

Abstract
Differential expression of human leukocyte antigen C (HLA-C) allotypes is mediated by the binding of a microRNA, miR-148a, to the 3′ untranslated region of some, but not all, HLA-C alleles. The binding results in lower levels of HLA-C expression, which is associated with higher levels of HIV-1 viral load among infected individuals. The alternative set of HLA-C alleles has several substitutions in the miR-148a binding site that prevent binding and HLA-C downregulation; these high-expression alleles associate with control of HIV-1 viral load. We show that the common ancestor of all extant HLA-C alleles was suppressed by miR-148a. Substitutions that prevent miR-148a binding arose by a sequence exchange event between an HLA-C allele and an HLA-B (MIM 142830) allele of a B∗07-like lineage. The event occurred 3–5 million years ago, resulting in an HLA-C variant that escape from miR-148a downregulation. We present evidence suggesting that selection played a role in the successful spread of the HLA-C escape alleles, giving rise to 7 of the 14 extant HLA-C lineages. Notably, critical peptide and KIR binding residues of the escape variants have selectively converged to resemble the sequence of their inhibited counterparts, such that the inhibited and escape groupings differ primarily by their levels of expression.
Main Text
The human leukocyte antigen C (HLA-C [MIM 142840]) locus is distinct relative to the other classical HLA class I loci in that it has relatively limited polymorphism,1 lower expression on the cell surface,2,3 and more extensive ligand-receptor interactions with killer cell immunoglobulin-like receptors (KIR).4 These characteristics have led to the notion that HLA-C has a relatively limited role in antigen presentation to T cells during the acquired immune response, but rather has evolved to serve a primary role in the innate immune response as a ligand for KIR. A SNP 35 Kb upstream of HLA-C (NG_002397.2: g.38352T>C; rs9264942; termed −35) was shown to be associated with levels of HLA-C mRNA transcripts5,6 and cell-surface expression,7 which was suggested to be the basis for its association with control of HIV viral load and disease progression.5,7 More recently, variation in the 3′ UTR of HLA-C, which is in strong linkage disequilibrium (LD) with −35, was shown to affect HLA-C cell-surface expression through differential regulation of HLA-C alleles by an miRNA, miR-148a.8 HLA-C alleles that escape miR-148a recognition have a single-bp deletion at position 263 downstream of the stop codon of HLA-C (NM_002117.4: c.∗263delG; rs67384697, representing the deletion [263del]) along with other closely linked variants (NM_002117.4: c.∗259C>T; NM_002117.4: c.∗261T>C; NM_002117.4: c.∗266C>T) in the miR-148a binding site. Allotypes encoded by these 263del alleles are expressed at a relatively high level on the cell surface. The 3′ UTR of the alternative set of HLA-C alleles has an insertion at position 263 (rs67384697G, representing the insertion [263ins]) and an intact miR-148a binding site, and expression of both mRNA and protein encoded by these alleles is downregulated by the miRNA.
The miRNA regulation of HLA-C indicates the potential for control of immune responses through expression. It is therefore of interest to examine the evolutionary history of this process in HLA-C alleles, to determine how escape from miRNA initially occurred, and to investigate whether selection might have influenced the spread of the escape variant. Here, we provide evidence that the target site through which miR-148a exerts control of HLA-C existed in the most recent ancestor of all extant HLA-C alleles, but this sequence differed extensively in the ancestor of extant HLA-B alleles, all of which escape miR-148a regulation. Our data indicate that some 3–5 million years ago (MYA), a genetic exchange occurred between an HLA-C allele and an HLA-B (MIM 142830) allele belonging to a B∗07-like lineage, resulting in the conversion of a short 3′ UTR tract of an HLA-C allele by the paralogous HLA-B region. The conversion event encompassed the miRNA binding site and rendered the escape of this novel HLA-C allele from miR-148a downregulation. The conversion event did not encompass the coding region, so functionally, this novel HLA-C allele differed from its parental HLA-C allele only by having higher expression levels. The chromosome carrying this variant successfully spread through human or proto-human populations, giving rise to multiple HLA-C lineages of different binding and functional propensities, suggesting some benefit of high HLA-C expression. Furthermore, many key variants of the antigen binding and KIR receptor binding regions of the escape HLA-C alleles resemble those found in miR-148a inhibited alleles, suggesting that selective convergence occurred to generate an array of escape alleles that resemble the inhibited counterparts in critical regions.
