

Abstract
Abnormally expanded polyglutamine domains are associated with at least nine neurodegenerative diseases, including Huntington’s disease. Expansion of the glutamine region facilitates aggregation of the impacted protein, and aggregation has been linked to neurotoxicity. Studies of synthetic peptides have contributed substantially to our understanding of the mechanism of aggregation, because the underlying biophysics of polyglutamine-mediated association can be probed independent of their context within a larger protein. In this report, interrupting residues were inserted into polyglutamine peptides (Q20), and the impact on conformational and aggregation properties was examined. A peptide with 2 alanine residues formed laterally-aligned fibrillar aggregates which were similar to the uninterrupted Q20 peptide. Insertion of 2 proline residues resulted in soluble, nonfibrillar aggregates, which did not mature into insoluble aggregates. In contrast, insertion of a β-turn template DPG rapidly accelerated aggregation and resulted in a fibrillar aggregate morphology with little lateral alignment between fibrils. These results are interpreted to indicate that (a) long-range nonspecific interactions lead to the formation of soluble oligomers, while maturation of oligomers into fibrils requires conformational conversion, and (b) that soluble oligomers dynamically interact with each other, while insoluble aggregates are relatively inert. Kinetic analysis revealed that the increase in aggregation caused by the DPG insert is inconsistent with the nucleation-elongation mechanism of aggregation featuring a monomeric β-sheet nucleus. Rather, the data support a mechanism of polyglutamine aggregation by which monomers associate into soluble oligomers, which then undergo slow structural rearrangement to form sedimentable aggregates.
Keywords: kinetic analysis, oligomers, peptide collapse, polyglutamine, nucleation-elongation
INTRODUCTION
The “expanded CAG” diseases are a collection of nine known neurodegenerative diseases, of which Huntington’s disease is the most common.1 These diseases derive their name from the abnormal expansion of CAG codons in disease-specific genes. Expression of these genes results in the production of proteins with expanded polyglutamine (polyQ) domains. Neuronal inclusions containing aggregates of the affected proteins are characteristic features of each of the CAG diseases.2, 3 Expansion of the polyQ region beyond a critical length results in a disease phenotype, with longer polyQ regions corresponding to a greater severity of symptoms and an earlier age of onset.4 The proteins implicated in the CAG diseases share no sequence, structural, or compositional homologies outside of the polyQ region,5 which itself varies in both length and location within the context of the affected proteins.6
Several studies have linked aggregation of polyQ-containing proteins to cellular toxicity. For example, overexpression of chaperone proteins slowed aggregation and decreased toxicity in both cellular and animal models.7–9 Both loss-of-normal-function and gain-of-toxic-function mechanisms have been posited to explain toxicity, but the case for the latter is stronger.10 Although evidence for the gain-of-toxic-function hypothesis is strong, the toxic species itself remains unresolved. Some researchers have suggested that insoluble polyQ aggregates are the toxic species,11, 12 others have found soluble oligomers to be cytotoxic,7, 13 and still others have suggested that a specific β-sheet conformation of the polyQ monomer is the most deleterious.14
Synthetic polyQ peptides have been gainfully employed as a model system for examining polyglutamine-mediated aggregation, in the absence of confounding contributions from the protein context into which the polyamino acid domain is placed.12, 15–21 PolyQ peptides recapitulate much of the relevant behavior of polyQ-containing proteins. For example, just as longer polyQ regions in disease-related proteins lead to earlier ages of onset in vivo,4 longer polyQ regions in synthetic peptides result in faster aggregation in vitro.11 Importantly, delivery of synthetic polyQ peptide aggregates to the nucleus causes cell death,12 a demonstration that toxicity does not require the disease-related protein.
Both experimental and theoretical studies have contributed to insights into the conformational ensemble adopted by polyQ monomers. Circular dichroism (CD) and NMR have both shown that polyQ peptide monomers lack regular secondary structural elements.22–25 Evidence exists to support polyQ peptides adopting both extended26 and collapsed27 conformations. Recent work in our lab indicated that the preference for collapsed or extended states is directly related to polyQ length; peptides with fewer than 16 residues were relatively well solvated and extended, whereas peptides with more than 16 residues preferred relatively compact conformations.19 This length-dependent pattern can be explained by considering that a preference for β-strand dihedrals, which tend to extend the polypeptide chain, competes with longer-range attractive contacts, which tend to collapse the chain and become more significant as the number of glutamines increases.28
Two mechanisms linking conformation of polyQ monomers to aggregation have been proposed. In an adaptation of the nucleation-elongation model, a thermodynamically unfavorable β-sheet monomer, in equilibrium with the bulk disordered monomer, is posited to serve as the nucleus.11, 29 Repeated rounds of addition of unfolded monomer to this nucleus produces fibrillar aggregates. A schematic of this model is presented in Fig. 1 (right-hand box). Indeed, monomer loss data for synthetic peptides containing from 28 to 47 glutamines were fitted to a kinetic model based on the nucleation elongation mechanism.11, 29 Recently, this mechanism has been modified to incorporate a change in nucleus size from monomer to tetramer as the number of glutamines decreases from 26 to 23.30 However, a number of alternative kinetic mechanisms fit these data equally well.31, 32 In an alternative mechanism, which we will call the association-conformational conversion model, disordered monomers self-associate via hydrophobic interaction into soluble oligomers. Self-association is proposed to increase with increasing polyQ length because increased intramolecular contacts reduce solvent-peptide hydrogen bonding and render the solvent effectively more poorer.19 Soluble oligomers could be considered as microphases with locally high peptide concentrations. Within the ‘liquid-like’ interior of the oligomers,32 conformational rearrangement leads to formation of β-sheet ‘nodes’ which then propagate through the oligomer, ultimately leading to formation of insoluble fibrillar aggregates with high β-sheet structure and little water.19 This mechanism is schematically depicted in Fig. 1 (left-hand box).
Figure 1.
