

Abstract
Familial biparental hydatidiform mole (FBHM) is the only known pure maternal-effect recessive inherited disorder in humans. Affected women, although developmentally normal themselves, suffer repeated pregnancy loss because of the development of the conceptus into a complete hydatidiform mole in which extraembryonic trophoblastic tissue develops but the embryo itself suffers early demise. This developmental phenotype results from a genome-wide failure to correctly specify or maintain a maternal epigenotype at imprinted loci. Most cases of FBHM result from mutations of NLRP7, but genetic heterogeneity has been demonstrated. Here, we report biallelic mutations of C6orf221 in three families with FBHM. The previously described biological properties of their respective gene families suggest that NLRP7 and C6orf221 may interact as components of an oocyte complex that is directly or indirectly required for determination of epigenetic status on the oocyte genome.
Main Text
Normal mammalian development requires biparental genetic contributions because of the phenomenon of genomic imprinting.1 Although at most genetic loci the paternal and maternal alleles are functionally equivalent and biallelically transcribed, a small subset of genes deviate from this general pattern. At these imprinted loci, different epigenetic modifications arise on the maternal and paternal alleles, resulting in differential gene expression (which may be temporally or spatially restricted2).
Although there is considerable diversity in the genomic architecture and physiological function of imprinted genes, some general observations may be made. (1) Many imprinted genes occur in clusters, which may include paternally, maternally, and biallelically expressed genes but which may be subject to coordinated control by locus control elements (imprinting control regions [ICRs]3). (2) Most imprinted loci depend for correct function on a primary epigenetic mark established on the maternal allele; only a small number of paternally-specified primary imprints have been identified.4 (3) In female mice, nuclear transplantation experiments have defined unique, locus-specific temporal windows during which an imprint can be established on maternal alleles as they pass through oogenesis.5
A variety of human developmental phenotypes result from incorrect imprint specification or dosage. In several cases, the phenotype resulting from a uniparental disomy (e.g., Prader-Willi syndrome [MIM 176270], maternal uniparental disomy 15) is closely mimicked by that due to an ICR mutation (e.g., that results in a genetically paternal 15q having a maternal-like epigenetic status and function).6 Such defects are cis-acting, the status of other imprinted chromosomal loci being unaffected.
Complete hydatidiform mole (CHM) represents an extreme example of a human developmental phenotype attributable to abnormal imprinting. In most CHM, the conceptus is wholly androgenetic in origin.7 This results in proliferation of the extraembryonic trophoblast, while embryonic development fails. CHM is generally a sporadic disorder, but a rare familial form of the disorder has been long recognized, in which affected women suffer recurrent molar pregnancies. Notably, in this familial form of the disorder the molar tissues are not androgenetic, but show a normal pattern of biparental diploid inheritance. This condition, familial biparental hydatidiform mole (FBHM [MIM 231090]) displays maternal-effect autosomal recessive inheritance.8
We and others9–13 have previously shown that FBHM is a consequence of a failure to establish maternal imprints at multiple genome-wide loci. This multilocus imprinting failure suggests that the genetic defect must be trans-acting. The autosomal recessive inheritance of FBHM is thus consistent with the idea that affected (homozygous mutant) mothers are deficient in a trans-acting gene product. This contrasts with the cis-acting effect and autosomal dominant inheritance pattern that characterize locus-specific ICR defects.2
In 2006, Slim and colleagues14 identified recessive mutations in NLRP7 (MIM 609661) as a cause of FBHM; subsequent reports have confirmed these findings and suggested that NLRP7 is mutated in the majority of FBHM families.11,13,15–17 However, we have previously shown that the original family (family L11,12) in which the FBHM imprinting defect was demonstrated is unlinked to the NLRP7 locus on chromosome 19. This and other reports of confirmed FBHM cases that do not harbor NLRP7 mutations support the existence of locus heterogeneity.11,15,17 Here, we show that mutations in C6orf221, a member of a rapidly evolving gene family specific to eutherian mammals, are a cause of FBHM.
All patient samples were obtained after written informed consent. Samples from family L, were obtained according to a protocol approved by the Leeds (East) Research Ethics Committee (reference 07/H1306/113). Other individuals analyzed provided samples following protocols approved by the institutional review board of the CHU Farhat Hached or by the Riverside Research Ethics Committee (RREC 2652).
In family L, (Figure 1), a multiply consanguineous family of Pakistani origin, two related women, individuals V:3 and IV:5, had clinically typical, recurrent CHM, confirmed in the index case to be biparental.12 We used these two individuals to perform a homozygosity scan using the Affymetrix Genome-Wide Human SNP Array 6.0. Genotype data were analyzed using AutoSNPa18 and IBDfinder software.19 Six regions of concordant homozygosity larger than 2 Mbp and encompassing a total genomic interval of 78 Mbp were shared by both affected women (Table S1, available online).
Figure 1.