Sequences of HLA-C were obtained from the survey of 9 consisted of about 4.5 kb covering HLA-C coding and flanking regions. Unless otherwise noted, phylogenetic reconstructions used here are based on these extended-length sequences. BLAST was used for searching DNA databases10 (and facilitating the exhaustive collection of homologous regions of HLA-C as well as great ape MHC-C and from HLA-B and great ape MHC-B). ClustalX11 was used for aligning the sequences. The BLAST-derived sequences are identified initially by accession codes and sequence descriptors. For descriptors that do not contain allelic identification, comparison with the IMGT/HLA database was used to identify and label sequences with appropriate HLA nomenclature. A sliding window (window length: 100 bp; step size: 25 bp) was used to calculate π across the entire alignment for each gene. The phylogeny of the sequences was reconstructed with MEGA 5.0 package12 and either neighbor-joining (NJ), maximum parsimony (MP), or maximum likelihood (ML) methods. For the NJ reconstruction, distances were estimated via Kimura's two-parameter method. A linearized ML tree of sequences based on a Tamura-Nei model generated by MEGA was used to date events following elimination of α1, α2, and a short divergent segment of the 3′UTR described below, which we refer to as the RHD (for region of high diversity).
HLA genotyping data from 30 diverse populations were generated previously.13 Worldwide HLA-C allele frequencies were also obtained from the International Histocompatibility Working Group (IHWG) anthropology section of the dbMHC database at NCBI.14 The Weblogo program15 was used to generate logograms of HLA-C polymorphic residues.
Several studies5,7,16 have identified genetic variants near, but outside of, the coding region of HLA-C that are markedly associated with outcomes to HIV infection. A sequencing survey9 indicates that a noncoding region at the 3′ end of the HLA-C alignment shows evidence of striking diversity among HLA-C alleles (Figure 1). In the short RHD segment of about 150 bp, which spans roughly 230–380 bp downstream of the stop codon, the nucleotide diversity in a collection of 45 distinct HLA-C alleles representing 13 distinct lineages reaches 0.08 substitutions per site. This is the highest level of diversity observed in the survey across 4.5 kb of aligned sequence spanning 982 bp 5′ to 688 bp 3′ of HLA-C. Given that full-length HLA-C alleles have a 3′ UTR of about 420 bp, this region of high diversity lies well within the 3′ UTR. This localized high diversity is not seen in the homologous 3′ UTR in a roughly comparable collection of HLA-B alleles where diversity reaches 0.01.
Figure 1.
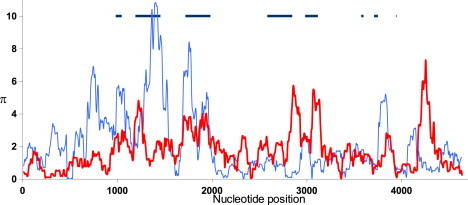
Nucleotide Diversity, π in HLA Genes
The ordinate shows π (%) measured in 100 bp windows across 4.6 kb of aligned sequences consisting of 45 HLA-C (red line) and 19 HLA-B (blue line) sequences. The horizontal axis indicates the nucleotide position in the alignment. The approximate positions and sizes of protein-coding regions are indicated by horizontal dark lines.
Closer examination reveals that the HLA-C alleles fall into two groups across the RHD in the 3′ UTR. HLA-C alleles are classified into 14 distinct lineages (represented by the initial two digits of the name). Alleles belonging to the C∗02, C∗05, C∗06, C∗08, C∗12, C∗15 and C∗16 lineages are nearly identical throughout the RHD from positions 230–380 and differ in at least ten positions from alleles of the C∗01, C∗03, C∗04, C∗07, C∗14 and C∗17 lineages. The former group of alleles is characterized by the deletion of position 263 in the 3′ UTR (263del) and neighboring substitutions that prevent miR-148a recognition, and we refer to these as “escape alleles.” The latter contain an intact miR-148a binding site and are therefore termed “inhibited” alleles. Some alleles of the inhibited group differ slightly from each other within the 3′ UTR RHD, but this accounts for very little of the overall diversity in this region and has no effect on miR-148a regulation.8 The primary cause of the high 3′ UTR diversity seen in Figure 1 is the differentiation of the HLA-C alleles into the two distinctive groups.