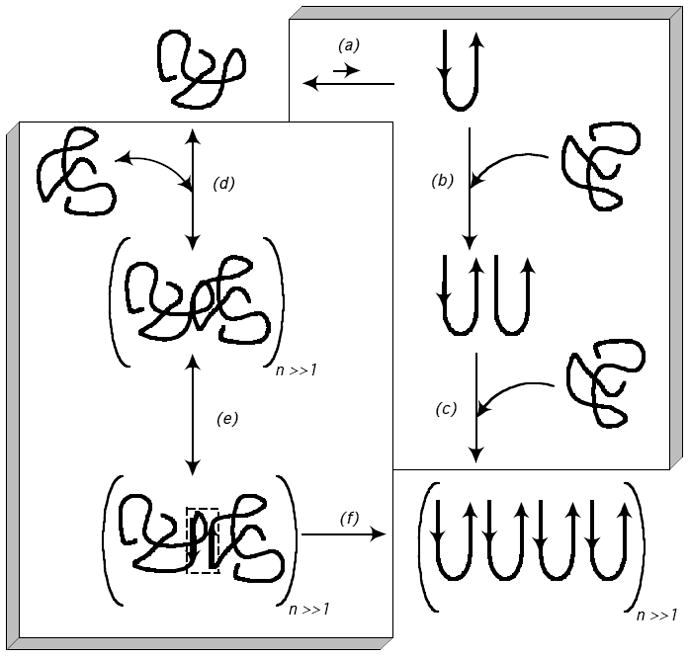
Proposed mechanisms of polyQ peptide aggregation from unfolded monomer to insoluble fibrillar aggregate. The box on the right-hand side outlines the nucleation-elongation mechanism (steps a-c), as described in e.g. references 11, 30. (a) Unfolded monomer is in rapid equilibrium with a thermodynamically unfavorable β-sheet nucleus. (b) β-sheet nucleus serves as a template for addition of unfolded monomer. (c) In repeated rounds of monomer addition, fibrils become elongated. The box on the left-hand side outlines the association-conformational conversion mechanism (steps d-f), as described in e.g. references 19, 32. (d) Monomers lacking regular secondary structure rapidly associate into large soluble oligomers, driven by hydrophobic interactions. (e) Within the large ‘liquid-like’ oligomers, conformational re-arrangement leads to formation of β-sheet nodes (indicated by the small dashed box). (f) β-sheet formation propagates throughout the oligomers, producing insoluble fibrillar aggregates.
Interruptions of the polyQ sequence have interesting effects on the aggregation behavior. Proline33–35 and histidine36 interruptions have been found to slow aggregation, although the impact of proline residues can be lessened if they are positioned such that they are easily incorporated into a β-turn.33 Conversely, peptides containing D-proline glycine, a β-turn template, display more rapid aggregation kinetics.33 Given these reports, we hypothesized that analysis of aggregation of polyQ peptides containing judiciously chosen interrupting residues provides an opportunity to gain further insights into the mechanism of polyQ aggregation.
In this study, we introduced interrupting residues into a polyQ peptide containing 20 glutamines and examined the impact of the interrupting residues on conformation and aggregation. The peptides are of the type K2WQ10XXQ10AK2, where XX represents the interrupting residues. The interrupting residues used include ProPro, AlaAla, and the β-turn template D-ProGly. Peptides will subsequently be referred to by their interrupting residues (PP, AA, and PG, respectively) or as Q20 for the uninterrupted control. We chose interrupting residues which we believed would have observable impacts on the kinetic pathway, in order to challenge and further refine that pathway. We also hoped to explore more thoroughly the connection between monomer conformation and aggregation, and the transition from soluble to insoluble aggregates. As two consecutive proline residues are expected to adopt an extended conformation,37 we predicted the PP peptide would fail to collapse, and subsequently fail to aggregate. At the other end of the spectrum, the β-turn template of the PG peptide should predispose it toward forming β-sheet structures that are characteristic of mature, insoluble aggregates.11 We wondered if imposition of a β-turn would yield a peptide effective at initiating new fibrils, or at adding to pre-existing aggregates. The double alanine interruption was chosen as a subtle perturbation to verify that not all interruptions to the glutamine region result in significant changes in aggregation behavior. Our results from this study offer new insights into and refinement of the mechanism of polyQ peptide aggregation.
RESULTS
Peptides of the type K2WQ10XXQ10AK2 were synthesized, where XX represents the interrupting residues. Flanking lysine residues were added to the polyQ core to increase solubility, and tryptophan was used for concentration determination and as a fluorescence donor for FRET measurements. The N-terminus was acetylated and the C-terminus was amidated to eliminate charge interactions of the termini. An additional set of peptides identical to the first was utilized for FRET studies, in which the FRET acceptor dansylated lysine was substituted for alanine.
Conformation of monomers
Circular dichroic (CD) spectra were collected for freshly-prepared solutions of Q20, AA, PP, and PG (Fig. 2). In all cases, spectra were collected on freshly prepared solutions, where peptides is virtually 100% monomeric (vide infra). The observed secondary structure can thus be interpreted as that of the monomer, although some, presumably minor, contribution from oligomers cannot be ruled out. spectra of AA and Q20 were virtually identical, indicating the AA insert had essentially no effect on the secondary structure. Both AA and Q20 displayed spectra characteristic of disordered peptides, consistent with other reports.15, 18, 24 The spectrum of PP differed significantly from that of Q20, with a stronger negative band around 200 nm and a less negative band around 222 nm. These changes are indicative of some sampling of poly-Pro II-like conformations in the PP peptide.38, 39 The spectrum of PG also differed significantly from that of Q20. We interpret the smaller negative peak at 212 nm and a weaker negative band at 200 nm to be indicative of increased β-turn content,40 as expected.
Figure 2.

CD spectra of interrupted polyQ peptides. Peptide stock solutions were diluted into a pH 7.4 phosphate buffer (10 mM buffer salts and 140 mM NaF) to a concentration of 40 μM peptide and filtered through a 0.45-μm membrane directly into a cuvette. Data were normalized to per-residue molar ellipticity.
We next examined the average extension of the interrupted polyQ peptides in solution using FRET, and report the data as the effective distance R between donor and acceptor (Table 1). All data were collected on freshly-prepared solutions at low concentration, where the peptides are virtually 100% monomer. AA was slightly more collapsed than Q20, especially given the two additional residues in AA. This decrease in R is consistent with a smaller R in peptides with a long polyalanine rather than polyglutamine core (manuscript in preparation). PP was the most extended of the peptides, but the difference between PP and AA was not statistically significant. The smallest value of R was observed with PG, consistent with the presence of a β-turn.
Table 1.
Length of the polyQ region determined by FRET. Fluorescence spectra of peptides with and without the fluorescence acceptor dansyl were collected and used to calculate the apparent distance between the fluorophores R. Four readings were taken for each peptide and reported as mean ± standard deviation.
| R (Å) | |
|---|---|
| Q20 | 19.7 ± 0.1 |
| PP | 19.9 ±0.2 |
| AA | 19.4 ±0.4 |
| PG | 18.3 ±0.4 |
Monomer loss and sedimentation kinetics
Samples were injected onto a calibrated size exclusion column (3–70 kDa). The elution profile was dominated by a large peak which we attributed to monomer. All the peptides ran larger than their true monomer molecular weights, because column calibration was based on folded proteins and it is well-known that unfolded polypeptides typically elute at an earlier time (larger hydrodynamic volume) than folded proteins of similar molecular weight. Specifically, PP monomer eluted at a molecular weight of 5764 Da (3587 expected), AA at 5437 (3534 expected), PG at 5350 (3546 expected), and Q20 at 5102 (3391 expected). The results were consistent with the FRET data showing PP to be the most extended and PG to be the most compact of the interrupted peptides.
