

Abstract
Anterior segment dysgenesis describes a group of heterogeneous developmental disorders that affect the anterior chamber of the eye and are associated with an increased risk of glaucoma. Here, we report homozygous mutations in peroxidasin (PXDN) in two consanguineous Pakistani families with congenital cataract-microcornea with mild to moderate corneal opacity and in a consanguineous Cambodian family with developmental glaucoma and severe corneal opacification. These results highlight the diverse ocular phenotypes caused by PXDN mutations, which are likely due to differences in genetic background and environmental factors. Peroxidasin is an extracellular matrix-associated protein with peroxidase catalytic activity, and we confirmed localization of the protein to the cornea and lens epithelial layers. Our findings imply that peroxidasin is essential for normal development of the anterior chamber of the eye, where it may have a structural role in supporting cornea and lens architecture as well as an enzymatic role as an antioxidant enzyme in protecting the lens, trabecular meshwork, and cornea against oxidative damage.
Main Text
Anterior segment dysgenesis (ASD) describes a group of ocular developmental disorders affecting the anterior chamber structures behind the cornea and in front of the lens, including the iris, trabecular meshwork, and ciliary body.1,2 These abnormalities can give rise to elevated intraocular pressure through obstruction of the trabecular meshwork drainage channels, increasing the risk of developing glaucoma.3 Examples of this phenotype include Axenfeld-Rieger syndrome due to dominant mutations in FOXC1 (MIM 601090)4,5 or PITX2 (MIM 601542),6,7 isolated aniridia caused by dominant mutations in PAX6 (MIM 607108),8,9 isolated trabeculodysgenesis resulting from recessive mutations in CYP1B1 (MIM 601771),10 and megalocornea associated with microspherophakia caused by recessive mutations in LTBP2 (MIM 602091).11,12 Some anterior segment anomalies are also associated with cataracts, because during development the anterior lens secretes factors that induce the differentiation of the cornea and trabecular meshwork.13,14 Examples of corneal opacity or microcornea with cataract include dominant mutations in CRYAA (MIM 123580),15 CRYBA4 (MIM 123631),16 CRYBB1 (MIM 600929),17 CRYBB2 (MIM 123620),18 CRYGC (MIM 123680),19 CRYGD (MIM 123690),15 GJA8 (MIM 600897),15,20–22 MAF (MIM 177075),15,23–25 or FOXE3 (MIM 601094)26–28 and a recessive mutation in CRYAA.29 These phenotypes may be expressed asymmetrically and can be highly variable, even between affected family members, because of genetic background and environmental factors.
We recently described genetic heterogeneity for recessively inherited congenital cataract-microcornea with corneal opacity (CCMCO) in three unrelated consanguineous families, MEP57, MEP60 and MEP68, from the Punjab province of Pakistan.30 We have since ascertained one additional Pakistani family, MEP59, with a history of poor vision that fits the description for CCMCO (Figure 1 and Table 1). Microcornea and corneal opacification, due to the sclera encroaching on the cornea, were the defining features for these families, with variable degrees of expression between affected members of the same family. Concurrently, through an epidemiological survey of blind schools in Cambodia,31 we also identified a consanguineous family, CA2, in which four affected individuals had severe developmental glaucoma associated with extensive corneal opacification and buphthalmos (Figure 2 and Table 1). In all the families described in this study, the patients did not have any systemic problems, or any neurological abnormalities, and the unaffected family members did not show any eye defects. The purpose of the current study was to identify the pathogenic mutations responsible for anterior segment dysgenesis in these families.
Figure 1.
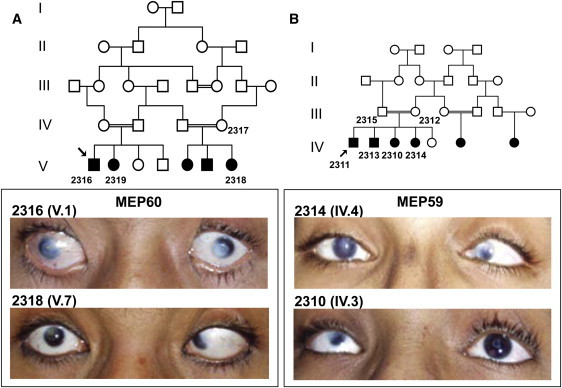
Clinical Features of the CCMCO Patients
(A and B) Pedigrees for MEP60 (A) and MEP59 (B). Individuals from whom DNA is available are numbered. Photos of the anterior segment of affected family members 2316 (V.1, aged 16 years) and 2318 (V.7, aged 8 years) from MEP60 as well as family members 2314 (IV.4, aged 18 years) and 2310 (IV.3, aged 14 years) from MEP59 are also shown. The left eye of 2310 (IV.3) is aphakic, and capsular condensation, the white ring that is visible, is present. Note variable central or peripheral corneal opacifications present in all cases, though most eyes have a well-defined corneoscleral junction. Strabismus in the amblyopic eye is also apparent. The clinical descriptions are presented in Table 1.
Table 1.