In order to determine whether the HLA-C escape and inhibited allelic groups remain distinct outside of the RHD, we made phylogenetic reconstructions for available complete HLA-C sequences covering about 4.5 kb.9 In addition to this HLA-C sequence data, we ran BLAST searches to find homologous full-length HLA-B17 and great ape MHC-B and MHC-C sequences for use in our alignment and phylogenetic analyses. To avoid effects of the RHD on our reconstruction, we removed this segment from the analysis. The tree of MHC-B and MHC-C sequences (Figure 2) indicates that both genes form separate clusters, where the escape and inhibited HLA-C groups cluster together and are distinct (100% bootstrap support) from HLA-B and great ape MHC-B sequences. Chimpanzee (Patr-C) and gorilla (Gogo-C) MHC-C alleles, all of which contain the miR-148a binding site, group with the inhibited and escape HLA-C alleles, separate from HLA-B (Figure 2). The MHC-C∗07 lineage shows evidence for trans-specific polymorphism between human and chimpanzee, indicating that it is sufficiently old to be shared with the nonhuman primate species. On the other hand, the HLA-C alleles of the escape group appear to be human specific. Furthermore, the escape alleles form a well-supported monophyletic cluster (97% bootstrap support) in this analysis that excludes the RHD. The monophyletic clustering of escape alleles indicates that the marked divergence seen in the RHD of extant HLA-C alleles (Figure 1) might be explained by an event that occurred in the common ancestor of the escape group. Although sequence information for the RHD region of the 3′ UTR of the HLA-C∗18 lineage is unavailable, all HLA-C∗18 alleles group closely to HLA-C∗04 alleles in phylogenetic analysis of the coding region (bootstrap support 74%, tree not shown), and we consider it likely that like the C∗04 lineage, it belongs to the miR-148a inhibited group.
Figure 2.
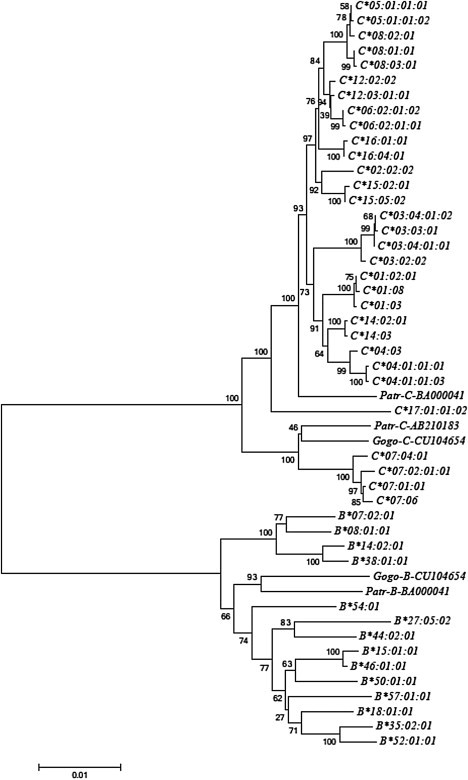
Neighbor-Joining Tree of Full-Length HLA-C and HLA-B over 4.5 kb of Aligned Sequence
The region of high diversity (152 bp) was excluded from the analysis, and pairwise elimination of indels was used. Numbers on nodes indicate the bootstrap recovery of that node in 500 replications, and the scale bar indicates the number of substitutions along a given branch length.