The area of the monomer peak was calculated for each injection and compared to the area of the monomer peak at day 0 to determine the percentage of peptide present as monomer (Fig. 3a). Monomer loss kinetics for Q20 and AA were almost identical. With PP, there was at most only a slight decrease in monomer, even after 50 days. In sharp contrast, the percentage monomer decreased rapidly for the PG peptide, reaching near zero within a few days.
Figure 3.

Aggregation kinetics of interrupted polyQ peptides. Peptides [PP (
 ), AA (
), AA (
 ), Q20 (△), and PG (
), Q20 (△), and PG (
 )] were diluted into PBSA at 40 μM, aliquoted into tubes, and incubated at 37°C. (A) Monomer loss kinetics. At the times indicated, a tube was centrifuged and the supernatant was analyzed by SEC. Percentage of monomer recovered is reported as the peak area of the monomer divided by the peak area of the monomer at time zero. (B) Sedimentation kinetics. Total soluble peptide was determined using a bichinchonicinic acid assay. Data are reported as the wt% of the peptide soluble, calculated from the ratio of the supernatant to the total concentration in an uncentrifuged sample. Lines connect data points as an aid to the eye.
)] were diluted into PBSA at 40 μM, aliquoted into tubes, and incubated at 37°C. (A) Monomer loss kinetics. At the times indicated, a tube was centrifuged and the supernatant was analyzed by SEC. Percentage of monomer recovered is reported as the peak area of the monomer divided by the peak area of the monomer at time zero. (B) Sedimentation kinetics. Total soluble peptide was determined using a bichinchonicinic acid assay. Data are reported as the wt% of the peptide soluble, calculated from the ratio of the supernatant to the total concentration in an uncentrifuged sample. Lines connect data points as an aid to the eye.
A sedimentation assay was used to study the growth of insoluble aggregates (Fig. 3b) by measuring the concentration of peptide remaining in the supernatant, which includes both monomer and soluble oligomers, as a function of time. Insoluble aggregates of Q20 and AA were observed after a lag of ~7 days or ~12 days, respectively. There was a delay in sedimentation of AA relative to Q20 during the first ~2 weeks, after which the two peptides tracked very closely to each other. PP did not form insoluble aggregates, even after 50 days. PG rapidly formed insoluble aggregates, with the percent soluble decreasing to less than 10% after 5 days of incubation. In general, the percentage soluble peptide closely paralleled, but slightly lagged behind, the percentage monomer determined from SEC (Fig. 3a).
Close examination of the chromatograms revealed a small peak that eluted before the large monomer peak (Fig. 4). The area of this peak was less than 1% of the area of the monomer peak (inset, Fig. 4a). The calculated molecular weights of the peaks based on column calibration were 14,760 Da for PP, 13,505 for AA, 12,482 for Q20, and 14,620 for PG, or roughly 4 times the expected molecular weights of the monomer. However, because the monomer itself ran larger than expected, multimers might also run larger than expected. Using the monomer elution values as reference, these peaks appear to contain dimers or trimers. Interestingly, this species was selectively removed once insoluble aggregates were observed by sedimentation assay. In the case of PP, the peak remained throughout the course of the experiment, as sedimentable aggregates were never detected. For both AA and Q20, the peak was present from the start, grew slightly at the 2 day mark, and subsequently decreased to near zero after 28 days. Monomer was readily detected at this time point, indicating that the intermediate peak was selectively removed from the sample. This phenomenon is even more pronounced in the PG sample, in which we observed the peak at only the 0 hour time point. These data suggest that these putative oligomers preferentially adsorb to insoluble aggregates.
Figure 4.

Close-ups of SEC chromatograms of interrupted polyQ peptides, focusing on peak eluting before the monomer. Peptides were diluted into PBSA at 40 μM, aliquoted into tubes, and incubated at 37°C. At the times indicated, the peptides were analyzed by SEC. Chromatograms are plotted as elution volume Ve divided by void volume Vo and adjusted to zero absorbance at Ve/Vo = 1.3 to correct for baseline drift. Data is displayed over a time of 0 to 28 days for PP, AA, and Q20, and 0 to 71 hours for PG. (a) PP. (b) AA. (c) Q20. (d) PG. The inset of (a) displays the full chromatogram for PP at day 0.
PG as a ‘seed’
PolyQ aggregation is promiscuous, in that polyQ peptides of different lengths are capable of aggregating together.16 Since PG aggregates rapidly, we explored whether it could accelerate the formation of insoluble aggregates when mixed with other polyQ peptides. Further, we reasoned that, if a β-sheet monomer did serve as a nucleus, then PG should act as a stabilized nucleus. If so, the addition of PG to Q20 should greatly increase the effective concentration of nuclei, eliminating the lag in the formation of insoluble aggregates and accelerating aggregation. We prepared solutions of Q20, AA, and PP at 35 μM, and added 5 μM PG. A 5 μM PG control and a mixture containing 40 μM Q20 and 40 μM PG were also prepared. The sedimentation behavior of the mixtures was examined over the course of 22 days (Fig. 5). Even at 5 μM, PG produced insoluble aggregates. In contrast, no insoluble aggregates were detected in the PP/PG mixture, even after 22 days. Compared to the pure solutions, the lag time before onset of sedimentation of AA and Q20 was reduced by spiking in 5 μM PG: insoluble aggregates were detected at 4–5 days, and subsequent aggregation occurred more rapidly.
Figure 5.

Sedimentation kinetics of interrupted polyQ peptides mixed with PG peptide. Peptides [PP (
), AA (
), Q20 (△)] were diluted into PBSA at 35 μM, spiked with 5 μM PG, and aliquoted into tubes. A 5 μM PG only control (
) and a mixture of 40 μM Q20 with 40 μM PG (▲) were also prepared in PBSA Samples were incubated at 37°C, and at the times indicated, a tube was centrifuged and the supernatant was analyzed for total soluble peptide using a bicinchoninic acid assay. Data are reported as the wt% of the peptide soluble, calculated from the ratio of the supernatant to the total concentration in an uncentrifuged sample. Lines connect data points as an aid to the eye.
Analysis with nucleation-elongation model
We explored whether the nucleation elongation model, in which a β-sheet monomer is postulated to be the nucleus,11 can account quantitatively for these results. The nucleation constant Kn describes the equilibrium between the nucleus and the bulk monomer, while k+ represents the elongation rate for both the nucleus and fibrils. The differential equations which govern monomer loss and the formation of fibrils are reported in the Supplemental Information. Data for monomer loss can be fitted to these coupled differential equations; however, if it is assumed that Kn[M] ≪ [F],29 only the lumped parameter Kn k+2 can be extracted from monomer loss data.11 Parameters were regressed using least-squares fitting of sedimentation data from 100% soluble to 15% soluble and numerical solution of the differential equations. Fitted values of Kn k+2 are displayed in Table 2. As a point of comparison, Chen et al. found Kn k+2 = 0.001128 M−2s−2 for Q28.11 Our values are reasonably consistent with this number, considering the expected increase in Kn k+2 with increasing glutamine length.11 Kn k+2 are similar for Q20 and AA, while Kn k+2 for PG is 543 times that of Q20. As PP was not found to form insoluble aggregates, no Kn k+2 value could be determined.
