

Abstract
The c-Abl tyrosine kinase localizes to the cytoplasm and plasma membrane in addition to the nucleus. However, there is little information regarding a role for c-Abl in the cytoplasm/plasma membrane compartments. Here we report that a membrane pool of c-Abl is activated by the growth factors PDGF and EGF in fibroblasts. The pattern and kinetics of activation are similar to growth factor activation of Src family kinases. To determine whether a link existed between activation of c-Abl and members of the Src family, we examined c-Abl kinase activity in cells that expressed oncogenic Src proteins. We found that c-Abl kinase activity was increased by 10- to 20-fold in these cells, and that Src and Fyn kinases directly phosphorylated c-Abl in vitro. Furthermore, overexpression of wild-type Src potentiated c-Abl activation by growth factors, and a kinase-inactive form of Src reduced this activation, showing that Abl activation by growth factors occurs at least in part via activation of Src kinases. Significantly, we show that c-Abl has a functional role in the morphological response to PDGF. Whereas PDGF treatment of serum-starved wild-type mouse embryo fibroblasts resulted in distinct linear or circular/dorsal membrane ruffling, c-Abl-null cells demonstrated dramatically reduced ruffling in response to PDGF, which was rescued by physiological re-expression of c-Abl. These data identify c-Abl as a downstream target of activated receptor tyrosine kinases and Src family kinases, and show for the first time that c-Abl functions in the cellular response to growth factors.
Keywords: c-Abl, Src, receptor tyrosine kinases, cytoskeleton
The c-Abl proto-oncogene encodes a 150-kD nonreceptor protein tyrosine kinase that is tightly regulated within the cell (Wang 1993; Pendergast 1996). c-Abl contains a catalytic domain, polyproline rich regions, and SH2 and SH3 domains that are involved in protein–protein interactions and may also regulate the kinase. Additionally, the carboxyl terminus of c-Abl contains nuclear localization and export signals, as well as F- and G-actin-binding domains (Van Etten et al. 1994; Wen et al. 1996; Taagepera et al. 1998). Mutant forms of c-Abl have been identified in murine, feline, and human leukemias. Most notably, the BCR–ABL oncoproteins (p185, p210, p230) have key roles in the development of three forms of human leukemia, acute lymphocytic (ALL), chronic myelocytic (CML), and chronic neutrophilic (CNL) leukemia, respectively (Melo 1996). Whereas the role of oncogenic forms of Abl in malignant phenotypes has been well studied, the biological function of c-Abl remains elusive. Recent work has suggested a role for the nuclear pool of c-Abl in response to DNA damage, and several reports have implicated the ATM and DNA-dependent protein kinases (PKs) as intermediates in this pathway (for review, see Wang 1998). It has been suggested that c-Abl has a role in the G1/S cell cycle arrest response to DNA damage (Yuan et al. 1996). However, subsequent studies have shown that the G1/S checkpoint response to ionizing radiation is unaltered in fibroblasts from c-Abl−/− mice (Liu et al. 1996), and in fibroblasts from mice doubly deficient in c-Abl and Arg, an Abl-related gene (Koleske et al. 1998). Therefore, although c-Abl is activated by ATM and DNA–PK, the biological significance of these events has yet to be determined.
In addition to the nucleus, c-Abl also localizes to the cytoplasm and plasma membrane and is associated with actin filaments (Van Etten et al. 1994). Recently, c-Abl has been shown to be activated following integrin engagement (Lewis et al. 1996); however, a biological role for this activation has not yet been reported. A role for c-Abl in the cytoskeleton is additionally supported by the finding that coexpression of c-Abl and the Abl-binding protein ALP1/amphiphysin II results in cytoskeletal changes in fibroblasts (Kadlec and Pendergast 1997). Furthermore, neuroepithelial cells derived from mice that are doubly deficient in c-Abl and Arg exhibit alterations in their actin cytoskeleton (Koleske et al. 1998). Moreover, cells transformed by constitutively activated BCR–ABL oncogenes exhibit increased motility on extracellular matrices and accelerated protrusion and retraction of pseudopodia (Salgia et al. 1997; Skorski et al. 1998). Taken together, these data suggest that c-Abl may have important functions outside of the nucleus, possibly to regulate cytoskeletal organization and/or cell movement. However, a functional role for c-Abl in the cytoplasm and membrane compartments has not yet been defined, and c-Abl has yet to be placed in any growth factor signaling pathway.
Platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) receptors are activated by ligand binding followed by dimerization and autophosphorylation (for review, see Heldin 1997; Hackel et al. 1999). Many SH2-containing proteins such as Ras–GAP (GTPase activating protein), Phospholipase C-γ (PLC-γ), Phosphatidylinositol-3′ (PI-3) kinase, and Grb-2 bind specific tyrosine phosphorylated sites in the activated receptors (for review, see Heldin 1997; Hackel et al. 1999). In addition, nonreceptor Src family tyrosine kinases bind the activated receptors via their SH2 domains and are subsequently tyrosine phosphorylated by the receptors (Kypta et al. 1990). Expression of a kinase-inactive Src or a SH3-domain mutant that lacks the PDGFβ receptor phosphorylation site inhibits PDGF-induced DNA synthesis (Twamley-Stein et al. 1993; Broome and Hunter 1996). Following growth factor stimulation, Src kinases translocate to the plasma membrane at actin-rich sites, possibly membrane ruffles (Fincham et al. 1996). Constitutively activated Src kinases localize to focal adhesions and are thought to play a role in cytoskeletal reorganization, focal adhesion turnover, and cell motility (Fincham and Frame 1998; Fincham et al. 1996). Significantly, activated Src kinases phosphorylate and/or form complexes with many of the same molecules targeted by activated forms of c-Abl (Bcr–Abl, v-Abl) such as paxillin (Weng et al. 1993; Salgia et al. 1995), Cbl (Andoniou et al. 1994; Dombrosky-Ferlan and Corey 1997), and focal adhesion kinase (FAK) (Cobb et al. 1994; Gotoh et al. 1995). These findings, together with previous data that suggest that c-Abl and oncogenic Abl forms such as BCR–ABL may be involved in cytoskeletal processes, led us to examine whether c-Abl might be involved in growth factor signaling, and whether c-Abl may play a role in the cytoskeletal response to growth factors.
Here, we report that the c-Abl tyrosine kinase is activated in response to growth factor stimulation through a mechanism that involves Src family kinases. Additionally, we demonstrate that c-Abl has a role in cytoskeletal reorganization in response to PDGF stimulation.
Results
c-Abl tyrosine kinase activity is activated by growth factor stimulation
To determine whether c-Abl is activated in response to mitogenic stimuli, we analyzed the effect of PDGF and EGF on c-Abl tyrosine kinase activity in murine fibroblast cell lines. Stimulation of NIH-3T3 cells with PDGF-BB resulted in activation of c-Abl kinase activity (utilizing c-Abl antibody K12) with peak activation (3.2- ± 0.55-fold) occurring after 5–10 min (Fig. 1A, left). Kinase activity remained elevated until 20 min when it decreased to twofold. c-Abl kinase activity also was elevated following PDGF stimulation in a second set of experiments utilizing a second c-Abl antibody (PEX4) that recognizes a distinct epitope in c-Abl (Fig. 1A, right). In contrast, EGF treatment of NIH-3T3 cells, which contain low levels of EGF receptors (Osherov and Levitzki 1994; Moro et al. 1998), did not cause increased c-Abl kinase activity (data not shown). Stimulation of 10T1/2 fibroblasts with PDGF-BB and/or EGF caused only minor increases in c-Abl activity (data not shown). However, EGF treatment of 10T1/2 fibroblasts stably overexpressing the EGF receptor (10T1/2–EGFR) resulted in activation of c-Abl tyrosine kinase activity (mean maximal activation 2.6- ± 0.15-fold) (Fig. 1b, left). Similar results were obtained in other experiments utilizing a second c-Abl antibody (PEX4) (mean maximal activation 3.1- ± 0.26-fold) (Fig. 1B., right) and a third antibody (Ab3) (data not shown). Little activation by PDGF was observed in 10T1/2–EGFR cells, which is likely due to the low levels of PDGF receptors in these cells (Fig. 1B, left). No kinase activity was observed in stimulated NIH-3T3 and 10T1/2–EGFR cells utilizing PEX4 preimmune serum, an irrelevant polyclonal antibody (Santa Cruz), or after preincubation of K12 antibody with blocking peptide (data not shown). In addition, phosphoaminoacid analysis showed that 32P incorporation into the GST–Crk substrate after immunoprecipitation with any of the three c-Abl antibodies occurred on tyrosine residues (data not shown). Thus, these data show that the c-Abl kinase is activated in response to PDGF and EGF treatment in cells that express sufficient levels of the receptors for these growth factors.
Figure 1.