Next, we considered the relationship of the HLA-C RHD to the homologous regions of genes closely related to HLA-C. Initial analyses indicated a substantially higher similarity of the RHD of escape alleles to the homologous region of HLA-B, rather than HLA-A (MIM 142800), HLA-G (MIM 142871) or HLA-H (MIM 235200) (the homologous region appears absent in HLA-E [MIM 143010] and HLA- F [MIM 143110]) (data not shown). We used BLAST to identify 50 HLA-B sequence submissions containing data for the region homologous to the RHD and covering 24 of the 36 known HLA-B lineages. Alignment of the RHD region of the 3′ UTR of HLA-C and HLA-B alleles (Figure 3) reveals that alleles of the escape group are strikingly similar to the HLA-B alleles in precisely those sites that differentiate escape from inhibited HLA-C alleles. Of the ten variant positions that differentiate escape from inhibited groups in the 152 bp 3′ UTR RHD (positions 259, 261, 263, 266, 294, 299, 300, 307, 335, and 336), up to nine are shared by the escape alleles and certain HLA-B alleles, and at least eight are shared by all escape and HLA-B alleles surveyed. Whereas the HLA-C escape and inhibited alleles differ markedly in this region, alleles representing seven of the 24 HLA-B lineages in our survey (the HLA-B∗07, HLA-B∗08, HLA-B∗40, HLA-B∗41, HLA-B∗48, HLA-B∗50, and HLA-B∗58 lineages) appear quite similar to alleles of the HLA-C escape group, differing only by a T>C substitution at position 266 and exclusively sharing a C>T substitution at position 261 with HLA-C escape alleles. When considered across the entire HLA-B sequence, some of these seven HLA-B lineages are relatively distinct and fall into four separate clades in phylogenetic reconstruction (data not shown). However, toward the 3′ end of the gene, the separate clades come together, sharing sequence features with the clade containing HLA-B∗07 and HLA-B∗08. We refer to these seven lineages as HLA-B∗07-like lineages. All other HLA-B alleles that we examined also show high similarity to the HLA-C escape alleles, including the deletion at position 263 within the miR-148a seed region. On the other hand, the inhibited HLA-C alleles are quite distinctive in the 3′ UTR and appear to have no similar counterparts among the HLA-B alleles for which sequence is available.
Figure 3.

Sequence Alignment of HLA-C and HLA-B Alleles in the 3′ UTR Region of High Diversity
For simplicity, identical sequences are reduced to a single representative. A dot indicates identity with the majority consensus, and a dash indicates a deletion. Sites are numbered according to distance from the stop codon of HLA-C. Nucleotides within the binding site of miR-148a are boxed.
Phylogenetic analysis of HLA-B and HLA-C alleles using only the 152 bp RHD in the 3′ UTR strengthens this view. There is strong (99%) bootstrap support for the grouping of escape alleles in a clade containing all HLA-B alleles but excluding inhibited HLA-C alleles (Figure 4). The dendrogram also indicates that the closest relatives of HLA-C escape alleles in this region are the seven previously identified HLA-B∗07-like lineages, although the bootstrap support (52%) is not significant. The presence of chimpanzee and gorilla MHC-C alleles in the clade containing the inhibited HLA-C alleles indicates that the ancestral condition is probably represented by inhibited alleles and that the substitutions that enable escape occurred later. A parsimonious explanation both for the resemblance of the HLA-C escape to HLA-B alleles in the RHD region and for the monophyly of escape alleles outside of the RDH region is the occurrence of a single conversion event in the common ancestor of the HLA-C escape alleles, with the donor being a sequence related to the HLA-B∗07-like group. Given that the B∗07 allele is in strong linkage disequilibrium with the C∗07 allele, which is not converted and remains miR-148a inhibited, the conversion event probably occurred between chromosomal homologs rather than between chromatid pairs. The boundaries of the conversion event cannot be precisely determined because many residues in the 3′ UTR are shared by HLA-C and HLA-B. However, by observing where substitution sharing between HLA-B and HLA-C escape alleles begins and ends, limits to the conversion event can be set. The 5′ limits of the exchange occur between position 230, where a deletion distinguishes HLA-C alleles (both escape and inhibited) from HLA-B alleles where a G is found; and position 259, where HLA-C escape and HLA-B alleles share a T while a C is found in inhibited HLA-C alleles. Likewise, the 3′ limits of the event occur between position 346, where an A is shared by HLA-B and HLA-C escape alleles but a G is found in inhibited HLA-C alleles; and position 383, where a T is found in all HLA-B alleles and a G in all examined HLA-C alleles. These limits set a maximum size of the conversion event at 152 bp and a minimum size at 88 bp.
Figure 4.