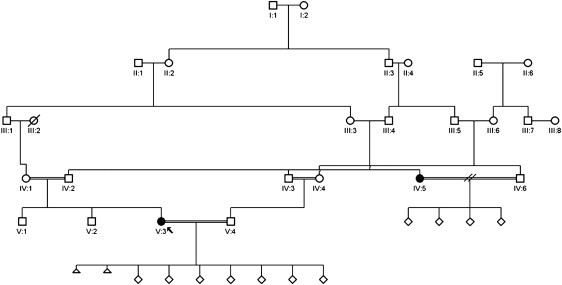
Pedigree of Family L
The proband is designated by the arrow; individuals V:3 and IV:5 are the affected individuals. CHM are displayed as clear diamonds. Miscarriages are displayed as small triangles.
To enrich for sequences of interest, we used the Agilent SureSelect Target Enrichment (Agilent, Santa Clara, CA, USA) in-solution method to capture RefSeq coding exons within the target interval identified in for family L. We designed biotinylated oligonucleotide baits by extracting all coding regions from the UCSC Genome Browser 8 for the 5451 unique RefSeq genes in the two loci. These regions were uploaded to Agilent's eArray software for automated oligonucleotide synthesis (in parallel with regions for seven other unrelated loci as part of a collaborative experiment). Three micrograms of genomic DNA was sheared and Illumina paired-end adapters were ligated according to Agilent's SureSelect Library Prep protocol version 1.0.1 (October 2009). Samples were size selected (200–300 bp) on an agarose gel followed by 12 cycles of PCR enrichment prior to hybridization to the SureSelect reagent for 24 hr at 65°C (following protocol version 1.0.1). A posthybridization amplification step was performed and samples were subsequently cleaned up with Ampure SPRI beads (Beckman-Coulter, High Wycombe, UK).
We denatured libraries with NaOH and diluted to a final concentration of 12 pM, 120 μl of which was hybridized onto a v5 single-read flow cell (Illumina, San Diego, CA, USA) according to the manufacturer's cluster station instructions. Samples were prepared for sequencing according to Illumina's standard amplification, linearization, blocking, and primer hybridization protocols. The flow cell was then transferred to the Illumina GAIIx for sequencing with an adapted single-read protocol for 80 cycles. Raw data files were processed by the Illumina pipeline (version 1.3.4) and sequence reads aligned to the human reference sequence (hg19/GRCh37) with Novoalign short-read alignment software (Novocraft Technologies, Selangor, Malaysia). Duplicate reads, resulting from PCR clonality or optical duplicates, and reads mapping to multiple locations were excluded from downstream analysis. Alignment files were further processed with the SAMtools program20 and the Genome Analysis Toolkit.21,22 Mean depth of coverage for targeted regions was 74.81, with 93.7% of target bases covered by at least five reads of sufficient base quality for variant calling (phred quality scores ≥ 17). Variants within the candidate regions were called in the VCF format using the Unified Genotyper and DINDEL23 functions of GATK. Variants were filtered using GATK on the basis of mapping quality, strand bias, and genotype quality.
Six hundred and forty-two variants matching known SNPs present in dbSNP131 were removed, leaving 15 functional variants (missense, stop loss, nonsense, indels and splice site) not annotated in dbSNP131. Of the 15 novel variants, seven were identified in at least one of the other samples captured and sequenced alongside the FBHM proband and was therefore likely to represent nonpathogenic variation.
The remaining eight variants all resulted in missense changes in their respective encoded proteins (Table S2). In order to identify potentially pathogenic variants, we used SIFT to identify variants likely to affect protein function.24 Only two variants were predicted to be damaging. The first of these was a valine to aspartate substitution (c.632T>A; p.Val211Asp) in TAAR8 (NM_053278.1), which encodes a trace amine-associated receptor. Members of the TAAR family are exclusively expressed in the olfactory epithelium, where they seem to play a chemosensory role related to the detection of social cues.25 The TAAR family displays an evolutionary pattern similar to that of olfactory receptors,26 and the presence of a common TAAR8 nonsense SNP (rs77605736, c.142G>T; p.Gly48∗) with an allele frequency of 8.3% suggests that this gene is dispensable in humans. This variant was therefore considered unlikely to be pathogenic.
The remaining predicted damaging variant alters the initiation codon of C6orf221 (NM_001017361.2) from ATG to ATT (c.3G>T) (Figure 2). The next available downstream ATG codon lies at residue 14 of the putative C6orf221 protein (predicted from reference sequence NM_001017361.2) and is in frame. The c.3G>T mutation segregated with the disease in family L and was not found in 328 ethnically matched control chromosomes.
Figure 2.
