

Abstract
The transcription factor Pho4 is phosphorylated and localized predominantly to the cytoplasm when budding yeast are grown in phosphate-rich medium and is unphosphorylated and localized to the nucleus upon phosphate starvation. We have investigated the requirements for nuclear import of Pho4 and find that Pho4 enters the nucleus via a nonclassical import pathway that utilizes the importin β family member Pse1/Kap121. Pse1 binds directly to Pho4 and is required for its import in vivo. We have defined the nuclear localization signal on Pho4 and demonstrate that it is required for Pse1 binding in vitro and is sufficient for PSE1-dependent import in vivo. Phosphorylation of Pho4 inhibits its interaction with Pse1, providing a mechanism by which phosphorylation may regulate import of Pho4 in vivo.
Keywords: Nuclear import, Pse1, Kap121, Pho4, phosphorylation
One way in which cells respond to extracellular signals is by modulating gene expression. This response requires the transfer of information from the plasma membrane, through the cytoplasm, and into the nucleus. Protein kinase cascades are commonly used to transduce extracellular signals, and they typically culminate in the phosphorylation of transcription factors. Phosphorylation of both transcription factors and kinases has been shown to result in regulation of their nuclear localization, suggesting that control of the subcellular localization of these proteins is important for the response to extracellular signals (for review, see Jans and Hubner 1996; Gaits et al. 1998; Khokhlatchev et al. 1998; Toone et al. 1998).
The subcellular localization of proteins can be controlled by regulating import into the nucleus and/or by regulating export from the nucleus. Nuclear import and export occur through the nuclear pore complex, a large macromolecular assembly of proteins embedded in the nuclear envelope. Transport is signal mediated, requires energy and physiological temperature, and is bidirectional. Targeting signals are recognized by soluble transport receptors, which then translocate with their cargo into or out of the nucleus (for review, see Nigg 1997).
The first targeting signal to be described was the SV40 large T antigen nuclear localization signal (NLS), consisting of a cluster of basic amino acids (Kalderon et al. 1984). The import receptor that recognizes the classical NLS is a heterodimer consisting of importin α and importin β. Importin α binds directly to the NLS (Gorlich et al. 1994; Weis et al. 1995), and importin β binds to importin α, the GTPase Ran, and the nuclear pore complex (Gorlich et al. 1995a; Moroianu et al. 1995; Rexach and Blobel 1995). A complex consisting of the NLS, importin α, and importin β assembles in the cytoplasm, docks at the nuclear pore, translocates across the pore, and then is disassembled in the nucleus (Newmeyer and Forbes 1988; Richardson et al. 1988; Gorlich et al. 1995b; Imamoto et al. 1995; Rexach and Blobel 1995; Moroianu et al. 1996).
Additional signals have been defined that target proteins from the nucleus to the cytoplasm (for review, see Nakielny and Dreyfuss 1997). The nuclear export signal (NES) contained in the HIV Rev protein and PKI, an inhibitor of cAMP dependent protein kinase A, is a small sequence rich in leucine residues (Wen et al. 1995). The leucine-rich NES is bound by the export receptor Crm1/Xpo1 and Ran–GTP in the nucleus. This complex translocates across the nuclear pore, and is disassembled in the cytoplasm (Fornerod et al. 1997; Fukuda et al. 1997; Kudo et al. 1997; Neville et al. 1997; Ossareh-Nazari et al. 1997; Stade et al. 1997).
A recent advance in the nuclear transport field is the discovery of multiple import and export pathways that utilize different transport receptors (for review, see Nakielny and Dreyfuss 1997; Weis 1998). This has led to the identification of a family of transport receptors related in sequence to importin β, with the amino terminus being most conserved in a ∼150 amino acid region required for binding to the small GTPase Ran (Gorlich et al. 1997). Searches of the yeast genome database revealed 13 proteins with significant homology to importin β (Gorlich et al. 1997). Several of these importin β family members have now been shown to be import or export receptors, and it has been postulated that the remainder are transport receptors defining novel import or export pathways. Many studies are focused on determining the functions of the different importin β family members.
Ran plays an essential role in nuclear import and export; it is thought that the nucleotide state of Ran is used to impart directionality to transport processes (Gorlich et al. 1996; Izaurralde et al. 1997). The regulators of Ran are compartmentalized within the cell; RanGAP, the Ran GTPase activating protein, is localized to the cytoplasm and RCC1, the guanine nucleotide exchange factor for Ran, is exclusively nuclear (Ohtsubo et al. 1989). The localization of RanGAP and RCC1 predicts that the GTP-bound form of Ran will predominate in the nucleus. The steep gradient of Ran–GTP between the nucleus and the cytoplasm controls assembly and disassembly of transport complexes. Ran–GTP binding by importin β family members facilitates release of import receptors from their cargo in the nucleus (Rexach and Blobel 1995; Schlenstedt et al. 1997). Ran–GTP binding by importin β family members involved in export is required for the formation of export complexes consisting of the export receptor, cargo, and Ran–GTP (Fornerod et al. 1997; Kutay et al. 1997, 1998). Thus, transport receptors utilize the Ran–GTP gradient as a marker for nuclear and cytoplasmic compartments.
Eukaryotic cells have taken advantage of the barrier between the cytoplasm and nucleus as a way to restrict access to the nucleus and to control the activity of regulatory proteins. Although there are many examples of regulated localization, for most systems it is not clear if import and/or export is regulated or how this regulation is achieved. Until recently, it was assumed that import into the nucleus was the step subject to regulation. The identification of multiple export pathways suggests that the localization of some proteins might be regulated at the level of export. A goal of the field is to understand the regulation of nuclear import and export. One major challenge is to understand how signal transduction pathways influence the import and export of regulatory proteins to control their subcellular localization in response to extracellular signals.
We are studying the regulation of Pho4, a yeast transcription factor whose localization is regulated by phosphorylation in response to changes in the extracellular concentration of inorganic phosphate (O’Neill et al. 1996). Pho4 is required for phosphate starvation-specific gene expression (for review, see Oshima 1997). When yeast are grown in phosphate-rich medium, Pho4 is phosphorylated by the Pho80–Pho85 cyclin–CDK complex (Kaffman et al. 1994) and is localized predominantly to the cytoplasm (O’Neill et al. 1996), resulting in transcriptional repression of phosphate starvation-specific gene expression. Upon phosphate starvation, the CDK inhibitor Pho81 inhibits Pho80–Pho85 (Schneider et al. 1994), leading to accumulation of unphosphorylated Pho4 in the nucleus and transcription of phosphate-responsive genes (O’Neill et al. 1996).
We are interested in understanding how phosphorylation of Pho4 regulates its subcellular localization in response to changes in extracellular inorganic phosphate. We find that Pho4 is imported into the nucleus via a nonclassical import pathway utilizing the importin β family member Pse1/Kap121. Pho4 is the first protein found to be imported exclusively by Pse1. We identify the Pho4 NLS and demonstrate that this region is both necessary for Pse1 binding in vitro and sufficient for PSE1-dependent nuclear import in vivo. Additionally, we demonstrate that the interaction between Pho4 and Pse1 is inhibited by phosphorylation, suggesting that import of Pho4 in vivo is regulated by phosphorylation. Our studies provide insight into the mechanism by which phosphorylation of Pho4 regulates its import, and serve as a paradigm for understanding other examples of regulated nuclear localization.