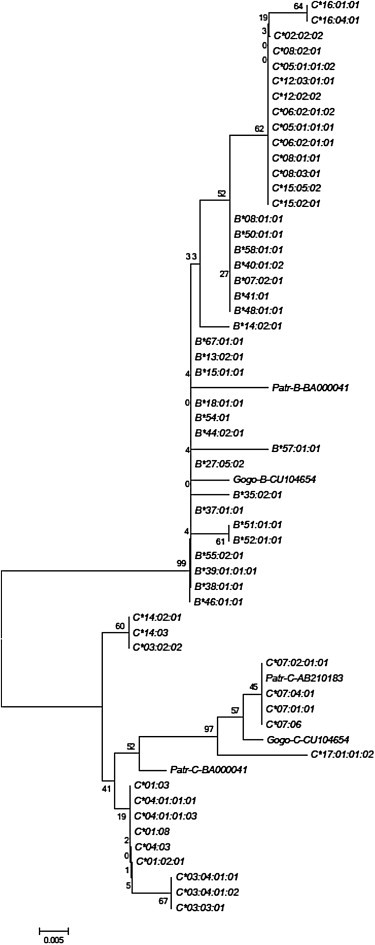
Neighbor-Joining Tree of Sequences of HLA-C and HLA-B for the 3′ UTR Region of High Diversity
Numbers on nodes indicate the bootstrap recovery of that node in 500 replications, and the scale bar indicates the number of substitutions along a given branch length. ML and MP trees have identical topology to the NJ tree.
To determine the time of occurrence of the conversion event, we generated a linearized ML tree of the MHC-C alleles (Figure 5). We eliminated sites likely to distort our date estimates (the α1, α2, and RHD regions). Taking the divergence of human and chimpanzee at 6 MYA19 and assuming that the most closely related pair of chimpanzee and human alleles approximate the speciation time (see20 for the rationale in estimating MHC divergence times), we estimate that the conversion event occurred about 3–5 MYA. This supports the notion that the event is specific to the human lineage. Examination of available nonhuman MHC-C alleles agrees with this interpretation because no nonhuman sequences resembling escape alleles in their 3′ UTR region have yet been found.
Figure 5.
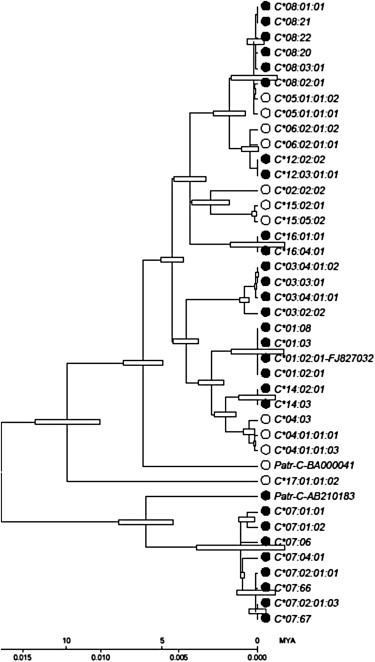
Linearized ML Tree of HLA-C Alleles, Region of High Diversity Excluded
The scale bar indicates the number of substitutions (below) and time (MY) for a given length of the tree. The time estimate assumes that chimpanzee (specifically Patr-AB210183) and human HLA-C∗07 alleles diverged as the species diverged about 6 MYA. The tree (log likelihood = −7378) is based on 3622 shared nucleotides after elimination of indels and the α1, α2, and RHD domains. Filled or open circles next to sequence names indicate, respectively, C1- or C2-type KIR-binding sites. Open horizontal bars represent the standard error estimate of the time of divergence of that node.
The conversion event that generated the escape alleles presumably occurred within a single allele, which diversified into the seven lineages of HLA-C escape alleles found today. We used the IHWG anthropology data in dbMHC to examine worldwide frequencies of HLA-C alleles on 14863 chromosomes typed in various populations. In this large survey, 81 different alleles occur, representing all 14 major lineages classified into 58 distinct sublineages. The frequency of escape alleles in worldwide populations is high, constituting an estimated 32.8% of HLA-C alleles worldwide. We further examined the frequency of escape alleles by using the survey of HLA-C allele frequencies13 from 30 diverse populations. Escape alleles were common (>20% frequency) in large population groups (Figure 6). No population showed extremely high (>70%) escape allele frequency, but a few isolated populations show lower frequencies of escape alleles. Among the 30 populations typed, only the Surui, Maya, and Micronesians have escape allele frequencies of less than 10%, but it is likely in these cases that low frequencies reflect drift or founder effects, because these populations show high homozygosity and low allelic diversity for HLA-C.