Table 2.
Kinetic parameters of interrupted polyQ peptides for nucleation elongation mechanism.
| Kn k+2 (M−2s−2) | |
|---|---|
| Q20 | 0.000235 |
| PP | N/A |
| AA | 0.000197 |
| PG | 0.127 |
The kinetic analysis of the nucleation elongation mechanism was expanded to apply to the mixtures of Q20 and PG. Differential equations for the concentrations of Q20 and PG monomer were derived while accounting for monomer and nuclei of both Q20 and PG, as described in Supplemental Information. Given that Knk+2 for PG is 543 times that of Q20, we considered two possibilities. First, we assumed that the higher value of Knk+2 for PG was entirely due to an increase in Kn; this would be true if PG were an especially efficient nucleus. Second, we examined the case where the increase in Knk+2 is attributable entirely to an increase in k+; this case would be true if PG is more efficient than Q20 at elongating nuclei and fibrils.
Model predictions were compared to sedimentation data for mixtures of 35 μM Q20 with 5 μM PG (Fig. 6a) and 40 μM Q20 with 40 μM PG (Fig. 6b). If the greater parameter value for PG is attributed entirely to an increase in Kn, the model predicts a much faster aggregation rate than what we observed experimentally. However, an increase in k+ yields predicted values that are similar to the experimental data (Fig. 6). Thus, if the data are fit to the nucleation elongation model, PG appears to accelerate aggregation primarily through an enhancement of the elongation rate constant, and not via stabilization of nucleation. This is the opposite effect than what one would expect if a β-sheet monomer were indeed the aggregation nucleus and elongation was by the disordered monomer. Thus, the nucleation-elongation mechanism of Fig. 1 is contradicted by this analysis.
Figure 6.

Nucleation elongation model applied to sedimentation kinetics of interrupted polyQ peptides mixed with PG peptide. The increase in the value of Kn k+2 for PG was applied strictly to Kn (dashed line) and strictly to k+2 (solid line). Data points are from (a) 35 μM Q20 with 5 μM PG (△) (b) 40 μM Q20 with 40 μM PG (▲).
Recently, Kar et al. proposed that, for shorter polyQ peptides, the nucleus is a β-sheet tetramer rather than monomer.30 We re-analyzed our data assuming a tetrameric nucleus; although the numerical values and units of the model parameters change, the conclusion does not (not shown).
Detection of soluble oligomers
In the association-conformational conversion mechanism (Fig. 1), soluble oligomers are proposed as intermediates in the aggregation pathway. Laser light scattering was used to directly test for the presence of soluble oligomers immediately after dilution into PBSA. The normalized scattered intensity, Inorm, is sensitive to an increase in the number concentration of oligomers and/or an increase in oligomer size. If no aggregates were present, Inorm would be expected to remain at 0, as it is in the stock solution (low pH, no salt) prior to dilution into PBSA. Inorm for Q20, AA and PP were all roughly the same, but still greater than zero, indicating the presence of soluble oligomers (Fig. 7a). For PG, Inorm was much greater than that of the other peptides, indicating the presence of a much larger number and/or size of aggregates. Measurements of the z-averaged hydrodynamic radius, Rhz (Fig. 7b), which is heavily weighted towards larger aggregates, revealed several interesting phenomena. The initial size of soluble oligomers was about the same in all samples (~50 nm). For AA and Q20 oligomers grew in size several-fold over the 2-hr measurement period. In contrast, PP oligomer size increased only slightly during the same time frame. Despite the large Inorm of PG (Fig. 7a), the oligomers themselves remained relatively small and stable in size. Thus, PG produced a larger population of oligomers that remained relatively small in size.
Figure 7.

Dynamic light scattering of interrupted polyQ peptides. Peptides [PP (
), AA (
), Q20 (△), and PG (
)] were diluted into PBSA at 40 μM and filtered through a 0.45-μm membrane directly into a light scattering cuvette. (a) Intensity of scattered light at 90°. Scattering due to the solvent was subtracted and results were normalized to the scattering intensity of toluene to account for changes in laser strength and aperture. (b) Apparent z-averaged hydrodynamic radius Rhz, as determined from cumulants analysis of autocorrelation data collected at 90° scattering angle. For all samples, the polydispersity index was typically 0.4, indicating a fairly broad size distribution.
Aggregate morphology
Peptide solutions (40 μM) were incubated for 50 days at 37°C and examined by transmission electron microscopy (TEM). Both Q20 (Fig. 8a) and AA (Fig. 8b) formed fibrillar aggregates. The fibrils showed a high degree of lateral alignment, as observed previously for polyQ peptides.19 PG aggregates (Fig. 8c) were also fibrillar but substantially less lateral alignment was observed compared to aggregates of Q20. Previous observations revealed that early polyQ aggregates lack the lateral alignment seen in mature aggregates.19 To see if the reduction in lateral alignment could be attributed to entanglement effects, PG was incubated at lower concentrations or shorter times. After 10 days of aggregation at 10 μM or 4 μM, and after 6 hours at 40 μM, the fibrils of PG again lacked much of the lateral alignment characteristic of the uninterrupted peptides (Supplemental Fig. 1). We interpret this to indicate that the PG interruption reduces interactions between individual fibrils compared to uninterrupted peptides. Finally, in striking contrast, globular structures of broad size distribution, including a few large agglomerates, were the dominant features in TEM images of PP (Fig. 8d); a few aggregates with a partially fibrillar morphology were also observed. The size of some of the aggregates in the TEM images is larger than the hydrodynamic radius determined by light scattering (Fig. 7b) because TEM images were taken after 50 days aggregation while light scattering data were collected for only 2 hours.
Figure 8.

TEM image of interrupted polyQ peptide aggregates. Samples were prepared at 40 μM in PBSA and then incubated at 37°C for 50 days prior to imaging. Images are representative of a large number of fields examined. The length of the white bar is 200 nm in all panels. (a) Q20. (b) AA. (c) PG. (d) PP.
DISCUSSION
Insights into polyQ aggregation pathway from interrupting peptides
Two competing mechanisms have been proposed to describe the pathway by which polyglutamine monomers are converted to insoluble fibrillar aggregates (Fig. 1). In the nucleation-elongation mechanism (steps (a), (b) and (c) of Fig. 1), a rare β-sheet monomer serves as nucleus.11 [This mechanism has recently been modified to incorporate a smooth increase in the size of the nucleus from monomer to tetramer as the number of glutamines in the polypeptide decreases from 26 to 23.30] Previous work in our group led to the suggestion of an alternative mechanism (steps (d), (e) and (f) of Fig. 1), in which polyQ peptides rapidly associate into soluble oligomers due to hydrophobically-driven interactions,19 then subsequent conformational rearrangement to β-sheet structure within the oligomer leads to the formation of insoluble fibrillar aggregates.
