

Abstract
Eukaryotes respond to the presence of unfolded protein in the endoplasmic reticulum (ER) by up-regulating the transcription of genes encoding ER protein chaperones, such as BiP. We have isolated a novel human cDNA encoding a homolog to Saccharomyces cerevisiae Ire1p, a proximal sensor for this signal transduction pathway in yeast. The gene product hIre1p is a type 1 transmembrane protein containing a cytoplasmic domain that is highly conserved to the yeast counterpart having a Ser/Thr protein kinase domain and a domain homologous to RNase L. However, the luminal domain has extensively diverged from the yeast gene product. hIre1p expressed in mammalian cells displayed intrinsic autophosphorylation activity and an endoribonuclease activity that cleaved the 5′ splice site of yeast HAC1 mRNA, a substrate for the endoribonuclease activity of yeast Ire1p. Overexpressed hIre1p was localized to the ER with particular concentration around the nuclear envelope and some colocalization with the nuclear pore complex. Expression of Ire1p mRNA was autoregulated through a process that required a functional hIre1p kinase activity. Finally, overexpression of wild-type hIre1p constitutively activated a reporter gene under transcriptional control of the rat BiP promoter, whereas expression of a catalytically inactive hIre1p acted in a trans-dominant-negative manner to prevent transcriptional activation of the BiP promoter in response to ER stress induced by inhibition of N-linked glycosylation. These results demonstrate that hIre1p is an essential proximal sensor of the unfolded protein response pathway in mammalian cells.
Keywords: Unfolded protein response, signal transduction, RNA processing, nuclear pore comples, glucose deprivation, glucose regulated proteins, Ire1/Ern1
The endoplasmic reticulum (ER) is an organelle specialized for protein folding and assembly of membrane proteins and of proteins destined for trafficking to lysosomes and the extracellular space. Newly synthesized lysosomal, secretory, and membrane proteins are translocated into the lumen of the ER that provides an oxidizing environment and contains a multitude of ER resident proteins that facilitate the folding process (for reviews, see Gething and Sambrook 1992; Hartl 1996). The transcription of many of the genes encoding ER resident proteins, such as BiP (immunoglobulin binding protein or GRP78; Munro and Pellham 1986), is upregulated in response to glucose deprivation (Lee 1987), in response to conditions that disrupt protein folding in the ER, and in response to the presence of unfolded or unassembled proteins in the ER (Lee 1987; Kozutsumi et al. 1988; Dorner et al. 1989). Thus, an unfolded protein response (UPR) exists in cells that detect unfolded protein in the ER lumen to transduce a signal(s) across the ER membrane to activate transcription of selective genes in the nucleus (Kozutsumi et al. 1988).
Although little is known about the mechanism of the UPR signal transduction pathway in higher eukaryotes, studies from the budding yeast, Saccharomyces cerevisiae, demonstrate the existence of a complex unique signaling pathway between these two organelles (Mori et al. 1992). Characterization of the promoters of the genes encoding ER resident proteins, for example, KAR2 (yeast BiP), demonstrated that they share a highly conserved cis-acting regulatory unfolded protein responsive element (UPRE), that is necessary and sufficient to mediate the response to unfolded protein in the ER (Cox et al. 1993; Mori et al. 1993). By use of genetic approaches, Ire1p/Ern1p, an ER type 1 transmembrane protein that contains a Ser/Thr protein kinase domain in its carboxyl terminus, was identified as the UPR proximal sensor that monitors the status of unfolded protein inside the ER lumen (Cox et al. 1993; Mori et al. 1993). Ire1p was originally identified as a gene required for inositol prototrophy in S. cerevisiae (Nikawa and Yamashita 1992). The kinase activity of Ire1p is essential to transmit the UPR signal from the ER to induce specific gene transcription in the nucleus (Mori et al. 1993; Shamu and Walter 1996). Cox and Walter (1996) subsequently reported that Ire1p directly regulated biosynthesis of Hac1p, a transcription factor that binds specifically to the UPRE. Recent studies demonstrate that HAC1 mRNA is synthesized as a precursor that is inefficiently translated. On activation of the UPR, Ire1p elicits an endonuclease activity that specifically cleaves an intron from HAC1 mRNA. Subsequently, the tRNA ligase Rlg1p is required to splice together the 5′ and 3′ cleaved fragments to yield a product that is efficiently translated (Cox and Walter 1996; Chapman and Walter 1997; Kawahara et al. 1997; Sidrauski et al. 1997). The increased level of Hac1p leads to the transcriptional activation of genes containing a UPRE.
Although the molecular mechanisms signaling the yeast UPR are well characterized, the mechanisms signaling the UPR in mammalian cells remain elusive. A conserved promoter region, the glucose-regulated core sequence, in several mammalian genes encoding for ER proteins was identified as a potential cis-acting regulatory element equivalent to the yeast UPRE (Resendez et al. 1988). Despite the sequence similarity between the mammalian glucose-regulated core sequence and the S. cerevisiae UPRE, no single element in this promoter region appears necessary and sufficient to mediate transcriptional induction as described for the UPRE in yeast cells. In addition, although transcriptional activation in response to conditions that disrupt protein folding in the ER correlates with changes in activities of protein kinases and phosphatases (Resendez et al. 1986; Koong et al. 1994a; Cao et al. 1995; Chen et al. 1998), a signaling molecule that responds to unfolded protein in the ER to induce transcription of the ER protein chaperone genes has not been identified. In this report we describe the identification and characterization of a human gene product that is equivalent to Ire1p of S. cerevisiae and functions as a proximal sensor for the UPR in mammalian cells.
Results
Isolation of complementary DNA encoding human Ire1p
To screen for a human homolog of S. cerevisiae IRE1, degenerate oligonucleotide primers were designed from the amino acid sequence (ISDFGLCK) in the kinase subdomain VII of S. cerevisiae IRE1 that was also conserved in a putative Caenorhabditis elegans IRE1 identified in the genbank, but was not present in other protein Ser/Thr protein kinases. The oligonucleotide was used in combination with a λgt 10 specific primer to amplify DNA fragments from a human fetal liver cDNA library. RH3 was isolated as a candidate clone containing a 270-bp PCR product that encoded for a portion of the catalytic domain of a novel human Ser/Thr protein kinase. The clone was used as a probe to screen for overlapping clones from a human fetal liver cDNA library (Fig. 1A). A 3.5-kb cDNA was assembled from overlapping clones that has a single ORF encoding 977 amino acid residues with a predicted molecular mass of 110 kD (Fig. 1C). One clone had a 106-bp putative 5′-untranslated region that did not contain either an ATG codon or an in frame termination codon upstream of the ATG codon having a favorable sequence context (CGCCATGCC) to serve as an initiation codon (Kozak 1987). In addition, immediately following the putative initiation codon was a sequence of residues that are predicted to serve as a signal peptide, having positively charged residues at the extreme amino-terminus followed by a core of hydrophobic residues and then turn-inducing residues (Pro and Gly) (Nielsen et al. 1997). We predict that signal cleavage occurs after Gly18. Finally, one clone (13-1) was identified that contained a 3′-untranslated region of 598 bp that contained multiple translation termination codons in all reading frames but did not contain a conserved polyadenylation signal, suggesting additional sequence exists within the 3′-untranslated region of the mRNA. On the basis of these observations, we believe that we have cloned the intact coding region for this putative kinase.
Figure 1.



Structure and amino acid sequence analysis of hIre1p. (A) Alignment and restriction map of overlapping complementary DNAs encoding human Ire1p. RH3 was the primary probe used to screen a human fetal liver cDNA library to obtain cDNA clones 3-1-1, 3-1.2, 8-1, 9-1,13-1, and 17-1. F14 was a 5′ RACE–PCR product amplified from RNA isolated from the human hepatoma cell line HepG2. The open bar represents the predicted ORF coding for hIre1p. (B) Domain organization of hIre1p. (Solid box) Potential signal sequence; (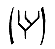 ) potential N-linked glycosylation site; (TM) a putative transmembrane region; (Linker) a region having no homology to known proteins; (S/T kinase) catalytic domain of Ser/Thr protein kinase; (RNase L) a domain having high homology to 2-5 oligo-(A)–dependent RNase. Percent identity to the corresponding domains of S. cerevisiae and C. elegans is indicated. (C) (H.s.) Amino acid sequence alignment of human Ire1p, (S.c.) S. cerevisiae Ire1p and (C.e.) its putative homologous protein from C. elegans. (Open boxes) The identical sequence; (shaded boxes) conserved residues; (dashes) gaps between residues to obtain maximum matching. Numbers are the position of the last amino acid. (▿) Potential signal peptide cleavage site; (•) invariant residues in protein kinase domain; (*) invariant Lys599 residue in kinase subdomain II. The glutamine rich cluster (I) and the serine rich cluster (II) in the linker region are also identified.
) potential N-linked glycosylation site; (TM) a putative transmembrane region; (Linker) a region having no homology to known proteins; (S/T kinase) catalytic domain of Ser/Thr protein kinase; (RNase L) a domain having high homology to 2-5 oligo-(A)–dependent RNase. Percent identity to the corresponding domains of S. cerevisiae and C. elegans is indicated. (C) (H.s.) Amino acid sequence alignment of human Ire1p, (S.c.) S. cerevisiae Ire1p and (C.e.) its putative homologous protein from C. elegans. (Open boxes) The identical sequence; (shaded boxes) conserved residues; (dashes) gaps between residues to obtain maximum matching. Numbers are the position of the last amino acid. (▿) Potential signal peptide cleavage site; (•) invariant residues in protein kinase domain; (*) invariant Lys599 residue in kinase subdomain II. The glutamine rich cluster (I) and the serine rich cluster (II) in the linker region are also identified.