Figure 6.
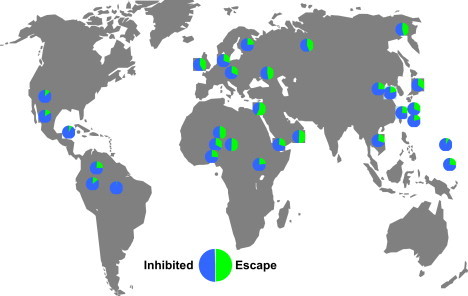
Worldwide Distribution of HLA-C Alleles According to miR-148a Binding Propensity
The alleles of 1480 individuals from 30 populations typed at HLA-C as described in13 are grouped by miR-148a binding propensity as escape or inhibited alleles.
Many polymorphisms and many of the functional motifs of HLA-C alleles occur in both escape and inhibited lineages. For example, the KIR ligand C1/C2 dichotomy is defined by the amino acid residues at positions 77 and 80 of the mature HLA-C protein18 (S77 with N80 defines C1, and N77 with K80 defines C2), and both C1 and C2 forms are found within the group of escape alleles, as well as in the group of inhibited alleles (Figure 5). We conclude that the C1/C2 dichotomy has arisen independently in both escape and inhibited lineages and that in escape lineages, the dichotomy has arisen within the last 3–5MY. Within the escape group, the frequency of C1 alleles (S77/N80) is 44%, whereas that of C2 (N77/K80) is 56%. The functional importance and, more importantly, the maintenance of these polymorphisms, located at KIR interaction sites of the HLA-C molecules, strongly suggest that selection plays a role in ensuring the functional diversity of alleles of the escape and inhibited groups.
Four clades of HLA-C alleles can be identified on the tree of HLA-C alleles (Figure 2). The splits leading to the C∗07 and the C∗17 lineages occur earlier than the split leading to the remaining inhibited and escape lineages. Thus, three groupings of inhibited alleles can be defined: (1) the C∗07 alleles; (2) the C∗17 alleles; and (3) the C∗01, C∗03, C∗04, and C∗14 alleles. We refer to the latter group as C∗X. The escape allelic lineages form a fourth group. We examined patterns of amino acid polymorphism by determining sequence logos for all unique alleles within each of the four defined groupings. Because we are interested in residues that characterize each lineage, we limit the analysis to these 30 sites that are the most polymorphic across the α1 and α2 domains of HLA-C. Figure 7 indicates the prevalence of amino acids in these most polymorphic positions. The extent of polymorphisms in the C∗07 and C∗17 lineages is limited. However, the sharing of polymorphisms between the inhibited C∗X and the escape lineages is striking. Polymorphisms at positions 9, 21, 24, 66, 73, 77, 80, and 90 of the α-1 domain as well as positions 94, 95, 97, 114, 116, 152, 156, and 163 of the α-2 domain are shared by both C∗X and escape groups. Within and flanking the KIR binding sites, polymorphisms at residues 73,77, 80, and 90 involve T/A, S/N, N/K, and D/A in both escape and inhibited lineages. The R/H and A/S polymorphisms of residues 21 and 24 are also seen in both lineage types. Additional shared polymorphisms are scattered along the peptide, as are sites that clearly differentiate lineage groups (e.g., S11 is found only in the C∗X group, H113 is limited to escape lineages). The extent of sharing shows that escape alleles have acquired many of the polymorphisms that are present in inhibited HLA-C lineages. Phylogenetic trees of HLA-C alleles based on the amino acid distances found for these 30 variable sites also contain clades consisting of both escape and inhibited lineages (not shown), indicating the close proximity of certain escape and inhibited lineages when measured at critical polymorphic sites. Both the high frequency of escape alleles and their extensive utilization of ancient polymorphisms in different lineages support the view that selection has been instrumental in the maintenance and diversification of escape lineages.