Results
Nuclear import of Pho4 is defective in a pse1-1 strain
Localization of Pho4 is regulated by phosphorylation in response to changes in the extracellular concentration of inorganic phosphate. A mechanistic understanding of the control of Pho4 localization by phosphorylation requires identification of the transport receptors that carry Pho4 into and out of the nucleus. To identify transport receptors required for import of Pho4, we examined the localization of Pho4 green fluorescent protein (GFP) in different yeast strains containing mutations in the importin β family members. In a wild-type strain, Pho4–GFP is predominantly cytoplasmic when cells are grown in phosphate-rich medium and is concentrated in the nucleus when yeast are starved for phosphate (Fig. 1). In a yeast strain harboring a temperature-sensitive mutation in the essential importin β family member PSE1 (Seedorf and Silver 1997), also referred to as KAP121 (Rout et al. 1997), we find that Pho4–GFP does not accumulate in the nucleus upon phosphate starvation (Fig. 1). The defect in Pho4 localization is observed even at the permissive temperature in a pse1-1 strain. Other strains with mutations in the importin β family members KAP104, LOS1, MTR10, SXM1, XPO1, and CSE1 (Hopper et al. 1980; Xiao et al. 1993; Kadowaki et al. 1994; Aitchison et al. 1996; Seedorf and Silver 1997; Stade et al. 1997) do not show a defect in localization of Pho4–GFP upon phosphate starvation (data not shown).
Figure 1.

Pho4–GFP does not accumulate in the nucleus in pse1-1 yeast. Pho4–GFP localization was monitored in PSE1+ (PSY580) or pse1-1 yeast grown at 25°C in either high- or no-phosphate medium.
The requirement of Pse1 for nuclear accumulation of Pho4 could reflect a role for Pse1 in transducing the phosphate starvation signal, or a role for Pse1 in import of Pho4. To determine if Pse1 is required for Pho4 import, we examined the localization of Pho4SA, a Pho4 mutant containing five serine-to-alanine substitutions at the sites of phosphorylation by Pho80–Pho85 (O’Neill et al. 1996). Pho4SA is localized to the nucleus of a wild-type strain even when phosphate is abundant (O’Neill et al. 1996). Thus, Pho4SA can be used to assess the requirements for Pho4 import that are independent of the function of the phosphate signal transduction pathway. In contrast to what is observed in wild-type yeast, Pho4SA–GFP localizes predominantly to the cytoplasm in a pse1-1 mutant strain (Fig. 2), indicating that Pse1 is required for the import of Pho4 into the nucleus.
Figure 2.

Nuclear import of Pho4SA–GFP is defective in pse1-1 yeast. The steady-state localization of Pho4SA–GFP and Pho4–GFP-NLS was monitored in PSE1+ KAP123+ (PSY580), pse1-1, or in kap123Δ yeast grown in high-phosphate medium at 25°C. Pho4SA–GFP is a mutant derivative of Pho4 that contains serine-to-alanine substitutions at all of the sites of phosphorylation by Pho80–Pho85 (O’Neill et al. 1996). Pho4–GFP NLS contains the SV40 NLS fused to the carboxyl terminus of Pho4–GFP.
Previous studies have suggested that Pse1 and a closely related importin β family member, Kap123, might have overlapping functions (Rout et al. 1997; Schlenstedt et al. 1997; Seedorf and Silver 1997). We wished to test if Kap123 also plays a role in the import of Pho4 by examining Pho4SA–GFP localization in a kap123Δ strain. We find no defect in the import of Pho4SA–GFP in a kap123Δ strain (Fig. 2), indicating that this importin β family member is not required for the import of Pho4.
If the mislocalization of Pho4 in a pse1-1 mutant strain reflects a defect in a specific import pathway rather than a defect that renders Pho4 incompetent for import, it should be possible to target Pho4 to the nucleus in a pse1-1 mutant strain by use of an alternate import pathway. Strains with mutations in either PSE1 or KAP123 are capable of importing a reporter protein targeted to the nucleus with an NLS derived from the SV40 large T antigen (Seedorf and Silver 1997). We fused the SV40 large T antigen NLS to Pho4–GFP (Pho4–GFP–NLS) and examined its localization in a wild-type strain, a pse1-1 strain, and a kap123Δ strain (Fig. 2). Pho4–GFP–NLS is localized to the nucleus in each of these strains, suggesting that the defect in Pho4 import in a pse1-1 mutant is the result of a defect in a specific import pathway. Taken together, these data suggest that Pse1 is the import receptor for Pho4.
Identification of the Pho4 NLS
To identify regions of Pho4 required for its import, we constructed several Pho4 deletion mutants as fusion proteins with GFP and analyzed their localization in a strain lacking PHO80. Pho4 cannot be phosphorylated in a pho80Δ strain, leading to its constitutive nuclear localization (Kaffman et al. 1994; O’Neill et al. 1996) and thereby simplifying analysis of the steady-state localization of the fusion proteins. The NLS of many transcriptional regulators resides within the DNA-binding domain (Williams et al. 1997; Latimer et al. 1998). Mutational analysis of Pho4 demonstrates that the Pho4 DNA binding domain (Fig. 3A) is neither necessary nor sufficient for nuclear localization in a pho80Δ strain (Pho42–247–GFP, Pho4248–312–GFP3, Fig. 3B). The smallest domain necessary for Pho4 import lies within amino acids 157–164 (Pho4Δ157–164–GFP, Fig. 3B); however, this small domain is not sufficient for nuclear import (Pho4156–171–GFP3, Fig. 3B). Amino acids 140–166 of Pho4 define the smallest domain of Pho4 that is both necessary (Pho4Δ141–165–GFP, Fig. 3B) and sufficient (Pho4140–166–GFP3, Fig. 3B,C) for nuclear import. These data demonstrate that Pho4 amino acids 140–166 function as an NLS in vivo (Fig. 3B,C). The Pho4140–166–GFP3 fusion protein is not imported into the nucleus in a pse1-1 mutant, indicating that this fusion protein is targeted to the nucleus via the PSE1-dependent import pathway (Fig. 3C). It is interesting to note that this domain of Pho4 contains a single site for phosphorylation by Pho80–Pho85 (Fig. 3D). Pho4140–166–GFP3 is localized to the nucleus in both high- and low-phosphate media (data not shown), presumably because the peptide NLS cannot be phosphorylated by the Pho80–Pho85 kinase. These studies demonstrate that Pho4 contains a single NLS, which is targeted to the nucleus in a PSE1-dependent manner.
Figure 3.




Pho4 amino acids 140–166 are necessary and sufficient for nuclear localization. (A) Structure and function of Pho4. A domain important for Pho4 transactivation is contained within Pho4 amino acids 1–109 (hatched box) (Ogawa and Oshima 1990). The Pho4 DNA-binding domain has been localized to amino acids 248–312 (shaded box) (Ogawa and Oshima 1990). The five sites on Pho4 phosphorylated by the kinase Pho80–Pho85 are at amino acids 100, 114, 128, 152, and 223 (O’Neill et al. 1996) and are shown with an asterisk (*). The Pho4 NLS lies within amino acids 140–166 (solid box). (B) Localization of Pho4–GFP derivatives. Pho4–GFP derivatives expressed in pho4Δ pho80Δ yeast were grown at 30°C in high-phosphate medium. Pho4 deletion mutants smaller than 27 kD were fused to three copies of GFP (GFP3), and those larger were fused to a single GFP. Localization of the Pho4–GFP derivatives is indicated [(N) nuclear; (C) cytoplasmic]. (C) Pho4 amino acids 140–166 are sufficient to target GFP3 to the nucleus in a PSE1-dependent manner. Localization of Pho4140–166–GFP3 was monitored in PSE1+ (pho4Δ pho80Δ) and in pse1-1 strains grown at 25°C in high-phosphate medium. (D) The Pho4 NLS resides within amino acids 140–166. The fourth phosphorylation site at Pho4 serine-152 is boxed.