Clinical Features of Patients with Homozygous PXDN Mutations
|
MEP60 |
MEP59 |
CA2 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2316 (V.1) | 2318 (V.7) | 2319 (V.2) | 2310 (IV.3) | 2311 (IV.1) | 2313 (IV.2) | 2314 (IV.4) | C (IV.3) | H (IV.8) | J (IV.10) | K (IV.11) | |
| Cornea | |||||||||||
| Corneal diameter OD;OS (mm) | 7; 7 | 7; 8 | smalla | 5; 9 | smalla | smalla | 8; 8 | largea | largea | smalla | largea |
| Vascularisation | − | − | − | − | − | − | − | ++ | ++ | ++ | ++ |
| Opacity | p>c | p>c | p>c | p; OD>OS | p | p | p | ++ | ++ | ++ | ++ |
| Central corneal thickness OD;OS (μm) | ND | ND | ND | ND | ND | ND | ND | 175;455 | a;a | a;828 | 332;277 |
| Globe | |||||||||||
| Microphthalmia | − | − | − | ? | ? | ? | ? | − | − | − | − |
| Buphthalmos | − | − | − | − | − | − | − | + | + | − | + |
| Intraocular pressure OD;OS (mm Hg) | ND | ND | ND | ND | ND | ND | ND | 45;37 | 19;8 | 30;19 | 25;10 |
| Iris | |||||||||||
| Irido-corneal adhesion | p | p | p | − | − | − | − | no view | no view | no view | no view |
| Iris Coloboma | − | − | − | − | − | − | − | no view | no view | no view | no view |
| Irido-lenticular adhesions | − | − | − | − | − | − | − | no view | no view | no view | no view |
| Iris hypoplasia | − | − | − | − | − | − | − | no view | no view | no view | no view |
| Lens | |||||||||||
| Kerato-lenticular touch | − | − | − | − | − | − | − | no view | no view | no view | no view |
| Cataract | + | + | + | + | + | + | + | no view | no view | no view | no view |
| Function | |||||||||||
| Nystagmus | − | − | − | − | − | − | − | ? | ? | ? | ? |
| Vision | CF | CF | CF | CFb | 6/60 | 6/60 | 6/19b | NPL/NPL | PL/NPL | NPL/PL | PL/PL |
All patients in families MEP60 and MEP59 had a normal fundal exam; no view of the fundus could be obtained of any affected members of CA2. The meanings of the symbols used in the table are as follows; +, present; ++, present in a severe form; −, absent; p, peripheral; c, central; OD, right eye; OS, left eye; ND, not done; CF, counting fingers; NPL, no perception of light; PL, perception of light.
Exact measurement unavailable.
With aphakic correction.
Figure 2.
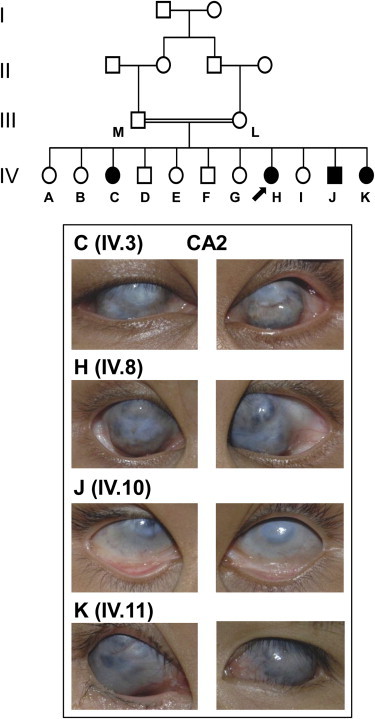
Clinical Features in Buphthalmic Patients
Pedigree for CA2. Individuals from whom DNA is available are depicted by a letter. Photos of the anterior segment of affected family members C (IV.3, aged 25 years), H (IV.8, aged 15 years), J (IV.10, aged 10 years), and K (IV.11, aged 8 years) are shown. Note that gross buphthalmos prevented accurate measurement of corneal diameter and that severe corneal opacification prevented both visualization of the anterior chamber or acquisition of accurate biometric data, with the exception of corneal thickness. The clinical descriptions are presented in Table 1.
This study was performed via a process approved by the Pakistan Medical Foundation, Lahore; the Leeds (East) Research Ethics Committee; the Royal Adelaide Hospital Research Ethics Committee; the Southern Adelaide Clinical Human Research Ethics Committee; and the National Ethics Committee for Human Research, Cambodia. Participants gave informed consent in accordance with the tenets of the Declaration of Helsinki. In all families studied herein the ocular abnormalities either were severe enough to be noted at birth or became apparent within the first year of life and did not progress thereafter. In the Pakistani cases, the anterior chambers were of near-normal depth with no obvious anterior capsular abnormality, though where clinically visible, the lenses were cataractous but with normal contour and size. Affected individuals had a normal fundus appearance observed through examination with an indirect ophthalmoscope. Peripheral blood was obtained and genomic DNA extracted according to standard procedures. In the Cambodian family, three patients (C, H, and K [IV.3, IV.8, and IV.11 in Figure 2]) displayed bilateral buphthalmos and severe opacification and vascularization of the cornea, with the fourth affected individual (J [IV.10 in Figure 2]) displaying severe corneal opacification. In three patients (C, J, and K [IV.3, IV.10, and IV.11 in Figure 2]), intraocular pressure, measured by iCare tonometry, was markedly elevated. The extent of corneal opacification prevented visualization of the anterior chamber or fundus and whether cataracts were present in all affected patients. Saliva samples were collected in OrageneDNA Collection Kits (DNA Genotek) for extraction according the manufacturer's protocol.
Homozygosity mapping and linkage analysis in Pakistani family MEP60 implicated two shared homozygous regions in all the affected members.30 The pathogenic mutation mapped within either a 6.7 Mb (14.1 cM) interval extending from 2pter to microsatellite marker D2S281, which contained 20 genes, or a 3.8 Mb (7.5 cM) region on chromosome 20 spanning D20S906 (1.5 Mb) to D20S835 (5.3 Mb) and containing 55 genes.30 Given the lack of any obvious candidate genes, we prepared a customized liquid-phase SureSelect Target Enrichment biotinylated cRNA bait, using the Agilent Technologies eArray platform. This reagent was designed against all coding exons and 20 bp regions flanking the exons from Refseq genes within the critical intervals on chromosomes 2 and 20. After merging of overlapping exons, 810 of 814 regions amounting to 135 kb DNA nucleotide sequence were covered, with only 120 bp missing due to homology with repetitive sequences. We then sheared 3 μg DNA from patient 2316 (V.1 in Figure 1A) by sonication, before ligation onto Illumina paired-end adapters according to standard procedures. This mixture was hybridized against the customized baits and eluted to capture the enriched DNA library, which was sequenced for 80 bp single-end reads on the Illumina Genome Analyzer GAIIx (Illumina).