A hydropathy plot of the deduced amino acid sequence revealed that it contained two stretches of hydrophobic residues: a leucine rich motif close to the amino terminus that could function as a signal sequence and a stretch of 21 consecutive hydrophobic residues lying approximately in the middle of the molecule that could provide a transmembrane domain. This suggested that the putative protein is a type 1 transmembrane protein with the kinase domain in the carboxyl terminus and with a single potential N-linked glycosylation site (Asn–Ala–Thr) in the luminal domain at residue 176 (Fig. 1B).
Searches of the protein sequence database suggested that the carboxy-terminal half of the protein could be divided into three domains, a linker region, the putative Ser/Thr protein kinase domain, and an RNase L-like domain homologous to 2′-5′ oligo (A)-dependent ribonuclease (Fig. 1B). The linker region is unique to the human protein and contains two subdomains rich in glutamine and serine residues, respectively. In contrast, the kinase domain displays high homology with both that of S. cerevisiae Ire1p and its putative counterpart from C. elegans, having conserved all 12 invariant residues that are present within the protein kinase superfamily (Fig. 1C; Hanks and Hunter 1995). The similarity among these three proteins also extends to the very carboxy-terminal RNase L domain (Fig. 1C; Zhou et al. 1993). The cytoplasmic domain of hIre1p, as well as its putative counterpart from C. elegans, do differ from yeast Ire1p in that they do not contain a potential nuclear localization signal and also lack an insertion of 30 hydrophilic amino acids between conserved kinase subdomains VIII and IX (Mori et al. 1993). In contrast, the amino-terminal half of the protein was less conserved with the C. elegans protein and displayed no significant similarity with S. cerevisiae Ire1p. On the basis of the homologies within the cytoplasmic domain, we refer to this putative human protein as human Ire1p (hIre1p).
hIRE1 is constitutively expressed in all tissues
The expression of endogenous hIRE1 was studied by Northern blot hybridization to 32P-labeled cDNA probes corresponding to either the luminal domain or the cytoplasmic domain of hIre1p. The hybridization patterns with these two probes were identical (Fig. 2). hIRE1 was ubiquitously expressed at low levels as detected by a single species of mRNA migrating at ∼8 kb in all tissues examined. On the basis of the size of endogenous hIRE1 mRNA together with its predicted translation product, we suggest that hIRE1 mRNA contains a long, ∼4 kb, 3′-untranslated region. Interestingly, the hIRE1 mRNA was most abundant in pancreatic tissue, suggesting that hIre1p might play a significant role in this particular organ.
Figure 2.

hIRE1 is ubiquitously expressed in human tissues. Northern blot analysis of poly(A)+ RNA isolated from various human tissues (Clontech) by hybridization with 32P-labeled-cDNA probes corresponding to the hIRE1 luminal domain, the hIRE1 cytoplasmic domain, or human β-actin cDNA. Exposure of the Ire1 autoradiograph was 24-fold longer than that of β-actin.
hIre1p displays kinase activity that is required to down-regulate its synthesis
To examine the biochemical properties of hIre1p, antibodies were raised in mice immunized with a GST–hIre1p fusion protein. Although this polyclonal antibody reacted with the antigen against which it was raised, the antibody did not detect endogenous Ire1p on immunoprecipitation from several human cell lines, suggesting that the level of endogenous hIre1p is extremely low (data not shown). To obtain sufficient amount of protein for characterization, hIre1p was overexpressed in COS-1 monkey cells by transient DNA transfection of the cDNA cloned in the expression vector pED (Kaufman et al. 1991). In addition, an expression vector encoding a kinase defective hIRE1 mutant was constructed in which the conserved lysine at residue 599 in the putative ATP binding site was substituted by alanine (pED–hIRE1 K599A) (Hanks and Quinn 1991). The expression of these proteins was monitored by immunoprecipitation of cell extracts from [35S]methionine and cysteine pulse-labeled cells by use of α-hIre1p antibody. As expected, both wild-type and K599A mutant hIre1p were expressed as 110-kD proteins (Fig. 3A). This protein product was not detected from mock-transfected cells or from pED vector-transfected cells (Fig. 3A; data not shown). Interestingly, the level of mutant K599A hIre1p synthesis in transfected COS-1 cells was ∼16 times higher than that of the wild-type hIre1p. The difference between wild-type and mutant hIre1p expression was not attributable to differences in transfection efficiency (determined by immunofluorescence) and different independent isolates of both plasmid DNAs yielded similar results. The increased synthesis of mutant hIre1p could account for the increased steady state level (∼10-fold from Fig. 3D), suggesting there is no significant difference in the rate of degradation.
Figure 3.
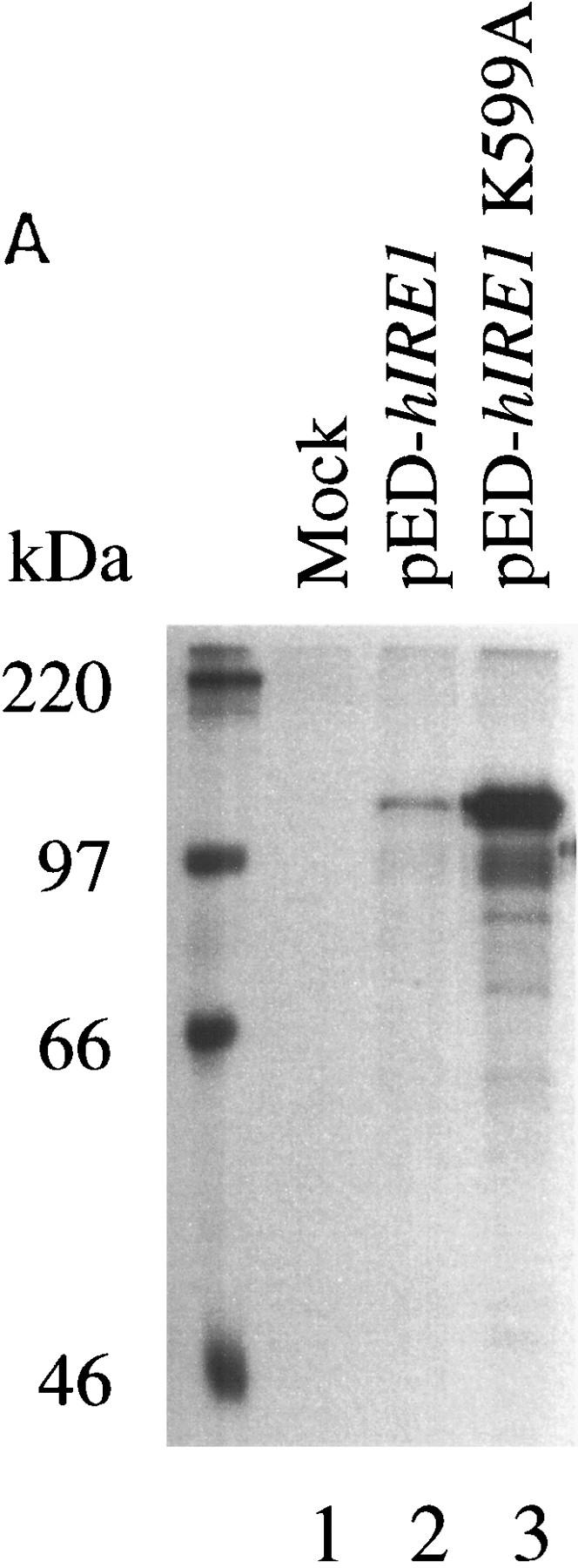

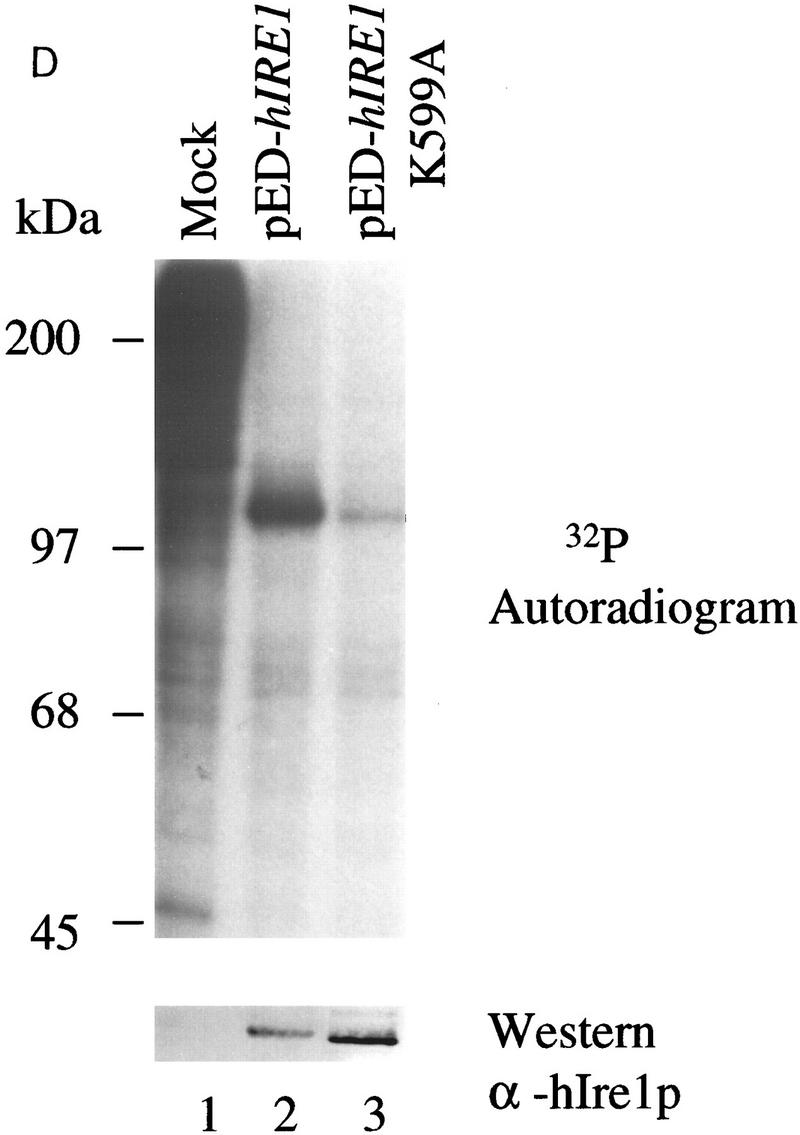
Overexpression of hIre1p in transiently transfected COS-1 monkey cells. (A) hIre1p expression in transfected COS-1 cells. COS-1 cells were transiently transfected with or without expression plasmids encoding wild-type hIre1p (pED–hIRE1) or its kinase defective mutant (pED–hIRE1 K599A). Transfected cells were pulse labeled with [35S]-methionine and cysteine for 15 min. Cell extracts were prepared and equal amounts were immunoprecipitated with α-hIre1p antibodies and analyzed by SDS-PAGE and autoradiography. (B) Expression of eIF-2α in cotransfected cells. COS-1 cells were mock transfected (lane 1) or cotransfected with pED–eIF-2α in the presence of pED (lane 2), pED–hIRE1 (lane 3), or pED–hIRE1 K599A (lane 4). The cells were pulse-labeled with [35S]methionine and cysteine for 15 min. Cell extracts were prepared and equal cpm of radiolabeled protein were analyzed directly by SDS-PAGE and autoradiography. (C) Functional hIre1p limits accumulation of hIRE1 mRNA. Total RNA was isolated from COS-1 cells transfected with pED, pED–hIRE1, or pED–hIRE1 K599A plasmid and treated in the presence (+) or absence (−) of cycloheximide. RNA samples (10 μg) were resolved in a formaldehyde–agarose gel, blotted onto nylon membrane, and hybridized with 32P-labeled hIRE1 cDNA probe. (Arrow) The hIRE1 transcript. (D) hIre1p has intrinsic kinase activity. Wild-type or K599A mutant hIre1p was immunoprecipitated from transiently transfected COS-1 cells. (Mock) Cells that did not receive plasmid DNA. The proteins were incubated in kinase buffer in the presence of [γ-32P]ATP at 30°C for 40 min. The proteins were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. (Top) Incorporation of [32P]phosphate into hIre1p determined by autoradiography; (bottom panel) the Ire1p protein level determined by estern blot analysis by use of α-hIre1p antibodies and alkaline phosphatase staining. The amount of K599A mutant hIre1p loaded onto the gel is one-third the amount of the immunoprecipitated proteins loaded for lanes 1 and 2. Therefore, the amount of steady-state K599A mutant hIre1p is ∼10-fold greater than the wild-type hIre1p.