Pho4 binds to Pse1 directly
If Pse1 is the import receptor for Pho4, we expect Pse1 to bind to Pho4. To test for interaction between Pho4 and Pse1, we purified a fusion protein from Escherichia coli consisting of Pho4 joined to two Protein A z domains (zz) derived from Protein A (Pho4WT–zz). Pho4WT–zz was immobilized on IgG Sepharose and incubated with extract derived from yeast expressing Pse1–GFP. The resin was washed, bound proteins were eluted from Pho4WT–zz, separated by SDS-PAGE, and analyzed by immunoblotting with anti-GFP antibodies. We find that Pho4WT–zz binds Pse1–GFP (Fig. 4B). As a control for the functional relevance of the Pho4–Pse1 interaction, we examined interaction of Pse1 with Pho4Δ157–164–zz, a Pho4 mutant that cannot be targeted to the nucleus (Figs. 3B and 4A). No detectable interaction is observed between Pho4Δ157–164–zz and Pse1–GFP (Fig. 4B, lane 3). However, both Pho4Δ157–164–zz and Pho4WT–zz interact to the same extent with the transcription factor Pho2, known to bind Pho4 (Magbanua et al. 1997), suggesting that both proteins are properly folded (data not shown). These data indicate that Pse1 and Pho4 interact and suggest that the interaction is functionally relevant for import of Pho4.
Figure 4.

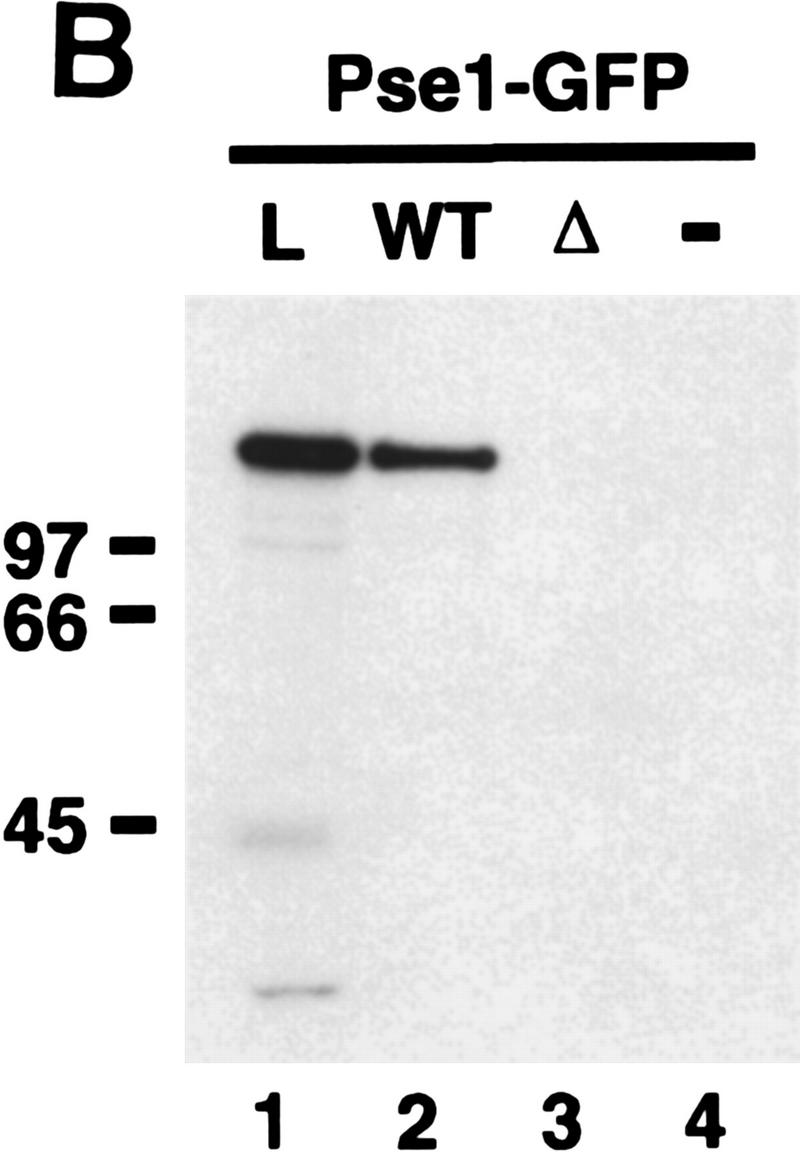

Pho4 binds to Pse1. (A) Amino acids 157–164 are required for nuclear localization of Pho4 in vivo. Pho4 lacking amino acids 157–164 fused to GFP (Pho4Δ157–164–GFP) was expressed in pho4Δ yeast grown in high- or no-phosphate medium at 30°C. (B) Pho4 associates with Pse1, but a mutant Pho4 lacking residues required for nuclear localization does not. Wild-type Pho4 (Pho4WT–zz) and Pho4 lacking amino acids 157–164 (Pho4Δ157–164–zz) were expressed as zz fusion proteins in E. coli. zz-containing proteins were purified with IgG–Sepharose beads and incubated with yeast extract containing Pse1–GFP. The resin was washed extensively and proteins bound to Pho4–zz were eluted with 1 m MgCl2. Eluted proteins were precipitated, separated on 7.8% SDS-PAGE, transferred to a PVDF membrane, and visualized by immunoblotting with anti-GFP monoclonal antibodies. (Lane 1) Three percent of the Pse1–GFP extract loaded (L); (lane 2) Pse1–GFP bound to Pho4WT–zz (WT); (lane 3) Pse1–GFP bound to Pho4Δ157–164–zz (Δ); (lane 4) Pse1–GFP bound to IgG–Sepharose beads (−). Half of the 1 m MgCl2 eluate was loaded onto the gel. (C) Pho4 associates with Pse1 but not with Kap123 or Sxm1. Pho4WT–zz and Pho4Δ157–164–zz immobilized on IgG–Sepharose beads and IgG–Sepharose beads alone were incubated with yeast extract expressing either Pse1–GFP, Kap123–GFP, or Sxm1–GFP. The resin was washed extensively, and proteins bound to Pho4–zz were eluted with 1 m MgCl2. Eluted proteins were precipitated, separated on 7.8% SDS-polyacrylamide gels, transferred to PVDF membrane, and visualized by immunoblotting with anti-GFP monoclonal antibodies. (B,C) (Lanes 1,5) About 3% of the importin–GFP loaded; (lanes 2,6) importin–GFP bound to Pho4WT–zz; (lanes 3,7) importin–GFP bound to Pho4Δ157–164–zz; (lanes 4,8) importin–GFP bound to the IgG–Sepharose control. Half of the 1 m MgCl2 eluate was loaded onto the gel.
To examine the specificity of the interaction between Pho4 and Pse1, we tested for interaction between Pho4 and the closely related importin β family members Sxm1 and Kap123 (Gorlich et al. 1997; Seedorf and Silver 1997). In yeast extract, neither Kap123–GFP nor Sxm1–GFP binds immobilized Pho4WT–zz (Fig. 4C). These results are consistent with the in vivo localization data (Fig. 2) and suggest that the interaction between Pho4 and Pse1 is specific for this importin β family member.
Next, we wished to determine if the interaction between Pse1 and Pho4 was direct, or mediated by another protein. To test for a direct interaction between Pho4 and Pse1, we incubated Pho4WT–zz with Pse1–6His, both purified from E. coli, in the presence of either yeast extract or bovine serum albumin. We find that Pho4WT–zz binds directly to Pse1–6His and that this interaction is not potentiated by proteins in the yeast extract, indicating that Pse1 directly recognizes Pho4 (Fig. 5).
Figure 5.