The raw data files were processed by the Illumina pipeline (version 1.3.4) and the sequencing reads were aligned to the human reference sequence (hg19/GRCh37) with Novoalign software (Novocraft Technologies). Alignments were processed in the SAM/BAM format32 with Picard and the Genome Analysis Toolkit (GATK)33 Java programs in order to correct alignments around indel sites and mark potential PCR duplicates. The mean depth of coverage for the targeted region was 242 reads; however, after duplicate removal the mean depth amounted to 79 reads, with minimum mapping and base phred quality scores of 20 and 17, respectively. Of the targeted bases, 92% were covered by at least five reads matching these criteria. Variants for the coding regions and the 20 bp flanking regions were reported in the VCF format through the use of the Unified Genotyper and DINDEL34 functions of GATK before filtering against variants recorded in dbSNP131. A total of 125 variants were detected, 118 of which were identified in dbSNP131. This left two intronic (not splice site) variants, four nonsynonymous variants, and a deletion, of which only two were unique to MEP60 and absent from seven unrelated DNA samples that were run in a parallel experiment (Table S1 available online); a homozygous G-to-C nucleotide change in the coding sequence of TGM6 (transglutaminase 6 [MIM 613900], NM_198994.2) (on chromosome 20) that substitutes a serine for an arginine residue (c.1995G>C [p.Arg665Ser]) and a homozygous deletion of a C nucleotide in the coding sequence of PXDN (peroxidasin [MIM 605158], NM_012293.1) (on chromosome 2) that creates a frameshift and premature truncation of the protein (c.2568delC [p.Cys857AlafsX5]). The functional impact of the TGM6 variant was assessed with the PolyPhen algorithm. With the use of a HumVar model, a score of 0.021 predicted that the TGM6 variant was benign. It was therefore considered likely that the truncating PXDN mutation was pathogenic. Presence of this mutation and segregation with CCMCO in family MEP60 (Figure 1A) was confirmed by direct sequencing of exon 17 of PXDN with oligonucleotide primer 17BF on an ABI3130xl Genetic Analyzer (Figure 3A), and this change was excluded from 170 ethnically matched control DNA samples (340 chromosomes). Primer pairs used in the PCR amplification and sequencing are depicted in Table S2.
Figure 3.
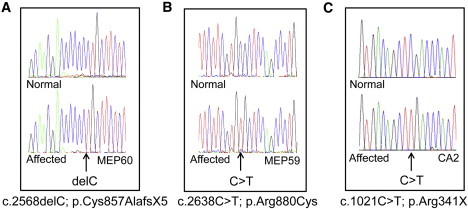
Genetic Analysis of CCMCO and Buphthalmic Patients
(A–C) Sequence chromatograms highlighting the PXDN mutation in affected patients from MEP60 (A), MEP59 (B), and CA2 (C) are shown against the wild-type sequence from a healthy individual.
Genetic analysis in two other Pakistani families who fit the diagnostic criteria for CCMCO, MEP57 and MEP68, failed to identify linkage to the 2pter interval harboring PXDN.30 However, another such family, MEP59, was linked to this region (Figure S1). Direct sequencing of PXDN in affected member 2311 (IV.1 in Figure 1B) identified a missense change in exon 17 that changed an arginine residue to a cysteine (c.2638C>T [p.Arg880Cys]) (Figure 3B). This variant scored 0.979 with PolyPhen under the HumVar model, suggesting that it is likely to be functionally damaging. Furthermore, the mutation changes a conserved arginine residue within the peroxidase domain and is likely to affect the three-dimensional structure of the protein because the new cysteine is within the atomic range for interacting with one of the cysteines in the native disulfide bond (Figure 4). This mutation was predicted to disrupt the enzymatic catalytic site and is therefore likely to be pathogenic. The mutation segregated with the disease in MEP59 (Figure 1B) as expected for recessive inheritance, following direct sequencing of exon 17 with oligonucleotide primer 17BF, and was absent from 340 ethnically matched chromosomes.
Figure 4.
Molecular Analysis of PXDN
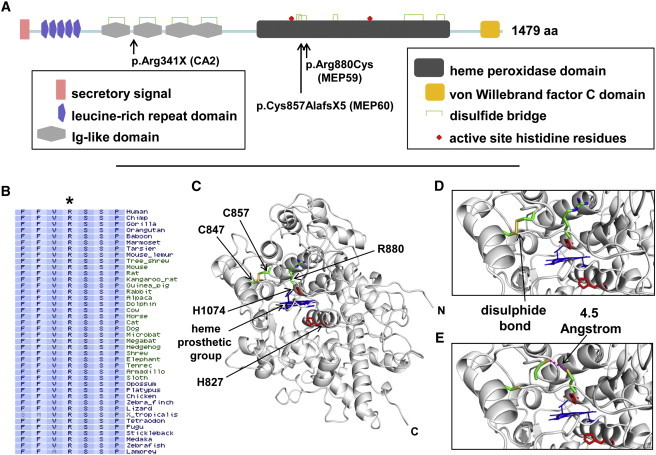
(A) Diagrammatic representation of PXDN. The protein sequence (NP_036425.1) was analyzed with the SMART program. The position of the domains in the protein, the disulfide bridges, and the active site histidine residues are shown. The positions of the mutations relative to the full-length 1479 amino acid protein are also highlighted.