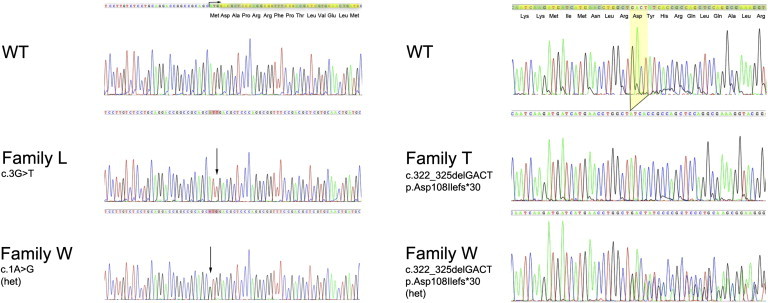
Mutations Identified in C6orf221 in Three Cases of FBHM
The following abbreviations are used: WT, wild-type; family L, c.3G>T homozygote; family T, c.322_325delGACT homozygote; family W, compound heterozygote c.1A>G + c.322_325delGACT.
To investigate whether the c.3G>T mutation results in the use of the downstream ATG at position 14, we used in vitro gene-transfer analysis. pcDNA3.1myc-His (Invitrogen, Renfrew, UK) expression constructs for wild-type and mutant proteins were created; restriction site-tagged cDNAs were amplified from IMAGE clone 40146866 template DNA, with 20 cycles of PCR with Pfu polymerase (Stratagene, Stockport, UK) under manufacturers' recommended conditions. The mutant construct was generated by use of the Quikchange mutagenesis kit (Stratagene, UK). Primer sequences for generation of the wild-type and mutant constructs are listed in Table S3.
We cultured HEK293 cells (ECACC, Salisbury, UK) in MEM, 5% fetal bovine serum, and 2 mM glutamine and transfected them with the vectors described above by using Lipofectamine 2000 (Invitrogen) according to the manufacturers' instructions. Whole-cell protein extracts were resolved with SDS-PAGE, and immunoblotted. Blots were probed with a commercial mouse monoclonal Myc antibody (Sigma-Aldrich, Irvine, UK) and with a custom-made rabbit polyclonal antibody raised against a peptide antigen (MDAPRRFPTLVQLMQC) containing residues 1–15 of human C6orf221. Figure 3 shows that as expected, a shortened protein is produced consequent upon the c.3G>T mutation. Also as expected, this mutant protein is not detected by the N-terminal C6orf221 antibody. These results are consistent with the initiation of the mutant protein at position 14.
Figure 3.
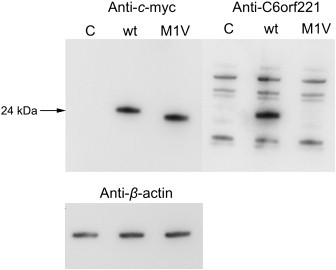
Effect of c.3G>T Mutation on C6orf221
HEK293 cells (ECACC, Salisbury, UK) were transfected with Myc-tagged wild-type or c.3G>T C6orf221 containing vectors. Whole-cell protein extracts were resolved with SDS-PAGE and immunoblotted. Blots were probed with a commercial mouse monoclonal c-Myc antibody (Sigma-Aldrich, Irvine, UK) and with a custom-made rabbit polyclonal antibody raised against a peptide antigen (MDAPRRFPTLVQLMQC) containing residues 1–15 of human C6orf221. The following abbreviations are used: C, negative control; Wt, wild-type protein; M1V, site directed mutagenesis produced a protein with the initiation codon mutation. In lanes 1, 2, and 3, the probe used is the anti-c-Myc antibody. The expected 24 kDa wild-type C6orf221 is detected in lane 2; in lane 4 the shorter protein is seen consequent upon the c.3G>T mutation. In lanes 6, 7, and 8, the probe used is the antibody raised against N-terminal peptide. In lane 7 the expected 24 kDa wild-type C6orf221 is detected. The peptide antibody fails to detect the protein produced from the vector containing the c.3G>T mutation run in lane 8. This is consistent with the predicted initiation at codon 14 as a result of the mutation.
We next analyzed probands from a further 14 cases of recurrent hydatidiform mole in which NLRP7 mutations had not been identified on sequencing. Conventional Sanger sequencing of C6orf221 was performed using PCR primers that flanked all coding exons of C6orf221. These were designed using the ExonPrimer program (Table S4). Direct sequencing was performed using the dideoxy-chain-termination method (ABI BigDye 3.0 system) on an ABI 3730 DNA Sequencer and sequences analyzed with Chromas v2.0 software. All mutations were verified bidirectionally.
In an affected individual of Tunisian origin (individual T1), we identified a homozygous 4 bp deletion (c.322_325delGACT; p.Asp108Ilefs∗30) in exon 2 of C6orf221 (Figure 2C). Details of this case have been reported previously, and the molar tissue has again been proven to be biparental.27 This mutation was not present in 208 Tunisian control chromosomes. It results in a frameshift at amino acid 108 and premature termination after 29 novel amino acid residues. The truncated protein thus lacks its normal 110 C-terminal amino acids, including the entire exon 3-encoded repeat motif (discussed below).