Pho4 interacts directly with Pse1. Pho4WT–zz bound to IgG–Sepharose beads was incubated with Pse1–6His purified from E. coli in the presence of either yeast extract or BSA. The resin was washed extensively and proteins bound to Pho4WT–zz were eluted with 1 m MgCl2. Eluted proteins were precipitated, separated on 7.8% SDS-PAGE, and visualized by staining with Coomassie blue. Approximately 50% of the Pse1–6His preparation consists of an amino-terminally truncated form of Pse1 (*). (Lane 1) Pse1–6His bound to Pho4WT–zz in the presence of whole cell extract (WCE); (lane 2) Pse1–6His bound to Pho4WT–zz in the presence of BSA; (lane 3) Pse1–6His bound to IgG– Sepharose beads alone in the presence of WCE; (lane 4) 4% of the load. Half of the 1 m MgCl2 eluate was loaded onto the gel.
The Pse1–Pho4 complex is disassembled by Ran–GTP
Studies of other import receptor-cargo interactions suggest that they form stable complexes in the cytoplasm, which are then dissociated in the nucleus by binding to Ran–GTP (Rexach and Blobel 1995; Moroianu et al. 1996; Schlenstedt et al. 1997). We wished to determine if the Pho4–Pse1 complex could be dissociated by incubation with the yeast Ran homolog, Gsp1. Pse1–6His was prebound to Pho4WT–zz and the Pse1–Pho4WT–zz complex was incubated with either Gsp1–GTP, Gsp1–GDP, or a buffer control. Pse1 dissociates from Pho4 when incubated with Gsp1–GTP, but not when incubated with Gsp1–GDP or buffer alone (Fig. 6). The amino-terminally truncated form of Pse1 (labeled as *), which lacks the Ran-binding domain (data not shown), does not dissociate from Pho4WT–zz after incubation with Gsp1–GTP (Fig. 6, lanes 4–6). These data are consistent with a model in which Pho4 binds Pse1 in the cytoplasm and is released in the nucleus by the binding of Gsp1–GTP to Pse1. These results provide further support for the functional relevance of the interaction between Pho4 and Pse1.
Figure 6.

Gsp1–GTP dissociates the Pho4–Pse1 complex. Purified Pse1–6His and Pho4WT–zz were incubated and the Pse1–Pho4WT–zz complex was purified with IgG–Sepharose beads. The resin was washed extensively, split into three equal parts, and incubated with Gsp1–GTP, Gsp1–GDP, or buffer alone. Proteins released during this treatment were collected as the eluate. The Pho4WT–zz resin was washed once with IgG buffer, and proteins bound to Pho4WT–zz were eluted with 1 m MgCl2 and precipitated (bound fraction). Proteins were separated on 10% SDS-PAGE and visualized by staining with Coomassie blue. Lanes 1,4) Gsp1–GTP; (lanes 2,5) Gsp1–GDP; (lanes 3,6) buffer alone. One-third of the eluate and one-half of the bound proteins were loaded onto the gel. The amino-terminally truncated form of Pse1–6His (*), which cannot bind Ran–GTP (data not shown), is not dissociated from Pho4WT–zz by incubation with Gsp1–GTP.
Phosphorylation of Pho4 regulates its association with Pse1
Because phosphorylation regulates the nuclear localization of Pho4, we sought to determine the effect of phosphorylation on the interaction of Pho4 with Pse1. Pho4WT–zz was purified from E. coli and either phosphorylated in vitro with a yeast extract containing the Pho80–Pho85 cyclin–CDK complex or mock phosphorylated by incubating with yeast extract in the absence of ATP. Pho4 can be efficiently phosphorylated in vitro and the sites of phosphorylation utilized in vitro and in vivo are indistinguishable (Kaffman et al. 1994). Unphosphorylated and phosphorylated Pho4WT–zz were purified with IgG Sepharose beads and incubated with yeast extract containing Pse1–GFP. The resin was washed, and proteins bound to Pho4WT–zz were eluted and analyzed by SDS-PAGE followed by immunoblotting with an anti-GFP antibody (Fig. 7A). We find that Pho4WT–zz interacts preferentially with Pse1 in its unphosphorylated state.
Figure 7.


Phosphorylated Pho4 has reduced affinity for Pse1. (A) Phosphorylation affects binding of Pho4 to Pse1 in an extract. Purified Pho4WT–zz and Pho4SA–zz, a mutant containing serine-to-alanine substitutions at the sites of phosphorylation, were phosphorylated or mock phosphorylated in vitro in an extract containing the Pho80–Pho85 cyclin–CDK complex. zz-Containing proteins were purified from the extract with IgG–Sepharose beads, washed extensively, and incubated with extract containing Pse1–GFP. The resin was washed and proteins bound to Pho4WT–zz and Pho4SA–zz were eluted with 1 m MgCl2. Eluted proteins were precipitated, separated on 7.8% SDS-PAGE, transferred to PVDF membrane, and visualized by immunoblotting with anti-GFP monoclonal antibodies (top). To ensure that the same amount of Pho4 was immobilized on IgG–Sepharose, Pho4–zz proteins were eluted from IgG–Sepharose with acetic acid, separated on a 7.8% SDS-polyacrylamide gel, and visualized by staining with Coomassie blue (bottom). (Lane 1) Mock-phosphorylated Pho4WT–zz (U); (lane 2) phosphorylated Pho4WT–zz (P); (lane 3) mock-phosphorylated Pho4SA–zz; (lane 4) phosphorylated Pho4SA–zz. Phosphorylation by Pho80–Pho85 causes Pho4WT–zz to migrate slower than unphosphorylated Pho4WT–zz in SDS-PAGE. (B) Phosphorylation affects Pho4 binding to purified Pse1. Pho4WT–zz was phosphorylated or mock phosphorylated and the immobilized proteins were incubated with Pse1–6His purified from E. coli. The resin was washed and proteins bound to Pho4WT–zz were eluted with 1 m MgCl2. Eluted proteins were precipitated, separated on a 7.8% SDS-polyacrylamide gel, and visualized by staining with silver (top). To ensure that the same amount of Pho4 was immobilized on IgG–Sepharose, Pho4WT–zz proteins were eluted from IgG–Sepharose with acetic acid, separated by 7.8% SDS-PAGE, and visualized by staining with Coomassie blue (bottom). (Lane 1) Mock-phosphorylated Pho4WT–zz; (lane 2) phosphorylated Pho4WT–zz; (lane 3) IgG–Sepharose beads alone (−). (*) Truncated form of Pse1.
As a control, we performed the same experiment with Pho4SA, a mutant Pho4 containing alanine substitutions at the sites of phosphorylation. We find that phosphorylated and mock-phosphorylated Pho4SA–zz have a similar affinity for Pse1–GFP, indicating that the difference in binding we observe between phosphorylated and unphosphorylated Pho4 is the result of specific phosphorylation at the sites utilized by Pho80–Pho85 (Fig. 7A, lanes 3,4). Moreover, phosphorylated and unphosphorylated Pho4WT–zz and Pho4SA–zz bind Pho85 with the same affinity (data not shown).
To determine if the preferential binding of unphosphorylated Pho4 to Pse1 is mediated by proteins in the yeast extract, we compared binding of purified Pse1–6His with purified phosphorylated and unphosphorylated Pho4WT–zz (Fig. 7B). We find that purified Pse1–6His binds preferentially to unphosphorylated Pho4, suggesting that phosphorylation of Pho4 reduces its intrinsic affinity for Pse1. These data suggest that phosphorylation of Pho4 regulates its import into the nucleus by preventing its interaction with Pse1.