The c-Abl tyrosine kinase is activated in response to PDGF and EGF stimulation in fibroblasts. (A) Subconfluent NIH-3T3 cells were serum starved and stimulated with PDGF-BB. c-Abl tyrosine kinase activity was measured by immunoprecipitation of c-Abl with K12 (left) or PEX4 (right) antibody followed by in vitro kinase assay using GST–Crk as a substrate (top). These data represent one of three independent experiments in which mean maximal activation was 3.2 ± 0.55. Cellular lysates were simultaneously analyzed for c-Abl protein levels by immunoblotting with anti-Abl antibody (8E9) (bottom). (B) Serum-starved murine 10T1/2 fibroblasts overexpressing the EGF receptor were stimulated with PDGF-BB or EGF. Kinase assays and immunoblots were performed as above and represent one of three independent assays with mean maximal c-Abl activation by EGF of 2.6- ± 0.15-fold with K12 antibody and of 3.1- ± 0.26-fold with PEX4 antibody. (C) 10T1/2 fibroblasts overexpressing the EGF receptor were serum starved, stimulated, and fractionated into nuclear, membrane/cytoskeletal, and soluble cytoplasmic fractions. Kinase assays were performed as above and represent one of three independent experiments with mean activation 1.7 ± 0.1 in the membrane fraction with PEX4 antibody.
Because c-Abl is maximally activated within 5 min of PDGF or EGF stimulation, we hypothesized that the cytoplasmic or membrane rather than nuclear pool of c-Abl must be activated. To test this hypothesis, we performed subcellular fractionation followed by c-Abl in vitro kinase assays on unstimulated and stimulated NIH-3T3 and 10T1/2–EGFR cells. In both cases, the majority of c-Abl protein was distributed between membrane/cytoskeletal (hereafter designated membrane) and nuclear fractions with little c-Abl protein detected in the soluble cytosolic fraction (Fig. 1C, bottom; data not shown). In both PDGF-stimulated NIH-3T3 cells (data not shown) and EGF-stimulated 10T1/2–EGFR cells, only the membrane pool of c-Abl became activated (Fig. 1C). This was confirmed by two different c-Abl antibodies (PEX4 and K12). Membrane fractions reproducibly gave increased c-Abl kinase activity after stimulation, whereas soluble cytosolic and nuclear pools did not. Western blots on the fractions with antibodies specific for the different cellular compartments showed that the fractions were pure (data not shown). For the first time, these data clearly show activation of membrane-associated c-Abl in response to extracellular signals.
Expression of activated Src kinases results in elevated c-Abl kinase activity
Like c-Abl, endogenous Src family kinases (Src, Fyn, and Yes in fibroblasts) are activated (two- to threefold) by PDGF in NIH-3T3 cells (Kypta et al. 1990), with peak activation at 5–10 min. Similar to the results obtained in Figure 1B, Src kinase activity is elevated in response to EGF only in cells that overexpress the EGF receptor (Osherov and Levitzki 1994). Therefore, c-Abl and Src kinases are activated by similar signals and with similar kinetics and both proteins are activated at the cell membrane. In addition, we have shown recently that constitutively activated Abl and Src kinases target the Abl-interacting protein-2 (Abi-2) for degradation by the ubiquitin-dependent proteasome machinery (Dai et al. 1998). The above findings, together with our observation that c-Abl is activated by the same stimuli and with similar kinetics as Src kinases, suggest that the activities of c-Abl and Src kinases may be linked.
To determine whether Src family kinases could activate c-Abl, we utilized BaF3 cell lines that stably contain v-src under the control of a zinc-inducible promoter. Treatment of BaF3–v-Src cells with zinc led to induction of v-Src protein that reached maximum levels at 6 hr (Fig. 2A, bottom). Treatment of vector control cells with zinc did not induce activation of c-Abl kinase activity (Fig. 2A, top). However, zinc treatment of BaF3 cells expressing v-Src resulted in increased c-Abl kinase activity that paralleled the induction of v-Src protein and peaked at 6 hr (5.7- ± 0.42-fold) (Fig. 2A). Incubation of c-Abl immunoprecipitates with the Src substrate Sam68 (Santa Cruz) resulted in no phosphorylation, demonstrating that the activity observed was c-Abl specific and was not Src (data not shown). In addition, no detectable activity was observed after immunoprecipitation with an irrelevant polyclonal antibody (Santa Cruz) or after preincubation of K12 antibody with blocking peptide (data not shown).
Figure 2.


c-Abl tyrosine kinase activity is elevated in cells that express oncogenic Src. (A) Murine BaF3 cells containing either vector alone (−) or zinc-inducible v-src (+) were treated with 75 μm zinc chloride. c-Abl tyrosine kinase activity was determined by immunoprecipitation with K12 antibody (top), and c-Abl protein level (8E9) (middle), or Src protein level (EC10) (bottom) were analyzed for each time point. (B) c-Abl and Src kinase activities from murine C3H10T1/2 fibroblasts expressing vector alone, wild-type chicken c-Src, or activated chicken v-Src were examined in triton-soluble and -insoluble lysates by immunoprecipitation with PEX4 (Abl) or GD11 (Src) antibodies. In vitro kinase assays were performed with GST–Crk (Abl) or Sam68 (Src) as substrates. Abl and c-Src/v-Src levels were determined as above. The assay represents one of three independent experiments with K12 or PEX4 antibody with mean increased c-Abl activity of 22.5- ± 9.6-fold (K12) as compared with vector control in triton-soluble lysates.
c-Abl activity was also examined in fibroblasts stably expressing oncogenic v-Src. Murine 10T1/2 fibroblasts expressing v-Src had substantially elevated c-Abl kinase activity as compared with cells containing the empty vector (neo) or wild-type chicken c-Src (Fig. 2B, top panels). Src in vitro kinase activity was high in 10T1/2 cells expressing wild-type or oncogenic v-Src (Fig. 2B, bottom panels). However, the majority of v-Src activity was retained in the triton-insoluble fraction, whereas most of the wild-type c-Src activity was present in the triton-soluble lysate (Fig. 2B, bottom). v-Src-expressing cells contained 10- to 22-fold higher c-Abl kinase activity (with PEX4 or K12 antibodies, respectively) as compared with wild-type Src cells (Fig. 2B, top left), and a portion of c-Abl activity was associated with triton-insoluble cytoskeletal/membrane structures (Fig. 2B, top right). This suggests that activation of c-Abl by oncogenic Src kinases may cause translocation of soluble cytoplasmic c-Abl to the triton-insoluble cytoskeleton or alternatively oncogenic Src may directly activate the membrane/cytoskeletal pool of c-Abl. Additionally, c-Abl was activated (6.3- ± 0.7-fold over wild type) in Rat1 cells expressing oncogenic chicken c-Src 523am, and c-Abl kinase activity also was retained in cytoskeletal/membrane structures in these cells (data not shown). These data show that oncogenic, but not wild-type Src, expression results in dramatic c-Abl activation.
c-Abl activation by growth factors is dependent on Src family kinases
Because expression of oncogenic Src resulted in c-Abl activation, we examined whether c-Abl activation by growth factors could be potentiated by expression of wild-type Src family kinases. Overexpression of wild-type c-Src in 10T1/2 cells has been shown to enhance the mitogenic responsiveness to EGF (Wilson et al. 1989) by hyperphosphorylating specific tyrosine residues on the receptor (Tice et al. 1999). Whereas c-Abl kinase activity was not elevated following PDGF and EGF treatment of untransfected 10T1/2 cells, PDGF treatment of 10T1/2 cells stably overexpressing wild-type chicken c-Src resulted in elevation of c-Abl kinase activity (Fig. 3A, left). Moreover, treatment of these cells with PDGF and EGF together resulted in an earlier peak of c-Abl activation (2 min) compared with each factor alone (Fig. 3A, right). Therefore, overexpression of c-Src facilitates growth factor activation of c-Abl. This finding suggests that c-Abl activation may occur, at least in part, via activation of Src family kinases.
Figure 3.

Activation of c-Abl by PDGF involves Src family kinases. (A) Serum-starved 10T1/2 cells stably expressing wild-type chicken c-Src were stimulated with PDGF-BB and/or EGF. Kinase assay represents one of three independent experiments with mean maximal activation 3.3 ± 0.4 for EGF/PDGF stimulation. (B) Spontaneously immortalized MEFs from Fyn−/−, Src+/−, and Yes−/− mice were infected with retrovirus containing either vector, wild-type mammalian c-Src, or kinase-inactive Src (K297M). c-Abl kinase assays were performed on stimulated and unstimulated cells after serum starvation with Ab3 anti-Abl antibody for immunoprecipitations. Src protein levels were determined by Western blot analysis with Src2 antibody. c-Abl levels were determined as above. c-Abl activation was decreased 40%–60% in cells expressing kinase-inactive Src as compared with vector in three independent experiments. (C) NIH-3T3 cells were infected with retroviruses containing either vector, wild-type, or a kinase-inactive form of Src (K297M). Kinase assays were performed as above with c-Abl K12 antibody. c-Abl activation was decreased 40%–75% in cells expressing kinase-inactive Src as compared with vector in three independent experiments. (D) Ph cells expressing wild-type (Wt), kinase-inactive (K−), or Src-binding mutant (F72/74) α/β chimeric PDGF receptors were serum starved, stimulated with PDGF-AA, and c-Abl kinase assays were performed as above with Pex4 antibody. c-Abl and Src/Fyn/Yes proteins levels were assessed as above. Protein levels of α/β chimeric receptors were determined by Western blot analysis with 80.8 antibody.