Figure 7.

Logogram of the Most Polymorphic Residues
The horizontal axis indicates the position of residues in the mature peptide of unique sequences of four groups of HLA-C alleles. The single-letter code is used to indicate amino acids, and colors indicate the physicochemical properties as polar (green), basic (blue), acidic (red), or hydrophobic (black). The height of the letter indicates the relative abundance of each amino acid at that position.
The data presented herein indicate that a single gene-conversion event occurring in the 3′ UTR of the HLA-C locus 3–5 MYA resulted in a lineage of HLA-C alleles that escape miRNA downregulation. Two observations indicate that the gene-conversion event leading to the escape lineage of HLA-C alleles almost certainly occurred only once. First, the escape feature is highly characteristic, involving a large segment of somewhere between 88 bp and 152 bp with multiple polymorphic sites that are shared between the escape HLA-C alleles and HLA-B, but which distinguish them from the inhibited HLA-C alleles. By this reasoning, the escape feature can be likened to a cladistic marker for which homoplasy is unlikely.21 Second, all extant HLA-C alleles containing the escape variant group monophyletically with high bootstrap support. The conversion event that led to escape alleles thus shows complete congruence with the phylogeny of the alleles that bear it, and descent from a single common ancestor provides the simplest explanation for the shared escape mechanism (Figure 2). Taken together, these data indicate that it is very unlikely and unnecessary to postulate that the same gene-conversion event would occur a second time involving the same ancestral HLA-C allele.
The escape HLA-C alleles are expressed at significantly higher levels on the cell surface than are those that are inhibited by miR-148a. These high-expression HLA-C alleles have accumulated a substantial number of functional coding-region variants that are also present in the group of inhibited HLA-C alleles that contain an intact miR-148a binding site, the ancestral form of the 3′ UTR. Among the shared variations are features involved in binding of KIR receptors as well as peptide-binding regions. The convergent evolution evident in the escape versus inhibited lineages provides a strong argument in favor of selection for the high-expression HLA-C allotypes, which must maintain the functional polymorphisms characteristic of low-expression HLA-C allotypes. High-expression HLA-C alleles are associated with control of HIV viral load and slower disease progression.7,8 It is possible that these same alleles have conferred some level of protection against infectious diseases throughout human history, resulting in an overall frequency in excess of 30% worldwide and their presence in every population sampled in this study. Alternatively, we presume that the inhibited alleles also provide benefit, as they have been maintained over the past several million years since the escape allele first arose.
The variation in miR-148a binding is unique to HLA-C as compared to HLA-B, where all alleles escape miR-148a binding, and HLA-A, where all alleles have an intact miR-148a binding site. This dimorphic feature of HLA-C adds to other unique characteristics of HLA-C, including relatively limited polymorphism and more extensive ligand-receptor interactions with KIR. Thus, the function of HLA-C alleles is dependent not only on their peptide specificities, but also on their expression levels as determined by polymorphic regulatory elements outside of the protein-coding region. This may be true for HLA-A and –B, as well, but through mechanisms that do not involve miR-148a. Efforts are being made to incorporate a more comprehensive view of HLA variability by considering such regions.9,17 A more inclusive view of HLA variability might be facilitated by extending the sequence length in the IMGT/HLA database to include sequences such as the 3′ UTR. Certainly, a greater understanding of the nature of the diversity at the HLA class I loci and the mechanisms by which the diversity occurs, as described herein, may provide clues to its involvement in health and disease.
