

Abstract
DNA-dependent protein kinase (DNA-PK), which is involved in DNA double-strand break repair and V(D)J recombination, is comprised of a DNA-targeting component termed Ku and an ∼465-kD catalytic subunit, DNA-PKcs. Although DNA-PK phosphorylates proteins in the presence of DSBs or other discontinuities in the DNA double helix in vitro, the possibility exists that it is also activated in other circumstances via its association with additional proteins. Here, through use of the yeast two-hybrid screen, we discover that the recently identified high affinity DNA binding protein C1D interacts with the putative leucine zipper region of DNA-PKcs. Furthermore, we show that C1D can interact with DNA-PK in mammalian cells and that C1D is a very effective DNA-PK substrate in vitro. Finally, we establish that C1D directs the activation of DNA-PK in a manner that does not require DNA termini. Therefore, these studies provide a function for C1D and suggest novel mechanisms for DNA-PK activation in vivo.
Keywords: DNA-PK, DNA repair, recombination, C1D, nuclear matrix
The DNA double-strand break (DSB) is one of the most dangerous and potentially lethal lesions that can occur within the cell. DNA DSBs can arise when DNA replication complexes encounter damage or secondary structures in the DNA template, by free radical damage as a consequence of oxidative metabolism, or are induced by ionizing radiation (IR) or radiomimetic chemicals. DNA DSBs are also generated as intermediate structures during V(D)J recombination, which mediates the rearrangement of immunoglobulin and T-cell receptor genes (for review, see Alt et al. 1992; Gellert 1992). Therefore, DSB repair is not only critical for maintaining genomic integrity but is also essential for the normal development of the immune system.
Genetic and biochemical complementation studies have revealed that DSB repair and V(D)J recombination are reliant on the DNA-dependent protein kinase (DNA-PK). DNA-PK is an abundant nuclear protein serine/threonine kinase that is activated in vitro by DNA DSBs and certain other perturbations in the DNA double helix (for review, see Anderson and Lees-Miller 1992; Jackson 1997; Smith et al. 1998). Biochemical studies have revealed that DNA-PK is a multiprotein complex consisting of a ∼465-kD catalytic subunit termed DNA-PKcs and a DNA-binding component called Ku, which is composed of two tightly associated polypeptides of ∼70 and ∼80 kD (Ku70 and Ku80, respectively; Dvir et al. 1993; Gottlieb and Jackson 1993). DNA-PK has been shown to phosphorylate a variety of proteins in vitro, including Hsp90, Sp1, SV40 T antigen, p53, serum response factor (SRF), c-Fos, c-Jun, and the carboxy-terminal domain of RNA polymerase II (Anderson and Lees-Miller 1992; Jackson 1997). Although Ku helps to recruit DNA-PKcs to DNA in vitro and is thus likely to be required for the physiological activation of DNA-PK at sites of DNA damage, there is evidence that, at least in vitro, DNA-PKcs can bind to DNA and exhibit kinase activity in the absence of Ku (Yaneva et al. 1997; Hammarsten and Chu 1998). The activation of DNA-PK by DNA DSBs suggests that it may function in vivo by recognizing recombination intermediates and/or DNA ends at sites of DNA damage. Defects in DNA-PK components cause radiosensitivity and an inability to perform V(D)J recombination (for review, see Jackson and Jeggo 1995; Jackson 1996; Lieber et al. 1997).
The biochemical characteristics of DNA-PK have led to several hypotheses regarding its function in vivo (Jackson and Jeggo 1995; Roth et al. 1995 and references therein). For example, it has been suggested that DNA-PK may align the broken DNA ends or interact directly with other components of the DNA repair machinery to target them to sites of DNA damage. Alternatively, or in addition, DNA-PK might function by modifying the activity of DNA repair factors by phosphorylation. It could also counteract the action of transcription factors or chromatin, which might otherwise interfere with the assembly of the DNA repair complex. Finally, and consistent with the fact that DNA-PKcs is related to several factors involved in DNA damage-induced cell cycle checkpoint control processes (Hartley et al. 1995; Keith and Schreiber 1995; Jackson 1997), DNA-PK activation could trigger DNA damage signaling cascades that ultimately impinge on the transcription, DNA replication, cell cycle, and/or apoptotic machineries.
Despite the substantial progress achieved in recent years in our understanding of DNA-PK, a number of issues concerning its physiological functions and mechanism of action remain outstanding. For example, although several proteins have been found to act as DNA-PK substrates in vitro, factors phosphorylated by DNA-PK in vivo have yet to be defined unequivocally. Clearly, the identification of physiological targets for DNA-PK would greatly facilitate investigations into the mechanisms and consequences of DNA-PK activation. In addition, it remains to be established whether there are situations in vivo in which DNA-PK activation does not involve free DNA ends and, if so, whether this is brought about by DNA-PK associating with additional proteins. Regarding this point, it should be noted that DNA-PK is present at ∼1 × 105 to 5 × 105 molecules per human cell, which is far in excess of the number of DNA DSBs generated by physiological doses of DNA damaging agents. Although one explanation for this is that large quantities of DNA-PK are required to allow the rapid detection and repair of DNA damage, an alternative model is that DNA-PK may have functions in addition to its roles in DNA DSB repair. A final crucial issue is that all assays for DNA-PK have so far employed naked DNA templates, whereas it is clear that DNA is complexed with histones and various nonhistone proteins in vivo. The question thus arises as to whether such chromatin components exert positive or negative influences on DNA-PK activation. Notably, in this regard the chromatin-associated high mobility group (HMG) proteins 1 and 2 have been shown to stimulate DNA-PK activity in vitro (Watanabe et al. 1994) and recent investigations have established that certain proteins required for heterochromatin-mediated gene silencing in yeast interact with Ku and are required for effective DNA DSB repair (Tsukamoto et al. 1997; Boulton and Jackson 1998).
As an approach to address some of the above issues, we have applied the yeast two-hybrid system (Fields and Song 1989) to screen for polypeptides that interact with DNA-PKcs. As reported herein, such an approach has led us to identify a novel protein, C1D, which interacts specifically with the potential leucine zipper (LZ) region of DNA-PKcs both in vitro and in vivo. We also show that C1D is a very efficient substrate for DNA-PK in vitro. Moreover, unlike most other DNA-PK substrates identified so far, which are phosphorylated by DNA-PK only in the presence of linear DNA, C1D can activate DNA-PK in the presence of a supercoiled plasmid DNA molecule. The potential implications of these results in regard to the biological roles of C1D and DNA-PK in DNA repair, V(D)J recombination, and other processes are discussed.
Results
Identification of proteins interacting with the putative LZ region of DNA-PKcs
The cloning of the cDNA for human DNA-PKcs has revealed that it encodes a protein of ∼465 kD that possesses a phosphatidylinositol 3-kinase (PIK) catalytic domain toward its carboxyl terminus (Hartley et al. 1995). Other than this, the only other significant feature that has been noted previously for the DNA-PKcs polypeptide is the presence of a potential LZ motif encompassing residues 1503–1538 (Fig. 1A). Because some LZs have well-documented roles in mediating protein–protein interactions, we decided to see whether we could identify factors that interact with this region of DNA-PKcs by the yeast two-hybrid screen approach. To this end, we generated the yeast expression plasmid pLexA–LZcs, which directs the synthesis of residues 1501–1602 of DNA-PKcs fused to the carboxy-terminal DNA-binding domain of the bacterial LexA protein (this fusion is referred to as pLexA–LZcs). pLexA–LZcs was then introduced into the yeast reporter strain L40 (Hollenberg et al. 1995) that contains both the HIS3 and lacZ genes under the control of LexA operators. Importantly, L40 containing pLexA–LZcs is His− and expresses no detectable β-galactosidase activity, indicating that the LZ region of DNA-PKcs is transcriptionally inert when fused to LexA.