To determine whether Src family kinases are required for growth factor activation of c-Abl, we transiently expressed wild-type mammalian c-Src or a c-Src kinase-inactive mutant (K297M) (Broome and Hunter 1996) into Fyn−/−, Yes−/−, and Src+/− immortalized mouse embryo fibroblasts (MEFs). Transient introduction of wild-type Src by retroviral infection resulted in elevated c-Abl activation in response to PDGF treatment, whereas expression of a kinase-inactive form of Src (K297M) caused a decrease in c-Abl activation (Fig. 3B). Similar results were obtained in NIH-3T3 cells (Fig. 3C). To determine whether residual activation of c-Abl following expression of the kinase-inactive Src was due to the presence of uninfected cells, or resulted from a Src-independent pathway to c-Abl activation, we tested the ability of c-Abl to be activated by a PDGF receptor mutant that lacked the Src-binding sites. Ph fibroblasts that lack endogenous PDGF-α receptors were transfected with wild-type, kinase-inactive, or a Src-binding mutant (F72/74) of a chimeric α/β PDGF receptor (DeMali and Kazlauskas 1998). The chimeric receptors contain the extracellular, transmembrane, and juxtamembrane domains of the PDGF-α receptor and the remainder of the intracellular domain is derived from the PDGF-β receptor. Therefore, the chimeric receptors are activated by PDGF-AA but signal via PDGF-β receptor pathways (DeMali and Kazlauskas 1998). PDGF-AA stimulation of Ph cells containing the wild-type chimeric receptor resulted in c-Abl activation (3.3- ± 0.3-fold), whereas no activation of c-Abl was observed after stimulation of cells containing a kinase-inactive mutant of the chimeric receptor (Fig. 3D). In cells expressing a mutant chimeric receptor that cannot bind Src family members (F72/74) activation of c-Abl by PDGF-AA is markedly reduced (1.4- ± 0.2-fold) (Fig. 3D). Taken together, these results indicate that growth factor stimulation leads to the activation of Src kinases that in turn activate c-Abl. Furthermore, activation of c-Abl by growth factors may occur through Src-dependent and Src-independent pathways.
Src family kinases phosphorylate c-Abl
To determine whether expression of Src family kinases results in c-Abl tyrosine phosphorylation, we coexpressed a kinase-inactive (K290R) form of c-Abl with mammalian activated forms of either Fyn or Src in COS cells. Coexpression of either activated Fyn or Src resulted in tyrosine phosphorylation of kinase-inactive c-Abl (Fig. 4A). In contrast, kinase-inactive c-Abl was not tyrosine phosphorylated in cells coexpressing the cytoplasmic tyrosine kinases Jak1 and Jak2 (Fig. 4B) despite dramatic induction of cellular tyrosine phosphorylation. These data support the notion that tyrosine phosphorylation of c-Abl by Src kinases is specific. To examine whether tyrosine phosphorylation of c-Abl by Fyn results in elevated c-Abl kinase activity, we transfected cells with activated Fyn, and analyzed endogenous c-Abl activity by in vitro kinase assay in control cells and Fyn-transfected cells (Fig. 4C). The c-Abl kinase was consistently activated four- to eightfold in cells that overexpressed activated Fyn. To determine whether Src family kinases could directly phosphorylate c-Abl, we produced Src and Fyn in baculovirus-infected Sf9 insect cells, and incubated Src or Fyn immunoprecipitates with various GST–Abl fragments. Src and Fyn phosphorylated the c-Abl SH2–SH1 K290R fragment, which contains the SH2 and kinase domains of c-Abl and is kinase inactive, but did not phosphorylate other portions of c-Abl containing the carboxy-terminal domain, SH3 domain, SH2 domain alone, or GST (Fig. 5A). These results show that Src and Fyn can directly phosphorylate the kinase domain of c-Abl and that increased tyrosine phosphorylation of c-Abl in the presence of Src kinases correlates with enhanced c-Abl tyrosine kinase activity.
Figure 4.
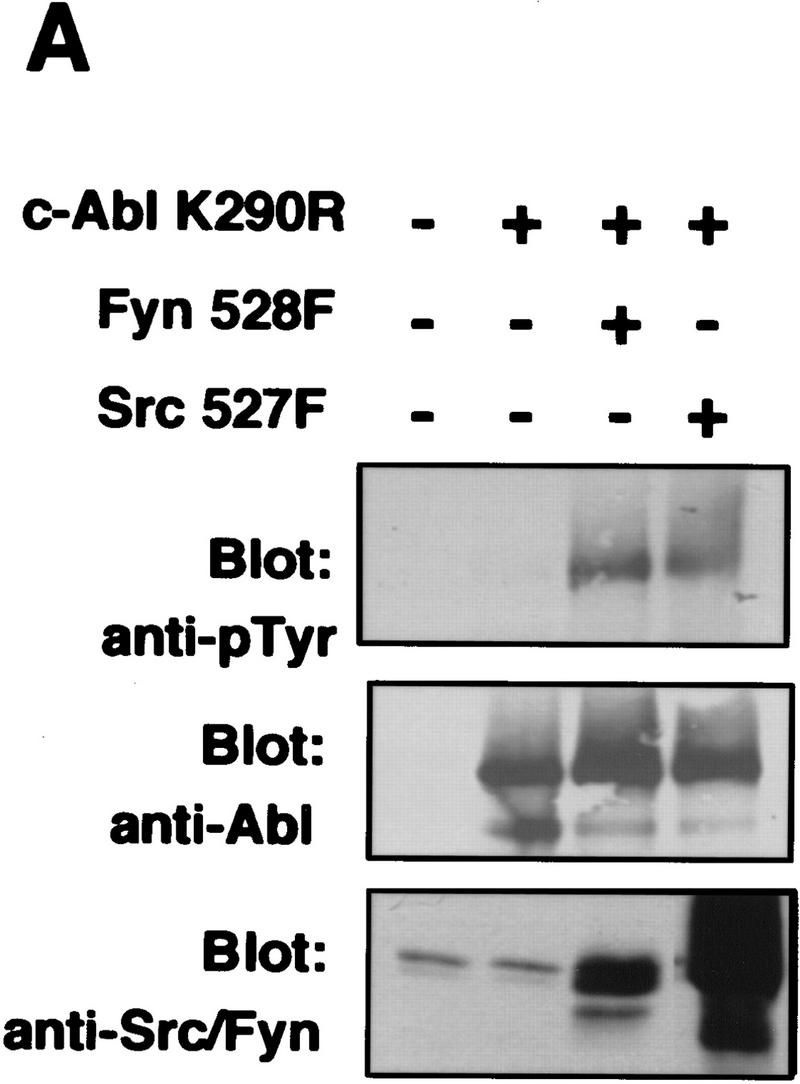
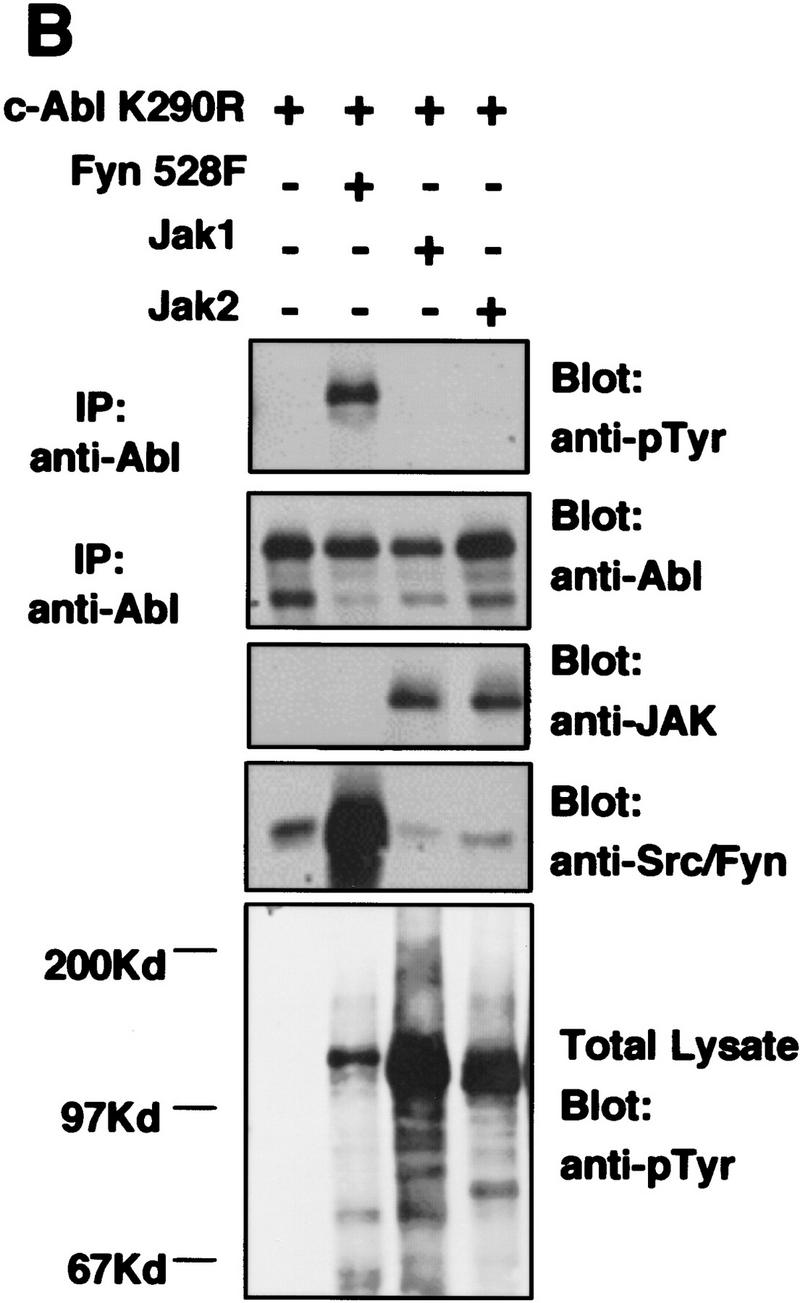
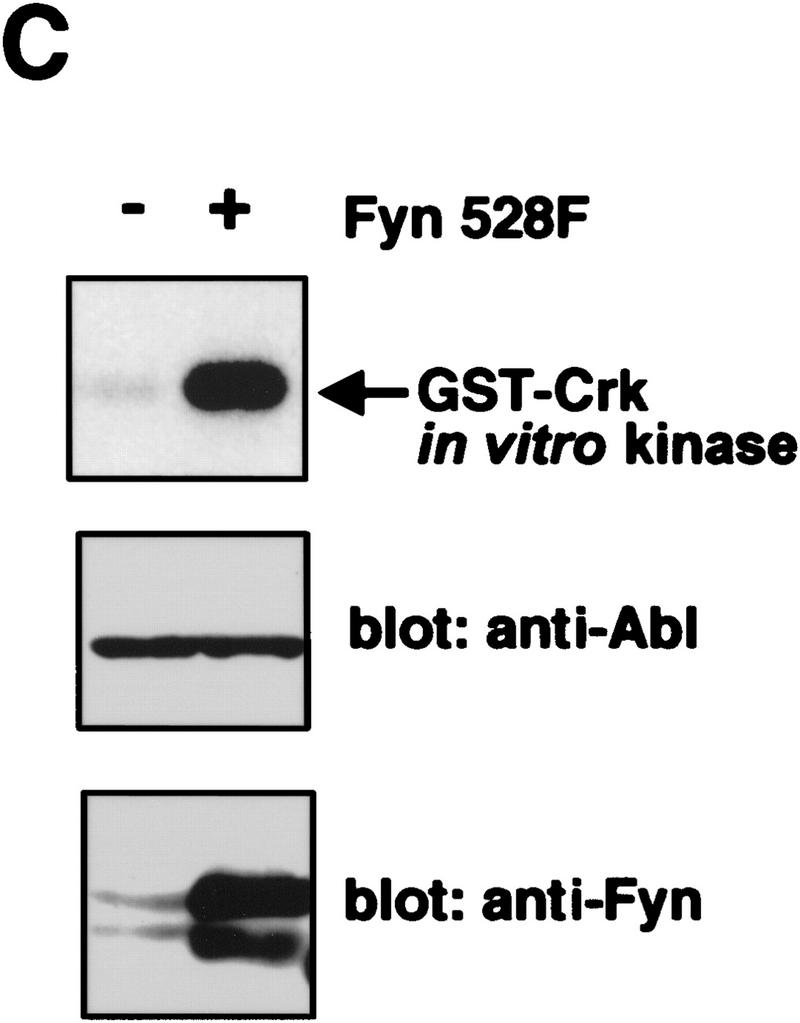
Expression of Src family kinases results in tyrosine phosphorylation and activation of c-Abl following transient transfection. (A,B) COS cells were mock transfected (no DNA) or transfected with kinase-inactive K290R c-Abl, activated mammalian Src family kinases (Fyn528F or Src527F), or JAK1/2. Immunoprecipitates were analyzed by Western blotting with Abl (8E9) or anti-pTyr (4G10) antibodies, and lysates were analyzed with anti-JAK (HR-785), or anti Src/Fyn (Src2) antibodies. (C) 293T cells were transfected with or without activated mammalian Fyn (528F), and the activity of endogenous c-Abl was analyzed by in vitro kinase assay. c-Abl and Fyn protein levels were analyzed as above.
Figure 5.