Discussion
The subcellular localization of Pho4 is regulated by phosphorylation in response to changes in the extracellular concentration of inorganic phosphate. Pho4 is phosphorylated and localized predominantly to the cytoplasm when yeast are grown in phosphate-rich medium, and it is unphosphorylated and concentrated in the nucleus when yeast are starved for phosphate. We wished to determine how phosphorylation of Pho4 regulates its nucleocytoplasmic transport and to identify the machinery that moves Pho4 into and out of the nucleus. We demonstrate here that Pse1 is the import receptor for Pho4 and that phosphorylation of Pho4 directly regulates its association with Pse1, providing a mechanism by which phosphorylation may regulate import of Pho4 in vivo.
Although previous studies have suggested a function for Pse1 in nucleocytoplasmic transport, its exact role is unclear. Pse1 is an essential gene, suggesting that it transports cargo whose proper localization is required for vegetative growth (Chow et al. 1992; Seedorf and Silver 1997). On the basis of the following data, Pse1/Kap121 was proposed to play a role in both ribosomal protein import and in mRNA export: (1) mRNA export is defective in the pse1-1 kap123Δ double mutant, but not the kap123Δ or pse1-1 mutant (Seedorf and Silver 1997); (2) the kap123Δ strain is defective in import of the ribosomal protein L25 (Rout et al. 1997; Schlenstedt et al. 1997) and that defect can be suppressed by overexpression of PSE1 (Rout et al. 1997); and (3) in yeast extract, Kap123 binds to the ribosomal protein L25 and Pse1 can bind to L25, but only in the absence of Kap123 (Rout et al. 1997; Schlenstedt et al. 1997). These data are consistent with Kap123 being an import receptor for ribosomal proteins and with Pse1 playing a partially redundant role in ribosomal protein import. The mRNA export defect observed in the pse1-1 kap123Δ double mutant may be indirect, as Pse1 clearly has a direct role in protein import.
We have demonstrated that nuclear import of Pho4 requires PSE1. Pho4 is the first cargo whose import is dependent exclusively on the function of Pse1. Our results, in combination with previous studies, allow us to ascribe unique and separate functions to Pse1 and Kap123, as the kap123Δ strain is not defective in Pho4 import, nor does Kap123 interact with Pho4. The essential role for Pse1 in transport remains unclear, because cells lacking Pho4 or exhibiting defects in Pho4 localization do not have growth defects when grown in rich medium. The identification of a 27 amino acid peptide within Pho4 that is targeted to the nucleus in a PSE1-dependent manner should facilitate the establishment of a consensus NLS recognized by Pse1. This consensus sequence may lead to the identification of other Pse1 cargoes, providing a better understanding of its essential role in vegetative growth.
We have shown that phosphorylation of Pho4 reduces its affinity for Pse1 and that this reduction in affinity is a direct consequence of phosphorylation and is not mediated by other proteins in yeast extract. Remarkably, the NLS in Pho4 contains a single site of phosphorylation by Pho80–Pho85. It will be of interest to determine the contribution of individual phosphorylation sites to the regulation of the interaction with Pse1. In addition, we wish to understand the structural effect that phosphorylation has on the ability of Pho4 to interact with Pse1.
The subcellular localization of many proteins, including the transcriptional regulators NF-AT, Mig1, Swi5, and Swi6, appears to be regulated by phosphorylation in response to either cell cycle position or extracellular signals (Moll et al. 1991; Sidorova et al. 1995; Beals et al. 1997; De Vit et al. 1997). The role of phosphorylation in regulation of localization is not understood at a mechanistic level in any of these cases. The cell cycle regulated localization and phosphorylation of the yeast transcription factor Swi5 has striking parallels with the regulation of Pho4. In both systems, phosphorylation of a transcription factor by a cyclin-CDK complex results in the localization of the transcription factor to the cytoplasm. For Swi5, a region of the protein containing sites of phosphorylation by the CDK Cdc28 has been shown to be both necessary and sufficient for cell cycle-regulated localization (Moll et al. 1991). However, it is unclear if the regulated localization of Swi5 reflects regulation of import and/or export. Additionally, it is not clear if phosphorylation of Swi5 regulates its association with a transport receptor, with an adapter protein, or with a nuclear or cytoplasmic anchor.
Phosphorylation of a fusion protein containing the SV40 large T antigen NLS has been shown to affect its nuclear import by modulating its interaction with importin α (Jans et al. 1991; Xiao et al. 1996; Hubner et al. 1997). However, these phosphorylation events have not been shown to occur in vivo, nor have they been observed to regulate the nuclear import of full-length SV40 large T antigen. Our work provides a simple demonstration of how phosphorylation of a protein by a physiologically relevant kinase modifies its association with a transport receptor. This change in binding affinity could regulate the rate of nuclear import, thereby leading to a change in the nucleocytoplasmic localization of the cargo.
Previously, a correlation between the phosphorylation state of a protein and its cytoplasmic compartmentalization has been interpreted as evidence for regulated import. The discovery of nuclear export pathways has led to the understanding that both nuclear import and/or export are potential points of regulation. The regulated localization of Pho4 in response to extracellular concentrations of inorganic phosphate is likely to reflect control of both import and export of Pho4. Data presented in this manuscript suggest that import of Pho4 is regulated by phosphorylation and our unpublished studies suggest that export of Pho4 also is regulated by phosphorylation. An understanding of how phosphorylation of Pho4 leads to a change in its subcellular localization will require an experimental dissection of the effect of phosphorylation on the rates of import and export.
We propose that phosphorylation of a transcription factor in response to an environmental cue acts as a molecular switch, inhibiting import and, at the same time, promoting export. Blocking import prevents further nuclear entry of a transcription factor, whereas enhancing export leads to a rapid elimination of residual nuclear activity. This type of molecular switch allows cells to utilize nucleocytoplasmic localization of a transcription factor as a way to regulate gene expression in a rapid and efficient way.
Materials and methods
Yeast strains and plasmids
Yeast strains were grown in YEPD or synthetic (SD) medium supplemented with amino acids (Sherman 1991) (except in phosphate starvation assays; see below). pho4Δ::TRP1 (EY0130) and pho4Δ::TRP1 pho80Δ::HIS3 (EY0219) yeast strains derived from K699 (Nasmyth et al. 1990) were generated by standard gene replacement techniques (Rothstein 1991). PSY580, pse1-1, kap123Δ have been described previously (Seedorf and Silver 1997). Yeast transformations were performed by the lithium acetate method essentially as described (Guthrie and Fink 1991).