Figure 1.


C1D interacts with the LZ region of DNA-PKcs. (A) Schematic diagram of DNA-PKcs. The kinase domain (hatched box) and the LZ region used as a bait in the yeast two-hybrid system are indicated. Mutations introduced into the LZ motif to produce constructs, Mut1LZcs and Mut2LZcs, are indicated by asterisks, and the leucine residues of the motif are underlined. (B) C1D fused to VP16 activation domain (Prey) was transformed into L40 strain together with LexA–LZcs, LexA, or other LexA fusion proteins (Lamin and Daughterless). Interactions were measured by β-galactosidase activity. (C) C1D fused to the carboxy-terminal DNA-binding region of LexA (Bait), and the LZcs was fused to the VP16 activation domain. (D) Mutations that disrupt the LZ motif abolish the DNA-PKcs—C1D two-hybrid interaction.
Strain L40 containing pLexA–LZcs was transformed with a human B-lymphocyte cDNA library in the vector pACT. A total of ∼1 × 106 transformants were obtained, of which, ∼1 × 103 were capable of growing on medium lacking histidine. Of these, 300 were found to produce elevated β-galactosidase activity. After elimination of false positives by use of other LexA fusion proteins (linked to the proteins Lamin and Daughterless) as baits, 100 transformants were identified as containing cDNAs that conferred a His+, LacZ+ phenotype in a fashion that was specific to L40 cells expressing the LexA–DNA-PKcs fusion protein. The cDNA expression vectors from 20 of these transformants were then rescued, and sequencing revealed that 17 contained overlapping sequences derived from the same cDNA. A search of the GenBank database revealed that this cDNA corresponds to that of a 16-kD polypeptide, termed C1D (GenBank accession no. X95592). C1D is a ubiquitously expressed nuclear protein that has been identified recently by screening a cDNA expression library with monoclonal antibodies raised against the residual polypeptides that remain attached to DNA even following aggressive purification methods that employ SDS, proteinase K, and phenol extraction (Nehls et al. 1998; U. Yavuzer and S.P. Jackson, unpubl.; also see Discussion). Notably, the amino acid sequence of C1D does not reveal significant homologies with any known functional protein domains nor to previously characterized proteins.
C1D interacts with the putative LZ region of DNA-PKcs specifically
To establish the specificity of the two-hybrid interaction between C1D and the LZ region of DNA-PKcs, the rescued C1D cDNA clone was transformed into L40 together with baits comprising the LexA DNA-binding domain alone, LexA–LZcs, LexA–Daughterless, or LexA–Lamin, and the level of activity from the integrated lacZ reporter was determined in each case. As shown in Figure 1B, C1D interacts with LexA–LZcs very strongly but not with LexA alone, LexA–Daughterless, nor LexA–Lamin. To confirm the specificity of the C1D–DNA-PKcs interaction further, we performed the two-hybrid experiment in reverse. Thus, C1D was fused to the carboxy-terminal DNA-binding region of LexA, and the region of DNA-PKcs containing the LZ was fused to the VP16 activation domain. Significantly, the introduction of plasmids expressing these two proteins into strain L40 results in significant β-galactosidase activity (Fig. 1C), whereas only background β-galactosidase activity is detected when LexA alone is tested for a two-hybrid interaction with VP16–LZcs. Therefore, C1D and the region encompassing the putative LZ motif of DNA-PKcs can interact irrespective of which acts as bait.
Because the region of DNA-PKcs used in the above studies comprises ∼100 amino acid residues and extends beyond the putative LZ motif, we wished to determine whether this motif is required for the interaction with C1D. To this end, two specific mutations were introduced into the LZ motif, and the ability of each of these mutated derivatives to interact with C1D in the two-hybrid assay was compared with that of the wild-type polypeptide. In one mutant, termed mut1LZcs, the second leucine residue within the LZ motif is mutated to a proline residue, whereas in the second mutant, mut2LZcs, the third leucine and preceding glutamic acid residue are mutated to proline and aspartic acid residues, respectively (Fig. 1A). Notably, as shown in Figure 1D, although C1D interacts efficiently with the wild-type DNA-PKcs derivative, no significant interaction is detected for each of the specific point mutations affecting the integrity of the LZ motif. These data therefore reveal that the two-hybrid interaction between DNA-PKcs and C1D requires an intact DNA-PKcs LZ motif and suggest strongly that this region mediates, at least in part, the interaction between these two factors.
C1D and DNA-PKcs interact in vitro
Next, we wanted to determine whether C1D is able to bind to DNA-PKcs in vitro. To this end, C1D was expressed in Escherichia coli as a fusion to GST, was purified by use of glutathione–agarose beads, and the immobilized GST–C1D fusion protein was then incubated with a protein fraction containing the DNA-PK holoenzyme. After extensive washing of the beads, the proteins bound to GST–C1D were resolved by SDS-PAGE followed by Western blotting and probing with an antibody specific for DNA-PKcs. As shown in Figure 2A, although no interaction is observed between DNA-PKcs and GST alone, a substantial proportion (∼10%) of the input DNA-PKcs is bound by GST–C1D. In contrast, when a control protein was employed consisting of a fusion of GST to the protein CBP, no interaction with DNA-PKcs was apparent (data not shown). Because the DNA-PKcs preparation used for the above assays also contained Ku, we were prompted to ascertain whether C1D binds DNA-PKcs alone or whether it can interact with the Ku–DNA-PKcs complex. In addition, because both C1D and Ku can bind to DNA, it was important to see whether the interaction detected was direct or was mediated indirectly via DNA that might have contaminated either the GST–C1D or DNA-PK samples. To address these issues, we used ethidium bromide, which disrupts protein–DNA interactions by intercalating into the DNA double helix (Lai and Herr 1992). Thus, GST–C1D was tested for interactions with DNA-PKcs and Ku in the presence or absence of ethidium bromide. Significantly, these studies revealed that whereas both DNA-PKcs and Ku bind to the GST–C1D beads in the absence of ethidium bromide, in the presence of ethidium bromide, only DNA-PKcs is retained (Fig. 2B). Taken together, these data indicate that the C1D–DNA-PKcs interaction is direct and does not appear to be mediated by or require a DNA cofactor. In contrast, C1D does not interact with Ku directly, although these factors can associate indirectly in the presence of DNA, presumably via their interactions with DNA-PKcs. Consistent with this, no interaction is observed between C1D and Ku in the absence of ethidium bromide in reactions that lack DNA-PKcs (data not shown). Finally, these results suggest strongly that DNA-PKcs is able to contact both C1D and Ku simultaneously.
Figure 2.