In a further nonconsanguineous FBHM patient of Asian origin (individual W1, case 20 of Wang et al.17), we found compound heterozygosity for two mutations, c.1A>G and the 4 bp c.322_325delGACT mutation in exon 2. Sequencing of cloned PCR products spanning exons 1 and 2 confirmed that these two mutations were on opposite alleles (data not shown.) Inspection of data from the 1000 Genomes Project alignment files showed that the c.1A>G mutation was absent from all 1127 individuals for which a genotype could be called, including 734 individuals with genotype phred scale quality scores of ≥ 30. The exon 2 deletion was similarly not found in 1145 individuals for whom a genotype could be called, including 739 at a minimum genotype phred scale quality score of 30.
Clinical details of these cases are found in Table S5. They are typical FBHM, indistinguishable clinically from those with NLRP7 mutations.
We next investigated the expression of C6orf221 by using RT-PCR on amplified cDNA derived from a developmental series of staged human ovarian follicles, oocytes, preimplantation embryos, and human blastocysts. Methods for sample origination, preparation, and validation have been described.28–30 Briefly, human ovarian follicles were isolated after enzymatic digestion and needle dissection31 from samples of frozen-thawed ovarian cortex obtained from a patient who was 29 at the time of ovarian tissue cryopreservation; cryopreservation was performed by slow freezing for fertility preservation.32 Ovarian follicles were staged according to size and morphology; samples were collected from the primordial, early primary, and primary stage, and pooled between 25–40 follicles per sample. Denuded, mature metaphase II (MII) oocytes were derived after 24 hr of in vitro maturation of immature germinal vesicle (GV) or metaphase I oocytes from patients undergoing infertility treatment by intracytoplasmic sperm injection at the assisted conception unit at Leeds General Infirmary. Additionally, denuded, GV-staged secondary oocytes were harvested from nonluteinized antral follicles of 5 mm diameter that were aspirated from two patients during immature oocyte recovery as previously described.28,29 All samples were obtained after informed consent under ethically approved protocols, which were licensed in the UK by the Human Fertilisation and Embryology Authority. Day 6–7 human blastocysts (total n = 10) that were either considered unsuitable for transfer and were donated for research or that were surplus to the patients' treatment requirements, had been in cryostorage, and were donated for research; we obtained the blastocysts with full patients' consent from the Assisted Conception Unit at Leeds General Infirmary and Bourn Hall Clinic, Cambridge, UK, under ethically approved protocols, which were licensed by the HFEA as previously described.30 All samples were washed in Ca2+ and Mg2+-free phosphate buffered saline at 4°C (Invitrogen) before being snap frozen in 50 μl lysis buffer (Dynal lysis buffer, Dynal, Merseyside, UK) supplemented with 5 μl of RNA Later (Ambion, Austin, TX, USA) per sample on ice, and mRNA was extracted using Oligo-dT magnetic beads (Dynal, Oslo, Norway). cDNA was generated using SMART amplification (Clontech, Oxford, UK) or a related cDNA amplification protocol33 with 1 μg each of forward and reverse primers (details in Table S4). In each case, reverse transcription was performed with Superscript II RNaseH-Reverse Transcriptase (Invitrogen) for 2 hr at 42°C. The cDNA was amplified by PCR with an additional 1 μg of each primer, 2 μl 50× Advantage 2 Polymerase (BD Clontech), in a thermal cycler for 32 cycles of 95°C for 45 s, 65°C for 6 min 45 s. All cDNAs generated were tested with the housekeeping gene (GAPDH).
Expression appeared to be maximal in germinal vesicle oocytes (GV) and then to tail off through metaphase II oocytes (MII) and to be undetectable following the completion of the oocyte to embryo transition34 (Figure 4). It may be noted that this temporal pattern of expression of C6orf221 in oocytes is similar to that of NLRP7 reported previously35 and is typical of the expression we and others have previously reported for oocyte-specific genes.29,36–38
Figure 4.
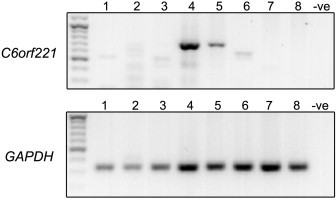
C6orf221 Expression in Human Ovarian Follicles, Oocytes, Preimplantation Embryos
Expression of C6orf221 and GAPDH was investigated by RT-PCR of cDNA amplified from human ovarian follicles (including granulosa/pregranulosa cells), mature oocytes (granulosa/pregranulosa cell free) and preimplantation embryos. DNA size standard: lane1, primordial follicles (n = 25–40); lane 2, early primary follicles (n = 25–40); lane 3, primary follicles (n = 25–40); lane 4, GV oocyte pool (n = 2); lane 5, MII oocyte pool (n = 5); lane 6, Morula pool (n = 5); lane 7, blastocyst pool 1 (n = 5); lane 8, blastocyst pool 2 (n = 5); negative control (-ve). Diameters used for follicle staging: primordial follicles 34–38 mm, early primary follicles 34–53 mm, primary follicles 52–62 mm, secondary follicles 62–86 mm. The upper panel shows expression of expected 716 bp long C6orf221 mRNA; the lower panel shows expression of the housekeeping gene GAPDH mRNA, used as a control.