The difference in the expression level between wild-type and the K599A mutant Ire1p protein in transfected COS-1 cells (Fig. 3A) lead to a speculation that hIre1p might autoregulate its expression. Alternatively, overexpression of wild-type hIre1p may inhibit general expression in the subpopulation of transiently transfected cells caused by a general toxicity. To address this possibility, COS-1 cells were cotransfected with another marker gene encoding the eukaryotic translation initiation factor eIF-2α subunit (pED–eIF-2α) with either pED–hIRE1 or pED–hIRE1 K599A. The transfected cells were metabolically pulse labeled with [35S]methionine and cysteine and protein synthesis was analyzed by SDS-PAGE of total cell extract samples. The presence of either hIre1p or its mutant (K599A) had no effect on the synthesis of eIF-2α, suggesting that wild-type hIre1p overexpression is not toxic and does not inhibit global gene expression in the transfected cells (Fig. 3B; cf. lane 3 and lanes 2 and 4).
To further investigate the mechanism for the reduced expression of wild-type hIre1p, we analyzed the level of plasmid derived hIRE1 mRNA from COS-1 transfected cells. Total RNA was prepared from COS-1 cells transfected with pED, pED–hIRE1 or pED–hIRE1 K599A and treated in the presence or absence of cycloheximide for 12 hr before harvesting RNA. Northern blot hybridization demonstrated that the level of hIRE1 K599A mRNA was 10 times higher than the wild-type hIRE1 mRNA derived from the transfected DNA (Fig. 3C). Inhibition of protein synthesis by cyclohexamide had no effect on the steady-state level of these mRNAs, suggesting that ongoing protein synthesis is not required to down-regulate hIRE1 mRNA. Taken together, we conclude that functional hIre1p down-regulates its own expression at the level of mRNA production and/or stability.
The deduced amino acid sequence of hIre1p suggested the presence of an intact catalytic Ser/Thr protein kinase domain. To demonstrate functional activity of the kinase, the capability for autophosphorylation was measured, because this activity correlates with the functional activity of yeast Ire1p (Welihinda and Kaufman 1996). The wild-type and mutant K599A hIre1p were immunoprecipitated from transfected COS-1 cells and incubated in kinase buffer with [γ32P]ATP. The proteins were then resolved by SDS-PAGE and transferred onto a nitrocellulose membrane prior to autoradiography and probing with α-hIre1p antibody. The wild-type hIre1p was efficiently autophosphorylated (Fig. 3D). The phosphorylation resulted in a slightly slower mobility as determined by Western blotting. Substitution of the conserved lysine residue in the putative ATP binding pocket with alanine significantly reduced the phosphorylation detected, especially when corrected for the greater amount of protein immunoprecipitated (Fig. 3D, cf. lanes 2 and 3). The low level of phosphorylation of this mutant Ire1p may result from either the presence of endogenous COS-1 cell-derived Ire1p or another kinase(s) in the immunoprecipitation reaction. Taken together, we conclude that hIre1p displays an intrinsic protein kinase activity.
hIre1p is a bifunctional enzyme having an endoribonuclease activity specific to yeast HAC1 mRNA
Sidrauski and Walter (1997) recently demonstrated that the cytoplasmic domain of yeast Ire1p exhibits a site-specific endoribonuclease activity capable of cleaving HAC1 mRNA at both the 5′ and 3′ splice site junctions in vitro. The proposed catalytic domain of RNase L displays greater sequence similarity to hIre1p than to the yeast Ire1p. This led to the hypothesis that hIre1p might exhibit a similar endoribonuclease activity and may be able to catalyze the same specific RNA cleavage as observed for yeast Ire1p. Because the identity of the mammalian HAC1 homolog is unknown, we determined whether yeast HAC1 mRNA could serve as a substrate to test for an endoribonuclease activity of hIre1p. A 550-nucleotide substrate derived from S. cerevisiae HAC1 mRNA that contained both the 5′ and 3′ splice site junctions was synthesized in vitro. Incubation of this substrate in the presence of a GST–yeast Ire1p fusion protein simultaneously cleaved the HAC1 mRNA substrate at the 5′ and 3′ splice site junctions, as shown previously by Sidrauski and Walter (1997). The cleavage generated three species of RNA products (corresponding to a 224-nucleotide 5′ exon, a 252-nucleotide intron, and a 74-nucleotide 3′ exon) and two intermediates (corresponding to a 476-nucleotide 5′ exon/intron and a 326-nucleotide intron/3′exon) (Fig. 4A, lanes 3,4). The cleavage was not observed in the absence of GST–Ire1p, or with control GST protein alone (Fig. 4A, lanes 1,2). Surprisingly, hIre1p isolated by immunoprecipitation from transfected COS-1 cells that over-express hIre1p was able to catalyze cleavage; however, only two species of RNA products were observed in this reaction (Fig. 4A, lanes 8,9). The two products appeared to be the same size as those derived from GST–yeast Ire1p-mediated cleavage at only the 5′ splice site junction (representing the 224- nucleotide 5′ exon and the 326-nucleotide intron/3′exon). Prolonged incubation of the substrate wit hIre1p did not generate the intron or the 3′ exon fragments (data not shown). In contrast, no cleavage was observed when the mutant K599A hIre1p was substituted for hIre1p (Fig. 4A, lanes 6,7). Taken together, we conclude that hIre1p exhibits endoribonuclease activity and its intrinsic kinase activity is required to elicit the endoribonuclease activity.
Figure 4.