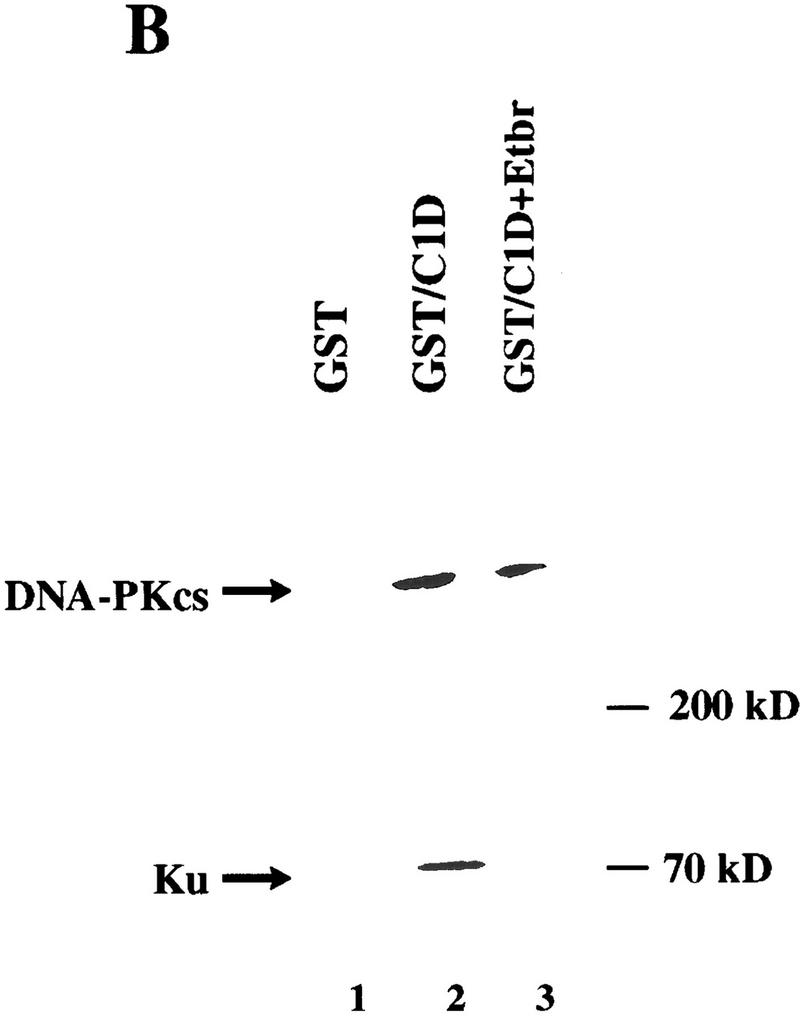
C1D and DNA-PKcs interact in vitro. (A) GST or GST–C1D (0.5 μg in each case) was incubated with 5 μg of purified DNA-PK and subjected to GST pull-down assays, and Western blots were performed with a DNA-PKcs antibody. Quantitation reveals that ∼10% of DNA-PKcs binds to GST–C1D in this experiment, suggestive of a 1:1 binding ratio. (B) GST pull-downs were performed as in A (for lane 3, the pull-down reaction included ethidium bromide), then proteins were immunoblotted with antibodies against Ku or DNA-PKcs.
C1D is an efficient substrate for DNA-PK in vitro
The interaction between DNA-PKcs and C1D suggested that C1D might represent a substrate for DNA-PK and/or might regulate DNA-PK activity. To address these issues, we expressed full-length C1D in E. coli as a His-tagged fusion protein. Notably, this protein on its own is highly insoluble under nondenaturing conditions, but can be obtained in soluble form by denaturing it with 8 m urea and then gradually refolding the protein by withdrawing the urea by dialysis in the presence of either linear or supercoiled plasmid DNA. Such a procedure results in soluble DNA-bound C1D (if DNA is not present in the dialysis procedure, essentially all C1D becomes insoluble; D. Werner and U. Yavuzer, unpubl.). Recombinant C1D purified and refolded by this method was recognized by rabbit polyclonal sera raised against either the His-tagged C1D or a GST–C1D fusion protein (see Materials and Methods). To determine whether C1D can serve as a substrate for DNA-PK, we incubated the His-tagged C1D that had been assembled onto a linear plasmid molecule with DNA-PK (comprising DNA-PKcs and Ku) in the presence of [γ32P]-ATP. These studies revealed that C1D is recognized very effectively as a DNA-PK substrate, with phosphorylation being of similar efficiency to the well-characterized DNA-PK substrate p53 (Fig. 3A, lanes 3,4). However, the GST–C1D fusion protein was not recognized as a substrate, presumably because of the target site(s) on C1D being obscured.
Figure 3.



C1D is a strong substrate for DNA-PK in vitro. (A) C1D refolded on a linear plasmid (C1D-L) or p53 was incubated with purified DNA-PK, and in vitro kinase assays were performed as described in Materials and Methods. Linear DNA was added to the reaction for lane 4. (B) C1D refolded on supercoiled plasmid (C1D-Sc) was incubated with purified DNA-PK as described above. Where indicated, ethidium bromide or LY294002 were incubated with proteins for 30 min before starting the kinase reaction. (C) As indicated, highly purified DNA-PKcs and/or Ku was incubated with C1D-Sc and kinase assays were performed.
C1D activates DNA-PK in the presence of supercoiled plasmid DNA
Previous studies have established that DNA-PK interacts with DNA ends and, therefore, is activated strongly by linear DNA but only weakly, if at all, by closed–circular plasmid DNA (Gottlieb and Jackson 1993; Suwa et al. 1994). In the course of our investigations into C1D phosphorylation by DNA-PK, we made the surprising observation that C1D assembled onto a supercoiled plasmid DNA (pKTMLP) also serves as a very effective DNA-PK substrate (Fig. 3B, lane 4). Furthermore, the inclusion of free supercoiled plasmid DNA does not result in an increase in C1D phosphorylation (Fig. 3B, lane 3). The phosphorylation of C1D is dependent on the presence of the DNA-PK preparation, because no C1D phosphorylation is obtained either with C1D alone or with C1D together with free plasmid DNA (Fig. 3B, lanes 1 and 7, respectively).
Although the above studies utilized highly purified DNA-PK, the possibility existed that C1D might be being phosphorylated not by DNA-PK but by a contaminating kinase in the DNA-PK preparation. To address this issue, we took advantage of the fact that the fungal metabolite wortmannin and the synthetic agent LY294002 have been shown to be potent inhibitors of DNA-PK but do not affect most characterized protein Ser/Thr kinases (G. Smith, N. Lakin, and S.P. Jackson, unpubl.). Significantly, the phosphorylation of C1D by DNA-PK preparations is abolished by the addition of either wortmannin (data not shown) or LY294002 (Fig. 3B, lane 5). In addition, phosphorylation is abrogated in the presence of ethidium bromide (Fig. 3B, lane 6). These data therefore provide strong support that the active C1D kinase is DNA-PK. Given that C1D interacts directly with DNA-PKcs but not Ku, we asked whether C1D could be phosphorylated by DNA-PKcs in the absence of Ku. As seen in Figure 3C, no phosphorylation of C1D is observed in the presence of DNA-PKcs or Ku alone, and phosphorylation is detected only when both proteins are present concurrently. Taken together, these data reveal that although C1D phosphorylation by DNA-PK is DNA dependent, it differs from other well-characterized DNA-PK-mediated phosphorylation events in that it can be promoted by supercoiled plasmid DNA (also, see below). Nevertheless, Ku is still required for DNA-PKcs to phosphorylate C1D that is bound to a supercoiled plasmid molecule.
C1D that is associated with supercoiled DNA can direct the phosphorylation of other polypeptides by DNA-PK
The above results reveal that C1D interacts with DNA-PKcs and show that DNA-PK can phosphorylate C1D which is bound to supercoiled plasmid DNA. To see whether C1D can induce DNA-PK to phosphorylate other substrates in the presence of supercoiled DNA and whether it can substitute for Ku in these events, we analyzed the effect of C1D that was bound to supercoiled DNA (C1D-Sc) on the ability of DNA-PK to phosphorylate a synthetic peptide derived from the amino-terminal transcriptional activation region of p53 (Anderson and Lees-Miller 1992; Finnie et al. 1995). As a control, a mutant peptide was used that is not phosphorylated by DNA-PK. Consistent with previous observations, addition of linear DNA to reactions containing both DNA-PKcs and Ku was found to result in strong kinase activity (Fig. 4A, lane 4), whereas the p53-derived peptide is phosphorylated poorly in reactions containing DNA-PKcs alone or containing DNA-PKcs in the presence of supercoiled DNA (Fig. 4A, lanes 1 and 2, respectively). Moreover, addition of purified Ku, the DNA-end-binding protein poly(ADP)–ribose polymerase (PARP), or bovine serum albumin (BSA) does not increase DNA-PK-mediated kinase activity in the presence of supercoiled DNA (Fig. 4, lanes 8–10, respectively). However, the inclusion of C1D-Sc in reactions containing DNA-PKcs results in a slight but significant increase in kinase activity compared with that obtained with DNA-PKcs alone, suggesting that C1D is able to activate DNA-PKcs for phosphorylation of other protein targets (Fig. 4A, cf. lanes 1 and 5). Most strikingly, addition of Ku to such reactions results in kinase activity as strong as that obtained with linear DNA (Fig. 4A, lanes 6,4).