Src family kinases phosphorylate c-Abl in the kinase (SH1) domain. (A) Src and Fyn were isolated from baculovirus lysates by immunoprecipitation with antibodies to c-Src (N16) or c-Fyn (Fyn3), and incubated with various GST–Abl fragments or GST alone. Uninfected lysate, immunoprecipitated with Fyn antibody and incubated with the Abl GST–SH2–SH1 K290R fragment demonstrated that the fragment was devoid of autokinase activity. The Src substrate Sam68 fragment was used as a positive control. Equivalent amounts of GST fragments were utilized as determined by Coomassie Blue staining (data not shown). (F) Fyn baculovirus; (S) Src baculovirus. Kinase assay is representative of three independent experiments. (B) The kinase inactive GST–Abl fragment used above was utilized as substrate for in vitro kinase assays. Reactions were performed in either the presence or absence of human c-Fyn, with cold ATP. Reaction products were analyzed by Western blotting for Abl, pTyr, or phospho-Abl.
Previous work on the crystal structure of the insulin receptor has identified potential tyrosine residues responsible for activating the kinase (Hubbard 1997) by stabilizing the active conformation of the protein. Tyrosine Y412 in c-Abl, the major autophosphorylation site (Muller et al. 1993), corresponds to this residue. We therefore hypothesized that activation of c-Abl by Src family kinases may be due, at least in part, to the ability of Src kinases to phosphorylate the c-Abl Y412 residue. To test this hypothesis, we raised an antibody to an Abl phosphopeptide phosphorylated at Y412. We used the Abl GST–SH2–SH1 K290R fragment in an in vitro kinase reaction containing unlabeled ATP, with or without Fyn. c-Abl was recognized by the c-Abl phospho-antibody only when phosphorylated by Fyn (Fig. 5B). This provides evidence that at least one of the residues phosphorylated by Fyn is the c-Abl autophosphorylation site and suggests that the mechanism of activation of c-Abl by Src kinases may involve tyrosine phosphorylation of a residue in the activation loop of c-Abl, allowing stabilization of an active conformation.
c-Abl function is required for cytoskeletal reorganization in response to PDGF
The findings that c-Abl is activated by growth factor signaling pathways that also activate Src family kinases, that Src kinases directly phosphorylate and activate c-Abl, and that activated Src and Abl are associated with the cytoskeleton (Van Etten et al. 1994; Fincham et al. 1996; Lewis et al. 1996), led us to examine whether c-Abl function was necessary for a proper morphological response to PDGF. Treatment of serum-starved fibroblasts with PDGF induces cytoskeletal reorganization in the form of membrane ruffling followed by stress-fiber formation, processes that are mediated by the small G-proteins Rac and Rho, respectively (Ridley et al. 1992). Actin reorganization mediates changes in cell shape associated with cell motility, migration, and chemotaxis in response to PDGF (Westermark et al. 1990; Wennstrom et al. 1994; Hooshmand-Rad et al. 1997). We treated serum-starved primary MEFs from Abl−/− and Abl+/+ siblings with PDGF for various times, and stained filamentous actin with rhodamine-conjugated phalloidin. Primary MEFs from Abl+/+ and Abl−/− mice cultured in 10% serum had thick, organized cytoplasmic actin stress fibers (Fig. 6A,B). Serum deprivation caused a decrease in stress fibers and increased actin condensation at the cell surface (cortical actin) in both Abl+/+ and Abl−/− MEFs (Fig 6C,D). PDGF treatment of Abl+/+ cells caused linear membrane ruffling that appeared as early as 10 min after stimulation, with peak ruffling occurring after 30 min (Fig. 6E). In contrast, Abl−/− cells showed very little response to PDGF treatment even after 30 min (Fig. 6F). In other experiments, stimulation of Abl+/+ fibroblasts with PDGF caused the formation of striking circular/dorsal ruffles (Fig. 6G). Circular/dorsal ruffles are hallmarks of PDGF stimulation and are thought to correlate with macropinocytosis and cell motility (Mellstrom et al. 1988; Eriksson et al. 1992). Dorsal/circular ruffles were numerous in Abl+/+ cells and were heavily actin rich (Fig. 6G). In contrast, Abl−/− cells contained markedly fewer (4.49- ± 0.9-fold in three different experiments) dorsal/circular ruffles, and the ruffles were much less obvious due to decreased actin content (Fig. 6H). Differences in the cytoskeletal response to PDGF were not due to decreased receptor expression as primary Abl+/+ and Abl−/− fibroblasts were found to express equivalent levels of both α and β PDGF receptors (data not shown). Furthermore, PDGF treatment resulted in elevation of the c-Abl tyrosine kinase activity in the primary Abl+/+ fibroblasts but not in the Abl−/− fibroblasts as measured by in vitro kinase assay with two different anti-Abl antibodies (data not shown).