(B, C, D, and E). Analysis of the PXDN missense mutation in MEP59. (B) Protein sequence conservation across mammalian and nonmammalian vertebrates using the Vertebrate Multiz Alignment and PhastCons Conservation package at the UCSC Genome Browser. The diagram shows the conserved arginine (R) residue in the normal sequence that is mutated to a cysteine in patients with CCMCO. (C) Ribbon representation of the peroxidase domain in PXDN. The structural model was derived by homology modeling using the M4T server.58,59 The bovine peroxidase (PDB 3q9k) and human myeloperoxidase (PDB 1d2v) were used as templates. Both templates and the peroxidase domain sequence shared more than 40% sequence identity, with over 90% coverage, thus ensuring structural similarity.60 The quality and stereochemistry of the model was assessed with ProSA-II and PROCHECK, and the figures were generated using PyMOL. The heme group is depicted in blue, and active site histidines (H824 and H1074) are in red. The disulfide bond between C847 and C857 is also shown. R880 (D) and C880 (E) are shown as stick representations and colored by atom type. Note that C880 is only 4.5 Angstroms away from C857, which is within atomic range for disulfide bond formation.
Independently of the above analysis, homozygosity mapping in the consanguineous Cambodian family CA2 also revealed linkage to 2pter (Figure S2). Each of the four affected siblings had a single region in common between the telomere and rs1821797 (5.7 Mb; 13.0 cM). Primers were designed for 218 amplicons covering the annotated exons and splice-site flanking regions within the linked interval (NCBI 36 and ENSEMBL release 54). PCR products from the proband were sequenced on an ABI3730xl DNA Analyzer according to standard protocols. The data were analyzed with Sequencher V4.09 (GeneCodes) by comparison with the reference sequence (NCBI/GenBank). In total, 110 variants were identified, 103 of which were recorded in dbSNP131. Of the seven variants remaining, only one mapped within an exon. This variant, in exon 10 of PXDN, was predicted to cause premature protein truncation and so is likely to be the pathogenic mutation in CA2 (c.1021C>T [p.Arg341X]) (Figure 3C). The mutation segregated with disease in family CA2 (Figure 2) following direct sequencing of exon 10 with oligonucleotide primer 10R and was absent from 58 ethnically matched control chromosomes.
The 23 exon human PXDN, also known as MG50 (melanoma-associated gene 50),35,36 KIAA0230,37 PRG2 (p53-responsive gene 2 protein)38 and VPO1 (vascular peroxidase 1),39 encodes a 1479 amino acid protein with a unique structure containing a mammalian peroxidase domain and motifs that are typically found in extracellular matrix proteins (Figure 4A). The peroxidase domain consists of a heme-binding cavity, critical catalytic residues, and a calcium binding site, similar to the known mammalian peroxidases myeloperoxidase (MPO), lactoperoxidase (LPO), eosinophil (EPX), and thyroid peroxidases (TPO).39 MPO, LPO, and EPX have a role in antimicrobial defense by oxidation of microbial proteins targeting them for degradation, and TPO is involved in catalyzing the iodination of thyroid hormones. The main catalytic function of mammalian peroxidasin is the metabolism of hydrogen peroxide, although the in vivo substrate for this reaction is currently unknown.39,40 The other motifs in peroxidasin consist of an amino-terminal secretory signal sequence, domains that are involved in ligand binding, including five leucine-rich repeat regions,41 four C2-type immunoglobulin-like motifs,42 and a carboxy-terminal von Willebrand factor type C domain. The latter contains multiple cysteine residues and is thought to be involved in forming large protein complexes with itself and other proteins containing the VWFC domain, such as thrombospondin and procollagen.43 It is interesting to note that although mammalian peroxidasin usually localizes to the endoplasmic reticulum, it is also secreted into the extracellular space following TGF-β-induced differentiation of fibroblasts into myofibroblasts, where it forms part of the fibril-like network with fibronectin as well as other extracellular matrix proteins.40 Caenorhabditis elegans peroxidasin (PXN-2) mutants have been shown to have aberrant extracellular matrix ultrastructure, in which muscles have detached from the epidermis, supporting a role for the normal protein in maintaining basement membrane integrity.44 It is also worth highlighting that siRNA knockdown of peroxidasin in cancer cells reduces the cells' ability to attach to laminin- and fibronectin-coated wells, further suggesting a role in cell adhesion to the extracellular matrix.45
Although the mammalian PXDN transcript is expressed in most tissues,38,40,46 the corresponding protein has only been shown to localize to the heart and its vascular wall.39 In light of our finding of only congenital eye defects associated with human PXDN mutations, we investigated the precise distribution of the normal peroxidasin protein in mouse embryonic and adult eyes by staining serial sections with a rabbit polyclonal antiserum against PXDN (Figure 5). In brief, whole mouse embryos at embryonic day 18.5 and adult eyes at postnatal day 60 were cryopreserved according to standard procedures. The cryosections (6 μm) were incubated with hematoxylin and eosin, anti-PXDN (Sigma-Aldrich), or a rabbit IgG isotype negative control (Invitrogen). Details regarding the specificity of anti-PXDN are found at the Human Protein Atlas. Bound primary antibody was detected with Alexa Fluor 488-conjugated secondary antibody (Invitrogen), and the nuclei were counterstained with DAPI (Invitrogen). Immunofluorescence was analyzed with an Eclipse TE2000-E inverted confocal microscope (Nikon Instruments). At both stages PXDN localized to the corneal epithelial layer, but for adult eyes there was also localization to the lens epithelium. PXDN may be involved in providing adhesive support to these structures. However, it may also be responsible, with other antioxidant enzymes, for metabolizing hydrogen peroxide (H2O2), superoxide anion (ċO2−), and hydroxyl free radicals (ċOH) generated through normal metabolism in response to cytokines or exposure to light and radiation (Figure 6).47 It is likely that peroxidasin functions as the main extracellular antioxidant enzyme whereas glutathione peroxidase has primarily an intracellular role.
Figure 5.