Figure 4.

C1D activates DNA-PK when bound to a supercoiled plasmid. (A) DNA-PK activity was tested by standard DNA-PK peptide assays (see Materials and Methods) in the presence of the indicated components. In reactions 8 and 9, PARP or BSA were used instead of C1D-Sc. A total of 800 ng of C1D-Sc was subjected to sequential phenol/chloroform extraction (φ/CHCl3) and the DNA was precipitated, resuspended in a small amount of water, and added into reaction 7 (this amount of DNA is in excess of the amount in 100 ng of C1D-Sc; lanes 5,6,10). Where indicated, 100 ng of excess supercoiled (Sc) or linear (L) DNA was added. (B) Proteins were incubated with the indicated antibodies for 30 min on ice and the kinase reaction was started by adding [γ32P]ATP. The anti-C1D antiserum used in this experiment was raised against the GST–C1D fusion and, therefore, is a distinct derivative from the recombinant His-tagged version of C1D used in the kinase assay. (C) DNA-PK peptide phosphorylation assays were conducted with the indicated components. To generate the Rxn DNA, a standard kinase assay was conducted containing C1D-Sc, DNA-PKcs, and Ku, then the DNA was retrieved by φ/CHCl3 extraction and precipitation.
Because C1D used in the above experiments had been refolded onto supercoiled plasmid DNA and because no additional DNA was used, we considered it a possibility that the DNA present in the C1D preparation had become degraded, and it was this degraded DNA that led to DNA-PK activation. To determine whether this was the case, the DNA present in the refolded C1D preparation was purified by sequential phenol and chloroform extractions. Analysis of such DNA by agarose gel electrophoresis revealed that it was still almost entirely supercoiled (data not shown). Moreover, when this recovered DNA is added to reactions containing Ku and DNA-PKcs, no significant stimulation of DNA-PK-mediated p53 peptide kinase activity is observed (Fig. 4A, lane 7). Therefore, the supercoiled DNA to which C1D was bound had not become degraded. To address the possibility that a nuclease might exist in C1D preparations and become active only under kinase assays conditions, we incubated C1D-Sc and DNA-PK together under kinase assay conditions (containing Mg2+) with a radiolabeled DNA fragment. No degradation of the DNA was detectable when it was subsequently analyzed alongside its untreated control by electrophoresis on a denaturing urea/polyacrylamide gel (data not shown). As a final control, we assembled a kinase reaction containing C1D-Sc, DNA-PKcs/Ku, incubated this under kinase assay conditions, and then extracted the DNA. As shown in Fig. 4C, this DNA (Rxn DNA) was unable to promote DNA-PK activation, revealing that it had not become degraded. Taken together, these results show that the C1D preparation used is devoid of detectable nuclease activity and reveal that DNA-PK activation ensues only when supercoiled plasmid DNA is complexed to C1D. In addition, they reveal that Ku is required for DNA-PKcs to be stimulated maximally by C1D-Sc. To confirm these conclusions, reactions containing DNA-PKcs and Ku together with C1D-Sc were preincubated for 30 min with either preimmune serum, anti-C1D antiserum, or anti-Ku antiserum, before being tested for phosphorylation of the p53-derived peptide (Fig. 4B). These studies revealed that whereas incubation with preimmune serum does not affect DNA-PK mediated kinase activity significantly, kinase activity is essentially abolished on incubation with either anti-C1D antiserum or with anti-Ku antiserum (the anti-C1D antiserum used here was raised against a different recombinant version of C1D than that used in the kinase assay; see legend to Fig. 4B for details). These results, therefore, reinforce the notion that both C1D and Ku are required for the efficient activation of DNA-PKcs by supercoiled DNA.
Because the above studies utilized a synthetic peptide derived from p53, and because p53 has been implicated recently as an in vivo target for DNA-PK (Shieh et al. 1997), we tested whether similar effects are obtained with the full-length p53 protein. Importantly, virtually no protein phosphorylation is observed in assays with either DNA-PK (DNA-PKcs plus Ku), p53, or C1D-Sc alone (Fig. 5A, lanes 1–3). Furthermore, and consistent with previous work, p53 phosphorylation by DNA-PK is very poor in the absence of added DNA (Fig. 5A, lane 5), and is stimulated strongly by the addition of linear DNA (lane 6) but not by supercoiled DNA (lane 7). Hence, in the absence of C1D, efficient phosphorylation of p53 by DNA-PK is dependent on the DNA in the assay being linear. In contrast, when supercoiled DNA is used that has been assembled with C1D, p53 is phosphorylated as efficiently as it is in the presence of linear DNA (Fig. 5A, lane 8; C1D is also phosphorylated under these conditions). Because addition of increasing amounts of supercoiled plasmid DNA or purified supercoiled DNA derived from the refolded C1D preparation (see above) does not cause any increase in p53 phosphorylation (data not shown), we conclude that the ability of DNA-PK to phosphorylate p53 in the presence of supercoiled DNA is dependent on C1D. In line with this and with results obtained with the peptide phosphorylation studies, the phosphorylation of the p53 protein is inhibited by preincubation with anti-C1D or anti-Ku serum but not with preimmune serum (Fig. 5B).
Figure 5.


C1D can activate DNA-PK to phosphorylate full-length p53, and this activation is C1D-dependent. (A) DNA-PK, C1D-Sc, and p53 were incubated as described in Materials and Methods. Where indicated, 100 ng of linear (L) or supercoiled (Sc) plasmid was included. (B) Assays were conducted as in A, except that proteins were incubated with the indicated antibodies before reactions were initiated by the addition of [γ-32P]ATP.
DNA-PK and C1D can interact in mammalian cells
In light of the above data, we wished to test whether DNA-PKcs and C1D are capable of interacting with one another in mammalian cells. Furthermore, because the interaction between DNA-PK and the protein c-Abl has been shown to be stimulated in response to DNA damage (Kharbanda et al. 1997), we wanted to determine whether this is also the case for the interaction between C1D and DNA-PKcs. Although initial experiments were designed to address an interaction between endogenous mammalian cell C1D and DNA-PKcs in coimmunoprecipitation assays, such studies were not feasible because C1D remains tightly bound to DNA during whole cell or nuclear extract preparation and, consequently, is not detectable by Western blotting of soluble cell extracts (Nehls et al. 1998; U. Yavuzer, unpubl.). To circumvent this problem, we expressed recombinant C1D in mammalian cells in anticipation that a fraction of the expressed protein would remain soluble following cell extract preparation. Thus, COS cells were transiently transfected with a vector expressing an HA epitope-tagged version of human C1D. After 48 hr, lysates were prepared from the transfected cells with or without prior exposure to 15 Gy of IR, and were immunoprecipitated with anti-DNA-PKcs, anti-HA, or preimmune sera. The immunoprecipitates were then analyzed by Western blotting with an anti-HA antibody or an anti-DNA-PKcs antiserum. As shown in Figure 6A, a protein of ∼22 kD is detected by the anti-HA antibody in anti-DNA-PKcs immunoprecipitates derived from transfected cells (Fig. 6A, lanes 2,4). In contrast, no such protein is evident when extracts of transfected cells are immunoprecipitated with preimmune serum (Fig. 6A, lanes 1,3), nor when extracts of untransfected cells are immunoprecipitated with the anti-DNA-PKcs antiserum (lane 6). When the blot was stripped and reprobed with an anti-C1D antiserum, the protein of ∼22 kD was again detected in anti-DNA-PKcs immunoprecipitates derived from transfected cells (data not shown). In light of these results, and because the predicted molecular mass of HA-tagged C1D is ∼22 kD, these data suggest strongly that the protein immunoprecipitated by the anti-DNA-PKcs antisera from extracts of transfected cells is C1D.