By evaluating the effects of appropriately chosen interrupting residues, we hoped to gain further insight into the mechanism of polyglutamine-mediated aggregation. The AA insert served to test whether disruption of the glutamine stretch intrinsically prevents (or accelerates) aggregation. We chose a PP insert with the expectation of stopping aggregation by forcing the peptide into an extended state, while the PG insert was expected to accelerate aggregation because it is a template for a β-turn.
The AA interruption has only a minor effect on conformation and aggregation, demonstrating that interruption of the glutamine region per se is not sufficient to prevent the transition to insoluble aggregates. Q20 and AA have nearly identical CD spectra (Fig. 2), with both lacking regular secondary structure. The distance between donor and acceptor was slightly lower for AA compared to Q20 (Table 1), suggesting that AA is slightly more compact than Q20, possibly due to the higher hydrophobicity of alanine. The fibrillar morphology observed for AA was virtually indistinguishable from that of Q20 (Fig. 8). Light scattering characterization (Fig. 7), as well as monomer-loss and sedimentation kinetics (Fig. 3) were very similar for AA and Q20. However, there were some small but intriguing differences. The lag phase for the transition to insoluble aggregates was slightly longer for AA than for Q20 (Fig. 3b), and the soluble oligomers grew to a slightly larger size (Fig. 7b). Conversion within the oligomer from disordered to β-sheet is presumably driven by energetic gains from hydrogen bonding and ordered hydrophobic interactions, but hindered by loss of conformational entropy. We speculate that the slight differences between AA and Q20 could be attributable to two factors: (1) the alanine side chain, unlike glutamine, does not participate in hydrogen bonding, and (2) efficient packing of the long glutamine side chain is a significant driving force favoring ordered aggregates that is relatively unimportant for the short alanine side chain.
The insertion of 2 proline residues modestly altered monomer conformation, and dramatically altered the aggregation properties. Circular dichroism (Fig. 2) revealed an increase in poly-Pro II-like structure in this peptide as compared to the Q20 control. From a comparison of SEC elution times and FRET R, we conclude that PP prefers to occupy slightly more extended states than Q20 or AA. However, the rigidity of the PP insert appears to be mostly a local effect, and does not have a large global effect on the average distance between the fluorophores located near the ends of the peptide. PP failed to form insoluble aggregates, and there was not much loss of monomer (Fig. 3). However, soluble aggregates were detected by light scattering for PP in PBSA. TEM of an aged sample revealed a population of globular aggregates of broad size distribution, along with a few fibrillar aggregates.. These data demonstrate that the PP insert has a profound effect on the aggregation kinetics of polyQ peptides. PP serves as a strong inhibitor of β-turn formation. If the nucleation-elongation mechanism were applicable, one would anticipate that PP would markedly reduce Kn, the equilibrium constant between β-sheet monomer and unfolded monomer (step (a) in Fig. 1), and therefore greatly inhibit any further self-association, because it could not readily adopt the β-sheet conformation required of the putative nucleus. However, the presence of globular oligomers (Fig. 7 and 8) indicates that the PP peptides still have a proclivity for associating with each other (step (d) in Fig. 1). We interpret this to indicate a specific conformation of the monomer is not necessary to initiate self-association. Instead, we hypothesize that PP strongly inhibits conversion of soluble oligomers to fibrillar aggregates (steps (e) and (f) of Fig. 1).
The D-proline-glycine insert PG was used as a β-turn template, shown by others to increase the rate of aggregation when inserted in an appropriate position to encourage β-sheet formation.33 Increased β-turn content was confirmed by CD (Fig. 2), and FRET revealed the peptide to be relatively compact, as would be expected. All techniques used to study aggregation kinetics revealed rapid aggregation, with almost complete loss of soluble material after only 5 days. Light scattering revealed the presence of many comparatively small soluble oligomers prior to the onset of insolubility (Fig. 7). If β-sheet content is indeed important for the aggregate to sediment, it makes sense that the PG oligomers would not grow large prior to becoming insoluble, as the oligomer does not need to undergo as significant a structural rearrangement from disordered to fibrillar (step (e)) as the other peptides. We hypothesized that, if the nucleation-elongation mechanism shown in Fig. 1 were applicable, then the β-turn template of PG should effectively increase Kn (step (a)), because PG should be able to readily adopt the putative nucleus conformation. Indeed, a small amount of PG added to Q20 increased the rate of sedimentation. However, a fit of the sedimentation data of Q20/PG mixtures to the nucleation-elongation model demonstrated that PG increases k+ but not Kn; in other words, PG acts not as an efficient nucleus, but rather as an effective ‘elongator’. One can interpret this result more readily in terms of the association-conformational conversion model: by dint of its β-turn template, PG serves as a β-sheet node and is an effective catalyst for conformational conversion to fibrils.
In summary, our experiments with polyQ peptides containing interrupting residues demonstrate the significance of monomer conformation in influencing aggregation kinetics. Our data are more consistent with the association-conformational conversion mechanism than with the nucleation-elongation mechanism. Rather than a monomeric β-sheet nucleus, we propose the formation of soluble oligomers through hydrophobically-driven self-association of monomers. These oligomers could be thought of as a microphase with a locally high concentration of peptides. For Q20 or AA oligomers, conversion to the fibrillar aggregate is a slow process, as many conformations may be sampled before a ‘β-sheet node’ is formed and then propagated through the entire oligomer. Sedimentation rate is increased in PG because the imposed β-turn predisposes this peptide to the mature aggregate structure, whereas sedimentation and fibril formation is inhibited in PP because this interruption prevents β-turns.
Aggregate-aggregate association
The mechanisms illustrated in Fig. 1 do not consider the possibility of aggregate-aggregate association, yet our data hint that these interactions exist. First, it is interesting to note in the SEC data the appearance of a small peak with an elution time corresponding to a small multimer (Fig. 4). Although the peak appears as a small multimer, it could be indicative of larger, loosely associated soluble oligomers that partially fall apart upon dilution or under shear stress on the SEC column. Or, it is possible that this peak corresponds to alternate monomeric conformers that adsorb weakly to the column, but we believe this is less likely given the normal peak shape. This species is present in all of the samples initially, but is selectively reduced once insoluble aggregates begin to form. These data suggest that the kinetic model should be amended to include an association step, in which soluble oligomers are efficiently recruited into insoluble aggregates. Second, previous work in our lab indicated that early polyQ aggregates have an overall linear morphology but appear somewhat disordered, while mature polyQ aggregates form well defined laterally aligned fibrils.19 For PG, well defined fibrils are visible early, but these fibrils do not laterally align as frequently. We hypothesize that laterally aligned fibrils arise as a result of oligomer-oligomer association, in parallel with the slow process of conversion (step (f) in Fig. 1) as aggregates sample different conformations on the way to a finalized fibrillar structure. The incorporation of associations between oligomers and aggregates into quantitative kinetic models is no small task, but some progress has been made.41
Our observations could lend support to the idea that the toxic species are soluble oligomers rather than insoluble aggregates, as indicated by a number of recent studies.42–44 During the process of conformational conversion, we hypothesize that the soluble oligomers interact with each other, resulting in lateral alignment of fibrils. Mature fibrils are more inert, as indicated by the lack of lateral alignment in PG aggregates. Other cellular components, such as transcription factors, are often found in inclusion bodies of polyQ aggregates.45 It seems likely that soluble oligomers would be more prone to interact with and recruit cellular components as they undergo structural rearrangement, compared to the mature aggregates which behave in a more inert manner. A study by Arrasate et al. noted that diffuse aggregates correlated with cell death while inclusion body formation reduced diffuse aggregates and was protective,46 lending credibility to the hypothesis that mature aggregates are more biologically inert than those which are actively forming.