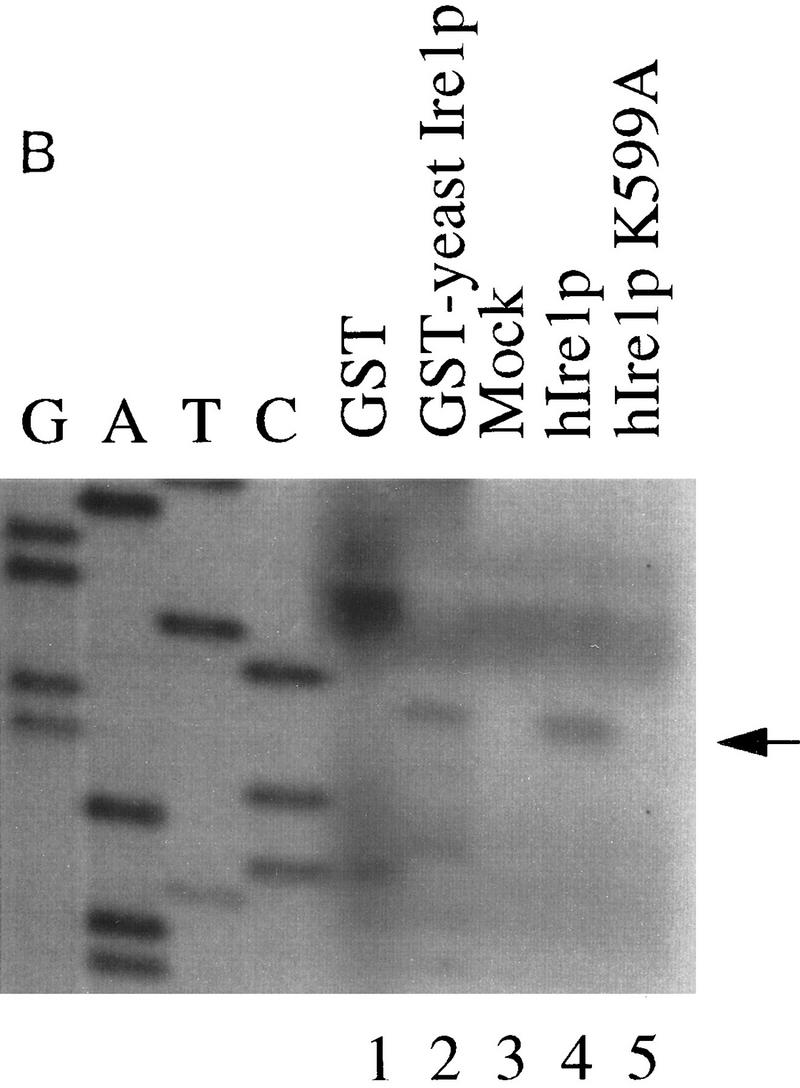
hIre1p is a site-specific endoribonuclease. (A) In vitro cleavage of yeast HAC1 mRNA by hIre1p. An in vitro-transcribed 32P-labeled HAC1 mRNA was incubated with E. coli-expressed GST or GST–Ire1p adsorbed to glutathione beads or with COS-1 cell-expressed hIre1p or hIre1p K599A protein adsorbed to protein A–Sepharose beads. After the indicated period of time, the cleavage products were analyzed by electrophoresis on a 5% denaturing polyacrylamide gel. Schemes on the left depict the predicted cleavage products. Numbers at right indicate predicted base pair size of RNA products expected based on yeast HAC1 mRNA cleavage by yeast Ire1p (Sidrauski and Walter 1997). (B) hIre1p cleaves yeast HAC1 mRNA at residue G661. The HAC1 RNA cleavage site was mapped using in vitro-transcribed HAC1mRNA after incubation with GST,GST–Ire1p, hIre1p, or hIre1p K599A as described in A. The products were reverse transcribed with Superscript II Reverse Transcriptase (Bethesda Research Labs) by use of oligonucleotide primer complementary to the intron of HAC1 RNA. Sequencing ladders on the left represent HAC1 DNA sequence determined with the same primer. (Arrow) Position of primer extended products.
To precisely map the hIre1p cleavage site in HAC1 mRNA, primer extension analysis was performed. An antisense oligonucleotide complementary to the HAC1 intron was used to reverse transcribe HAC1 mRNA cleaved by either Escherichia coli expressed GST-yeast Ire1p or hIre1p expressed and immunoprecipitated from transfected COS-1 cells. The same primer was also used to determine the nucleotide sequence of the HAC1 gene (Fig. 4B). The length of primer-extended products derived from HAC1 mRNA cleaved with GST–yIre1p and hIre1p were identical, indicating that both GST–yeast Ire1p and hIre1p cleave HAC1 mRNA at the same position at the predicted 5′exon/intron junction (Fig. 4B, cf. lanes 2 and 4). Comparison of these two extended products with the HAC1 DNA sequence ladder indicated that they both were terminated after the guanine at residue 661. In contrast, this primer-extended product was not observed in the reverse transcription reactions of HAC1 mRNA incubated with control GST from E. coli or with control immunoprecipitated protein from either pED–hIRE1 K599A- or mock-transfected cells.
hIre1p is an ER membrane protein preferentially localized to the nuclear envelope
ER resident glycoproteins that do not transit to the Golgi complex have high mannose-containing oligosaccharides that are sensitive to digestion by endoglycosidase H. The presence of a single potential amino-linked glycosylation site in the amino-terminal domain of hIre1p was used to determine the subcellular localization of hIre1p. hIre1p immunoprecipitated from extracts prepared from metabolically labeled transfected cells treated with tunicamycin, a drug that inhibits addition of N-linked core oligosaccharides, displayed a slightly reduced molecular mass compared with Ire1p isolated from untreated cells, suggesting the absence of the single N-linked core oligosaccharide (Fig. 5; cf. lanes 1 and 2). Treatment of immunoprecipitated hIre1p with endoglycosidase H decreased the molecular mass of the labeled hIre1p to that comparable with unglycosylated hIre1p isolated from tunicamycin treated cells (Fig. 5, cf. lanes 2 and 4). In addition, deletion of the carboxy-terminal 462 amino acid residues (the putative cytosolic domain) generated a protein that contained an N-linked oligosaccharide (data not shown). These results support localization of hIre1p to the ER, which is consistent with the predicted topology of hIre1p having its amino termini in the ER lumen, similar to yeast Ire1p (Mori et al. 1993).
Figure 5.

hIre1p contains high-mannose core oligosaccharides. Transfected COS-1 cells that overexpress hIre1p were pulse labeled with [35S]methionine and cysteine for 15 min in the presence (lane 2) or absence (lane 1) of tunicamycin and cell extracts were prepared. In parallel, cells pulse labeled 15 min in the absence of tunicamycin were incubated 3 hr in medium containing excess unlabeled methionine and cysteine before harvesting cell extracts. The 35Slabeled hIre1p was immunoprecipitated from cell extracts and analyzed by SDS-PAGE. Prior to SDS-PAGE, immunoprecipitated samples were incubated in the absence (lanes 1–3) or presence (lane 4) of endoglycosidase H.
Confocal laser-scanning immunofluorescence microscopy was used to identify the hIre1p subcellular localization. COS-1 cells were transiently transfected with the wild-type hIRE1 expression plasmid and cells were double labeled with mouse antibody specific to hIre1p and rabbit antibody specific to GRP94, a resident protein of the ER. The immune complexes were visualized by secondary antibody conjugated with fluorescein isothiocyanate (FITC) or rhodamine, respectively (Fig. 6A). It was not possible to detect staining of hIre1p in nontransfected cells, possibly because of its low level of expression. In contrast, wild-type hIre1p was detected in transfected cells as perinuclear fluorescence and appeared similar to the fluorescence pattern observed for endogenous GRP94. Although the fluorescence patterns of the two proteins were similar, analysis of the merged images suggested that hIre1p was preferentially localized close to the nuclear membrane. Significantly greater staining was observed in cells transfected with the hIRE1 mutant K599A expression plasmid, consistent with its greater level of expression (Fig. 6B). The mutant hIre1p protein was also preferentially localized to the perinuclear region indicating that the kinase activity is not required for this localization. To determine if hIre1p was localized to the nuclear envelope, the fluorescence pattern of wild-type hIre1p was compared with that of endogenous RanGAP1 protein, a component of the nuclear pore complex (Mahajan et al. 1997). Unlike hIre1p, which was localized to the ER membrane throughout the cytoplasm, RanGAP1 protein exhibited a specific nuclear rim fluorescence staining pattern. Interestingly, when the two fluorescence images were merged, a subpopulation of hIre1p colocalized with the nuclear pore complex protein RanGAP1 (Fig. 6C).
Figure 6.