Figure 6.


C1D and DNA-PKcs interact in mammalian cells. (A) Lysates from untransfected (lane 6) or HA–C1D-transfected COS cells (lanes 1–5) were immunoprecipitated with preimmune (PI), anti-DNA-PKcs, or anti-HA antisera. For lanes 3 and 4, the cells were treated with 15 Gy of ionizing irradiation before lysate preparation. Western blotting was performed by an anti-HA antibody. The higher band is the IgG light chain (25 kD). (B) Lysates from transfected and untransfected cells were immunoprecipitated with the antisera described in A, and the Western blot was developed by anti-DNA-PKcs antibody.
To further verify the interaction between DNA-PKcs and C1D detected above, we conducted the immunoprecipitation experiments in reverse by analyzing immunoprecipitates of C1D for the presence of DNA-PKcs. Notably, DNA-PKcs is recovered in anti-HA immunoprecipitates of cells transfected with the HA–C1D expression construct (Fig. 6B, lane 2) but is not evident in preimmune immunoprecipitates of transfected cell extracts or in anti-HA immunoprecipitates from extracts of untransfected cells (Fig. 6B, lanes 1 and 4, respectively). Taken together, these data indicate that C1D interacts with DNA-PKcs in unfractionated mammalian cell extracts. Significantly, however, and in contrast to the situation reported for the interaction between DNA-PKcs and c-Abl, no significant change in the association between C1D and DNA-PKcs is evident in response to IR (Fig. 6A, cf. lanes 2 and 4).
C1D is induced in response to IR
Although DNA damage has been shown to activate the kinase activity of DNA-PK in vitro, the molecular mechanisms underlying activation of DNA-PK in vivo have yet to be established. One possibility is that the interaction between C1D and DNA-PKcs plays some role in the regulation of DNA-PK activity in vivo in response to DNA damage. No difference in the association between DNA-PK and C1D in response to DNA damage was detected in the transfection assays described above. However, because C1D expression in these experiments was under the control of a heterologous promoter, any potential effects of IR on C1D expression may have been overlooked. Therefore, we analyzed C1D levels in total cell lysates by Western immunoblot analysis either before or after exposing cells to 15 Gy of IR. As shown in Figure 7A (left), following IR treatment there is robust induction in the levels of a protein of ∼18 kD (the predicted size of endogenous C1D) that is recognized by an antiserum raised against recombinant His-tagged C1D. In contrast, no significant induction of this protein was observed in response to UV irradiation. To confirm that the protein induced by IR is C1D, the blot was stripped and reprobed with another C1D antibody raised against a GST–C1D fusion protein (α-C1D 2). As seen in Figure 7A (right), the same ∼18 kD protein band is recognized by this second antibody, providing strong evidence that it is C1D. To determine whether the induction of C1D is at the mRNA level, RNA prepared from irradiated and nonirradiated cells was analyzed by quantitative RT PCR. As shown in Figure 7B, C1D mRNA levels are barely detectable in nonirradiated cells but are induced ∼10-fold on exposing cells to IR. Significantly, no difference is observed in the level of GAPDH expression between irradiated and nonirradiated cells. Thus, both C1D protein and mRNA are increased in response to γ-irradiation.
Figure 7.


C1D is induced in response to IR. (A) Total cell lysate from cells with or without prior exposure to 15 Gy of IR was resolved by SDS-PAGE and transferred onto nitrocellulose. Western blotting was performed by antibodies raised against a His-tagged C1D (α-C1D 1) or GST–C1D (α-C1D 2) fusion proteins. (B) C1D transcript expression in nonirradiated and irradiated cells. RT–PCR was performed on RNA extracted from irradiated or nonirradiated cells, and cDNA amplified by primers described in Materials and Methods. GAPDH was used as an internal control.
Discussion
Previous work on DNA-PK has revealed that its catalytic activity in vitro is stimulated markedly by the presence of free DNA ends. Although DNA-PKcs itself has been shown recently to have an inherent capability to bind to and be activated by DNA DSBs (Yaneva et al. 1997; Hammarsten and Chu 1998), efficient activation of the enzyme in vitro requires the DNA end-binding protein Ku. In line with DNA-PKcs and Ku functioning in concert in vivo, the loss of either component leads to defects in DNA DSB repair and V(D)J recombination in cell lines and animal systems. These observations suggest strongly that DNA-PK is recruited to DSBs in vivo. The results presented in this manuscript reveal an alternative mechanism by which DNA-PK catalytic activity may be regulated. Specifically, we show that DNA-PK can be activated in the absence of DSBs through association with the recently identified ∼18-kD-cellular protein termed C1D. This suggests that alternative mechanisms may exist for DNA-PK activation in vivo.
Using the yeast two-hybrid approach to screen a human B-lymphocyte library, we have discovered that C1D binds specifically to the putative LZ motif of DNA-PKcs. Furthermore, we have demonstrated the importance for the DNA-PKcs LZ motif by showing that point mutations of key residues within it severely inhibit the two-hybrid interaction with C1D. In addition, we have established that full-length derivatives of DNA-PKcs and C1D can interact in vitro and in vivo, either as purified proteins or when they are present in unfractionated mammalian cell extracts. Significantly, the binding of DNA-PKcs to C1D appears to be direct, as evidenced by the fact that it does not require DNA and is not disrupted by the addition of ethidium bromide. In contrast, no direct interaction between C1D and Ku is detectable by the assays that we have used, although Ku does appear to associate with C1D indirectly via DNA-PKcs in the presence of DNA. The apparent ability of DNA-PKcs to contact C1D and Ku simultaneously is consistent with the fact that the region of DNA-PKcs that interacts with C1D is distinct from that which has been shown recently to bind Ku (Shengfang et al. 1997).
C1D was identified initially as an antigen that remains associated with DNA even after treatment with harsh denaturants. When expressed in bacteria, C1D is highly insoluble in the absence of ionic detergents such as SDS, or chaotropic agents such as urea or guanidine hydrochloride. However, if C1D is solubilized in the presence of 8 m urea and the urea is then removed slowly by dialysis, C1D is able to refold onto DNA that is provided. By taking advantage of this property of C1D, we have shown that it is a very good substrate for DNA-PK mediated phosphorylation. Furthermore, we have found that DNA-PK is able to phosphorylate C1D not only when it has been refolded onto a linear DNA molecule but also when C1D is bound to a supercoiled plasmid. Several lines of evidence indicate that the activation of DNA-PK in this latter instance requires C1D. First, when the supercoiled DNA used to refold C1D was retrieved by repeated phenol/chloroform extractions and was then analyzed by agarose gel electrophoresis, it was still found to exist almost exclusively in supercoiled form. Second, this retrieved DNA was unable to activate DNA-PK in phosphorylation assays containing either a synthetic peptide or the full-length p53 protein. Thus, the DNA that had been associated with C1D had not sustained nicks or DSBs that could have allowed it to activate DNA-PK directly. Third, we have not detected any nuclease activities in our C1D preparations that are activated on incubation under kinase assay conditions. Finally, the stimulation of DNA-PK activity by C1D in association with supercoiled DNA is inhibited by antibodies raised against C1D but not by any of the preimmune antisera we have tested.