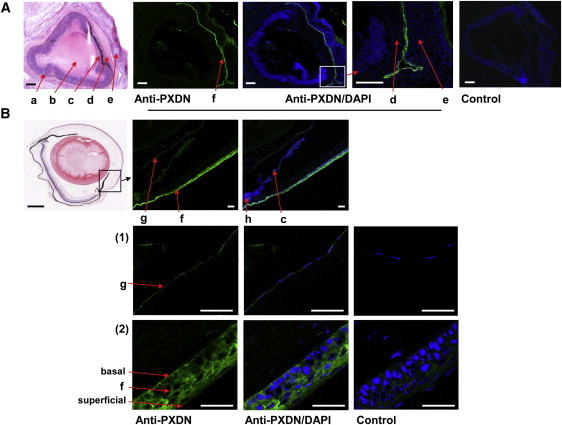
Localization of PXDN in Embryonic and Adult Eyes
(A) Cryopreserved sagittal sections of embryonic mouse eyes at E18.5 stained with hematoxylin and eosin, anti-PXDN, or rabbit IgG negative control. a, retina; b, lens; c, iris; d, cornea; e, lower eyelid. Note PXDN immunoreactivity, depicted as green fluorescence at the corneal and conjunctival epithelium (f) between the corneal stroma (d) and the eyelid (e). Scale bar represents 100 μm.
(B) Sagittal cryosections of adult mouse eyes at postnatal day 60 stained with hematoxylin and eosin (scale bar represents 500 μm), anti-PXDN, or rabbit IgG negative control (scale bar represents 100 μm). The green fluorescence corresponding to PXDN immunoreactivity is abundant in the lens epithelium (g) and the corneal epithelium (f) but absent from the ciliary body (h) and iris (c). Magnified images of (1) the lens epithelium and (2) the corneal epithelium are also shown. The basal and superficial layers of the corneal epithelium are indicated. Scale bar represents 50 μM.
Figure 6.
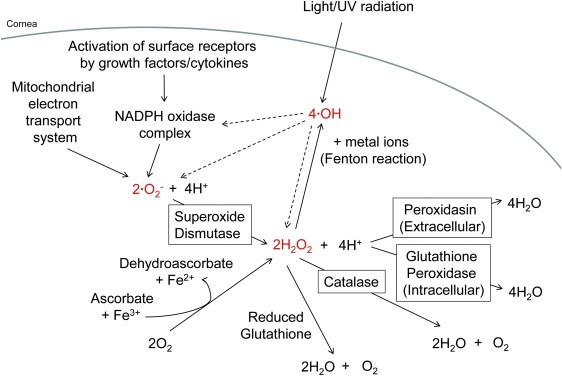
The Generation and Metabolism of Reactive Oxygen Intermediates in the Anterior Chamber
The reactive oxygen intermediates, hydrogen peroxide (H2O2), superoxide anion (ċO2−), and hydroxyl free radicals (ċOH) are highlighted in red. Within the cornea and lens epithelial cells, ċO2− is generated from the mitochondrial electron transport system and the activation of surface receptors by growth factors or cytokines. H2O2 is produced either by the metabolism of ċO2− by superoxide dismutase or by the molecular reaction of oxygen with ascorbate in the presence of ferric ions, and ċOH results from light and ultraviolet radiation as well as from the reaction of H2O2 with metal ions in the Fenton reaction. In terms of decomposition, H2O2 can be inactivated nonenzymatically by reduced glutathione or enzymatically by catalase, glutathione peroxidase, or peroxidasin.
The precise mechanisms by which mutations in PXDN lead to the abnormal lens, corneal, and glaucoma phenotypes are unknown. It is possible that the absence of PXDN in the cornea and lens epithelial layers causes a buildup of reactive oxygen intermediates that indiscriminately oxidize proteins and lipids, causing them to form insoluble aggregates that lead to corneal clouding and cataractogenesis.48–51 Although a peroxidasin knockout mouse is not available, examination of the lens in mice lacking glutathione peroxidase showed that they are indeed more sensitive to oxidative stress and eventually develop cataracts.52,53 Furthermore, a buildup of reactive oxygen intermediates in the aqueous humor is also likely to affect the development and differentiation of anterior segment structures and has previously been implicated in glaucoma pathogenesis.54 For example, enzymes induced by oxidative stress are increased and antioxidant protective mechanisms decreased in the aqueous humor of glaucoma patients,55 and of all the anterior segment structures, the trabecular meshwork is the most sensitive to oxidative damage.56,57
In summary, we have shown that homozygous mutations in PXDN cause a spectrum of ocular anterior segment dysgenesis phenotypes, ranging from congenital cataract-microcornea with mild to moderate corneal clouding to developmental glaucoma with buphthalmos, severe opacification, and vascularization of the cornea. The observed buphthalmos is highly suggestive of a period of severe ocular hypertension during the perinatal period. The common etiology of the two phenotypes was surprising, though this may be due to genetic background or environmental factors rather than a direct consequence of the mutations observed. These findings reveal that PXDN is important for normal development of the cornea and lens, where it may have a role in structural support as well as a functional role as an antioxidant protecting the lens, trabecular meshwork, and cornea from oxidative damage. Further work will be required to determine the precise role of PXDN in normal ocular development and the mechanism by which the mutations cause the diverse phenotypic consequences. Our results will also contribute toward a more accurate classification for anterior segment conditions based on genetic as well as phenotypic diagnosis.