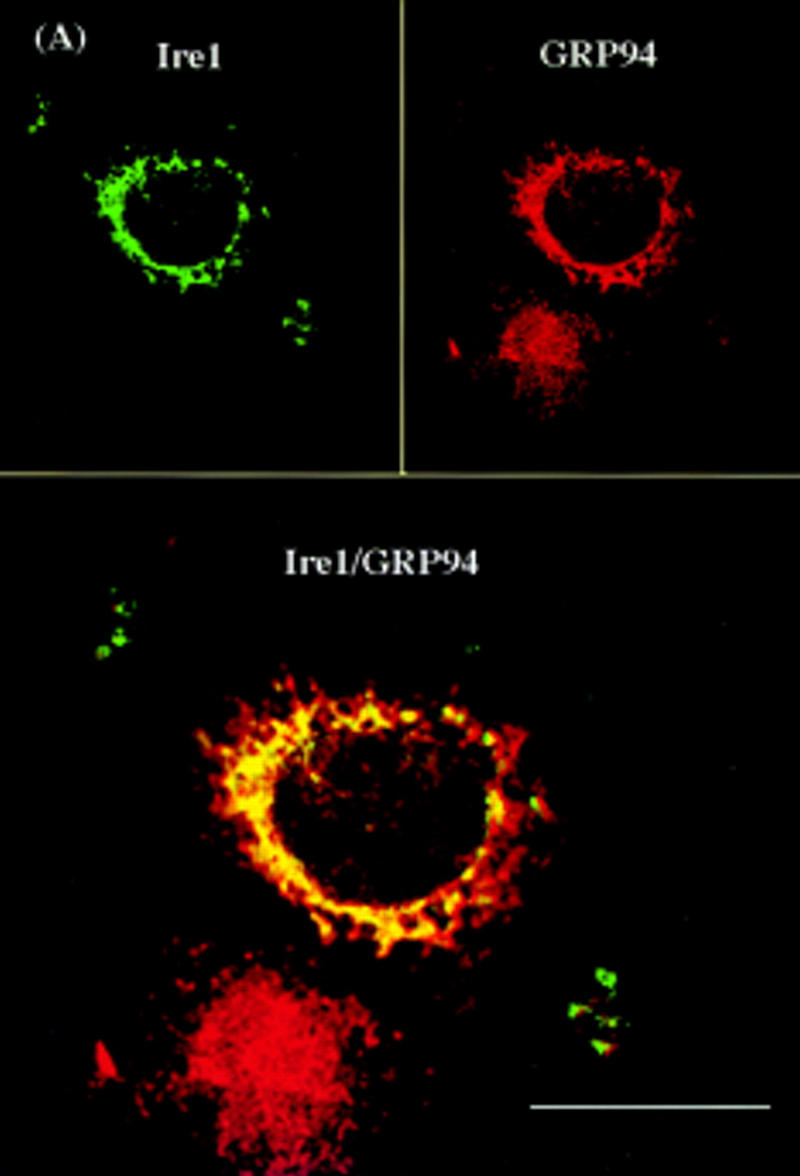
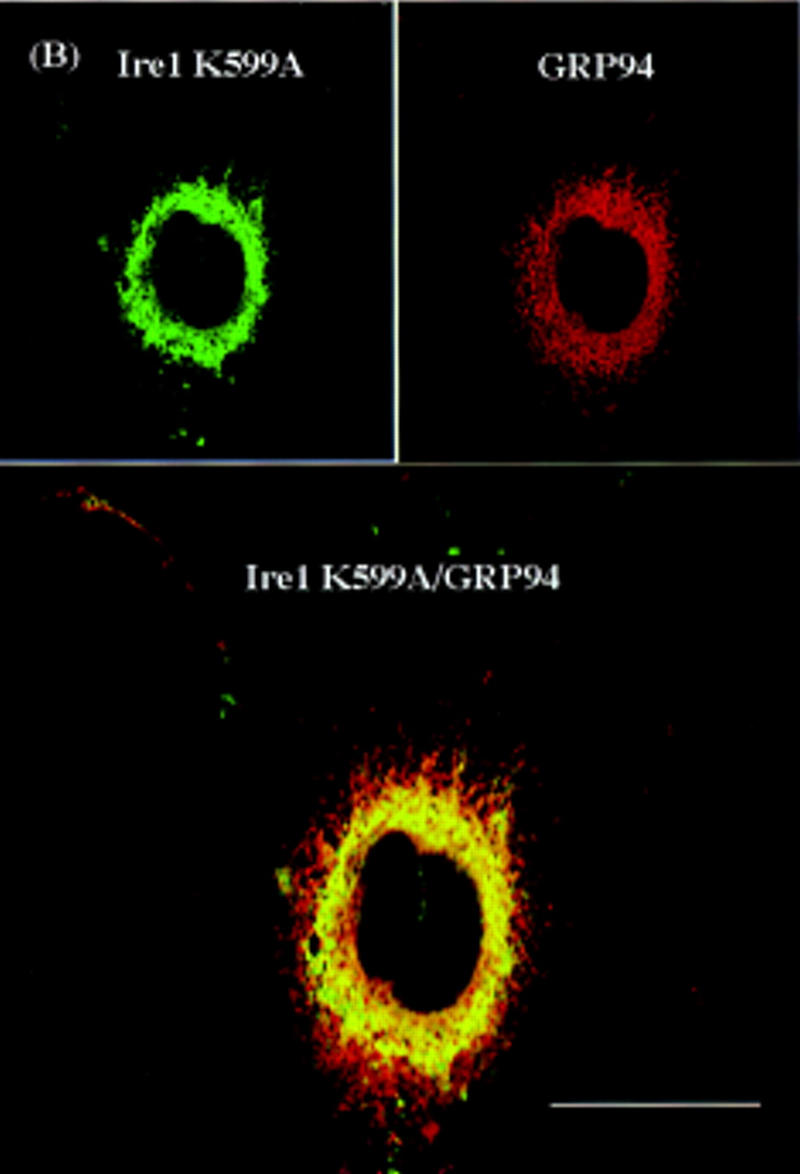
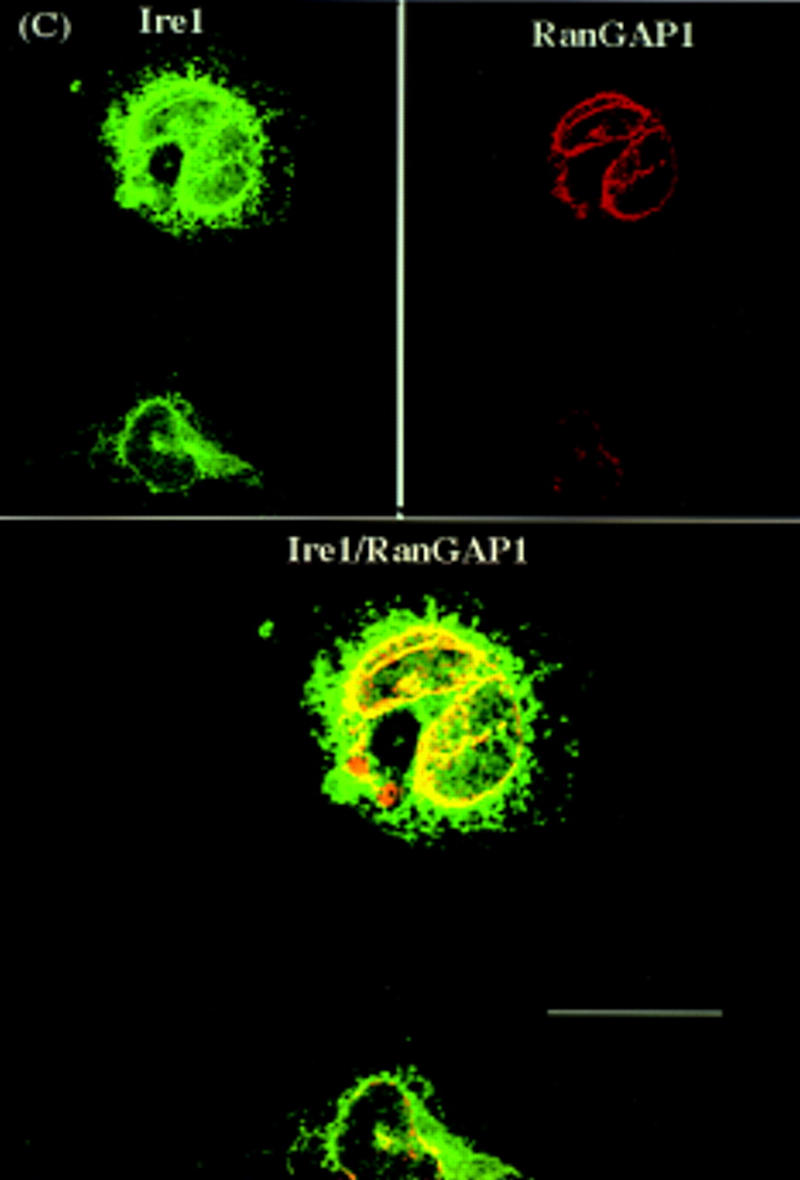
Confocal laser scanning fluorescence microscopy of hIre1p expressed in COS-1 cells. The subcellular localization of hIre1p in transfected COS-1 cells was determined by immunofluorescence with mouse α-hIre1p. (A) COS-1 cells transfected with wild-type, or (B) K599A mutant IRE1 expression plasmids were double labeled with mouse α-hIre1p and rabbit α-GRP94. (C) COS-1 cells transfected with wild-type IRE1 expression plasmid were double labeled with mouse α-hIre1p and guinea pig α-RanGAP1. Secondary antibodies used were either rhodamine-conjugated goat α-rabbit or rhodamine-conjugated goat α-guinea pig (red) in the presence of fluorescein-conjugated goat α-mouse (green). The images were merged where colocalization is shown in yellow. Cells were viewed and digitally photographed with a Bio-Rad confocal fluorescence microscope. Bar, 25 μm.
Overexpression of kinase-defective K599A hIre1p blocks the unfolded protein response in mammalian cells
The previous data demonstrate that hIre1p displays several features similar to those of S. cerevisiae Ire1p. To directly test whether hIre1p plays an essential role in the UPR in mammalian cells, a mammalian reporter plasmid was constructed by inserting a 0.5-kb fragment of the rat BiP promoter, including the cis-acting element capable of mediating the UPR (Chang et al. 1987), upstream from a luciferase-coding region. The UPR reporter plasmid was cotransfected with either wild-type or the mutant K599A hIre1p expression vectors into COS-1 cells. The luciferase activity reflects the activation of the UPR in these cells. Luciferase activity was detected in extracts from cells transfected with the reporter plasmid alone and this activity was further increased threefold by treatment of the cells with tunicamycin (Fig. 7). Cotransfection of the reporter plasmid with pED–hIRE1 K599A prevented induction of the UPR on tunicamycin (Tm) treatment. The inability to elicit the response suggests that the mutant hIre1p can act as a trans-dominant-negative kinase to down-regulate endogenous Ire1p. In contrast, overexpression of wild-type hIre1p caused constitutive induction of the UPR that was not further increased by treatment with tunicamycin. These results are consistent with those reported previously for expression of yeast Ire1p in S. cerevisiae (Mori et al. 1993; Shamu and Walter 1996) and support the conclusion that hIre1p is a proximal sensor of the UPR pathway in mammalian cells.
Figure 7.

hIre1p-dependent induction of UPR in mammalian cells. The activation of the unfolded protein response was measured by cotransfection of COS-1 cells with a luciferase reporter plasmid under the control of the rat BiP promoter, RSV–β-gal and either pED–hIRE1 or pED–hIRE1 K599A plasmid DNAs. At 60 hr post-transfection, the cells were treated with 10 μg/ml tunicamycin for 6 hr. The luciferase activity was determined from triplicate independent transfection experiments and was normalized to β-galactosidase activity to correct for transfection efficiency.