Figure 6.
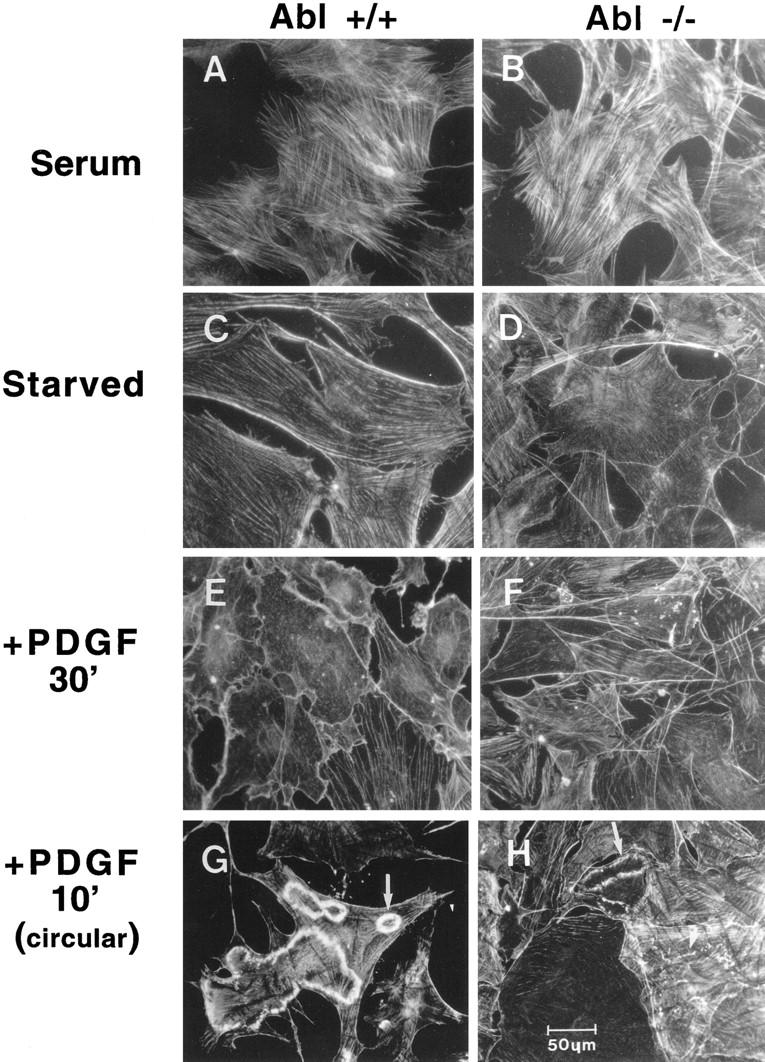
PDGF-induced actin reorganization is altered in c-Abl-null primary fibroblasts. (A–F) c-Abl+/+ and Abl−/− MEFs from matched littermates at the same passage (3–5), were maintained either in 10% FBS, serum starved, or serum starved followed by stimulation with PDGF-BB for the indicated times. Rhodamine–phalloidin-stained cells were visualized on an Olympus microscope utilizing a 20× objective. Pictures are representative of three independent experiments. (G,H) In a second set of experiments, striking dorsal/circular ruffles were observed after PDGF stimulation of primary MEFs. Pictures are representative of three independent experiments utilizing two independent isolates of Abl−/− cells. Circular ruffles were counted in fifty −20× power fields from three experiments. Abl- null fibroblasts had 4.49- ± 0.9-fold fewer circular ruffles. Circular ruffles in Abl−/− cells contained less actin and were less apparent.
To confirm that the defective cytoskeletal response to PDGF observed in the Abl−/− MEFs was due to the absence of c-Abl, we generated spontaneously immortalized Abl−/− fibroblasts reconstituted with Abl or vector alone using a bicistronic GFP vector followed by multiple rounds of cell sorting with FACS. Western blot analysis showed that Abl protein levels in c-Abl-reconstituted cells (Abl+) approximated those observed in primary Abl+/+ MEFs (data not shown). It was critical to obtain levels of c-Abl comparable with endogenous c-Abl in the reconstituted Abl−/− MEFs as previous work has shown that c-Abl overexpression causes growth arrest (Sawyers et al. 1994). The c-Abl kinase was activated by PDGF in the c-Abl-reconstituted cells (data not shown). Abl-reconstituted cells demonstrated heavy cell-surface actin redistribution after 5 min of PDGF treatment (Fig. 7C), followed by dramatic circular ruffling that occurred at 10 min and was maintained at 30 min (Fig. 7E). By 1-hr poststimulation, actin rings decreased and were totally absent after 2 hr. In contrast, Abl-null cells transfected/sorted with GFP vector alone showed a minimal cytoskeletal response at all time points (Fig 7B,D,F). A few actin rings (6.0- ± 1.8-fold fewer than in Abl+ cells) were observed, but the rings were less obvious because of decreased actin content (Fig. 7F). Introduction of c-Abl K290R, a kinase-inactive form of c-Abl, did not rescue the membrane ruffling defect of Abl−/− β cells (data not shown). These data suggest that the kinase activity of c-Abl is necessary for its cytoskeletal effects. In summary, Abl−/− fibroblasts have a reduced ability to reorganize the cytoskeleton following PDGF stimulation, and this defect is rescued by re-expression of physiological levels of c-Abl.
Figure 7.
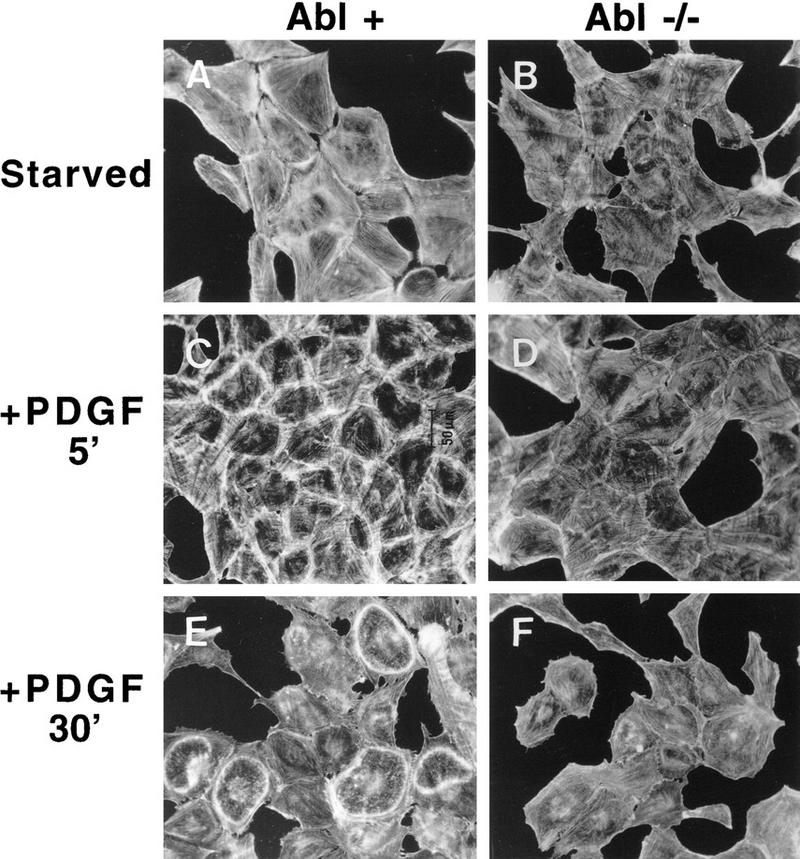
Reintroduction of c-Abl into c-Abl−/− immortalized MEFs rescues the actin cytoskeletal reorganization defect in response to PDGF. Spontaneously immortalized Abl−/− cells containing either GFP vector alone or a bicistronic GFP–c-Abl vector were stimulated with PDGF-BB or unstimulated and stained with rhodamine–phalloidin. Pictures are representative of three independent experiments. Abl-null fibroblasts exhibited 6.63- ± 2.0-fold fewer ruffles compared with Abl-reconstituted cells.
Discussion
Our findings demonstrate for the first time that the membrane pool of c-Abl is activated in response to growth factor stimulation in mammalian cells. The activation of c-Abl by growth factors occurs, at least in part, via activation of Src kinases. This conclusion is supported by our data showing that overexpression of wild-type Src potentiates Abl activation by growth factors, a kinase-inactive form of Src reduces Abl activation, a PDGF receptor mutant that lacks Src-binding sites is defective in its ability to induce c-Abl activation, and by the finding that oncogenic Src kinases phosphorylate and activate c-Abl. Taken together, these data suggest that c-Abl activation by PDGF is partially dependent on Src tyrosine kinase activity. Incomplete inhibition of c-Abl activation by PDGF in the presence of Src dominant-negative molecules and residual c-Abl activation by a Src-binding mutant of the PDGF receptor point to the existence of a Src-independent pathway to c-Abl activation by growth factor receptors. Significantly, dual activation of Src kinases and c-Abl in response to growth factor stimulation increases the range of protein targets and sequence-specific sites that are phosphorylated following activation of receptor tyrosine kinases.