Taken together, the available data suggest a model whereby DNA-PK can be activated in the absence of DNA ends. In this model, the interaction between C1D and DNA-PKcs serves as a mechanism to target DNA-PKcs to a DNA molecule. However, this complex appears to be inactive catalytically, because we have observed no phosphorylation of C1D or other proteins when DNA-bound C1D is incubated with DNA-PKcs alone. Rather, DNA-PKcs activity is only triggered when Ku subsequently enters the complex. One explanation for the demand for Ku despite the lack of DNA ends is that C1D alters the conformation of DNA-PKcs so that it is able to bind to Ku without DNA ends. Alternatively, it is possible that the binding of C1D to DNA alters the DNA structure so that it is now able to interact with Ku, and consequently, trigger the ability of Ku to bind and activate DNA-PKcs. In this model, the inhibition of kinase activity mediated by antibodies that recognize C1D or Ku could be through them, preventing the assembly of the multiprotein complex, and the inhibition effected by ethidium bromide could be brought about by disruption of the Ku–DNA and/or C1D–DNA interactions. Finally, active DNA-PK may then phosphorylate various target proteins, including C1D itself. Although the consequences of C1D phosphorylation by DNA-PK are currently unclear, it is tempting to speculate that this activates C1D for some additional function(s), or dissociates the complex between C1D and DNA-PKcs once its function is complete.
What could be the physiological role(s) for the interaction between C1D and DNA-PKcs and the model for DNA-PK activation described above? One potential clue is provided by the fact that C1D binds to DNA in an extremely stable fashion, as evidenced by the fact that it remains DNA-associated even after rigorous extraction procedures. Highly stable nonhistone proteins that are bound to DNA tightly have been identified in various systems, including insects, plants, and mammalian cells (Krauth and Werner 1979; Capesius et al. 1980; Bodnar et al. 1983; Avramova and Tsanev 1987). Although the biological functions for these proteins are still largely obscure, it is noteworthy that some have been reported to be associated with highly repetitive DNA sequences and to be involved in targeting a subset of genomic DNA to the nuclear matrix (Neuer and Werner 1985; Neuer-Nitsche et al. 1988; Werner and Neuer-Nitsche 1989; Pfutz et al. 1992). This is of particular interest in regard to the results described herein, as the nuclear matrix is thought to provide the structural framework for nuclear/chromatin organization, and has been shown to associate with several nuclear metabolic processes including DNA replication, transcription, RNA splicing, topoisomerase activity, nucleotide excision repair, and DNA DSB repair (Berezney 1984; Cockerill and Garrard 1986; Nelson et al. 1986; Verheijen et al. 1988; Jackson 1991; Kaufman and Shaper 1991; Yasui et al. 1991, 1994; Korte and Yasui 1993; Johnston and Bryant 1994; Koehler and Hanawalt 1996).
By operational definitions, C1D is a nuclear matrix-associated protein and can be detected in nuclear matrix preparations by Western immunoblots and immunofluorescence. C1D is also induced by γ-irradiation. Therefore, it is possible that C1D targets DNA-PK to the nuclear matrix and matrix-associated DNA in response to DNA damage. Once DNA-PK is activated, it is possible that it may then allow proteins involved directly in DNA repair to perform their functions. Alternatively, or in addition, C1D might target DNA-PK to the nuclear matrix constitutively. Evidence in support of the notion that DNA-PK function is linked to the nuclear matrix comes from the observation that the radiosensitive cell line xrs5, which is deficient in Ku80, exhibits irregularly shaped nuclei, has nuclear envelope abnormalities, and has altered nuclear matrix attachment regions with respect to wild-type cells (Yasui et al. 1991, 1994; Korte and Yasui 1993). Furthermore, this cell line has also been shown to be defective specifically in the joining of matrix attached DNA DSBs (Johnston and Bryant 1994). This suggests that the targeting of both DNA-PK and DNA to the nuclear matrix may play a crucial role in DNA DSB repair processes. Studies into interactions between DNA-PK and C1D, and possibly also with other matrix associated proteins, should help address this important issue.
Materials and methods
Cloning, mutagenesis, and DNA sequencing
pLexA–LZcs was generated by cloning into the SpeI site of pBTM116PS, the region of the DNA-PKcs cDNA corresponding to nucleotide residues 4560–4860 (containing a natural 5′ and a polylinker-derived 3′ XbaI sites). pBTM116PS was derived from plasmid pBTM116 (obtained from S. Elledge, HHMI, Baylor College of Medicine, Houston, TX) by modifying the frame of the cloning site and the addition of polystop codons. To construct the wild-type LZ region of DNA-PKcs as a VP16 fusion (LZcs–VP16), nucleotide residues 4381–4860 were amplified by PCR with a 5′ NotI site and XhoI–NotI sites on 3′ end, and cloned into pVP16 (obtained from S. Hollenberg) as well as pBlueskript SK+ (pBSK+) as a NotI fragment. Mut1LZcs–VP16 was isolated in two steps. First, DNA-PKcs nucleotide residues 4560–4860 were isolated by PCR with mismatch primers placing an XbaI site at the 5′ end and XhoI and NotI sites at the 3′ end, then replacing this by the XbaI–XhoI fragment of wild-type LZcs in (pBSK+). Second, the whole fragment containing the entire LZ region of DNA-PKcs in which the second leucine is mutated to a proline was cut out as a NotI fragment and cloned into pVP16. Mut2LZcs–VP16 was isolated as a linker-scanner mutation. Nucleotide residues 4601–4860 were isolated by PCR with primers placing BamHI and NotI sites on the 5′ and 3′ ends, respectively. Residues 4381–4601 were isolated by PCR with a 5′ NotI and 3′ BamHI site at each end and both fragments were cloned into NotI site in pVP16 allowing the two fragments to join at the BamHI site in the middle, which mutates the third leucine to a proline and the preceding glutamic acid to aspartic acid. For bacterial and mammalian C1D expression constructs, the ORF of C1D was isolated by PCR with primers placing suitable restriction sites to the 5′ and 3′ ends and cloning intoexpression plasmids. Bacterial expression plasmids are: pGEX (Pharmacia), pQE30 (Qiagen expression systems). For the mammalian expression construct, an HA epitope (a peptide derived from influenza hemagglutinin protein)-tagged expression vector pCMV 5′2N3T (Lavender et al. 1997) was used. Sequencing of PCR fragments, point mutations, and cDNA inserts from positive clones of the two-hybrid screen was performed with an automatic sequencer by the dideoxy termination method. Sequence comparisons were conducted by GenBank database searches.
Yeast two-hybrid screen
A human B-lymphocyte cDNA library in pACT was used (Harper et al. 1993). The other components of the system were provided by S. Hollenberg. Saccharomyces cerevisiae L40 (MATa trp1 leu2 his3 LYS2:lexA–HIS3 URA3::lexA–lacZ) was grown at 30°C in YPD medium (1% yeast extract, 2% polypepton, and 2% glucose), was sequentially transformed with plexA–LZcs and human B-lymphocyte cDNA library by the lithium acetate method. Double transformants were plated on yeast dropout medium lacking Trp, Leu, His, Lys, and uracil and were grown at 30°C for 3 days. Colonies were then transferred on Whatman 40 filters to test for β-galactosidase activity [with an X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) colony filter assay]. Positive clones were rescued and tested for specificity by transformation into L40 either with plexA–LZcs or control plasmids plexA–Lamin, plexA–Daughterless, or plexA alone.
Bacterial expression and purification of C1D
Expression of GST (C1DpGEXTK2) or His-tagged C1D (C1DpQE30) fusion proteins was induced for 4 hr by adding 2 mm IPTG (isopropyl-β-O-thiogalactopyroniside) and the fusion proteins were isolated from bacterial lysates by affinity chromatography by use of glutathione–agarose beads (Sigma) or Ni–NTA agarose (Qiagen) columns, respectively. C1DpQE30 fusion protein was purified under denaturing conditions in the presence of 8 m Urea and dialyzed against 0.1× TE in the presence of linear or supercoiled plasmid DNA. Antibodies against the recombinant proteins were raised in rabbits by standard procedures (Harlow and Lane 1988).