Discussion
We have identified and characterized a gene product, hIre1p, that we propose functions as the proximal sensor for the mammalian UPR. The following criteria support that hIre1p is a functional human homolog of the yeast Ire1p. First, hIre1p and yeast Ire1p are both type 1 transmembrane proteins in which the carboxy-terminal domains are 34% identical at the amino acid level. Although the amino-terminal halves of these two proteins have extensively diverged, the amino-terminal half of hIre1p is 37% identical to a C. elegans putative gene product having a similar domain organization as hIre1p. The cytoplasmic domain of hIre1p contains all the conserved subdomains present in Ser/Thr protein kinases and a carboxy-terminal tail that displays greater homology to human RNase L than S. cerevisiae Ire1p. Second, hIre1p displayed both intrinsic kinase activity measured by autophosphorylation capability and an endoribonuclease activity that specifically cleaved the 5′ splice site of S. cerevisiae HAC1 mRNA at the same nucleotide, guanine 661, as the S. cerevisiae Ire1p. Third, overexpressed hIre1p was specifically localized to the ER, with particular concentration around the nuclear envelope. Fourth, overexpression of wild-type hIre1p constitutively activated a marker gene under control of the rat BiP promoter. Finally, overexpression of a catalytically inactive kinase mutant K599A completely prevented induction by tunicamycin, a treatment that promotes accumulation of unfolded protein in the ER. Although hIre1p can directly elicit a signal cascade to result in activation of the BiP promoter, we do not know whether there are additional components that either function in parallel to, or in concert with, hIre1p. For example, there may be different Ire1p homologs that respond to different stimuli within the ER. A search of the human genbank identified a 400-nucleotide expressed sequence tag that has extensive homology to hIre1p in the RNase L domain. Further studies are required to elucidate wheher this encodes another Ire1p homolog. In this respect, it is interesting that the human and yeast Ire1p genes have extensively diverged in their luminal domains. This might suggest that the ligands responsible for activation of the S. cerevisiae and human kinases are not conserved. The extensive homology over their cytoplasmic domains and the ability of hIre1p to specifically cleave the 5′ splice site of S. cerevisiae HAC1 mRNA indicates that the downstream events in signaling from Ire1p in human and yeast are very conserved and likely function in a mechanistically similar manner. These observations strongly support the existence of a similar unique splicing reaction that will be specific to substrates, including a possible HAC1 mRNA homolog, in mammalian cells.
hIre1p has an intrinsic autophosphorylation and endoribonuclease activity. Previous studies on the yeast Ire1p demonstrated that the endoribonuclease activity required an adenine nucleotide as a cofactor. Because a nonhydrolyzable analog AMP–PNP and ADP stimulated the activity as well as ATP, it was concluded that the endoribonuclease activity of yeast Ire1p did not require the kinase activity. We demonstrated that mutant K599A hIre1p had defective kinase activity as well as endoribonuclease activity, suggesting that autophosphorylation is required to elicit the endoribonuclease activity, possibly by phosphorylation of residues within its endoribonuclease domain. It is possible that the GST–Ire1p fusion protein studied by Sidrauski and Walter (1997) was autophosphorylated on expression as a dimeric protein in E. coli. We propose that the hIre1p endoribonuclease activity requires autophosphorylation as well as an adenine nucleotide. However, at present we cannot rule out that the K599A mutant Ire1p is defective in endoribonuclease activity as a consequence of altered nucleotide binding, and not necessarily the result of a requirement for autophosphorylation. hIre1p was able to cleave the yeast HAC1 mRNA substrate at the identical 5′ splice site as yeast Ire1p. However, our primer extension analysis identified the 5′ cleavage site was after guanine 661, in contrast to previous primer extension analysis that identified cytosine 660 as the 5′ cleavage site (Sidrauski and Walter 1997), but consistent with the recent characterization of in vivo spliced products derived from mutated templates (Kawahara et al. 1998). Whereas our primer extension analysis was compared with DNA sequencing ladder derived from a reaction by use of the same primer for extension, Sidrauski and Walter (1997) used a different primer and this may explain the discrepancy. Although hIre1p efficiently cleaved the 5′ splice site of yeast HAC1 mRNA, there was no detectable cleavage at the 3′ splice site. This is consistent with our observations that hIre1p was not able to complement S. cerevisiae deleted of IRE1 (data not shown). The complete cleavage of HAC1 mRNA at both splice sites is required for the UPR function in yeast (Sidrauski and Walter 1997; Kawahara et al 1998). The inability for hIre1p to cleave the yeast HAC1 mRNA 3′ splice site was surprising because Kawahara et al. (1998) recently demonstrated that the sequence requirements for the 5′ and 3′ splice site cleavages within HAC1 mRNA by yeast Ire1p are remarkably similar. However, there were a couple of nucleotide differences in cleavage specificity identified between the 5′ and 3′ splice sites, particularly the +1 position and the +5 position with respect to the site of cleavage. Therefore, the hIre1p cleavage specificity for the 3′ splice site may have diverged from the yeast Ire1p. Alternatively, there may be another homolog of hIre1p that displays a different cleavage specificity restricted to the 3′ splice site of a human HAC1 mRNA homolog, and the two nucleolytic events required to release the intron may require a heterodimer, of which each subunit has unique specificity to catalyze cleavage at either the 5′ or 3′ splice site. This latter possibility is observed in the cleavage specificity of yeast tRNA endonuclease, in which two subunits are required, each having its own active site that recognizes either the 5′ or 3′ splice site of precursor tRNA molecules (Trotta et al. 1997).
Expression of wild-type hIre1p was ∼16-fold reduced compared with K599A catalytically inactive mutant hIre1p. The reduced expression of the wild-type kinase was not the result of a general toxicity or transcriptional inhibition specific to the promoter used in the expression vector, because expression of a cotransfected cDNA, eIF-2α, contained within the same expression vector was not reduced in the presence of the wild-type hIre1p kinase expression vector. Analysis of mRNA demonstrated that the wild-type kinase also had a corresponding decrease in the steady state level of mRNA compared with the mRNA encoding the K599A mutant Ire1p. Because the K599A mutant hIRE1 mRNA had only 2 base changes compared with the wild-type hIRE1 mRNA, we expect that the reduced steady-state level of hIRE1 mRNA is a consequence of activated hIre1p kinase activity. It is possible that the expression of wild-type Ire1p is limited because of a specific autoregulatory process in which the endoribonuclease activity of activated Ire1p cleaves its own mRNA, resulting in its degradation. Experiments are presently in progress to test this intriguing hypothesis. The specific feedback on hIRE1 mRNA suggests that the biosynthesis of hIre1p is tightly controlled. Stringent regulation of Ire1p synthesis may be necessary for cell survival as overproduction of Ire1p leads to constant activation of the UPR pathway and retardation of cell growth (Shamu and Walter 1996).
Although the extremely low levels of endogenous Ire1p precluded its direct visualization in mammalian cells, interestingly overexpressed wild-type Ire1p was preferentially localized to a subcompartment within the ER, with particular concentration around the nuclear envelope. In addition, a portion of hIre1p was colocalized with RanGAP1, a protein associated with the nuclear pore complex. This raises the intriguing possibility that hIre1p might be a component of the nuclear pore complex. This localization would be ideal if hIre1p-dependent RNA splicing is coupled with nucleocytoplasmic transport of substrate RNA molecules. In S. cerevisiae, the tRNA ligase, Rlg1p, mediates ligation of the HAC1 mRNA cleaved substrate (Sidrauski et al. 1997) and is also localized to the nucleoplasmic side of the nuclear pore (Simos et al. 1996).
Induction of glucose regulated proteins (GRPs), including BiP and GRP94, protects cells from death induced by calcium release from the ER (Morris et al. 1997), oxidative stress (Gomer et al. 1991), and anticancer treatments such as adriamycin and topoisomerase inhibitors (Shen et al. 1987; Hughes et al. 1989). Conversely, inhibition of GRP induction increases sensitivity to death in response to calcium release from the ER (Li et al. 1991, 1992), oxidative stress (Gomer et al. 1991), hypoxia (Koong et al. 1994b), and T cell mediated cytotoxicity (Sugawara et al. 1993). Therefore, elucidating the signaling mechanism(s) by which cells respond to ER stress may have important therapeutic implications. The remarkable conservation between yeast Ire1p and hIre1p functional activities suggests the existence of a human homolog to yeast HAC1 that may exhibit selective mRNA cleavage and ligation by a human homolog of S. cerevisiae tRNA ligase gene RLG1. Furthermore, additional components of this pathway may also be conserved. For example, in S. cerevisiae the transcriptional coactivator complex having histone acetyltransferase activity composed of Gcn5p, Ada2p, and Ada3p is required for maximal transcriptional induction of the KAR2 promoter (Welihinda et al. 1997). In addition, Ada5p, another component of this complex, is absolutely required to elicit the UPR (Welihinda et al. 1997). An interaction between Gcn5p and Ire1p was demonstrated (Welihinda et al. 1997) and suggests that the nucleoplasmic domain of Ire1p, localized to the nuclear envelope, may serve as a nucleation site for assembly of a multisubunit transcriptional activator complex required for transcriptional activation of genes under control of the UPRE. Human homologs for several of these transcriptional coactivator gene products have been identified (Candau et al. 1996), and it is likely that these products also participate in transcriptional activation of the ER stress-responsive genes in higher eukaryotes. In addition, we have recently described a Ser/Thr protein phosphatase of the PP2C gene family that is required to turn off activated Ire1p signaling in response to unfolded protein (Welihinda et al. 1998). Future studies should elucidate the existence of additional mammalian counterparts also involved in the UPR signaling pathway that may provide specific targets for pharmacological intervention in different disease states.