Expression of oncogenic Src kinases results in dramatically increased c-Abl activity. These results suggest that c-Abl also may have a role in Src-mediated transformation. Following shift to the permissive temperature, a temperature-sensitive form of v-Src rapidly translocates from the perinuclear region to focal adhesions (Fincham et al. 1996), where it catalyzes focal adhesion turnover (Fincham and Frame 1998). v-Src induces deregulated cell growth, cell rounding, and decreased cell attachment, which are dependent on reorganization of the actin cytoskeleton. Because c-Abl activity is increased in the presence of v-Src, this suggests that c-Abl may have a role in one or more of these events. Future experiments will determine whether c-Abl is required for Src transformation and whether cytoskeletal changes induced by oncogenic forms of Src are altered in the absence of c-Abl function.
We have shown that c-Abl has a functional role in PDGF signaling. Abl function is necessary for linear and circular/dorsal ruffling, which are both dependent on reorganization of actin. Activation of PI-3-kinase has been shown to be necessary for the membrane ruffling response to PDGF (Wennstrom et al. 1994). Therefore, we determined whether activation of a PI-3-kinase pathway is altered in Abl−/− cells as compared to Abl+ cells. In both cell lines, the PI-3-kinase target Akt was efficiently activated within 30 sec of PDGF stimulation, peaking at 5 min as assessed using an Akt phosphoantibody (New England Biolabs) (data not shown). The extent and kinetics of activation were identical in Abl+ and Abl −/− cells. The kinetics of Akt activation, which were faster than that observed for c-Abl , suggest that c-Abl may be downstream of PI-3-kinase/Akt or in a parallel pathway.
Circular/dorsal ruffling is associated with macropinocytosis (internalization of solutes and membrane components) (Veithen et al. 1996), a process that occurs prior to cell movement. Interestingly, an oncogenic form of Abl (v-Abl) has been shown previously to activate Rac-dependent pinocytosis (Renshaw et al. 1996), and v-Src also induces constitutive macropinocytosis (Veithen et al. 1996). Additionally, cells transformed by the activated Bcr–Abl tyrosine kinase exhibit increased motility on extracellular matrices and accelerated protrusion and retraction of pseudopodia (Salgia et al. 1997; Skorski et al. 1998). Taken together, these data implicate the c-Abl tyrosine kinase in the regulation of cell shape and cell movement.
In addition to affecting cytoskeletal reorganization in response to PDGF, c-Abl also appears to influence entry of quiescent cells into S phase following PDGF stimulation (data not shown). We observed a consistent 4-hr delay in S-phase entry of spontaneously immortalized Abl−/− cells in response to PDGF after serum deprivation as compared with those reconstituted with c-Abl by use of [3H]thymidine incorporation assays and FACS analysis (data not shown). Primary and spontaneously immortalized Abl−/− MEFs divide equally well as their Abl+/+ or Abl-reconstituted counterparts under serum conditions. In addition, there are no morphological differences between Abl-null and Abl-containing cells. These data suggest that Abl−/− cells have an altered mitogenic as well as cytoskeletal response to PDGF.
Recently, integrins have been shown to associate with PDGF and EGF receptors, and have been implicated in ligand-independent activation of the receptors as integrin engagement can potentiate chemotactic and mitogenic responses of the receptor tyrosine kinases (Schneller et al. 1997; Moro et al. 1998; Wang et al. 1998). Additionally, integrin activation is necessary for S-phase entry in response to EGF and serum and is required for cell survival mediated by the extracellular matrix (Moro et al. 1998). Because c-Abl kinase activity is increased by both growth factors (Fig. 1) and integrins (Lewis et al. 1996), and c-Abl has a role in the cytoskeletal response to PDGF (Figs. 6 and 7), c-Abl may link mitogenic and adhesive signals to actin reorganization, thus affecting growth factor-mediated cell movement.
A role for Abl in the regulation of cytoskeletal dynamics is supported by genetic evidence in Drosophila. Drosophila (D)-Abl has been shown to regulate axonal outgrowth (a process that involves actin polymerization). Embryos that lack abl and one of a number of other genes such as disabled (dab; Gertler et al. 1989, 1993), fascilin I (Elkins et al. 1990), prospero (Gertler et al. 1993), fax (Hill et al. 1995), notch (Giniger 1998), and armadillo (Loureiro and Peifer 1998) exhibit defects in axonal outgrowth. More recently, it has been demonstrated that loss of D-Abl alone produces axonal defects (Wills et al. 1999). A significant link between D-Abl and the regulation of the actin cytoskeleton has been provided by the finding of genetic interactions between D-Abl and the Drosophila profilin gene (chickadee, Wills et al. 1999). Loss-of-function mutants of D-Abl and profilin result in an identical growth-cone-arrest phenotype for specific motor axons, and produce similar axonal abnormalities in the Drosophila central nervous system (Wills et al. 1999). These findings support the hypothesis that Abl and profilin function together to promote axon outgrowth. Whereas profilin has been implicated as both an inhibitor and activator of actin polymerization (Theriot and Mitchison 1993), recent reports support a requirement for profilin in actin polymerization in vivo. A profilin mutant that is defective in actin binding suppresses the formation of filopodia induced by N-WASP and activated Cdc42 (Suetsugu et al. 1998). Our findings indicate that like profilin, the Abl tyrosine kinase has a positive role in the reorganization of the actin cytoskeleton in mammalian cells.
Recent findings have revealed a role for the mammalian Abl tyrosine kinases in neurulation (Koleske et al. 1998). Mice that are doubly deficient for c-Abl and Arg die during embryogenesis and exhibit delayed neural tube closure. Significantly, the neuroepithelium of the double-mutant homozygotes displays a disorganized actin network. These findings suggest that the Abl tyrosine kinases affect neurulation through regulation of the actin cytoskeleton.
Thus, c-Abl activation in response to growth factors and its effects on cytoskeletal reorganization may underlie the phenotypes observed in flies and mice with Abl mutations. Activation of Abl kinases may occur following engagement of cell-adhesion molecules and receptor tyrosine kinases in multiple cell types, and the activated Abl kinases may function to link the membrane-bound receptors to signaling cascades that affect cell morphology and motility. Our findings have opened the door to examine these possibilities.
Materials and methods
Reagents and cell culture
10T1/2 and Rat1 cells containing either vector, wild-type chicken c-src, or oncogenic src, as well as Src antibodies EC10 and GD11, were kindly provided by S.J. and J.T. Parsons (University of Virginia, Charlottesville) and have been described previously (Wilson et al. 1989). Mammalian c-src and c-fyn constructs were provided by Andrey Shaw (Washington University, St. Louis, MO), and the MIGR1 construct was provided by Warren Pear (University of Pennsylvania, Philadelphia). BaF3 cells expressing zinc-inducible v-src were provided by T.M. Gilmer (GlaxoWellcome, Inc., Research Triangle Park, NC) and were maintained as described (Dai et al. 1998). Retroviral wild-type and dominant-negative src constructs (pMSVneo–pp60src, Src–K297M) were obtained from Martin Broome (SUGEN, Inc., South San Francisco, CA). Fyn−/−, Yes−/−, and Src+/− spontaneously immortalized MEFs were obtained from Paul Stein (Wistar Institute, Philadelphia, PA). Fyn baculovirus was kindly provided by Sara Courtneidge (SUGEN, Inc., South San Francisco, CA). Src baculovirus was the gift of Frank McCormick and Robin Clark (ONYX Pharmaceuticals, Richmond, CA). Wild-type and null (Abl2 mutation) MEFs from matched littermates were provided by Victor Tybulewicz (MRC, London, UK). MEFs, Cos, and 293T cells were maintained in DMEM supplemented with 10% FBS. For growth factor stimulation assays, 10T1/2 cells were serum starved for 48 hr in 0.25% FBS, and NIH-3T3 cells were starved 16 hr in 0.1% calf serum. Ph cells containing mutant PDGF receptors were maintained as described (DeMali and Kazlauskas 1998). Ph cells were stimulated with 50 ng/ml PDGF-AA [Upstate Biotechnology, Inc. (UBI)], and all other cells were stimulated with 150 ng/ml EGF (Boehringer-Mannheim) or 12.5 ng/ml PDGF-BB (UBI). Wild-type and dominant-negative Src retrovirus was produced in 293T cells as described (Dai et al. 1998).
Abl-null MEFs were immortalized by culture of a mass population through senescence. Murine c-Abl cDNA was cloned into the EcoRI site of the retroviral bicistronic GFP vector MIGR1 (Pear et al. 1998). MIGR1–c-Abl or MIGR1 alone was introduced by retroviral infection into spontaneously immortalized Abl null MEFs. GFP-positive cells were selected by multiple rounds of FACS cell sorting. Cells that expressed high levels of c-Abl were unable to divide or divided very slowly (Sawyers et al. 1994), and therefore were counter selected. After three to four rounds of sorting, a population of cells expressing physiological levels of c-Abl was obtained. Only cells that expressed physiological c-Abl levels were employed in the experiments described here.
