparison of database search strategies for high precursor mass accuracy MS/MS data. J Proteome Res. 2010;9(2):1138–43. doi: 10.1021/pr900816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005 0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 35.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 36.Chu LJ, Chen MC, Setter J, Tsai Y, Yang H, Fung X, Ting YS, Shaffer SA, Taylor GK, von Haller PD, Goodlett D, Ng WV. New Structural Proteins of Halobacterium salinarum Gas Vesicle Revealed by Comparative Proteomics Analysis. J Proteome Res. 2010 doi: 10.1021/pr1009383. [DOI] [PubMed] [Google Scholar]
- 37.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 38.Rangaswamy V, Altekar W. Ketohexokinase (ATP:D-fructose 1-phosphotransferase) from a halophilic archaebacterium, Haloarcula vallismortis: purification and properties. J Bacteriol. 1994;176(17):5505–12. doi: 10.1128/jb.176.17.5505-5512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rawal N, Kelkar SM, Altekar W. Alternative routes of carbohydrate metabolism in halophilic archaebacteria. Indian J Biochem Biophys. 1988;25(6):674–86. [PubMed] [Google Scholar]
- 40.Johnsen U, Selig M, Xavier KB, Santos H, Schonheit P. Different glycolytic pathways for glucose and fructose in the halophilic archaeon Halococcus saccharolyticus. Arch Microbiol. 2001;175(1):52–61. doi: 10.1007/s002030000237. [DOI] [PubMed] [Google Scholar]
- 41.Johnsen U, Schonheit P. Novel xylose dehydrogenase in the halophilic archaeon Haloarcula marismortui. J Bacteriol. 2004;186(18):6198–207. doi: 10.1128/JB.186.18.6198-6207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falb M, Muller K, Konigsmaier L, Oberwinkler T, Horn P, von Gronau S, Gonzalez O, Pfeiffer F, Bornberg-Bauer E, Oesterhelt D. Metabolism of halophilic archaea. Extremophiles. 2008;12(2):177–96. doi: 10.1007/s00792-008-0138-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lechner J, Wieland F. Structure and biosynthesis of prokaryotic glycoproteins. Annu Rev Biochem. 1989;58:173–94. doi: 10.1146/annurev.bi.58.070189.001133. [DOI] [PubMed] [Google Scholar]
- 44.Goldman S, Hecht K, Eisenberg H, Mevarech M. Extracellular Ca2(+)-dependent inducible alkaline phosphatase from extremely halophilic archaebacterium Haloarcula marismortui. J Bacteriol. 1990;172(12):7065–70. doi: 10.1128/jb.172.12.7065-7070.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wieland F, Paul G, Sumper M. Halobacterial flagellins are sulfated glycoproteins. J Biol Chem. 1985;260(28):15180–5. [PubMed] [Google Scholar]
- 46.Dassarma S, Fleischmann EM, Robb FT, Place AR, Sowers KR, Schreier HJ. Archaea - A laboratory mannual (Halophiles) Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1995. [Google Scholar]
- 47.Schmid AK, Reiss DJ, Pan M, Koide T, Baliga NS. A single transcription factor regulates evolutionarily diverse but functionally linked metabolic pathways in response to nutrient availability. Mol Syst Biol. 2009;5:282. doi: 10.1038/msb.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.