In bovine ovary, an antibody to the N-terminal peptide (MASPKRFPTLVQLEQC) of the apparent bovine ortholog (NP_001017361.2) showed a pattern of staining restricted to oocyte cytoplasm (Figure 5) in primary and secondary follicles. In antral follicles the staining is again cytoplasmic and takes on a punctate appearance.
Figure 5.
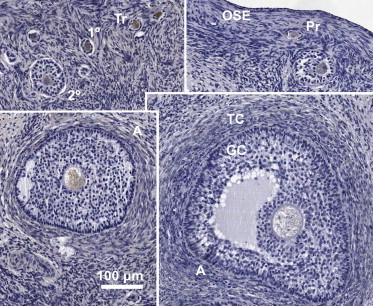
Immunohistochemical Staining of Bovine Ovary for C6orf221 Ortholog
The primary antibody was raised against the 15 N-terminal amino acids (MASPKRFPTLVQLEQ) of predicted bovine protein XP_002690064.1. (computationally predicted gene, C9H6orf221, Bos taurus ES-cell-associated transcript). The following abbreviations are used: OSE, ovarian surface epithelium; Pr, primordial (nongrowing) follicle; Tr, transitional follicle; 1°, primary follicle; 2°, secondary follicle; A, antral follicle; GC, granulosa cells; TC, thecal cells. Protein is diffusely distributed throughout the cytoplasm of oocytes in the primary and secondary follicles. In the antral follicles localization is again restricted to oocyte cytoplasm, but the staining pattern now takes on a more punctate appearance. There is no staining in surrounding granulosa or stromal cells.
C6orf221 is a member of a 100 kb cluster containing four related genes, located at ∼74 Mb on human chromosome 6.39 The other members of the family are KHDC1 (KH domain containing 1 [MIM 611688]), DPPA5 (developmental pluripotency associated 5 [MIM 611111]) and OOEP (oocyte expressed protein [MIM 611689]). The human gene order and orientation is (<KHDC1 < DPPA5 C6orf221 > < OOEP). Members of this gene family display a mostly oocyte- or early embryo-specific expression pattern.39 They are characterized structurally by an N-terminal KH domain (K homology domain, some examples of which are known to be RNA binding).40 It should be noted that there is some ambiguity regarding nomenclature for members of this gene family in various mammalian species. In the evolutionary analysis by Pierre et al.,39 primate C6orf221 is referred to as ECAT1. In this study, Pierre et al. concluded that Ecat1 has been lost from the rodent genome, coincident with a disruption of synteny between the Dppa5/Ooep cluster (on Mmu9 at ∼78.22 Mb) and Khdc1 (on Mmu1 21.35 Mb). However, Ecat1 transcripts were actually originally identified in mouse ES cells (Mm.157658, also known as Filia).41 In the NCBI37/mm9 build, Mmu Ecat1/Filia can be found on Mmu9 at 72.95 Mb, more than 5 Mb distant from Dppa5/Ooep. This makes it somewhat uncertain whether are not Ecat1/Filia and C6orf221 are true orthologs. In the rabbit and cow genomes, the synteny and convergent transcriptional orientation of ECAT1/C6orf221 and OOEP are maintained, making orthology with the human genes clearer. Sequence alignment of the human, bovine, rabbit, and mouse genes (Figure S1) similarly leaves the orthology of Mmu Ecat1 with the other three genes uncertain because although closely similar, it is the most divergent of the four.
Regardless of precise cross-species orthology, the KHDC1/DPPA5/C6orf221(ECAT1)/OOEP cluster appears to be specific to eutherian mammals, in which these genes have evolved rapidly.39 Interestingly, the same is also true for the NLRP7/NLRP2 group of genes, within which cross-species orthology is again difficult to establish.11,35,42 Indeed, these are evolutionary features characteristic of a number of reproduction-related gene clusters.42 We also note, however, the existence of an additional, much more highly conserved gene related to OOEP, C12orf66, which unlike the members of the KHDC1/DPPA5/C6orf221(ECAT1)/OOEP cluster, is present in nonmammalian vertebrates.
The KH domain of the KHDC1/DPPA5/C6orf221(ECAT1)/OOEP family is encoded by the first two of three coding exons, followed by a variable C terminus. In the ECAT1/C6orf221 genes, the C-terminal domain encoded by exon 3 displays an interesting ∼12 aa repeat motif, with the size of different species' proteins varying considerably, due to variation in the number of repeats. In bovine C6orf221, for example, there are 20 almost perfect tandem repeats of the sequence EAATQRSPGAAR. In human C6orf221, seven much less homogeneous TQRS-containing repeats are present, while the rabbit protein only contains three. Murine Ecat1/Filia also has 18 12 aa repeats of a slightly different sequence in this position.37 Closely related repeat motifs could not be identified in any other gene product, despite exhaustive search.