Materials and methods
Cloning of human IRE1 cDNA
A degenerate antisense oligonucleotide [5′-(TC)TT(AG)CTIT(AG)ICC(AG)AA(AG)TCIG(AT)IAT 3′] was designed from the conserved amino acid sequence in kinase subdomain VII (ISDFGLCK) between S. cerevisiae IRE1/ERN1 and its putative homolog from C. elegans. Inosine was incorporated into positions to minimize degeneracy and improve stability upon hybridization (Sambrook et al. 1989). This primer and a λgt10-specific primer were used to amplify sequences from a human fetal liver cDNA library (Clontech). Total PCR products were ligated into the TA cloning vector (Invitrogen) and transformed into E. coli DH5α. A candidate clone, RH3, that showed highest homology to the yeast IRE1 and its counterpart gene in C. elegans was subsequently used to screen the λgt10 human fetal liver cDNA library by standard procedures (Sambrook et al. 1989). The 5′ end of the hIRE1 (F14) was obtained by 5′ RACE PCR (Bethesda Research Labs) by use of template RNA isolated from the human hepatoma cell line HepG2. Each cDNA fragment was subcloned into pBluescript II SK(−) plasmid (Stratagene) at the EcoRI site. All cDNA fragments were sequenced from both directions by the dideoxynucleotide sequencing method (Sequenase, Amersham).
pBluescript-13-1 is a recombinant plasmid containing the largest ORF of hIre1p but lacking the 5′-end fragment. To assemble the full-length hIRE1 cDNA, the 0.3-kb PCR product including the initiator methionine was amplified from pBluescript-F14 by use of two primers: 1058G (5′-GCTCTAGAACCATGCCGGCCCGGCGGCT-3′) and 865G (5′-AGGCTGCCATCATTAGGATCT-3′) and Vent DNA polymerase (New England Biolabs). The 0.3-kb PCR product was introduced into clone 17-1 by overlap-extension PCR by use of two primers: 1058G and 9241B (5′-CATTGATGTGCATCACCTTCCTC-3′) to yield a 0.7-kb PCR product. The 0.7-kb fragment was digested with XbaI, located upstream to the first ATG introduced by PCR, and BamHI. The fragment was ligated to pBluescript-17-1 at the same restriction endonuclease sites to yield pBluescript-17-1/5′. The 0.9-kb XbaI–SacII fragment from pBluescript-17-1/5′ was ligated to pBluescript-13-1 at the same sites to yield pBluescript-hIRE1. The 3.5-kb XbaI–EcoRI hIRE1 cDNA was subcloned into the XbaI site of the mammalian expression vector, pED (pEDΔC) (Kaufman et al. 1991) to yield pED–hIRE1.
Site-directed mutagenesis
The conserved lysine residue at position 599 in kinase subdomain II was mutated by a PCR-based method by use of Vent DNA polymerase (New England Biolabs). The MstI and PvuI fragment from pED–hIRE1 was replaced with the homologous fragment containing mutated sequence (AAG → GCG) to yield pED–hIRE1 K599A. The mutation was confirmed by DNA sequencing.
Antibody production
The 1.6-kb cDNA encoding the entire cytoplasmic domain of hIre1p (amino acid residues 460–977) was generated by PCR amplification by use of primer 168G (5′-CGGAATTCATCACCTATCCCCTGAGCATG-3′), 169G (5′-CGGAATTCTCAGAGGGCGTCTGGAGTCA-3′), and Vent DNA polymerase (New England Biolabs). To make GST-hIre1p fusion protein, the PCR product was inserted in-frame into pGEX–1γT (Pharmacia) at the EcoRI site and then transformed into E. coli DH5α. The fusion protein was produced and purified as described by Frangioni and Neel (1993) except that the induction was performed at 30°C. The purified GST–cytoplasmic hIre1p fusion protein was repeatedly injected into mice as described by Harlow and Lane (1988). Sera collected from tail bleed was used for determining the titer by Western blot analysis. When optimal titer was obtained, the mice were injected with the sarcoma cell line S180 to induce ascites fluid that was directly used (Harlow and Lane 1988).
Transient DNA transfection and analysis
COS-1 monkey cells were transfected as described previously (Kaufman 1997). Briefly, cells were plated the day before transfection. Cells were transfected with 2 μg/ml of pED–hIRE1 or pED–hIRE1 K599A plasmid DNA by the diethylaminoethyl–dextran method for 6 hr. The cells were fed with fresh medium at 36 hr post-transfection. Total cell extract was prepared from the transfected cells at 60 hr post-transfection by use of NP-40 lysis buffer (1% NP-40, 50 mm Tris-HCl at pH 7.5, 150 mm NaCl, 0.05% SDS) supplemented with 1 mm PMSF, 40 μg/ml aprotinin, and 20 μg/ml leupeptin. For metabolic labeling (unless otherwise specified), the transfected cells were labeled with [35S]methionine and cysteine (1000 Ci/mmole, Amersham Corp.) for 15 min before harvesting cells. For immunoprecipitation, cell extract was preabsorbed with protein A Sepharose. The precleared lysate was subsequently incubated with α-hIre1p for 14 hr at 4°C and then incubated with rabbit α-mouse IgG antibodies for 1 hr. The immune complexes were adsorbed with protein A Sepharose and successively washed with PBS containing Triton X-100 at 1%, 0.1%, and 0.05%. Samples were analyzed by SDS-PAGE under reducing conditions and autoradiography. Band intensities were quantified by the NIH Image 1.55b program.
Northern blot analysis
Poly(A)+ RNA isolated from various human tissues (Clontech) was hybridized with 32P-labeled, 1.5-kb NsiI–PvuI fragment of hIRE1 cDNA corresponding to the luminal domain of hIre1p, the 0.9-kb EcoRI insert fragment of pBluescript-9-1 corresponding to the cytoplasmic domain of hIre1p, or 2-kb human β-actin cDNA (Clontech). The hybridization was performed in ExpressHyb Hybridization buffer according to the manufacturer’s instructions (Clontech).
Total RNA from transfected COS-1 cells were prepared by use of TRIzol reagent (Bethesda Research Labs). RNA (10 μg) was resolved in 1% formaldehyde agarose gel and blotted onto Hybond nylon membrane (Amersham). Blots were hybridized with 32P-labeled 0.9-kb EcoRI fragment of hIRE-1 pBluescript-9-1 as described (Sambrook et al.1989).
In vitro phosphorylation and Western blotting
Immunoprecipitated protein from transfected COS-1 cells was incubated in kinase buffer }50 mm Tris-HCl at pH 7.4, 150 mm NaCl, 1 mm MnCl2, 1 mm MgCl2, 1 mm Na2MoO4, 2 mm NaF, 1 mm dithiothreitol and 10 μCi [γ32P]ATP (6000 Ci/mmole, Amersham Corp.)} at 30°C for 40 min. The protein samples were resolved by electrophoresis on an SDS–10% polyacrylamide gel and then transferred onto a nitrocellulose membrane. The membrane was probed with mouse α-hIre1p antibody followed with goat α-mouse antibody conjugated with alkaline phosphatase. The phosphorylation was quantified by autoradiography.
Confocal immunofluorescence microscopy
Immunofluorescence staining was followed as described by Paterson et al. (1995). Briefly, COS-1 cells were plated onto coverslips and transfected with pED–hIRE1 plasmid as described above. At 60 hr post-transfection, the transfected cells were stained with mouse α–GST–hIre1p and either rabbit α-GRP94 (generously provided by Dr. Michael Green, St. Louis University, MO) or guinea pig α-RanGAP1 (generously provided by Dr. Frauke Melchior, Scripps Institute, San Diego, CA). The cells were then incubated with secondary antibodies (goat α-mouse IgG conjugated with fluorescein isothiocyanate and goat α-rabbit or goat α-guinea pig conjugated with rhodamine) (Boehringer Mannheim), washed, and mounted onto slides with Prolong mounting (Molecular Probes). The fluorescence images were examined with a confocal laser scanning fluoresence microscope (Bio-Rad MRC 600).
In vitro cleavage of HAC1 mRNA
The procedure followed was described previously by Sidrauski and Walter (1997). Briefly, a 550-bp fragment of HAC1 DNA fragment flanking the intron region (Mori et al. 1996) was PCR amplified from S. cerevisiae genomic DNA and subcloned into pBluescript II SK (−) plasmid (Stratagene) at PstI and XhoI sites (pBluescript–HAC1). HAC1 mRNA was transcribed in vitro from XhoI-digested pBluescript–HAC1 by use of T7 RNA polymerase (Boehringer Mannheim) in the presence of [α32P]UTP (3000 Ci/mmole, Amersham Corp.). The RNA was resolved by electrophoresis in a 5% denaturing polyacrylamide gel and the 32P-labeled HAC1 mRNA was purified as described (Sidrauski and Walter 1997) and dissolved in endonuclease buffer (20 mm HEPES, 1 mm DTT, 10 mm MgOAc, 50 mm KOAc, and 2 mm ATP). Purified RNA (3 × 104 cpm) was added to the immunoprecipitated hIre1p, hIre1p K599A, or 0.5 μg GST-cytoplasmic Ire1p (Welihinda and Kaufman 1996) in final volume 100 μl reaction. After incubating at 30°C for the indicated time, the reaction was terminated by extraction with phenol/chloroform, precipitated with ethanol, and analyzed by electrophoresis on 5% denaturing polyacrylamide gel. Gels were dried prior to autoradiography.
Primer extension
The procedure was followed as described by Sambrook et al. (1989).
Luciferase assay
To construct the pBiP-luciferase reporter plasmid, the promoter region of rat BiP gene including the putative unfolded protein response element (nucleotides −457–+33; Chang et al. 1987) was amplified by PCR and subcloned into the KpnI and HindIII sites of pGL3-basic vector (Promega). Each 10 cm plate of COS-1 cells was cotransfected with pBiP-luciferase reporter plasmid (2 μg), RSVβ-gal (2 μg, generously provided by Dr. Gary Nabel, University of Michigan, Ann Arbor) and pED–hIRE1 or pED–hIRE1 -K599A (4 μg each) by the calcium phosphate procedure (Chen and Okayama 1988). At 60 hr post-transfection, cells were treated with or without 10 μg/ml of tunicamycin for 6 hr. Preparation of the cell lysate, β-galactosidase assays and luciferase assays were performed according to the manufacturer s instructions (Promega). The luciferase activity was normalized to β-galactosidase activity.