Effect of flanking lysines
Flanking lysines are commonly used to facilitate synthesis and initial solubility of polyQ peptides.11, 18, 22, 30 We have previously shown that the loss of charge on the flanking lysines of Q20, obtained via an increase in pH, results in a more compact monomer and greater extent of aggregation.19 Thus, the conformational properties of the monomer as well as the aggregation kinetics are influenced by repulsive electrostatic interactions of the flanking lysines. It is possible for example that polyQ peptides lacking charged flanking lysines could become insoluble without conversion to β-sheet or formation of fibrillar aggregates. Thus, our conclusions are therefore strictly applicable only for our peptide design.
Comparison to simulations
Recent simulations 47, 48 of polyQ conformational equilibria and aggregation suggest that the formation of oligomers may indeed be an important step on the pathway by which polyQ aggregation occurs. Work by Vitalis et al. demonstrated that the free energy penalty associated with forming structures with high β-content actually increases with increasing chain length.47 This result contradicts the expectations of the nucleation elongation model, which asserts that the formation of the monomeric nucleus should become more favorable as the number of glutamines increases.29 Instead, as the chain length increased, Vitalis et al. observed increased stability of disordered globular monomers, increased formation of disordered dimers from the monomers, and increased stability of the dimers. Although the simulation only looked at dimer formation, results could be extrapolated to oligomer formation. Our observation of soluble oligomers in the PP peptide corroborates the idea that oligomers arise from nonspecific association rather than demanding a specific conformation of the monomer. Subsequent simulations showed that when conformational constraints toward high β-content were applied, no increase in the spontaneity of dimer formation was observed. Rather, there was an increase in the formation of canonical backbone-driven β-sheet structures at intermolecular interfaces of the dimers.48 Essentially, peptides conformationally constrained to have high β-content form dimers which are more similar to mature aggregates. Our results with the PG peptide, which rapidly formed mature aggregates, corroborate the conclusions of this simulation.
Comparison to previously published experimental data
Kar et al. recently reported studies of the kinetics of aggregation of polyQ peptides of similar, but not identical, design (Ac-K2QnK2-NH2), where n varies from 18 to 37.30 A direct comparison is difficult, but our sedimentation data are generally reasonably consistent with theirs. For example, they report for Q25 at 47 μM, a 5-day lag before significant sedimentation, about 80% soluble after 8 days, and about 30% soluble after 12 days. This is somewhat faster kinetics than our data for Q20 at a similar concentration (Fig. 3b), where we observed an ~7 day lag, 12 days to reach 80% soluble, and 30 days to reach 30% soluble. In contrast to our results, these researchers looked for, but did not detect, any soluble oligomers in their preparations prior to the onset of sedimentation. The reasons for this difference are unclear, but could be attributed to differences in peptide design, in sample preparation, and/or instrument sensitivity. These researchers interpreted their data in terms of a nucleation-elongation mechanism, with the nucleus size changing from monomer to tetramer as the number of glutamines changed from 27 to 23. Recently, Vitalis and Pappu have demonstrated that kinetic data calculated from a model similar to the association-conformational conversion mechanism can be fitted to the nucleation-elongation model equations,32 and we have shown that monomer loss kinetic data cannot be used alone to uniquely define a specific aggregation mechanism to the exclusion of other mechanisms.31 More detailed quantitative analysis of kinetic data, to include not only monomer loss but also aggregate size, number and growth rates, will be required to definitively discriminate among various proposed mechanism.
Inhibitor design
Our data support the potential for using polyQ peptides with interrupting residues as inhibitors of fibrillogenesis and cytotoxicity. Past studies have revealed that proline-containing polyQ proteins can act as inhibitors of aggregation when mixed with polyQ proteins.34 The polyQ regions of the PP peptide could interact with polyQ regions of fibril forming peptides and sequester them into globular aggregates. Indeed, our sedimentation assay failed to detect any insoluble aggregates when PP was mixed with PG. It is possible that PG may have been recruited into soluble aggregates of the PP peptide, thereby preventing the formation of insoluble aggregates. If soluble oligomers are indeed the most deleterious species, one might expect that proline inserts would increase toxicity. This was not the case in a study by Popiel et al.,34 which noted both decreased aggregation and toxicity in COS-7 cells expressing polyQ-GFP constructs when the polyQ region was interrupted by proline residues. We speculate that the answer to this apparent contradiction may lie in the cellular environment and the impact that proline inserts have on the stability of soluble oligomers. A study by Schiffer et al.7 noted that chaperones reduce both aggregation and toxicity of polyQ proteins, while also concluding that soluble oligomers are cytotoxic. This implies that chaperones must act on the soluble oligomers themselves or on a precursor to them. Thus, in a cellular environment, proline interruptions may alter the aggregation pathway enough to allow chaperones to act more efficiently, preventing both toxicity and the formation of insoluble aggregates.
MATERIALS AND METHODS
Peptide synthesis and purification
All materials were from Fisher Scientific (Pittsburgh, PA) except where indicated. Peptides were synthesized using standard Fmoc solid phase methods on a Protein Technologies Inc. Symphony synthesizer. Alanine, glycine, proline, D-proline, glutamines with a Trityl side chain protecting group, and lysines and tryptophan with a Boc side chain protecting group were purchased from Novabiochem (Gibbstown, NJ). Dansylated lysine was purchased from Anaspec (San Jose, CA). Peptides were synthesized and purified as described previously.19 Peptide identity was confirmed by MALDI-TOF mass spectrometry, which yielded molecular weights of 3586.9 (PP, 3585.9 theoretical), 3533.8 (AA, 3533.8 theoretical), 3390.7 (Q20, 3391.6 theoretical), and 3545.8 (PG, 3545.8 theoretical). For the dansylated peptides, molecular weights were 3876.0 (PP-D, 3875.9 theoretical), 3824.0 (AA-D, 3823.8 theoretical), 3680.8 (Q20-D, 3681.6 theoretical), and 3836.0 (PG-D, 3835.8 theoretical).