Acknowledgments
We wish to thank the families for participating in this study and the Wellcome Trust (grant 090224), The Sir Jules Thorn Charitable Trust (grant 09/JTA), Yorkshire Eye Research (grant 022), The Ophthalmic Research Institute of Australia, and The Eye Foundation and Sight for All for providing financial support. K.P.B. and J.E.C. are research fellows of the National Health and Medical Research Council of Australia. Z.I.A.A. is funded by a scholarship from the Egyptian Government. We would also like to thank Graeme Black (School of Medicine, University of Manchester, Manchester, UK), Veronica van Heyningen (Human Genetics Unit, Western General Hospital, Edinburgh, UK), Colin E. Willoughby (Centre for Vision Science, Queen's University, Belfast, Northern Ireland), Ordan J. Lehmann (Department of Ophthalmology, University of Alberta, Edmonton, Canada), and Juan Carlos Zenteno (Instituto de Oftalmologia, “Conde de Valenciana,” Mexico City, Mexico) for sending DNA samples from patients with anterior segment dysgenesis for mutation screening.
Contributor Information
Jamie E. Craig, Email: jamie.craig@flinders.edu.au.
Manir Ali, Email: medma@leeds.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent Earray, https://earray.chem.agilent.com/earray/
Ensembl, http://www.ensembl.org/index.html/
GATK, http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
Gene Cards, http://www.genecards.org/
Homozygosity Mapper, http://www.homozygositymapper.org/
Human Protein Atlas, http://www.proteinatlas.org/
Novoalign, http://www.novocraft.com/main/index.php
Online Mendelian Inheritance in Man, http://www.omim.org/
PDB, http://www.rcsb.org/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
PROCHECK, http://www.ebi.ac.uk/thornton-srv/software/PROCHECK/
SAM/BAM, http://sourceforge.net/apps/mediawiki/samtools/index.php?title=SAM_FAQ
SUPERLINK, http://bioinfo.cs.technion.ac.il/superlink-online/
UCSC, http://genome.ucsc.edu/
References
- 1.Idrees F., Vaideanu D., Fraser S.G., Sowden J.C., Khaw P.T. A review of anterior segment dysgeneses. Surv. Ophthalmol. 2006;51:213–231. doi: 10.1016/j.survophthal.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Sowden J.C. Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond.) 2007;21:1310–1318. doi: 10.1038/sj.eye.6702852. [DOI] [PubMed] [Google Scholar]
- 3.Gould D.B., John S.W.M. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum. Mol. Genet. 2002;11:1185–1193. doi: 10.1093/hmg/11.10.1185. [DOI] [PubMed] [Google Scholar]
- 4.Nishimura D.Y., Swiderski R.E., Alward W.L.M., Searby C.C., Patil S.R., Bennet S.R., Kanis A.B., Gastier J.M., Stone E.M., Sheffield V.C. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet. 1998;19:140–147. doi: 10.1038/493. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann O.J., Ebenezer N.D., Jordan T., Fox M., Ocaka L., Payne A., Leroy B.P., Clark B.J., Hitchings R.A., Povey S. Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am. J. Hum. Genet. 2000;67:1129–1135. doi: 10.1016/s0002-9297(07)62943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semina E.V., Reiter R., Leysens N.J., Alward W.L.M., Small K.W., Datson N.A., Siegel-Bartelt J., Bierke-Nelson D., Bitoun P., Zabel B.U. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 7.Perveen R., Lloyd I.C., Clayton-Smith J., Churchill A., van Heyningen V., Hanson I., Taylor D., McKeown C., Super M., Kerr B. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest. Ophthalmol. Vis. Sci. 2000;41:2456–2460. [PubMed] [Google Scholar]
- 8.Jordan T., Hanson I., Zaletayev D., Hodgson S., Prosser J., Seawright A., Hastie N., van Heyningen V. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1992;1:328–332. doi: 10.1038/ng0892-328. [DOI] [PubMed] [Google Scholar]
- 9.Sale M.M., Craig J.E., Charlesworth J.C., FitzGerald L.M., Hanson I.M., Dickinson J.L., Matthews S.J., Heyningen Vv V., Fingert J.H., Mackey D.A. Broad phenotypic variability in a single pedigree with a novel 1410delC mutation in the PST domain of the PAX6 gene. Hum. Mutat. 2002;20:322. doi: 10.1002/humu.9066. [DOI] [PubMed] [Google Scholar]
- 10.Stoilov I., Akarsu A.N., Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 1997;6:641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- 11.Ali M., McKibbin M., Booth A., Parry D.A., Jain P., Riazuddin S.A., Hejtmancik J.F., Khan S.N., Firasat S., Shires M. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 2009;84:664–671. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Désir J., Sznajer Y., Depasse F., Roulez F., Schrooyen M., Meire F., Abramowicz M. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur. J. Hum. Genet. 2010;18:761–767. doi: 10.1038/ejhg.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beebe D.C., Coats J.M. The lens organizes the anterior segment: specification of neural crest cell differentiation in the avian eye. Dev. Biol. 2000;220:424–431. doi: 10.1006/dbio.2000.9638. [DOI] [PubMed] [Google Scholar]
- 14.Thut C.J., Rountree R.B., Hwa M., Kingsley D.M. A large-scale in situ screen provides molecular evidence for the induction of eye anterior segment structures by the developing lens. Dev. Biol. 2001;231:63–76. doi: 10.1006/dbio.2000.0140. [DOI] [PubMed] [Google Scholar]
- 15.Hansen L., Yao W., Eiberg H., Kjaer K.W., Baggesen K., Hejtmancik J.F., Rosenberg T. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest. Ophthalmol. Vis. Sci. 2007;48:3937–3944. doi: 10.1167/iovs.07-0013. [DOI] [PubMed] [Google Scholar]