FBHM is currently the only known human maternal-effect recessive disorder. Maternal-effect recessives have been known since the 1980s in Drosophila, but only much more recently in mammals. Interestingly, one of the first maternal-effect genes identified in mice was Mater/Nlrp543. Mater/Nlrp5 transcripts are expressed in the growing oocyte, but the maternally-encoded protein product is required early in embryogenesis. Mater/Nlrp5 protein associates specifically with Ecat1/Filia protein to form a complex, which in the early embryo becomes preferentially localized in those cells destined to form extraembryonic structures.37 This subcortical maternal complex (SCMC) also includes the product of another maternal-effect gene related to Filia/Ecat1, namely Floped/Ooep. There is evidence that the SCMC may be identical to the oocyte ultrastructural organelles known as cytoplasmic lattices (CPLs), since both the SCMC and CPLs are absent from oocytes of Floped/Ooep null female mice.44
These observations, together with the fact that mutations of C6orf221/ECAT1 and of NLRP7 have identical phenotypes, make it likely that NLRP7 and C6orf221 participate in a similar complex during human oogenesis and/or early embryogenesis. Because of the rapid evolution of the members of both these gene families, precise inferences based on presumed orthology should be made with caution. Also, the substantial differences in female reproductive physiology among mammalian groups must be borne in mind. Although mutations of mouse Filia/Ecat1 and human C6orf221 both display maternal-effect recessive inheritance, the phenotypes are not identical: Filia/Ecat1 null female mice produce embryos with reduced viability because of mitotic spindle dysfunction and chromosomal aneuploidy.45 Although general inferences concerning the biology of human NLRP7 and C6orf221 may be drawn from the mouse studies, it is therefore likely that a detailed picture will require the study of human oocytes and embryos.
Acknowledgments
The authors thank the families for participating in this study. This work was supported by grants from Wellbeing of Women (to E.S), the Sir Jules Thorn Award for Biomedical Research (to C.A.J., E.S., G.R.T., and D.T.B.). J.D. and H.M.P. are supported by funding from the Medical Research Council (grant number G0701388).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent Technologies eArray, https://earray.chem.agilent.com/earray/
AutoSNPa, http://dna.leeds.ac.uk/autosnpa/
ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer.html
IBDfinder, http://dna.leeds.ac.uk/ibdfinder/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Reik W., Walter J. Evolution of imprinting mechanisms: the battle of the sexes begins in the zygote. Nat. Genet. 2001;27:255–256. doi: 10.1038/85804. [DOI] [PubMed] [Google Scholar]
- 2.Horsthemke B. Mechanisms of imprint dysregulation. Am. J. Med. Genet. C. Semin. Med. Genet. 2010;154C:321–328. doi: 10.1002/ajmg.c.30269. [DOI] [PubMed] [Google Scholar]
- 3.Buiting K., Saitoh S., Gross S., Dittrich B., Schwartz S., Nicholls R.D., Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995;9:395–400. doi: 10.1038/ng0495-395. [DOI] [PubMed] [Google Scholar]
- 4.Thorvaldsen J.L., Bartolomei M.S. SnapShot: imprinted gene clusters. Cell. 2007;130:958. doi: 10.1016/j.cell.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 5.Obata Y., Kono T. Maternal primary imprinting is established at a specific time for each gene throughout oocyte growth. J. Biol. Chem. 2002;277:5285–5289. doi: 10.1074/jbc.M108586200. [DOI] [PubMed] [Google Scholar]
- 6.Sutcliffe J.S., Nakao M., Christian S., Orstavik K.H., Tommerup N., Ledbetter D.H., Beaudet A.L. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat. Genet. 1994;8:52–58. doi: 10.1038/ng0994-52. [DOI] [PubMed] [Google Scholar]
- 7.Kajii T., Ohama K. Androgenetic origin of hydatidiform mole. Nature. 1977;268:633–634. doi: 10.1038/268633a0. [DOI] [PubMed] [Google Scholar]
- 8.Van den Veyver I.B., Al-Hussaini T.K. Biparental hydatidiform moles: a maternal effect mutation affecting imprinting in the offspring. Hum. Reprod. Update. 2006;12:233–242. doi: 10.1093/humupd/dmk005. [DOI] [PubMed] [Google Scholar]