Sample preparation
Lyophilized peptides were disaggregated using a protocol similar to that developed by others.22 Briefly, peptide was incubated overnight in a 1:1 solution of TFA and HFIP. Solvent was evaporated under a gentle flow of N2 and the peptides were redissolved in water adjusted to pH 3 with TFA. Peptide stock solutions were filtered through a 0.45 μm filter, aliquoted, snap frozen in dry ice and ethanol and stored at −80°C. Concentrations of stock solutions (typically ~200 μM) were determined as described previously.19 Prior to each experiment, a vial was thawed and centrifuged at 19,500 rcf for 30 min, and then the supernatant (top 75%) was removed and used immediately.
Circular dichroism (CD)
Peptide stock solutions were diluted into phosphate buffer (10 mM K2HPO4/KH2PO4, 140 mM NaF, pH 7.4) to a concentration of 40 μM peptide. Freshly-prepared samples were filtered through a 0.45 μm membrane immediately before transfer to a 1 mm cell. CD spectra were collected using an Aviv 202SF circular dichroism spectrometer at 37°C immediately after filtration. Solvent spectra were collected and subtracted. We did not see any measurable change in concentration before and after filtration.
Fluorescence resonance energy transfer (FRET)
We inserted a fluorescent donor (Trp) and acceptor (dansyl) at each end of the polyQ stretch, and assumed the Forster radius R0 = 21.0 Å for the Trp-dansyl pair.49–51 Peptide samples were diluted to 5 μM in pH 7.4 PBSA (10 mM NaH2PO4/Na2HPO4, 0.14 M NaCl, and 3 mM NaN3). Low concentrations of the peptides were used to help ensure that the sample remained monomeric during the FRET measurement. Fluorescence spectra were obtained with a QuantaMaster Series spectrofluorometer (PTI, Inc., Birmingham, NJ), with excitation at 295 nm and emission spectra recorded from 310 to 550 nm. Measurements were made 4 times and at steady state. FRET efficiency E was calculated as52
| (1) |
where FDA is the donor (tryptophan) fluorescence with the acceptor (dansyl) group present, and FD is the donor fluorescence with the acceptor group absent. The values of FDA and FD in Eq. (1) were determined by integrating the peak area of the emission scan, with background subtracted, from 330–370 nm. The measured energy transfer represents an average <E> for the conformational ensemble of the peptides, and so the values of R are reported as an averaged distance between the fluorophores, as described in detail elsewhere.19
Size exclusion chromatography (SEC)
Peptides were diluted into PBSA to a concentration of 40 μM, distributed into sample tubes in 180 μL aliquots, and incubated at 37°C. At regular time intervals, a sample tube was withdrawn and centrifuged at 19,500 rcf for 30 min. The supernatant (top 75%) was removed and analyzed by SEC on a Superdex 75 PC 3.2/30 column (Pharmacia), with a molecular weight range of 3–70 kDa, using a Waters 625LC system. The mobile phase (PBS, pH 7.4) flow rate was set to 0.1 mL/min, an overfilled 50 μL sample loop was used to apply a standard volume of sample, and peaks were detected by absorbance at 280 nm. The column was calibrated with a set of molecular weight protein standards.
Sedimentation assay
Samples were prepared and centrifuged as described in the previous section and the supernatant was analyzed for the concentration of soluble peptide with a BCA (Pierce, Rockford IL) assay in the microplate format, using a BioTek EL 800 Universal
Microplate Reader and following the manufacture’s protocol. Samples were run in triplicate. All concentrations are reported as a percentage of the concentration of an uncentrifuged aliquot. For the mixed sedimentation assay, the same procedure was followed, but peptides were diluted into PBSA to a concentration of 35 μM, and 5 μM PG was added, or 40 μM Q20 and 40 μM PG was added. A 5 μM PG only sample was also produced.
Laser light scattering
Samples were diluted into PBSA that had been filtered twice with a 0.22 μm filter, then immediately filtered again through a 0.45 μm filter directly into a clean light scattering cuvette held at 37°C. The total scattered light intensity at 90° as well as the autocorrelation function were collected over time and analyzed to obtain the normalized intensity, Inorm, and the apparent hydrodynamic radius, Rhz, as described in detail elsewhere.19
Transmission electron microscopy (TEM)
Peptides at 40 μM were prepared in PBSA and incubated for 6 h, 10 days, or 50 days at 37°C. A drop of sample was applied to a pioloform coated grid and stained with methylamine tungstate stain, then imaged with a Philips CM120 scanning transmission electron microscope.
Supplementary Material
Acknowledgments
This work was supported by grant CBET-085 2278 from the National Science Foundation and grant 5T32 GM-08349 (RHW) from the National Institute of Health. We gratefully acknowledge the technical assistance of Dr. Gary Case, Dr. Randy Massey, and the UW Biotechnology Center mass spectrometry staff. CD data were obtained at the University of Wisconsin - Madison Biophysics Instrumentation Facility, which was established with support from the University of Wisconsin - Madison and grants BIR-9512577 (NSF) and S10 RR13790 (NIH).