Acknowledgments
We would like to thank members of the Carrington group and Ram Savan for informative discussion. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Web Resources
The URLs for data presented herein are as follows:
dbMHC, Diversity/Anthropology Component, http://www.ncbi.nlm.nih.gov/gv/mhc/ihwg.cgi?cmd=page&page=AnthroMain
WebLogo, http://weblogo.berkeley.edu/
References
- 1.Zemmour J., Parham P. Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C. J. Exp. Med. 1992;176:937–950. doi: 10.1084/jem.176.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCutcheon J.A., Gumperz J., Smith K.D., Lutz C.T., Parham P. Low HLA-C expression at cell surfaces correlates with increased turnover of heavy chain mRNA. J. Exp. Med. 1995;181:2085–2095. doi: 10.1084/jem.181.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snary D., Barnstable C.J., Bodmer W.F., Crumpton M.J. Molecular structure of human histocompatibility antigens: the HLA-C series. Eur. J. Immunol. 1977;7:580–585. doi: 10.1002/eji.1830070816. [DOI] [PubMed] [Google Scholar]
- 4.Bashirova A.A., Martin M.P., McVicar D.W., Carrington M. The killer immunoglobulin-like receptor gene cluster: tuning the genome for defense. Annu. Rev. Genomics Hum. Genet. 2006;7:277–300. doi: 10.1146/annurev.genom.7.080505.115726. [DOI] [PubMed] [Google Scholar]
- 5.Fellay J., Shianna K.V., Ge D., Colombo S., Ledergerber B., Weale M., Zhang K., Gumbs C., Castagna A., Cossarizza A. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stranger B.E., Forrest M.S., Clark A.G., Minichiello M.J., Deutsch S., Lyle R., Hunt S., Kahl B., Antonarakis S.E., Tavaré S. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas R., Apps R., Qi Y., Gao X., Male V., O'hUigin C., O'Connor G., Ge D., Fellay J., Martin J.N. HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 2009;41:1290–1294. doi: 10.1038/ng.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkarni S., Savan R., Qi Y., Gao X., Yuki Y., Bass S.E., Martin M.P., Hunt P., Deeks S.G., Telenti A. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;28:295–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y., Deng Z., O'hUigin C., Wang D., Gao S., Zeng J., Yang B., Jin S., Zou H. Characterization and polymorphic analysis of 4.5 kb genomic full-length HLA-C in the Chinese Han population. Tissue Antigens. 2011;78:102–114. doi: 10.1111/j.1399-0039.2011.01710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 11.Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011 doi: 10.1093/molbev/msr121. Published online May 4, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Single R.M., Martin M.P., Gao X., Meyer D., Yeager M., Kidd J.R., Kidd K.K., Carrington M. Global diversity and evidence for coevolution of KIR and HLA. Nat. Genet. 2007;39:1114–1119. doi: 10.1038/ng2077. [DOI] [PubMed] [Google Scholar]
- 14.Meyer D., Singe R.M., Mack S.J., Lancaster A., Nelson M.P., Erlich H., Fernandez-Vina M., Thomson G. Single Locus Polymorphism of Classical HLA Genes. In: Hansen J.A., editor. Vol. 1. IHWG Press; Seattle, WA: 2007. pp. 653–704. (Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatability Workshop and Conference). [Google Scholar]
- 15.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pereyra F., Jia X., McLaren P.J., Telenti A., de Bakker P.I., Walker B.D., Ripke S., Brumme C.J., Pulit S.L., Carrington M., International HIV Controllers Study The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y., Deng Z., Zou H., Wang D., He L., Wei T. Cloning and sequencing HLA-A and -B genomic DNA and analyzing polymorphism in regulatory regions in Chinese Han individuals. Yi Chuan. 2010;32:685–693. doi: 10.3724/sp.j.1005.2010.00685. [DOI] [PubMed] [Google Scholar]
- 18.Moretta A., Bottino C., Vitale M., Pende D., Biassoni R., Mingari M., Moretta L. Receptors for HLA class-I molecules in human natural killer cells. Ann. Rev. Immunol. 1996;14:619–648. doi: 10.1146/annurev.immunol.14.1.619. [DOI] [PubMed] [Google Scholar]
- 19.Glazko G.V., Nei M. Estimation of divergence times for major lineages of primate species. Mol. Biol. Evol. 2003;20:424–434. doi: 10.1093/molbev/msg050. [DOI] [PubMed] [Google Scholar]
- 20.Satta Y., O'hUigin C., Takahata N., Klein J. The synonymous substitution rate of the major histocompatibility complex loci in primates. Proc. Natl. Acad. Sci. USA. 1993;90:7480–7484. doi: 10.1073/pnas.90.16.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriener K., O'hUigin C., Klein J. Alu elements support independent origin of prosimian, platyrrhine, and catarrhine Mhc-DRB genes. Genome Res. 2000;10:634–643. doi: 10.1101/gr.10.5.634. [DOI] [PubMed] [Google Scholar]