Cell lysis and immunoblot analysis
Cellular lysates were prepared in Triton lysis buffer (Nehme et al. 1997) or kinase lysis buffer containing 150 mm NaCl, 10 mm sodium phosphate (pH 7), and 1% Triton X-100, which was supplemented with inhibitors (1 mm sodium orthovanadate, 1 mm PMSF, 25 mm sodium fluoride, and 1 μg/ml leupeptin, aprotinin, and pepstatin). Total protein (20 μg) was separated on polyacrylamide gels and immunoblots were incubated with c-Abl (8E9, Pharmingen), Fyn (15, Santa Cruz), chicken Src/v-Src (EC10), mammalian Src/Fyn/Yes (Src2, Santa Cruz), 80.8 anti-PDGFR-α (extracellular domain) (DeMali and Kazlauskas 1998), or pTyr (4G10, UBI) primary antibodies. For phospho-Abl blotting, a peptide containing phosphorylated Abl Y412 was produced ([H]CRLMTGDTpTyr,TAHA-[NH2]), and used to raise a polyclonal phospho-Abl antibody.
Immunoprecipitation and in vitro kinase assays
Endogenous c-Abl, chicken c-Src/v-Src, or c-Fyn was immunoprecipitated from 100 μg of cellular lysates or baculovirus insect cell lysates with anti-c-Abl antibodies (K12-amino-terminal, Santa Cruz; PEX4-carboxyl terminus; Ab3-carboxyl terminus, Oncogene Science), anti-Src (GD11) or anti-Fyn (Fyn3, Santa Cruz) antibody, and protein A– or G–Sepharose for 2 hr at 4°C. Kinase assays were performed as described (Nehme et al. 1997) using a stringent eight-wash protocol following immunoprecipitation, including two SDS–RIPA washes. The substrate GST–Crk (0.5 μg) or Sam68 (amino acids 331–443; Santa Cruz; 1 μg) was utilized for Abl or Src kinase assays, respectively, and 1 μg of GST–Crk, Sam68 (amino acids 331–443), GST alone, GST–Abl SH3 (amino acids 41–132), SH2–SH1 K290R (amino acids 137–671), SH2 alone (amino acids 132–230), or Abl carboxyl terminus (amino acids 739–1149) were used for in vitro phosphorylation by Src kinases. Triton-insoluble pellets from cells expressing oncogenic forms of Src were solubilized in RIPA buffer (50 mm Tris at pH 7.5, 150 mm NaCl, 1% Triton X-100, 1.5 mm MgCl2, 1 mm EGTA) and diluted into Triton lysis buffer for immunoprecipitation/in vitro kinase assay. GST–Abl proteins were described previously (Pendergast et al. 1991; Dai and Pendergast 1995). Phosphorylated proteins were quantified utilizing a PhosphorImager 445SI (Molecular Dynamics).
Subcellular fractionation
Stimulated and unstimulated NIH-3T3 or 10T1/2–EGFR cells were fractionated as described (Wang et al. 1994) using Dounce homogenization. Nuclear pellets were extracted with buffer D (20 mm HEPES–KOH at pH 7.6, 25% glycerol, 0.5 m NaCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA) supplemented with protease and phosphatase inhibitors, whereas membrane pellets were dissolved in RIPA buffer supplemented with protease and phosphatase inhibitors. Membrane fractions may also contain insoluble cytoskeletal-associated proteins, whereas nuclear fractions may contain perinuclear-associated structures. Equal amounts of protein from the various fractions were immunoprecipitated in Triton lysis buffer. The lower levels of c-Abl activation observed following cell fractionation compared with that obtained after total cell lysis may be due to protein denaturation following mechanical disruption of the cells, and to the increased amount of time required for subcellular fractionation prior to measurement of the c-Abl kinase activity.
Actin filament staining
MEFs were plated on coverslips, and 50% confluent wells were serum starved in 0.25% FBS for 3 days. Cells were stimulated for various amounts of time with PDGF-BB, and washed in ice-cold PBS. Cells were fixed for 10 min in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100 for 5 min, stained with rhodamine-conjugated phalloidin (Molecular Probes 1:100) for 10 min, and mounted in antifade solution (50% glycerol, 50% PBS, 25 mg/ml triethlenediamine).
Acknowledgments
We thank Sara Courtneidge, Tona Gilmer, Sarah Parsons, Thomas Parsons, Victor Tybulewicz, Frank McCormick, Robin Clark, Andrey Shaw, Warren Pear, Paul Stein, and Martin Broome for kindly providing reagents. We thank Anthony Means, Xiao Fan Wang, Joe Nevins, Patricia Zipfel, Kevin Courtney, and Mike Datto for reviewing the manuscript. This work was supported by the National Cancer Institute Grant CA70940 to A.M.P. and by a Glaxo-Wellcome Collaborative Research Program in Cancer Research Award. A.M.P. is a Whitehead Scholar and a Scholar of the Leukemia Society of America.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pende014@mc.duke.edu; FAX (919) 681-7148.
References
- Andoniou CE, Thien CBF, Langdon WY. Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO J. 1994;13:4515–4523. doi: 10.1002/j.1460-2075.1994.tb06773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome MA, Hunter T. Requirement for c-Src catalytic activity and the SH3 domain in platelet-derived growth factor BB and epidermal growth factor mitogenic signaling. J Biol Chem. 1996;271:16798–16806. doi: 10.1074/jbc.271.28.16798. [DOI] [PubMed] [Google Scholar]
- Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable association of pp60 src and pp59 fyn with the focal adhesion-associated protein tyrosine kinase, pp125 FAK. Mol Cell Biol. 1994;14:147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Pendergast AM. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes & Dev. 1995;9:2569–2582. doi: 10.1101/gad.9.21.2569. [DOI] [PubMed] [Google Scholar]
- Dai Z, Quackenbush RC, Courtney KD, Grove M, Cortez D, Reuther GW, Pendergast AM. Oncogenic Abl and Src tyrosine kinases elicit the ubiquitin-dependent degradation of the Abi family of proteins by a Ras-independent mechanism. Genes & Dev. 1998;12:1415–1424. doi: 10.1101/gad.12.10.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMali KA, Kazlauskas A. Activation of Src family members is not required for the platelet-derived growth factor B receptor to initiate mitogenesis. Mol Cell Biol. 1998;18:2014–2022. doi: 10.1128/mcb.18.4.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrosky-Ferlan PM, Corey SJ. Yeast two-hybrid in vivo association of the Src kinase Lyn with the proto-oncogene product Cbl but not with the p85 subunit of PI 3-kinase. Oncogene. 1997;14:2019–2024. doi: 10.1038/sj.onc.1201031. [DOI] [PubMed] [Google Scholar]
- Elkins T, Zinn K, McAllister L, Hoffmann FM. Genetic analysis of a Drosophila neural cell adhesion molecule: Interaction of fasciclin I and Abelson tyrosine kinase mutations. Cell. 1990;60:565–575. doi: 10.1016/0092-8674(90)90660-7. [DOI] [PubMed] [Google Scholar]
- Eriksson A, Siegbahn A, Westermark B, Heldin CH, Claesson-Welsh L. PDGF-α and β-receptors activate unique and common signal transduction pathways. EMBO J. 1992;11:543–550. doi: 10.1002/j.1460-2075.1992.tb05085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham VJ, Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 1998;17:81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham VJ, Unlu M, Brunton VG, Pitts JD, Wyke JA, Frame MC. Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J Cell Biol. 1996;135:1551–1564. doi: 10.1083/jcb.135.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Benet RL, Clark MJ, Hoffman FM. Drosophila abl tyrosine kinase in embryonic CNS axons: A role in axonogenesis is revealed through dosage-sensitive interactions with disabled. Cell. 1989;58:103–113. doi: 10.1016/0092-8674(89)90407-8. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Hill KK, Clark MJ, Hoffman FM. Dosage-sensitive modifiers of Drosophila abl tyrosine kinase function: prospero a regulator of axonal outgrowth, and disabled a novel tyrosine kinase substrate. Genes & Dev. 1993;7:441–453. doi: 10.1101/gad.7.3.441. [DOI] [PubMed] [Google Scholar]
- Giniger E. A role for Abl in Notch signaling. Neuron. 1998;20:667–681. doi: 10.1016/s0896-6273(00)81007-7. [DOI] [PubMed] [Google Scholar]
- Gotoh A, Miyazawa K, Ohyashiki K, Tauchi T, Boswell HS, Broxmeyer HE, Toyama K. Tyrosine phosphorylation and activation of focal adhesion kinase (p125 FAK) by BCR-ABL oncoprotein. Exp Hematol. 1995;23:1153–1159. [PubMed] [Google Scholar]
- Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: Critical mediators of multiple receptor pathways. Curr Opin Cell Biol. 1999;11:184–189. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- Heldin CH. Simultaneous induction of stimulatory and inhibitory signals by PDGF. FEBS Lett. 1997;410:17–21. doi: 10.1016/s0014-5793(97)00318-9. [DOI] [PubMed] [Google Scholar]