ABBREVIATIONS
- BCA
bicinchoninic assay
- CD
circular dichroism
- FRET
fluorescence resonance energy transfer
- HFIP
hexafluoroisopropanol
- HPLC
high-pressure liquid chromatography
- PBSA
phosphate-buffered saline with azide
- SEC
size exclusion chromatography
- TEM
transmission electron microscopy
- TFA
trifluoroacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 2.Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- 3.Wanker EE. Protein aggregation and pathogenesis of Huntington’s disease: Mechanisms and correlations. Biol Chem. 2000;381:937–942. doi: 10.1515/BC.2000.114. [DOI] [PubMed] [Google Scholar]
- 4.Gusella JF, MacDonald ME. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 5.Albrecht M, Golatta M, Wullner U, Lengauer T. Structural and functional analysis of ataxin-2 and ataxin-3. Eur J Biochem. 2004;271:3155–3170. doi: 10.1111/j.1432-1033.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- 6.Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schiffer NW, Broadley SA, Hirschberger T, Tavan P, Kretzschmar HA, Giese A, Haass C, Hartl FU, Schmid B. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J Biol Chem. 2007;282:9195–9203. doi: 10.1074/jbc.M607865200. [DOI] [PubMed] [Google Scholar]
- 8.Jana NR, Tanaka M, Wang GH, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 9.Carmichael J, Chatellier J, Woolfson A, Milstein C, Fersht AR, Rubinsztein DC. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc Natl Acad Sci U S A. 2000;97:9701–9705. doi: 10.1073/pnas.170280697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 11.Chen SM, Ferrone FA, Wetzel R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–356. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 14.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, Goto Y, Naiki H, Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 15.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 16.Slepko N, Bhattacharyya AM, Jackson GR, Steffan JS, Marsh JL, Thompson LM, Wetzel R. Normal-repeat-length polyglutamine peptides accelerate aggregation nucleation and cytotoxicity of expanded polyglutamine proteins. Proc Natl Acad Sci U S A. 2006;103:14367–14372. doi: 10.1073/pnas.0602348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen SM, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry (N Y) 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 18.Lee CC, Walters RH, Murphy RM. Reconsidering the mechanism of polyglutamine peptide aggregation. Biochemistry. 2007;46:12810–12820. doi: 10.1021/bi700806c. [DOI] [PubMed] [Google Scholar]
- 19.Walters RH, Murphy RM. Examining polyglutamine peptide Length: A connection between collapsed conformations and increased aggregation. J Mol Biol. 2009;393:978–992. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers - their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen SM, Wetzel R. Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 2001;10:887–891. doi: 10.1110/ps.42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–272. doi: 10.1016/s0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 24.Altschuler EL, Hud NV, Mazrimas JA, Rupp B. Random coil conformation for extended polyglutamine stretches in aqueous soluble monomeric peptides. J Pept Res. 1997;50:73–75. doi: 10.1111/j.1399-3011.1997.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 25.Klein FAC, Pastore A, Masino L, Zeder-Lutz G, Nierengarten H, Oulad-Abdeighani M, Altschuh D, Mandel J, Trottier Y. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: Towards a length-dependent toxicity gradient. J Mol Biol. 2007;371:235–244. doi: 10.1016/j.jmb.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Singh VR, Lapidus LJ. The intrinsic stiffness of polyglutamine peptides. J Phys Chem B. 2008;112:13172–13176. doi: 10.1021/jp805636p. [DOI] [PubMed] [Google Scholar]
- 27.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc Natl Acad Sci U S A. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Digambaranath JL, Campbell TV, Chung A, McPhail MJ, Stevenson KE, Zohdy MA, Finke JM. An accurate model of polyglutamine. Proteins. 2011;79:1427–1440. doi: 10.1002/prot.22970. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharyya AM, Thakur AK, Wetzel R. Polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction. Proc Natl Acad Sci U S A. 2005;102:15400–15405. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernacki JP, Murphy RM. Model discrimination and mechanistic interpretation of kinetic data in protein aggregation studies. Biophys J. 2009;96:2871–2887. doi: 10.1016/j.bpj.2008.12.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vitalis A, Pappu RV. Assessing the contribution of heterogeneous distributions of oligomers to aggregation mechanisms of polyglutamine peptides. Biophys Chem. 2011 doi: 10.1016/j.bpc.2011.04.006. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci U S A. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popiel HA, Nagai Y, Onodera O, Inui T, Fujikake N, Urade Y, Strittmatter WJ, Burke JR, Ichikawa A, Toda T. Disruption of the toxic conformation of the expanded polyglutamine stretch leads to suppression of aggregate formation and cytotoxicity. Biochem Biophys Res Commun. 2004;317:1200–1206. doi: 10.1016/j.bbrc.2004.03.161. [DOI] [PubMed] [Google Scholar]
- 35.Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM. Single homopolypeptide chains collapse into mechanically rigid conformations. Proc Natl Acad Sci U S A. 2009;106:12605–12610. doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jayaraman M, Kodali R, Wetzel R. The impact of ataxin-1-like histidine insertions on polyglutamine aggregation. Protein Eng Des Sel. 2009;22:469–478. doi: 10.1093/protein/gzp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson JS, Richardson DC. Principles and patterns of protein conformation. In: Fasman G, editor. Prediction of protein structure and principles of protein conformation. Plenum Press; New York: 1989. pp. 43–75. [Google Scholar]
- 38.Sahoo H, Roccatano D, Hennig A, Nau WM. A 10-angstrom spectroscopic ruler applied to short polyprolines. J Am Chem Soc. 2007;129:9762–9772. doi: 10.1021/ja072178s. [DOI] [PubMed] [Google Scholar]
- 39.Darnell GD, Derryberry J, Kurutz JW, Meredith SC. Mechanism of cis-inhibition of polyQ fibrillation by polyP: PPII oligomers and the hydrophobic effect. Biophys J. 2009;97:2295–2305. doi: 10.1016/j.bpj.2009.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perczel A, Hollosi M, Foxman BM, Fasman GD. Conformational-analysis of pseudocyclic hexapeptides based on quantitative circular-dichroism (CD), NOE, and X-Ray data - the pure CD spectra of Type-I and Type-II Beta-Turns. J Am Chem Soc. 1991;113:9772–9784. [Google Scholar]
- 41.Li Y, Roberts CJ. Lumry-Eyring nucleated-polymerization model of protein aggregation kinetics. 2. Competing growth via condensation and chain polymerization. J Phys Chem B. 2009;113:7020–7032. doi: 10.1021/jp8083088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 43.Schiffer NW, Ceraline J, Hartl FU, Broadley SA. N-terminal polyglutamine-containing fragments inhibit androgen receptor transactivation function. Biol Chem. 2008;389:1455–1466. doi: 10.1515/BC.2008.169. [DOI] [PubMed] [Google Scholar]
- 44.Diguet E, Petit F, Escartin C, Cambon K, Bizat N, Dufour N, Hantraye P, Deglon N, Brouillet E. Normal aging modulates the neurotoxicity of mutant Huntingtin. PLoS One. 2009;4:e4637. doi: 10.1371/journal.pone.0004637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freiman RN, Tjian R. Perspectives: Neurodegeneration - A glutamine-rich trail leads to transcription factors. Science. 2002;296:2149–2150. doi: 10.1126/science.1073845. [DOI] [PubMed] [Google Scholar]
- 46.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 47.Vitalis A, Wang X, Pappu RV. Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J Mol Biol. 2008;384:279–297. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vitalis A, Lyle N, Pappu RV. Thermodynamics of beta-sheet formation in polyglutamine. Biophys J. 2009;97:303–311. doi: 10.1016/j.bpj.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunn BM, Pham C, Raney L, Abayasekara D, Gillespie W, Hsu A. Interaction of alpha-dansylated peptide inhibitors with porcine pepsin - Detection of complex-formation by Fluorescence Energy-Transfer and chromatography and evidence for a 2-step binding scheme. Biochemistry (N Y) 1981;20:7206–7211. doi: 10.1021/bi00528a023. [DOI] [PubMed] [Google Scholar]
- 50.Wu PG, Brand L. Resonance Energy-Transfer - Methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 51.Gustiananda M, Liggins JR, Cummins PL, Gready JE. Conformation of prion protein repeat peptides probed by FRET measurements and molecular dynamics simulations. Biophys J. 2004;86:2467–2483. doi: 10.1016/S0006-3495(04)74303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.dos Remedios CG, Moens PDJ. In: Resonance energy transfer in proteins. Andrews DL, Demidov AA, editors. Chichester, UK: Wiley; 1999. pp. 1–64. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.