- 9.Fisher R.A., Hodges M.D., Rees H.C., Sebire N.J., Seckl M.J., Newlands E.S., Genest D.R., Castrillon D.H. The maternally transcribed gene p57(KIP2) (CDNK1C) is abnormally expressed in both androgenetic and biparental complete hydatidiform moles. Hum. Mol. Genet. 2002;11:3267–3272. doi: 10.1093/hmg/11.26.3267. [DOI] [PubMed] [Google Scholar]
- 10.El-Maarri O., Seoud M., Coullin P., Herbiniaux U., Oldenburg J., Rouleau G., Slim R. Maternal alleles acquiring paternal methylation patterns in biparental complete hydatidiform moles. Hum. Mol. Genet. 2003;12:1405–1413. doi: 10.1093/hmg/ddg152. [DOI] [PubMed] [Google Scholar]
- 11.Hayward B.E., De Vos M., Talati N., Abdollahi M.R., Taylor G.R., Meyer E., Williams D., Maher E.R., Setna F., Nazir K. Genetic and epigenetic analysis of recurrent hydatidiform mole. Hum. Mutat. 2009;30:E629–E639. doi: 10.1002/humu.20993. [DOI] [PubMed] [Google Scholar]
- 12.Judson H., Hayward B.E., Sheridan E., Bonthron D.T. A global disorder of imprinting in the human female germ line. Nature. 2002;416:539–542. doi: 10.1038/416539a. [DOI] [PubMed] [Google Scholar]
- 13.Kou Y.C., Shao L., Peng H.H., Rosetta R., del Gaudio D., Wagner A.F., Al-Hussaini T.K., Van den Veyver I.B. A recurrent intragenic genomic duplication, other novel mutations in NLRP7 and imprinting defects in recurrent biparental hydatidiform moles. Mol. Hum. Reprod. 2008;14:33–40. doi: 10.1093/molehr/gam079. [DOI] [PubMed] [Google Scholar]
- 14.Murdoch S., Djuric U., Mazhar B., Seoud M., Khan R., Kuick R., Bagga R., Kircheisen R., Ao A., Ratti B. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat. Genet. 2006;38:300–302. doi: 10.1038/ng1740. [DOI] [PubMed] [Google Scholar]
- 15.Muhlstein J., Golfier F., Rittore C., Hajri T., Philibert L., Abel F., Beneteau C., Touitou I. The spectrum of NLRP7 mutations in French patients with recurrent hydatidiform mole. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011;157:197–199. doi: 10.1016/j.ejogrb.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 16.Qian J., Deveault C., Bagga R., Xie X., Slim R. Women heterozygous for NALP7/NLRP7 mutations are at risk for reproductive wastage: report of two novel mutations. Hum. Mutat. 2007;28:741. doi: 10.1002/humu.9498. [DOI] [PubMed] [Google Scholar]
- 17.Wang C.M., Dixon P.H., Decordova S., Hodges M.D., Sebire N.J., Ozalp S., Fallahian M., Sensi A., Ashrafi F., Repiska V. Identification of 13 novel NLRP7 mutations in 20 families with recurrent hydatidiform mole; missense mutations cluster in the leucine-rich region. J. Med. Genet. 2009;46:569–575. doi: 10.1136/jmg.2008.064196. [DOI] [PubMed] [Google Scholar]
- 18.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 19.Carr I.M., Sheridan E., Hayward B.E., Markham A.F., Bonthron D.T. IBDfinder and SNPsetter: tools for pedigree-independent identification of autozygous regions in individuals with recessive inherited disease. Hum. Mutat. 2009;30:960–967. doi: 10.1002/humu.20974. [DOI] [PubMed] [Google Scholar]
- 20.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albers C.A., Lunter G., MacArthur D.G., McVean G., Ouwehand W.H., Durbin R. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 25.Liberles S.D., Buck L.B. A second class of chemosensory receptors in the olfactory epithelium. Nature. 2006;442:645–650. doi: 10.1038/nature05066. [DOI] [PubMed] [Google Scholar]
- 26.Vallender E.J., Xie Z., Westmoreland S.V., Miller G.M. Functional evolution of the trace amine associated receptors in mammals and the loss of TAAR1 in dogs. BMC Evol. Biol. 2010;10:51. doi: 10.1186/1471-2148-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landolsi H., Rittore C., Philibert L., Missaoui N., Hmissa S., Touitou I., Gribaa M., Yacoubi M.T. Screening for NLRP7 mutations in familial and sporadic recurrent hydatidiform moles: report of 2 Tunisian families. Int. J. Gynecol. Pathol. 2011;30:348–353. doi: 10.1097/PGP.0b013e31820dc3b0. [DOI] [PubMed] [Google Scholar]
- 28.Huntriss J., Hinkins M., Oliver B., Harris S.E., Beazley J.C., Rutherford A.J., Gosden R.G., Lanzendorf S.E., Picton H.M. Expression of mRNAs for DNA methyltransferases and methyl-CpG-binding proteins in the human female germ line, preimplantation embryos, and embryonic stem cells. Mol. Reprod. Dev. 2004;67:323–336. doi: 10.1002/mrd.20030. [DOI] [PubMed] [Google Scholar]