- 16.Zhou G., Zhou N., Hu S., Zhao L., Zhang C., Qi Y. A missense mutation in CRYBA4 associated with congenital cataract and microcornea. Mol. Vis. 2010;16:1019–1024. [PMC free article] [PubMed] [Google Scholar]
- 17.Willoughby C.E., Shafiq A., Ferrini W., Chan L.L., Billingsley G., Priston M., Mok C., Chandna A., Kaye S., Héon E. CRYBB1 mutation associated with congenital cataract and microcornea. Mol. Vis. 2005;11:587–593. [PubMed] [Google Scholar]
- 18.Wang K.J., Wang B.B., Zhang F., Zhao Y., Ma X., Zhu S.Q. Novel beta-crystallin gene mutations in Chinese families with nuclear cataracts. Arch. Ophthalmol. 2011;129:337–343. doi: 10.1001/archophthalmol.2011.11. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L., Fu S., Ou Y., Zhao T., Su Y., Liu P. A novel nonsense mutation in CRYGC is associated with autosomal dominant congenital nuclear cataracts and microcornea. Mol. Vis. 2009;15:276–282. [PMC free article] [PubMed] [Google Scholar]
- 20.Devi R.R., Vijayalakshmi P. Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea. Mol. Vis. 2006;12:190–195. [PubMed] [Google Scholar]
- 21.Vanita V., Singh J.R., Singh D., Varon R., Sperling K. A novel mutation in GJA8 associated with jellyfish-like cataract in a family of Indian origin. Mol. Vis. 2008;14:323–326. [PMC free article] [PubMed] [Google Scholar]
- 22.Hu S., Wang B., Zhou Z., Zhou G., Wang J., Ma X., Qi Y. A novel mutation in GJA8 causing congenital cataract-microcornea syndrome in a Chinese pedigree. Mol. Vis. 2010;16:1585–1592. [PMC free article] [PubMed] [Google Scholar]
- 23.Jamieson R.V., Perveen R., Kerr B., Carette M., Yardley J., Heon E., Wirth M.G., van Heyningen V., Donnai D., Munier F., Black G.C. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum. Mol. Genet. 2002;11:33–42. doi: 10.1093/hmg/11.1.33. [DOI] [PubMed] [Google Scholar]
- 24.Vanita V., Singh D., Robinson P.N., Sperling K., Singh J.R. A novel mutation in the DNA-binding domain of MAF at 16q23.1 associated with autosomal dominant “cerulean cataract” in an Indian family. Am. J. Med. Genet. A. 2006;140:558–566. doi: 10.1002/ajmg.a.31126. [DOI] [PubMed] [Google Scholar]
- 25.Hansen L., Eiberg H., Rosenberg T. Novel MAF mutation in a family with congenital cataract-microcornea syndrome. Mol. Vis. 2007;13:2019–2022. [PubMed] [Google Scholar]
- 26.Semina E.V., Brownell I., Mintz-Hittner H.A., Murray J.C., Jamrich M. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum. Mol. Genet. 2001;10:231–236. doi: 10.1093/hmg/10.3.231. [DOI] [PubMed] [Google Scholar]
- 27.Iseri S.U., Osborne R.J., Farrall M., Wyatt A.W., Mirza G., Nürnberg G., Kluck C., Herbert H., Martin A., Hussain M.S. Seeing clearly: the dominant and recessive nature of FOXE3 in eye developmental anomalies. Hum. Mutat. 2009;30:1378–1386. doi: 10.1002/humu.21079. [DOI] [PubMed] [Google Scholar]
- 28.Doucette L., Green J., Fernandez B., Johnson G.J., Parfrey P., Young T.L. A novel, non-stop mutation in FOXE3 causes an autosomal dominant form of variable anterior segment dysgenesis including Peters anomaly. Eur. J. Hum. Genet. 2011;19:293–299. doi: 10.1038/ejhg.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan A.O., Aldahmesh M.A., Meyer B. Recessive congenital total cataract with microcornea and heterozygote carrier signs caused by a novel missense CRYAA mutation (R54C) Am. J. Ophthalmol. 2007;144:949–952. doi: 10.1016/j.ajo.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Khan K., Al-Maskari A., McKibbin M., Carr I.M., Booth A., Mohamed M., Siddiqui S., Poulter J.A., Parry D.A., Logan C.V. Genetic heterogeneity for recessively inherited congenital cataract microcornea with corneal opacity. Invest. Ophthalmol. Vis. Sci. 2011;52:4294–4299. doi: 10.1167/iovs.10-6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sia D.I.T., Muecke J., Hammerton M., Ngy M., Kong A., Morse A., Holmes M., Piseth H., Hamilton C., Selva D. A survey of visual impairment and blindness in children attending four schools for the blind in Cambodia. Ophthalmic Epidemiol. 2010;17:225–233. doi: 10.3109/09286586.2010.489250. [DOI] [PubMed] [Google Scholar]
- 32.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albers C.A., Lunter G., Macarthur D.G., McVean G., Ouwehand W.H., Durbin R. Dindel: Accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiler S.R., Taylor S.M., Deans R.J., Kan-Mitchell J., Mitchell M.S., Trent J.M. Assignment of a human melanoma associated gene MG50 (D2S448) to chromosome 2p25.3 by fluorescence in situ hybridization. Genomics. 1994;22:243–244. doi: 10.1006/geno.1994.1374. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell M.S., Kan-Mitchell J., Minev B., Edman C., Deans R.J. A novel melanoma gene (MG50) encoding the interleukin 1 receptor antagonist and six epitopes recognized by human cytolytic T lymphocytes. Cancer Res. 2000;60:6448–6456. [PubMed] [Google Scholar]
- 37.Nagase T., Ishikawa K., Nakajima D., Ohira M., Seki N., Miyajima N., Tanaka A., Kotani H., Nomura N., Ohara O. Prediction of the coding sequences of unidentified human genes. VII. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res. 1997;4:141–150. doi: 10.1093/dnares/4.2.141. [DOI] [PubMed] [Google Scholar]