- Hill KK, Bedian V, Juang JL, Hoffmann FM. Genetic interactions between the Drosophila Abelson (Abl) tyrosine kinase and failed axon connections (Fax), a novel protein in axon bundles. Genetics. 1995;141:595–606. doi: 10.1093/genetics/141.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshmand-Rad R, Claesson-Welsh L, Wennstrom S, Yokote K, Siegbahn A, Heldin CH. Involvement of phosphotidylinositide 3′-kinase and Rac in platelet-derived growth factor-induced actin reorganization and chemotaxis. Exp Cell Res. 1997;234:434–441. doi: 10.1006/excr.1997.3636. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec L, Pendergast AM. The amphiphysin-like protein 1 (ALP1) interacts functionally with the cABL tyrosine kinase and may play a role in cytoskeletal regulation. Proc Natl Acad Sci. 1997;94:12390–12395. doi: 10.1073/pnas.94.23.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- Kypta RM, Goldberg Y, Ulug ET, Courtneidge SA. Association between the PDGF receptor and members of the src family of tyrosine kinases. Cell. 1990;62:481–492. doi: 10.1016/0092-8674(90)90013-5. [DOI] [PubMed] [Google Scholar]
- Lewis JM, Baskaran R, Taagepera S, Schwartz MA, Wang JYJ. Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc Natl Acad Sci. 1996;93:15174–15179. doi: 10.1073/pnas.93.26.15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZG, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JYJ. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- Loureiro J, Peifer M. Roles of Armadillo, a Drosophila catenin during central nervous system development. Curr Biol. 1998;8:622–632. doi: 10.1016/s0960-9822(98)70249-0. [DOI] [PubMed] [Google Scholar]
- Mellstrom K, Heldin CH, Westermark B. Induction of circular membrane ruffling on human fibroblasts by platelet-derived growth factor. Exp Cell Res. 1988;177:347–359. doi: 10.1016/0014-4827(88)90468-5. [DOI] [PubMed] [Google Scholar]
- Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–2384. [PubMed] [Google Scholar]
- Moro L, Venturino M, Bozzo C, Silengo L, Altruda F, Beguinot L, Tarone G, Defilippi P. Integrins induce activation of the EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998;17:6622–6632. doi: 10.1093/emboj/17.22.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AJ, Pendergast AM, Parmar K, Havlik MH, Rosenberg N, Witte ON. En bloc substitution of the Src homology region 2 domain activates the transforming potential of the c-Abl protein tyrosine kinase. Proc Natl Acad Sci. 1993;90:3457–3461. doi: 10.1073/pnas.90.8.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme A, Baskaran R, Aebi S, Fink D, Nebel S, Cenni B, Wang JYJ, Howel SB, Christen RD. Differential induction of c-Jun NH2-terminal kinase and c-Abl kinase in DNA mismatch repair-proficient and-deficient cells exposed to cisplatin. Cancer Res. 1997;57:3253–3257. [PubMed] [Google Scholar]
- Osherov N, Levitzki A. Epidermal-growth-factor-dependent activation of the Src-family kinases. Eur J Biochem. 1994;225:1047–1053. doi: 10.1111/j.1432-1033.1994.1047b.x. [DOI] [PubMed] [Google Scholar]
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving p210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- Pendergast AM. Nuclear tyrosine kinases: From Abl to WEE1. Curr Opin Cell Biol. 1996;8:174–181. doi: 10.1016/s0955-0674(96)80063-9. [DOI] [PubMed] [Google Scholar]
- Pendergast AM, Muller AJ, Havlik MH, Clark R, McCormick F, Witte ON. Evidence for regulation of the human Abl tyrosine kinase by a cellular inhibitor. Proc Natl Acad Sci. 1991;88:5927–5931. doi: 10.1073/pnas.88.13.5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw MW, Lea-Chou E, Wang JYJ. Rac is required for v-Abl tyrosine kinase to activate mitogenesis. Curr Biol. 1996;6:76–83. doi: 10.1016/s0960-9822(02)00424-4. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Salgia R, Li J-L, Lo SH, Brunkhorst B, Kansas GS, Sobhany ES, Sun Y, Pisick E, Hallek M, Ernst T, Tantravahi R, Chen LB, Griffin JD. Molecular cloning of human paxillin, a focal adhesion protein phosphorylated by p210 BCR-ABL. J Biol Chem. 1995;270:5039–5047. doi: 10.1074/jbc.270.10.5039. [DOI] [PubMed] [Google Scholar]
- Salgia R, Li J-L, Ewaniuk DS, Pear W, Pisick E, Burky SA, Ernst T, Sattler M, Chen LB, Griffin JD. BCR/ABL induces multiple abnormalities of cytoskeletal function. J Clin Invest. 1997;100:46–57. doi: 10.1172/JCI119520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers CL, McLaughlin J, Goga A, Havlik M, Witte O. The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell. 1994;77:121–131. doi: 10.1016/0092-8674(94)90240-2. [DOI] [PubMed] [Google Scholar]
- Schneller M, Vuori K, Ruoslahti E. αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO J. 1997;16:5600–5607. doi: 10.1093/emboj/16.18.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorski T, Nieborowska-Skorska M, Wlodarski P, Wasik M, Trotta M, Kanakaraj P, Salomoni P, Antonyak M, Martinez R, Majewski M, Wong A, Perussia B, Calabretta B. The SH3 domain contributes to BCR/ABL-dependent leukemogenesis in vivo: Role in adhesion, invasion and homing. Blood. 1998;91:406–418. [PubMed] [Google Scholar]
- Suetsugu S, Miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 1998;17:6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JYJ, Hope TJ. Nuclear-cytoplasmic shuttling of c-Abl tyrosine kinase. Proc Natl Acad Sci. 1998;95:7457–7462. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. The three faces of profilin. Cell. 1993;75:835–838. doi: 10.1016/0092-8674(93)90527-w. [DOI] [PubMed] [Google Scholar]
- Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twamley-Stein GM, Pepperkok R, Ansorge W, Courtneidge SA. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc Natl Acad Sci. 1993;90:7696–7700. doi: 10.1073/pnas.90.16.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains and bundling activity. J Cell Biol. 1994;124:325–340. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veithen A, Cupers P, Baudhuin P, Courtoy PJ. v-Src induces constitutive macropinocytosis in rat fibroblasts. J Cell Sci. 1996;109:2005–2012. doi: 10.1242/jcs.109.8.2005. [DOI] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three dimensional basement membrane breast cultures: A different perspective in epithelial biology. Proc Natl Acad Sci. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JYJ. Abl tyrosine kinase in signal transduction and cell cycle regulation. Curr Opin Genet Dev. 1993;3:35–43. doi: 10.1016/s0959-437x(05)80338-7. [DOI] [PubMed] [Google Scholar]
- ————— Cellular responses to DNA damage. Curr Opin Cell Biol. 1998;10:240–247. doi: 10.1016/s0955-0674(98)80146-4. [DOI] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Xua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Wen ST, Jackson PK, Van Etten RA. The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J. 1996;15:1583–1595. [PMC free article] [PubMed] [Google Scholar]
- Weng Z, Taylor JA, Turner CE, Brugge JS, Seidel-Dugan C. Detection of Src homology 3-binding proteins including paxillin, in normal and v-Src transformed Balb/c 3T3 cells. J Biol Chem. 1993;268:14956–14963. [PubMed] [Google Scholar]
- Wennstrom S, Siegbahn A, Yokote K, Arvidsson A, Heldin C, Mori S, Welsh L. Membrane ruffling and chemotaxis transduced by the PDGF-β receptor require the binding site for phosphotidylinositol 3′ kinase. Oncogene. 1994;9:651–660. [PubMed] [Google Scholar]
- Westermark B, Siegbahn A, Heldin CH, Claesson-Welsh L. B-type receptor for platelet-derived growth factor mediates a chemotactic response by means of ligand-induced activation of the receptor protein-tyrosine kinase. Proc Natl Acad Sci. 1990;87:128–132. doi: 10.1073/pnas.87.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills Z, Marr L, Zinn K, Goodman CS, VanVactor D. Profilin and the Abl tyrosine kinase are required for motor axon outgrowth in the Drosophila embryo. Neuron. 1999;22:291–299. doi: 10.1016/s0896-6273(00)81090-9. [DOI] [PubMed] [Google Scholar]
- Wilson LK, Luttrell DK, Parsons JT, Parsons SJ. pp60 c-src tyrosine kinase, myristylation, and modulatory domains are required for enhanced mitogenic responsiveness to epidermal growth factor seen in cells overexpressing c-src. Mol Cell Biol. 1989;9:1536–1544. doi: 10.1128/mcb.9.4.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZM, Huang Y, Whang Y, Sawyers C, Weichelbaum R, Kharbanda S, Kufe D. Role of c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382:272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]