GST–pull-down assays
Purified DNA-PKcs, Ku, or the DNA-PK holoenzyme was incubated for 2 hr with fusion protein bound to glutathione-coupled agarose beads (Hagemeier et al. 1994). Precipitates were washed several times with NETN (20 mm Tris at pH 8.0, 100 mm NaCl, 1 mm EDTA and 0.5% NP-40) and were subjected to SDS-PAGE and Western blotting. The GST–CBP protein (CPB1; Bannister and Kouzarides 1995) used as a control contained amino acid residues 461–662 of CBP.
Kinase assays
Two nanograms of purified DNA-PKcs together with 5 ng of Ku or 60 ng of DNA-PK holoenzyme was incubated for 2 min with 100 ng of C1DpQE refolded on a linear or a supercoiled DNA in the presence of 20 μl of Z′ buffer (25 mm HEPES–KOH at pH 7.9, 50 mm KCl, 10 mm MgCl2, 20% glycerol, 0.1% NP-40, 1 mm DTT, and 200 μm ATP) and the reaction was started by adding 10 μCi of [γ-32P]ATP (6000 Ci/mmole). Following incubation at 30°C for 20 min, phosphorylated proteins were subjected to 20% SDS-PAGE and visualized by autoradiography. When used, antibodies, ethidium bromide (200 μg/ml), or LY294002 (2.5 μm final concentration), were incubated on ice with the proteins for 30 min before starting kinase reactions. The amount of excess linear DNA was 100 ng. DNA-PK peptide phosphorylation assays were conducted as described previously (Finnie et al. 1995). Relative DNA-PK activity was calculated by subtracting the cpm incorporated into mutant peptide from the cpm incorporated into wild-type peptide, then dividing this by cpm incorporated by mutant peptide.
Cell culture and transient transfections
COS 7 (African green monkey kidney), HeLa, or NIH-3T3 cells were maintained in DMEM supplemented with 10% fetal calf serum and grown at 37°C with 10% CO2 as monolayers. Transfections were performed by the calcium phosphate precipitation technique. The precipitate was left on the cells for 8–10 hr before a glycerol shock (15% glycerol for 1 min) was given. Cells were then washed twice with PBS and fresh medium was added and cells were harvested 48 hr after transfection.
Coimmunoprecipitations
Five micrograms of HA–C1D was transfected into 1 × 108 COS or NIH-3T3 cells as described above. Forty-eight hours following transfection, cells were harvested and lysed in lysis buffer (150 mm NaCl with 0.2 % NP-40). Cells that were subjected to 15 Gy of ionizing irradiation were incubated for 1 hr at 37°C before harvesting. Lysates were incubated with appropriate sera for 1 hr on ice and bound to protein A– or G–Sepharose for 4 hr, rocking gently at 4°C. Precipitates were then washed three times with the lysis buffer (15 min each), separated by 15% SDS-PAGE, and blotted onto nitrocellulose. Western blotting was performed with anti-HA monoclonal antibody (12CA5, Boehringer) or a polyclonal antiserum raised against the full-length DNA-PKcs and immunoreactive bands were revealed by an ECL kit (Amersham) according to the manufacturer’s instructions.
RT–PCR
Total RNA from irradiated or nonirradiated cells was prepared as described (Sambrook et al. 1989), and 1 μg of RNA was used to reverse transcribe according to the enzyme manufacturer’s instructions (Boehringer Mannheim). The cDNA was then amplified with the primers 5′-ATGGCAGGTGAAGAAATT and 3′-ACTTTTACTTTTTCCTTT (C1D) that generates a 420-bp fragment of the C1D cDNA, and the primers 5′-CCACAGTCCATGCCATCACTG and 3′-CGCTGTTGAAGTCAGAGGAGA for a 340-bp fragment of the human GAPDH cDNA. Each PCR cycle consisted of incubation periods of 30 sec at 95°C, 1 min at 55°C, and 1 min at 72°C. PCR products were then analyzed on a 2% agarose gel. To ensure that the PCR reaction was quantitative, aliquots of each PCR reaction were analyzed every 4 cycles, between 12 and 32 cycles.
Acknowledgments
We thank members of the S.P.J. laboratory for their support. Many thanks also to C.R. Goding and T. Kouzarides for their valuable comments and to Steve Elledge and Stan Hollenberg for yeast two-hybrid system components. U.Y. was supported by a Special Fellowship from the Leukemia Society of America, and this work was funded by grants from the Cancer Research Campaign.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
C1D is apparently identical to the protein SUN-CoR, which has recently been identified as a putative transcriptional corepressor (Zamir et al. 1997 Proc. Natl. Acad. Sci. 94: 14400–14405).
Footnotes
E-MAIL spj13@mole.bio.cam.ac.uk; FAX (01233) 334089.
References
- Alt FW, Oltz EM, Young F, Gorman J, Taccioli G, Chen J. VDJ recombination. Immunol Today. 1992;13:306–314. doi: 10.1016/0167-5699(92)90043-7. [DOI] [PubMed] [Google Scholar]
- Anderson CW, Lees-Miller SP. The nuclear serine/threonine protein kinase DNA-PK. Crit Rev Eukaryot Gene Expr. 1992;2:283–314. [PubMed] [Google Scholar]
- Avramova Z, Tsanev R. Stable DNA protein complexes in eukaryotic chromatin. J Mol Biol. 1987;196:437–440. doi: 10.1016/0022-2836(87)90704-2. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J. 1995;14:4758–4762. doi: 10.1002/j.1460-2075.1995.tb00157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezney R. Organization and function of the nuclear matrix. Chrom Non-Histone Proteins. 1984;4:119–179. [Google Scholar]
- Bodnar JW, Jones CJ, Coombs DH, Pearson GD, Ward DC. Proteins tightly bound to HeLa cell DNA at nuclear matrix attachment sites. Mol Cell Biol. 1983;3:1567–1579. doi: 10.1128/mcb.3.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capesius I, Krauth W, Werner D. Proteinase K-resistant and alkali-stably bound proteins in higher plant DNA. FEBS Lett. 1980;110:184–186. doi: 10.1016/0014-5793(80)80068-8. [DOI] [PubMed] [Google Scholar]
- Cockerill PN, Garrard NT. Chromosomal loop anchorage of immunoglobin genes occurs adjacent to enhancers via DNA elements possesing topoisomerase-II sites. Cell. 1986;44:273–282. doi: 10.1016/0092-8674(86)90761-0. [DOI] [PubMed] [Google Scholar]
- Dvir A, Stein LY, Calore BL, Dynan WS. Purification and characterization of a template-associated protein kinase that phosphorylates RNA polymerase II. J Biol Chem. 1993;268:10440–10447. [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Finnie NJ, Gottlieb TM, Blunt T, Jeggo P, Jackson SP. DNA-PK activity is absent in xrs-6 cells; implications for site-specific recombination and DNA double-strand break repair. Proc Natl Acad Sci. 1995;92:320–324. doi: 10.1073/pnas.92.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M. Molecular analysis of V(D)J recombination. Annu Rev Genet. 1992;22:425–446. doi: 10.1146/annurev.ge.26.120192.002233. [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- Hagemeier C, Caswell R, Hayhurst G, Sinclair J, Kouzarides T. Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J. 1994;13:2897–2903. doi: 10.1002/j.1460-2075.1994.tb06584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten O, Chu G. DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci. 1998;95:525–530. doi: 10.1073/pnas.95.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. Immunizations; pp. 53–137. [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 CDK-interacting protein CIP1 is a potent inhibitor of G1 Cyclin-dependent kinases. Cell. 1993;75:805–806. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hartley KO, Gell D, Smith GCM, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, Jackson SP. DNA-dependent protein kinase catalytic subunit: A relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell. 1995;82:849–856. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Sternglanz R, Cheng PF, Weintraub H. Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two hybrid system. Mol Cell Biol. 1995;15:3813–3822. doi: 10.1128/mcb.15.7.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA. Structure-function relationships in Eukaryotic nuclei. BioEssays. 1991;13:1–10. doi: 10.1002/bies.950130102. [DOI] [PubMed] [Google Scholar]