- 38.Horikoshi N., Cong J., Kley N., Shenk T. Isolation of differentially expressed cDNAs from p53-dependent apoptotic cells: activation of the human homologue of the Drosophila peroxidasin gene. Biochem. Biophys. Res. Commun. 1999;261:864–869. doi: 10.1006/bbrc.1999.1123. [DOI] [PubMed] [Google Scholar]
- 39.Cheng G., Salerno J.C., Cao Z., Pagano P.J., Lambeth J.D. Identification and characterization of VPO1, a new animal heme-containing peroxidase. Free Radic. Biol. Med. 2008;45:1682–1694. doi: 10.1016/j.freeradbiomed.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Péterfi Z., Donkó A., Orient A., Sum A., Prókai A., Molnár B., Veréb Z., Rajnavölgyi E., Kovács K.J., Müller V. Peroxidasin is secreted and incorporated into the extracellular matrix of myofibroblasts and fibrotic kidney. Am. J. Pathol. 2009;175:725–735. doi: 10.2353/ajpath.2009.080693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobe B., Kajava A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 42.Halaby D.M., Mornon J.-P. The immunoglobulin superfamily: an insight on its tissular, species, and functional diversity. J. Mol. Evol. 1998;46:389–400. doi: 10.1007/pl00006318. [DOI] [PubMed] [Google Scholar]
- 43.Hunt L.T., Barker W.C. von Willebrand factor shares a distinctive cysteine-rich domain with thrombospondin and procollagen. Biochem. Biophys. Res. Commun. 1987;144:876–882. doi: 10.1016/s0006-291x(87)80046-3. [DOI] [PubMed] [Google Scholar]
- 44.Gotenstein J.R., Swale R.E., Fukuda T., Wu Z., Giurumescu C.A., Goncharov A., Jin Y., Chisholm A.D. The C. elegans peroxidasin PXN-2 is essential for embryonic morphogenesis and inhibits adult axon regeneration. Development. 2010;137:3603–3613. doi: 10.1242/dev.049189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tauber S., Jais A., Jeitler M., Haider S., Husa J., Lindroos J., Knofler M., Mayerhofer M., Pehamberger H., Wagner O. Transcriptome analysis of human cancer reveals a functional role of heme oxygenase-1 in tumor cell adhesion. Mol Cancer. 2010;9:200. doi: 10.1186/1476-4598-9-200. Published online July 28 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Homma S., Shimada T., Hikake T., Yaginuma H. Expression pattern of LRR and Ig domain-containing protein (LRRIG protein) in the early mouse embryo. Gene Expr. Patterns. 2009;9:1–26. doi: 10.1016/j.gep.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Berthoud V.M., Beyer E.C. Oxidative stress, lens gap junctions, and cataracts. Antioxid. Redox Signal. 2009;11:339–353. doi: 10.1089/ars.2008.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spector A., Roy D. Disulfide-linked high molecular weight protein associated with human cataract. Proc. Natl. Acad. Sci. USA. 1978;75:3244–3248. doi: 10.1073/pnas.75.7.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zigler J.S., Jr., Huang Q.L., Du X.Y. Oxidative modification of lens crystallins by H2O2 and chelated iron. Free Radic. Biol. Med. 1989;7:499–505. doi: 10.1016/0891-5849(89)90025-7. [DOI] [PubMed] [Google Scholar]
- 50.Babizhayev M.A., Costa E.B. Lipid peroxide and reactive oxygen species generating systems of the crystalline lens. Biochim. Biophys. Acta. 1994;1225:326–337. doi: 10.1016/0925-4439(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 51.Cejková J., Stípek S., Crkovská J., Ardan T., Pláteník J., Cejka C., Midelfart A. UV Rays, the prooxidant/antioxidant imbalance in the cornea and oxidative eye damage. Physiol. Res. 2004;53:1–10. [PubMed] [Google Scholar]
- 52.Spector A., Kuszak J.R., Ma W., Wang R.R., Ho Y., Yang Y. The effect of photochemical stress upon the lenses of normal and glutathione peroxidase-1 knockout mice. Exp. Eye Res. 1998;67:457–471. doi: 10.1006/exer.1998.0548. [DOI] [PubMed] [Google Scholar]
- 53.Reddy V.N., Giblin F.J., Lin L.-R., Dang L., Unakar N.J., Musch D.C., Boyle D.L., Takemoto L.J., Ho Y.-S., Knoernschild T. Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Invest. Ophthalmol. Vis. Sci. 2001;42:3247–3255. [PubMed] [Google Scholar]
- 54.Izzotti A., Bagnis A., Saccà S.C. The role of oxidative stress in glaucoma. Mutat. Res. 2006;612:105–114. doi: 10.1016/j.mrrev.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 55.Ferreira S.M., Lerner S.F., Brunzini R., Evelson P.A., Llesuy S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am. J. Ophthalmol. 2004;137:62–69. doi: 10.1016/s0002-9394(03)00788-8. [DOI] [PubMed] [Google Scholar]
- 56.Saccà S.C., Pascotto A., Camicione P., Capris P., Izzotti A. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005;123:458–463. doi: 10.1001/archopht.123.4.458. [DOI] [PubMed] [Google Scholar]
- 57.Izzotti A., Saccà S.C., Longobardi M., Cartiglia C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Invest. Ophthalmol. Vis. Sci. 2009;50:5251–5258. doi: 10.1167/iovs.09-3871. [DOI] [PubMed] [Google Scholar]
- 58.Fernandez-Fuentes N., Rai B.K., Madrid-Aliste C.J., Fajardo J.E., Fiser A. Comparative protein structure modeling by combining multiple templates and optimizing sequence-to-structure alignments. Bioinformatics. 2007;23:2558–2565. doi: 10.1093/bioinformatics/btm377. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez-Fuentes N., Madrid-Aliste C.J., Rai B.K., Fajardo J.E., Fiser A. M4T: a comparative protein structure modeling server. Nucleic Acids Res. 2007;35(Web Server issue) doi: 10.1093/nar/gkm341. W363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baker D., Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.