- 29.Huntriss J., Hinkins M., Picton H.M. cDNA cloning and expression of the human NOBOX gene in oocytes and ovarian follicles. Mol. Hum. Reprod. 2006;12:283–289. doi: 10.1093/molehr/gal035. [DOI] [PubMed] [Google Scholar]
- 30.Huntriss J., Woodfine K., Huddleston J.E., Murrell A., Rutherford A.J., Elder K., Khan A.A., Hemmings K., Picton H. Quantitative analysis of DNA methylation of imprinted genes in single human blastocysts by pyrosequencing. Fertil. Steril. 2011;95:2564–2567. doi: 10.1016/j.fertnstert.2011.04.035. e1–e8. [DOI] [PubMed] [Google Scholar]
- 31.Newton H., Picton H., Gosden R.G. In vitro growth of oocyte-granulosa cell complexes isolated from cryopreserved ovine tissue. J. Reprod. Fertil. 1999;115:141–150. doi: 10.1530/jrf.0.1150141. [DOI] [PubMed] [Google Scholar]
- 32.Newton H., Aubard Y., Rutherford A., Sharma V., Gosden R. Low temperature storage and grafting of human ovarian tissue. Hum. Reprod. 1996;11:1487–1491. doi: 10.1093/oxfordjournals.humrep.a019423. [DOI] [PubMed] [Google Scholar]
- 33.Wang E., Miller L.D., Ohnmacht G.A., Liu E.T., Marincola F.M. High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q.T., Piotrowska K., Ciemerych M.A., Milenkovic L., Scott M.P., Davis R.W., Zernicka-Goetz M. A genome-wide study of gene activity reveals developmental signaling pathways in the preimplantation mouse embryo. Dev. Cell. 2004;6:133–144. doi: 10.1016/s1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- 35.Zhang P., Dixon M., Zucchelli M., Hambiliki F., Levkov L., Hovatta O., Kere J. Expression analysis of the NLRP gene family suggests a role in human preimplantation development. PLoS ONE. 2008;3:e2755. doi: 10.1371/journal.pone.0002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayward B.E., De Vos M., Judson H., Hodge D., Huntriss J., Picton H.M., Sheridan E., Bonthron D.T. Lack of involvement of known DNA methyltransferases in familial hydatidiform mole implies the involvement of other factors in establishment of imprinting in the human female germline. BMC Genet. 2003;4:2. doi: 10.1186/1471-2156-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohsugi M., Zheng P., Baibakov B., Li L., Dean J. Maternally derived FILIA-MATER complex localizes asymmetrically in cleavage-stage mouse embryos. Development. 2008;135:259–269. doi: 10.1242/dev.011445. [DOI] [PubMed] [Google Scholar]
- 38.Zheng P., Dean J. Oocyte-specific genes affect folliculogenesis, fertilization, and early development. Semin. Reprod. Med. 2007;25:243–251. doi: 10.1055/s-2007-980218. [DOI] [PubMed] [Google Scholar]
- 39.Pierre A., Gautier M., Callebaut I., Bontoux M., Jeanpierre E., Pontarotti P., Monget P. Atypical structure and phylogenomic evolution of the new eutherian oocyte- and embryo-expressed KHDC1/DPPA5/ECAT1/OOEP gene family. Genomics. 2007;90:583–594. doi: 10.1016/j.ygeno.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Grishin N.V. KH domain: one motif, two folds. Nucleic Acids Res. 2001;29:638–643. doi: 10.1093/nar/29.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitsui K., Tokuzawa Y., Itoh H., Segawa K., Murakami M., Takahashi K., Maruyama M., Maeda M., Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 42.Tian X., Pascal G., Fouchécourt S., Pontarotti P., Monget P. Gene birth, death, and divergence: the different scenarios of reproduction-related gene evolution. Biol. Reprod. 2009;80:616–621. doi: 10.1095/biolreprod.108.073684. [DOI] [PubMed] [Google Scholar]
- 43.Tong Z.B., Gold L., Pfeifer K.E., Dorward H., Lee E., Bondy C.A., Dean J., Nelson L.M. Mater, a maternal effect gene required for early embryonic development in mice. Nat. Genet. 2000;26:267–268. doi: 10.1038/81547. [DOI] [PubMed] [Google Scholar]
- 44.Tashiro F., Kanai-Azuma M., Miyazaki S., Kato M., Tanaka T., Toyoda S., Yamato E., Kawakami H., Miyazaki T., Miyazaki J. Maternal-effect gene Ces5/Ooep/Moep19/Floped is essential for oocyte cytoplasmic lattice formation and embryonic development at the maternal-zygotic stage transition. Genes Cells. 2010;15:813–828. doi: 10.1111/j.1365-2443.2010.01420.x. [DOI] [PubMed] [Google Scholar]
- 45.Zheng P., Dean J. Role of Filia, a maternal effect gene, in maintaining euploidy during cleavage-stage mouse embryogenesis. Proc. Natl. Acad. Sci. USA. 2009;106:7473–7478. doi: 10.1073/pnas.0900519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.