pACPHO4–GFP (EB0347) was constructed as follows. First, the PHO4 promoter (nucleotides −323 to −1 upstream of the ATG) was amplified by PCR and subcloned into pRS316–GFP, which contains GFPS65T (Heim et al. 1995) cloned into the EcoRI and BamHI sites of pRS316 (Sikorski and Hieter 1989), to generate pPHO4pr–GFP (EB0346). The entire PHO4 coding region amplified by PCR was then subcloned in front of GFP in pRS316–GFP as a BglII–EcoRI fragment to generate pACPHO4–GFP. pACPHO4SA–GFP (EB0356) is identical to pACPHO4–GFP except that there are five serine-to-alanine substitutions at the sites of phosphorylation by Pho80–Pho85 (O’Neill et al. 1996). pPHO4–GFP3 (EB0757) was constructed by amplifying two additional copies of the complete GFPS65T gene which were cloned into pPHO4–GFP as MfeI–EcoRI fragments. pPHO4–GFP derivatives [pPHO42–247–GFP (EB0350), pPHO42–94–GFP3 (EB0824), pPHO4248–312–GFP3 (EB0825), pPHO4Δ95–162–GFP (EB0364), pPHO4Δ172–192–GFP (EB0365), pPHO4Δ141–165–GFP (EB0823), pPHO4Δ157–164–GFP (EB0383), pPHO495–166–GFP3 (EB0832), pPHO4140–171–GFP3 (EB0834), pPHO4156–171–GFP3 (EB0835), pPHO4140–166–GFP3 (EB0836)] were constructed by amplifying PHO4 from pAC312 with the appropriate primers and subcloning the resulting fragments into either pACPHO4–GFP or pPHO4–GFP3, replacing full-length PHO4. Further information on the construction of PHO4–GFP derivatives is available upon request. pT7–PHO4WT–zz (EB0801) was generated by amplifying zz from pTL27 (Lafontaine and Tollervey 1996) and was used to replace the EcoRI–BamHI fragment containing GFP in pACPHO4–GFP. The NcoI–BamHI fragment containing Pho4WT–z was then cloned into pET16b (Novagen) to generate pT7–PHO4WT–zz. pT7–PHO4SA–zz (EB0814) is identical to pT7–PHO4WT–zz except that there are five serine-to-alanine substitutions at the sites of phosphorylation by Pho80–Pho85 (O’Neill et al. 1996). pT7–PHO4Δ157–164–zz (EB0769) was generated by first subcloning the EcoRI–BamHI fragment containing zz from pPHO4–zz into pPHO4Δ157–164–GFP. Next, PHO4Δ157–164–zz was subcloned as an NcoI–BamHI fragment into pET16b (Novagen). pPSE1–GFP (pPS1069), pSXM1–GFP (pPS1117), and pKAP123–GFP (pPS1070) have been described previously (Seedorf and Silver 1997). pPSE1–6His (EB0815) was constructed by PCR amplifying PSE1 from the yeast genome (Promega) and subcloning the fragment into the BamHI site of pQE60 (Qiagen) to generate carboxy-terminally His-tagged Pse1. pSH101-1 (a gift from Shai Shaham, University of California, San Francisco) consists of GSP1 fused to three copies of the myc epitope tag under the control of the GSP1 promoter in pRS303 (Sikorski and Hieter 1989). pT7–myc–GSP1 was constructed by amplifying mycGSP1 from pSH101-1 and cloning the amplified fragment into the NdeI and XhoI sites of pAED4. All constructs were verified by sequencing except pPSE1–6His which was shown to complement a pse1Δ mutant (data not shown).
Phosphate starvation assay and microscopy
Yeast was grown overnight at 25°C in SD medium supplemented with amino acids (high-phosphate medium) and then diluted and grown to an OD600 of 0.1–0.3. One milliliter of this culture was pelleted, and the pellet was resuspended in 50 μl of high-phosphate medium for steady-state high-phosphate localization of GFP derivatives. Two microliters of the high-phosphate culture was placed on a microscope slide and GFP localization in live cultures was monitored by direct fluorescence. The remaining yeast culture was pelleted by centrifugation and washed twice with 10 ml of water, once with 1 ml of water, and resuspended in no-phosphate medium (SD dropout medium with potassium chloride replacing monobasic potassium phosphate) to an OD600 of 0.1–0.3. Yeast was grown in medium lacking phosphate for 30–45 min to starve the yeast for phosphate. One milliliter of phosphate-starved culture was pelleted and resuspended in 50 μl of no phosphate medium. Two microliters of the starved culture was place on a microscope slide and GFP localization was monitored. All images documenting GFP localization were collected with a CCD camera using identical settings (Photometrics).
Recombinant protein expression and purification
Pse1 was expressed in E. coli with a 6His tag at the carboxyl terminus. Bacterial cells (strain SG13009) containing the pPSE1–6His expression vector were grown in LB + 100 μg/ml carbenicillin + 25 μg/ml kanamycin to an OD600 of 0.6, and expression was induced with 1 mm IPTG for 3 hr at room temperature. Cells were resuspended in lysis buffer (30 mm Tris-Cl at pH 8, 500 mm NaCl, 0.05% Tween 20, and 2 mm β-mercaptoethanol) containing 30 mm imidazole and protease inhibitors (1 mm PMSF, 2 mm benzamidine, 1 μg/ml pepstatin A, 1 μg/ml leupeptin), lysed by sonication, and cleared by centrifugation at 17,000 rpm in an SS34 rotor for 20 min. The lysate containing the Pse1–6His fusion protein was loaded on a 1 ml of chelating HiTrap column (Pharmacia) at 1 ml/min in lysis buffer containing 30 mm imidazole and protease inhibitors and eluted with a 30 to 1000 mm imidazole gradient. Eluted proteins were dialyzed against 100 mm NaCl, 30 mm Tris-Cl (pH 8). After dialysis, sorbitol was added to a final concentration of 250 mm and the purified protein was frozen in small aliquots and stored at −80°C. Approximately 90% of the purified material contains an equal mixture of full-length Pse1–6His and a truncation product lacking part of the amino terminus. To express Myc–Gsp1, E. coli (strain BL21) containing pT7–mycGsp1 was grown in LB + 100 μg/ml carbenicillin to an OD600 of 0.6, and expression was induced with 0.4 mm IPTG for 3 hr at room temperature. Cell pellet from 3 liters of culture was resuspended in 40 ml of D-50 (20 mm KH2P04 at pH 6.6, 10% glycerol, 2 mm MgCl2, 50 mm KCl, 1 mm DTT, 5 μm GTP) containing protease inhibitors (1 mm PMSF, 2 mm benzamidine, 1 μg/ml pepstatin A, 1 μg/ml leupeptin), sonicated, and spun for 20 min in an SS34 rotor at 17,000 rpm. Extract was loaded onto a 10-ml SP Sepharose HR (Pharmacia) column at 2 ml/min and eluted with a 50 to 1000 mm KCl gradient. Fractions containing Myc–Gsp1 were precipitated with 60% ammonium sulfate, resuspended in 1.5 ml of F-50 (30 mm HEPES–KOH at pH 7.7, 8.7% glycerol, 2 mm MgCl2, 5 μm GTP, 50 mm KCl) and loaded onto a Sephacryl S100 (Pharmacia) column equilibrated in F-50. Fractions containing Myc–Gsp1 were concentrated on a 2-ml Bio-Scale Q column (Bio-Rad), and the purified protein (>95% pure) was frozen in small aliqouts. To produce Pho4–zz fusion proteins, E. coli (strain BL21) containing the Pho4–zz expression vector was grown to OD600 of 0.6 in LB + 100 μg/ml carbenicillin and induced with 0.4 mm IPTG for 3 hr at room temperature. Cell pellet from 1 liter of culture was resuspended in 15 ml of IgG buffer (50 mm Tris-Cl at pH 7.5, 150 mm NaCl, 0.05% Tween 20, 5 mm MgCl2), sonicated, and spun for 1 hr at 60,000g. Sucrose was added to a final concentration of 250 mm, and the high-speed supernatant was frozen in 0.5-ml aliquots. Pho4–zz proteins were purified by diluting this high-speed supernatant to ∼2 mg/ml in buffer B 0.1 (20 mm PIPES–KOH at pH 6.8, 1 mm EDTA, 10% glycerol, 100 mm NaCl) plus protease inhibitors (1 mm PMSF, 2 mm benzamidine, 1 μg/ml pepstatin A, 1 μg/ml leupeptin) and loaded onto a 1 ml SP Sepharose HiTrap column (Pharmacia) at 1 ml/min and eluted with a 100–1000 mm NaCl gradient. Fractions containing Pho4–zz (∼90% pure) were then frozen in small aliqouts.