Acknowledgments
We thank Chris Edwards and Joe Nowak for assitance in confocal microscopy and Drs. Michael Green and Frauke Melchior for providing antibodies. We thank Dr. Michael Uhler for critial review of the manuscript and Joseph Nowak for assistance in its preparation. W.T. was supported by the Thai Ministry of Science, Technology, and Environment.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
The sequence information described in the paper has been submitted to GenBank under accession no. AF059198.
Footnotes
E-MAIL kaufmanr@umich.edu; FAX (313) 763-9323.
References
- Candau R, Moore PA, Wang L, Barley N, Ying CY, Rosen CA, Berger SL. Identification of human proteins functionally conserved with the yeast putative adaptors ADA2 and GCN5. Mol Cell Biol. 1996;16:593–602. doi: 10.1128/mcb.16.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Zhou Y, Lee AS. Requirement of tyrosine- and serine/threonine kinase in the transcriptional activation of the mammalian grp78/BiP promoter by thapsigargin. J Biol Chem. 1995;270:494–502. doi: 10.1074/jbc.270.1.494. [DOI] [PubMed] [Google Scholar]
- Chapman RE, Walter P. Translational attenuation mediated by an mRNA intron. Curr Biol. 1997;7:850–859. doi: 10.1016/s0960-9822(06)00373-3. [DOI] [PubMed] [Google Scholar]
- Chang SC, Wooden SK, Nakaki T, Kim YK, Lin AY, Kung L, Attenello JW, Lee AS. The rat gene encoding the 78-kilodalton glucose-regulated protein (GRP78): Its regulatory sequences and the effect of glycosylation on its expression. Proc Natl Acad Sci. 1987;84:680–684. doi: 10.1073/pnas.84.3.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CA, Okayam H. Calcium phosphate-mediated gene transfer: A highly efficient system for stably transforming cells with plasmid DNA. BioTechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- Chen K, Chen L, Haung H, Lieu C, Chang Y, Chang MD, Lai Y. Involvement of p38 Mitogen-activated protein kinase signaling pathway in the rapid induction of the 78-kDa glucose-regulated protein in 9L rat tumor cells. J Biol Chem. 1998;273:749–755. doi: 10.1074/jbc.273.2.749. [DOI] [PubMed] [Google Scholar]
- Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:394–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose regulated proteins in butyrate treated CHO cells. J Biol Chem. 1989;264:20602–20607. [PubMed] [Google Scholar]
- Gething MJ, Sambrook JF. Protein folding in the cell. Nature. 1992;355:34–44. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Gomer CJ, Ferrario A, Rucker N, Wong S, Lee AS. Glucose regulated protein induction and cellular resistance to oxidative stress mediated by porphyrin photosensitization. Cancer Res. 1991;51:6574–6579. [PubMed] [Google Scholar]
- Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM. Protein kinase catalytic domain sequence database: Identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1988. [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hughes CS, Shen JW, Subject JR. Resistance to etoposide induced by three glucose-regulated stresses in Chinese hamster ovary cells. Cancer Res. 1989;49:4452–4454. [PubMed] [Google Scholar]
- Kaufman RJ, Davies MV, Wasley LC, Michnick D. Improved vectors for stable expression of foreign genes in mammalian cells by use of the untranslated leader sequence from EMC virus. Nucleic Acids Res. 1991;19:4485–4490. doi: 10.1093/nar/19.16.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Overview of vector design for mammalian gene expression. Methods Mol Biol. 1997;62:287–300. doi: 10.1385/0-89603-480-1:287. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress- induced mRNA splicing permits synthesis of transcription factor HAC1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8:1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Unconventional splicing of HAC1/ ERN4 mRNA required for the unfolded protein response. Sequence-specific and non-sequential cleavage of the splice sites. J Biol Chem. 1998;273:1802–1807. doi: 10.1074/jbc.273.3.1802. [DOI] [PubMed] [Google Scholar]
- Koong AC, Auger EA, Chen EY, Giaccia AJ. The regulation of GRP78 and messenger RNA levels by hypoxia is modulated by protein kinase C activators and inhibitors. Radiat Res. 1994a;138:860–863. [PubMed] [Google Scholar]
- Koong AC, Chen EY, Lee AS, Brown JM, Giaccia AJ. Increased cytotoxicity of chronic hypoxic cells by molecular inhibition of GRP78 induction. Int J Radiat Oncol Biol Phys. 1994b;28:661–666. doi: 10.1016/0360-3016(94)90191-0. [DOI] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8248. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook JF. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- Lee AS. Coordinated regulation of a set of genes by glucose and calcium ionophores in mammalian cells. Trends Biochem Sci. 1987;12:20–30. [Google Scholar]
- Li XA, Lee AS. Competitive inhibition of a set of endoplasmic reticulum protein genes (GRP78, GRP94, and Erp72) retards cell growth and lowers viability after ionophore treatment. Mol Cell Biol. 1991;11:3446–3453. doi: 10.1128/mcb.11.7.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LJ, Li X, Ferrario A, Rucker N, Liu ES, Wong S, Gomer CJ, Lee AS. Establishment of a Chinese hamster ovary cell line that expresses grp78 antisense transcripts and suppresses A23187 induction of both GRP78 and GRP94. J Cell Physiol. 1992;153:575–582. doi: 10.1002/jcp.1041530319. [DOI] [PubMed] [Google Scholar]
- Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- Mori K, Sant A, Kohno K, Normington K, Gething MJ, Sambrook JF. A 22-bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J. 1992;11:2583–2593. doi: 10.1002/j.1460-2075.1992.tb05323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Ma W, Gething MJ, Sambrook JF. A transmembrane protein with a cdc2+/CDC28- related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T. Signaling from endoplasmic reticulum to nucleus: Transcriptional factor with a basic-luecine zipper motif is required for the unfolded protein-response pathway. Genes Cells. 1996;1:803–317. doi: 10.1046/j.1365-2443.1996.d01-274.x. [DOI] [PubMed] [Google Scholar]
- Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997;272:4327–4334. doi: 10.1074/jbc.272.7.4327. [DOI] [PubMed] [Google Scholar]
- Munro S, Pelham HR. An Hsp 70-like protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46:291–300. doi: 10.1016/0092-8674(86)90746-4. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht HJ, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Nikawa J, Yamashita S. IRE1 encodes a putative protein kinse containing a membrane-spanning domain and is required for inositol prototrophy in Saccharomyces cerevisiae. Mol Microbiol. 1992;6:1441–1446. doi: 10.1111/j.1365-2958.1992.tb00864.x. [DOI] [PubMed] [Google Scholar]
- Paterson H, Adamson P, Roberson D. Microinjection of epitope-tagged Rho family cDNAs and analysis by immunolabeling. Methods Enzymol. 1995;256:162–173. doi: 10.1016/0076-6879(95)56021-1. [DOI] [PubMed] [Google Scholar]
- Resendez E, Jr, Ting J, Kim KS, Wooden SK, Lee AS. Calcium ionophore A23187 as a regulator of gene expression in mammalian cells. J Cell Biol. 1986;103:2145–2152. doi: 10.1083/jcb.103.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resendez EJ, Wooden SK, Lee AS. Identification of highly conserved regulatory domains and protein binding site in the promoter of the rat and human genes encoding the stress-inducible 78-kilodalton glucose regulated protein. Mol Cell Biol. 1988;8:4579–4584. doi: 10.1128/mcb.8.10.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989. [Google Scholar]
- Shamu CE, Walter P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15:3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Shen J, Hughes C, Chao C, Cai J, Bartels C, Gessner T, Subjeck J. Coinduction of glucose-regulated proteins and doxorubicin resistance in Chinese hamster cells. Proc Natl Acad Sci. 1987;84:3278–3282. doi: 10.1073/pnas.84.10.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski K, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Sidrauski K, Cox JS, Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87:405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- Simos G, Tekotte H, Grosjean H, Segref A, Tollervey D, Hury EC. Nuclear pore proteins are involved in the biosynthesis of functional tRNA. EMBO J. 1996;15:2270–2284. [PMC free article] [PubMed] [Google Scholar]
- Sugawara S, Takeda K, Lee A, Dennert G. Suppression of stress protein GRP78 induction in tumor B/C10ME eliminates resistance to cell mediated cytotoxicity. Cancer Res. 1993;53:6061–6005. [PubMed] [Google Scholar]
- Trotta CR, Miao F, Arn EA, Stevens SW, Ho CK, Rauhut R, Abeson JN. The yeast tRNA splicing endonuclease: A tetrameric enzyme with two active site subunits homologous to the Archaeal tRNA endonucleases. Cell. 1997;89:849–858. doi: 10.1016/s0092-8674(00)80270-6. [DOI] [PubMed] [Google Scholar]
- Welihinda AA, Kaufman RJ. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p(Ern1p) are required for kinase activation. J Biol Chem. 1996;271:18181–18187. doi: 10.1074/jbc.271.30.18181. [DOI] [PubMed] [Google Scholar]
- Welihinda AA, Tirasophon W, Green SR, Kaufman RJ. Gene induction in response to unfolded protein in the endoplasmic reticulum is mediated through Ire1p kinase interaction with a transcriptional coactivator complex containing Ada5p. Proc Nati Acad Sci. 1997;94:4289–4294. doi: 10.1073/pnas.94.9.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welihinda AA, Tirasophon W, Green SR, Kaufman RJ. Protein Serine/Thronine phosphatase, Ptc2p, negatively regulates the unfolded protein response by dephosphorylating Ire1p kinase. Mol Cell Biol. 1998;18:1967–1977. doi: 10.1128/mcb.18.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Hassel BA, Silverman RH. Expression cloning of 2-5A- dependent RNase: A uniquely regulated mediator of interferon action. Cell. 1993;72:753–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]