- Jackson SP. DNA damage detection by DNA dependent protein kinase and related enzymes. Cancer Surv. 1996;28:261–279. [PubMed] [Google Scholar]
- ————— DNA-dependent protein kinase. Int J Biochem Cell Biol. 1997;29:935–938. doi: 10.1016/s1357-2725(97)00006-x. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Jeggo PA. DNA double-strand break repair and V(D)J recombination: Involvement of DNA-PK. Trends Biochem Sci. 1995;20:412–415. doi: 10.1016/s0968-0004(00)89090-8. [DOI] [PubMed] [Google Scholar]
- Johnston PJ, Bryant PE. A component of DNA double-strand break repair is dependent on the spatial orientation of the lesions within the higher order structures of chromatin. Int J Radiat Biol. 1994;66:531–536. doi: 10.1080/09553009414551571. [DOI] [PubMed] [Google Scholar]
- Kaufman SH, Shaper JH. Association of topoisomerase II with the hepatoma cell nuclear matrix: The role of intermolecular disulfide bond formation. Exp Cell Res. 1991;192:511–523. doi: 10.1016/0014-4827(91)90071-2. [DOI] [PubMed] [Google Scholar]
- Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Pandey P, Jin S, Inoe S, Bharti A, Yuan Z, Welchselbaum R, Weaver D, Kufe D. Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature. 1997;386:732–735. doi: 10.1038/386732a0. [DOI] [PubMed] [Google Scholar]
- Koehler DR, Hanawalt PC. Recruitment of damaged DNA to the nuclear matrix in hamster cells following ultraviolet irradiation. Nucleic Acids Res. 1996;24:2877–2884. doi: 10.1093/nar/24.15.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte CC, Yasui LS. Morphological characterization of the radiation sensitive cell line, xrs-5. Scanning Microsc. 1993;7:943–951. [PubMed] [Google Scholar]
- Krauth W, Werner D. Analysis of the most tightly bound proteins in eukaryotic DNA. Biochim Biophys Acta. 1979;564:390–401. doi: 10.1016/0005-2787(79)90030-3. [DOI] [PubMed] [Google Scholar]
- Lai JS, Herr W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc Natl Acad Sci. 1992;89:6958–6962. doi: 10.1073/pnas.89.15.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavender P, Vandel L, Bannister AJ, Kouzarides T. The HMG-box transcription factor HBP1 is targeted by the pocket proteins and E1A. Oncogene. 1997;14:2721–2728. doi: 10.1038/sj.onc.1201243. [DOI] [PubMed] [Google Scholar]
- Lieber MR, Grawunder U, Wu X, Yaneva M. Tying loose ends: Roles of Ku and DNA-dependent protein kinase in the repair of double-strand breaks. Curr Opin Genet Dev. 1997;7:99–104. doi: 10.1016/s0959-437x(97)80116-5. [DOI] [PubMed] [Google Scholar]
- Nehls P, Keck T, Greferath R, Spiess E, Glaser T, Rothbarth K, Stammer H, Werner D. cDNA cloning, recombinant expression and characterization of polypeptides with exceptional DNA affinity. Nucleic Acids Res. 1998;26:1160–1166. doi: 10.1093/nar/26.5.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WG, Pienta KJ, Barrack ER, Coffey DS. The role of nuclear matrix in the organization and function of DNA. Annu Rev Biophys Biophys Chem. 1986;115:457–465. doi: 10.1146/annurev.bb.15.060186.002325. [DOI] [PubMed] [Google Scholar]
- Neuer B, Werner D. Screening of isolated DNA for sequences released from anchorage sites in nuclear matrix. J Mol Biol. 1985;181:15–25. doi: 10.1016/0022-2836(85)90321-3. [DOI] [PubMed] [Google Scholar]
- Neuer-Nitsche B, Lu X, Werner D. Functional role of a highly repetitive DNA sequence in anchorage of the mouse genome. Nucleic Acids Res. 1988;16:8351–8360. doi: 10.1093/nar/16.17.8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfutz M, Gileadi O, Werner D. Identification of human satellite DNA-sequences associated with chemically resistant nonhistone polypeptide adducts. Chromosoma. 1992;101:609–617. doi: 10.1007/BF00360538. [DOI] [PubMed] [Google Scholar]
- Roth DB, Lindahl T, Gellert M. How to make ends meet. Curr Biol. 1995;5:496–499. doi: 10.1016/s0960-9822(95)00101-1. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. Isolation of total RNA from mammalian cells; pp. 7.6–7.11. [Google Scholar]
- Shieh S-Y, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shengfang J, Kharbanda S, Mayer B, Kufe D, Weaver DT. Binding of Ku and c-Abl at the kinase homolgy region of DNA-dependent protein kinase catalytic subunit. J Biol Chem. 1997;272:24763–24766. doi: 10.1074/jbc.272.40.24763. [DOI] [PubMed] [Google Scholar]
- Smith GCM, Divecha N, Lakin ND, Jackson SP. The DNA-dependent protein kinase and related proteins. Biochem Soc Symp. 1998;64:87–100. [PubMed] [Google Scholar]
- Suwa A, Hirakata M, Takeda Y, Jesch SA, Mimori T, Hardin JA. DNA-dependent protein kinase (Ku protein-p350 complex) assembles on double-stranded DNA. Proc Natl Acad Sci. 1994;91:6904–6908. doi: 10.1073/pnas.91.15.6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Kato J, Ikeda H. Silencing factors participate in DNA repair and recombination in Saccharomyces cerevisiae. Nature. 1997;388:900–903. doi: 10.1038/42288. [DOI] [PubMed] [Google Scholar]
- Verheijen R, Van Venrooij W, Ramaekers F. The nuclear matrix: Structure and composition. J Cell Sci. 1988;90:11–36. doi: 10.1242/jcs.90.1.11. [DOI] [PubMed] [Google Scholar]
- Watanabe F, Shirakawa H, Yoshida M, Tsukada K, Teraoka H. Stimulation of DNA-dependent protein kinase activity by high mobility group protein-1 and protein-2. Biochem Biophys Res Com. 1994;202:736–742. doi: 10.1006/bbrc.1994.1992. [DOI] [PubMed] [Google Scholar]
- Werner D, Neuer-Nitsche B. Site specific location of covalent DNA-polypeptide complexes in the chicken genome. Nucleic Acids Res. 1989;17:6005–6015. doi: 10.1093/nar/17.15.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: Biochemical and atomic-force microscopy studies. EMBO J. 1997;16:5098–5112. doi: 10.1093/emboj/16.16.5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui LS, Ling-Indeck L, Johnson-Wint B, Fink T, Molsen D. Changes in the nuclear structure in the radiation-sensitive CHO mutant cell, xrs-5. Radiat Res. 1991;127:269–277. [PubMed] [Google Scholar]
- Yasui LS, Fink TJ, Enrique AM. Nuclear scaffold organization in the X-ray sensitive Chinese hamster mutant cell line, xrs-5. Int J Radiat Biol. 1994;65:185–192. doi: 10.1080/09553009414550221. [DOI] [PubMed] [Google Scholar]