Yeast extract preparation
Yeast strain Y57 was grown in 300 ml of synthetic high-phosphate medium to an OD600 of 1, harvested, and washed with 20 ml of H2O. Cells were resuspended in 2 ml of IgG buffer containing protease inhibitors (1 mm PMSF, 2 mm benzamidine, 1 μg/ml pepstatin A, 1 μg/ml leupeptin) and transferred to a 2-ml screw-cap Eppendorf tube containing ∼1 ml of 0.5-mm acid-washed glass beads. Trapped air was removed and cells were lysed in a minibead beater (BioSpec Products) by use of 5 × 1-min strokes with a 1-min resting period on ice. The supernatant was transferred to a new Eppendorf tube and cleared by spinning 2 × 20 min at 4°C in a microcentrifuge at 14,000 rpm and filtering through a 0.22-μm filter. Glycerol was added to a final concentration of 10%, and the extract was frozen at −80°C in small aliqouts.
Binding assay
For the assay of Pho4WT–zz and Pho4Δ157–164–zz binding to Pse1–GFP, Sxm1–GFP, and Kap123–GFP, saturating amounts of bacterial lysate from either Pho4WT–zz or Pho4Δ157–164–zz were bound to 25 μl of IgG–Sepharose by incubating for 90 min at 4°C. The resin with bound Pho4–zz (∼4 mg/ml) was washed with IgG buffer plus protease inhibitors (1 mm PMSF, 2 mm benzamidine, 1 μg/ml pepstatin A, 1 mg/ml leupeptin), followed by IgG buffer containing 1 m of NaCl, and then re-equilibrated with IgG buffer. Immobilized Pho4–zz proteins were incubated for 3 hr at 4°C with 1.65 mg of total protein from a Y57 extract expressing one of the importin β family members tagged with GFP. The resin was washed extensively with IgG buffer and proteins bound to Pho4–zz proteins were eluted with IgG buffer containing 1 m MgCl2 and concentrated with methanol–chloroform precipitation. Pho4WT–zz proteins were eluted with 0.5 m HOAc (pH 3.4). For binding Pho4WT–zz to purified Pse1–6His, Pho4WT–zz was purified from bacterial lysate as described above and incubated with 50 μg of purified Pse1–6His in the presence of either 1.65 mg of total protein from a Y57 extract or 1.65 mg of bovine serum albumin (BSA) for 3 hr at 4°C. Samples were processed as described above. For binding of phosphorylated and unphosphorylated Pho4WT–zz to Pse1–GFP in a yeast extract, 30 μg of immobilized phosphorylated or unphosphorylated Pho4WT–zz was incubated with 1.65 mg of protein from a Y57 extract containing Pse1–GFP, and samples were processed as described above. For binding of Pho4WT–zz to purified Pse1, 30 μg of immobilized phosphorylated or unphosphorylated Pho4WT–zz was purified away from the yeast extract by incubating with 1 m MgCl2 for 10 min at room temperature, re-equilibrating with IgG buffer, and incubating further with 10 nm purified Pse1–6His in the presence of 10% glycerol and 2 mg/ml BSA for 1 hr at room temperature. The resin was washed extensively with IgG buffer, eluted with 1 m MgCl2, and concentrated with methanol–chloroform precipitation.
In vitro phosphorylation of Pho4–zz proteins
Thirty micrograms of purified Pho4WT–zz or Pho4SA–zz was incubated with ∼1 mg of yeast whole-cell extract containing HA–Pho80 expressed from the GPD promoter for 1 hr at room temperature in the presence of phosphatase inhibitors (80 mm β-glycerophosphate, 10 mm NaF, 10 nm calyculin A), and protease inhibitors (1 mm PMSF, 2 mm benzamidine, 1 μg/ml leupeptin, 1 μg/ml pepstatin A), either in the presence of an ATP regenerating system (1 mm ATP, 5 mm creatine phosphate, 50 μg/ml creatine kinase), or in the absence of an ATP regenerating system (mock phosphorylation). The reaction was placed on ice, diluted with IgG buffer to ∼3 mg/ml, and incubated with 25 μl of IgG–Sepharose beads for 2 hr at 4°C. Bound Pho4–zz proteins were purified by extensive washes with IgG buffer followed by a wash with IgG buffer containing 1 m NaCl and then re-equilibrated in IgG buffer for binding studies.
Dissociation of the Pho4–Pse1 complex by Gsp1–GTP
Sixty micrograms of purified Pse1–6His and 60 μg of Pho4WT–zz were bound in a 240-μl reaction for 1 hr at 4°C. Complex containing Pse1–6His bound to Pho4WT–zz was purified by incubating the reaction with 35 μl of IgG–Sepharose beads for 2 hr at 4°C. The resin containing immobilized Pho4WT–zz bound to Pse1 was washed extensively with IgG buffer and split into three equal parts. One-third of the immobilized Pho4WT–zz bound to Pse1 was incubated with 10 μg of purified Myc–Gsp1 loaded with GTP, one-third with 10 μg of purified Myc–Gsp1 loaded with GDP, and one-third with buffer alone for 20 min at room temperature. Proteins released during this incubation period were collected (eluate), the resin was washed once with 0.5 ml of IgG buffer, bound proteins were eluted with 1 m MgCl2, and concentrated with methanol–chloroform precipitation. Proteins were separated on a 10% SDS-polyacrylamide gel and visualized by staining with Coomassie blue.
Nucleotide loading of Myc–Gsp1 was performed as follows. Purified Myc–Gsp1 (0.67 mg/ml) was incubated in the presence of 20 mm EDTA, 2 mm GTP or GDP, and 2 mm DTT for 1 hr at room temperature. MgCl2 was added to a final concentration of 50 mm and the reaction was incubated further on ice for 20 min. Unbound nucleotides were removed by loading the sample over a Bio-Spin P-6 spin column (Bio-Rad) equilibrated in F-50 and the Myc–Gsp1 was frozen in small aliquots.
Acknowledgments
We thank Pamela Silver for the pse1-1, kap123Δ, sxm1Δ, and PSY580 yeast strains, John Aitchison for the kap104ts strain, Molly Fitzgerald-Hayes for the cse1 mutant strain, and Karsten Weis and Katrin Stade for xpo1-1. We thank Pamela Silver for the plasmids expressing Pse1–GFP, SXM1–GFP and Kap123–GFP, Karsten Weis for pTL27 and Shai Shaham for pSH101-1. We also thank Karsten Weis and members of the E.K.O. and Guthrie laboratories for their critical comments on this work. This work was supported by a National Science Foundation Presidential Faculty Fellow Award and a grant from the David and Lucile Packard Foundation (to E.K.O.). A.K. was supported by the Medical Scientist Training Program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL oshea@biochem.ucsf.edu; FAX (415) 502-4315.
References
- Aitchison JD, Blobel G, Rout MP. Kap104p: A karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science. 1996;274:624–627. doi: 10.1126/science.274.5287.624. [DOI] [PubMed] [Google Scholar]
- Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes & Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- Chow TY, Ash JJ, Dignard D, Thomas DY. Screening and identification of a gene, PSE-1, that affects protein secretion in Saccharomyces cerevisiae. J Cell Sci. 1992;101:709–719. doi: 10.1242/jcs.101.3.709. [DOI] [PubMed] [Google Scholar]
- De Vit MJ, Waddle JA, Johnston M. Regulated nuclear translocation of the Mig1 glucose repressor. Mol Biol Cell. 1997;8:1603–1618. doi: 10.1091/mbc.8.8.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;39:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Gaits F, Degols G, Shiozaki K, Russell P. Phosphorylation and association with the transcription factor atf1 regulate localization of Spc1/Sty1 stress-activated kinase in fission yeast. Genes & Dev. 1998;12:1464–1473. doi: 10.1101/gad.12.10.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Laskey RA, Hartmann E. Isolation of a protein that is essential for the first step of nuclear protein import. Cell. 1994;79:767–778. doi: 10.1016/0092-8674(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Kostka S, Kraft R, Dingwall C, Laskey RA, Hartmann E, Prehn S. Two different subunits of importin cooperate to recognize nuclear localization signals and bind them to the nuclear envelope. Curr Biol. 1995a;5:383–392. doi: 10.1016/s0960-9822(95)00079-0. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Vogel F, Mills AD, Hartmann E, Laskey RA. Distinct functions for the two importin subunits in nuclear protein import. Nature. 1995b;377:246–248. doi: 10.1038/377246a0. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Pante N, Kutay U, Aebi U, Bischoff FR. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996;15:5584–5594. [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Dabrowski M, Bischoff FR, Kutay U, Bork P, Hartmann E, Prehn S, Izaurralde E. A novel class of RanGTP binding proteins. J Cell Biol. 1997;138:65–80. doi: 10.1083/jcb.138.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. Methods in enzymology. 1991;199:186. [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hopper AK, Schultz LD, Shapiro RA. Processing of intervening sequences: A new yeast mutant which fails to excise intervening sequences from precursor tRNAs. Cell. 1980;19:741–751. doi: 10.1016/s0092-8674(80)80050-x. [DOI] [PubMed] [Google Scholar]
- Hubner S, Xiao CY, Jans DA. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J Biol Chem. 1997;272:17191–17195. doi: 10.1074/jbc.272.27.17191. [DOI] [PubMed] [Google Scholar]
- Imamoto N, Tachibana T, Matsubae M, Yoneda Y. A karyophilic protein forms a stable complex with cytoplasmic components prior to nuclear pore binding. J Biol Chem. 1995;270:8559–8565. doi: 10.1074/jbc.270.15.8559. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Kutay U, von Kobbe C, Mattaj IW, Gorlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 1997;16:6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jans DA, Hubner S. Regulation of protein transport to the nucleus: Central role of phosphorylation. Physiol Rev. 1996;76:651–685. doi: 10.1152/physrev.1996.76.3.651. [DOI] [PubMed] [Google Scholar]
- Jans DA, Ackermann MJ, Bischoff JR, Beach DH, Peters R. p34cdc2-mediated phosphorylation at T124 inhibits nuclear import of SV- 40 T antigen proteins. J Cell Biol. 1991;115:1203–1212. doi: 10.1083/jcb.115.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki T, Chen S, Hitomi M, Jacobs E, Kumagai C, Liang S, Schneiter R, Singleton D, Wisniewska J, Tartakoff AM. Isolation and characterization of Saccharomyces cerevisiae mRNA transport-defective (mtr) mutants. J Cell Biol. 1994;126:649–659. doi: 10.1083/jcb.126.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffman A, Herskowitz I, Tjian R, O’Shea EK. Phosphorylation of the transcription factor PHO4 by a cyclin-CDK complex, PHO80-PHO85. Science. 1994;263:1153–1156. doi: 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb MH. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- Kudo N, Khochbin S, Nishi K, Kitano K, Yanagida M, Yoshida M, Horinouchi S. Molecular cloning and cell cycle-dependent expression of mammalian CRM1, a protein involved in nuclear export of proteins. J Biol Chem. 1997;272:29742–29751. doi: 10.1074/jbc.272.47.29742. [DOI] [PubMed] [Google Scholar]
- Kutay U, Bischoff FR, Kostka S, Kraft R, Gorlich D. Export of importin alpha from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90:1061–1071. doi: 10.1016/s0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Kutay U, Lipowsky G, Izaurralde E, Bischoff FR, Schwarzmaier P, Hartmann E, Gorlich D. Identification of a tRNA-specific nuclear export receptor. Mol Cell. 1998;1:359–369. doi: 10.1016/s1097-2765(00)80036-2. [DOI] [PubMed] [Google Scholar]
- Lafontaine D, Tollervey D. One-step PCR mediated strategy for the construction of conditionally expressed and epitope tagged yeast proteins. Nucleic Acids Res. 1996;24:3469–3471. doi: 10.1093/nar/24.17.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latimer M, Ernst MK, Dunn LL, Drutskaya M, Rice NR. The N-terminal domain of IkappaB alpha masks the nuclear localization signal(s) of p50 and c-Rel homodimers. Mol Cell Biol. 1998;18:2640–2649. doi: 10.1128/mcb.18.5.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magbanua JP, Ogawa N, Harashima S, Oshima Y. The transcriptional activators of the PHO regulon, Pho4p and Pho2p, interact directly with each other and with components of the basal transcription machinery in Saccharomyces cerevisiae. J Biochem (Tokyo) 1997;121:1182–1189. doi: 10.1093/oxfordjournals.jbchem.a021713. [DOI] [PubMed] [Google Scholar]
- Moll T, Tebb G, Surana U, Robitsch H, Nasmyth K. The role of phosphorylation and the CDC28 protein kinase in cell cycle- regulated nuclear import of the S. cerevisiae transcription factor SWI5. Cell. 1991;66:743–758. doi: 10.1016/0092-8674(91)90118-i. [DOI] [PubMed] [Google Scholar]
- Moroianu J, Hijikata M, Blobel G, Radu A. Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: Alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc Natl Acad Sci. 1995;92:6532–6536. doi: 10.1073/pnas.92.14.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu J, Blobel G, Radu A. Nuclear protein import: Ran-GTP dissociates the karyopherin alphabeta heterodimer by displacing alpha from an overlapping binding site on beta. Proc Natl Acad Sci. 1996;93:7059–7062. doi: 10.1073/pnas.93.14.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S, Dreyfuss G. Nuclear export of proteins and RNAs. Curr Opin Cell Biol. 1997;9:420–429. doi: 10.1016/s0955-0674(97)80016-6. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Adolf G, Lydall D, Seddon A. The identification of a second cell cycle control on the HO promoter in yeast: Cell cycle regulation of SW15 nuclear entry. Cell. 1990;62:631–647. doi: 10.1016/0092-8674(90)90110-z. [DOI] [PubMed] [Google Scholar]
- Neville M, Stutz F, lee L, Davis LI, Rosbash M. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr Biol. 1997;7:767–775. doi: 10.1016/s0960-9822(06)00335-6. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Forbes DJ. Nuclear import can be separated into distinct steps in vitro: Nuclear pore binding and translocation. Cell. 1988;52:641–653. doi: 10.1016/0092-8674(88)90402-3. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nucleocytoplasmic transport: Signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- Ogawa N, Oshima Y. Functional domains of a positive regulatory protein, PHO4, for transcriptional control of the phosphatase regulon in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:2224–2236. doi: 10.1128/mcb.10.5.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo M, Okazaki H, Nishimoto T. The RCC1 protein, a regulator for the onset of chromosome condensation locates in the nucleus and binds to DNA. J Cell Biol. 1989;109:1389–1397. doi: 10.1083/jcb.109.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill EM, Kaffman A, Jolly ER, O’Shea EK. Regulation of PHO4 nuclear localization by the PHO80-PHO85 cyclin-CDK complex. Science. 1996;271:209–212. doi: 10.1126/science.271.5246.209. [DOI] [PubMed] [Google Scholar]
- Oshima Y. The phosphatase system in Saccharomyces cerevisiae. Genes Genet Syst. 1997;72:323–334. doi: 10.1266/ggs.72.323. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nayari B, Bacheline F, Dargemmt C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Rexach M, Blobel G. Protein import into nuclei: Association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell. 1995;83:683–692. doi: 10.1016/0092-8674(95)90181-7. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Mills AD, Dilworth SM, Laskey RA, Dingwall C. Nuclear protein migration involves two steps: Rapid binding at the nuclear envelope followed by slower translocation through nuclear pores. Cell. 1988;52:655–664. doi: 10.1016/0092-8674(88)90403-5. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: Integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Rout MP, Blobel G, Aitchison JD. A distinct nuclear import pathway used by ribosomal proteins. Cell. 1997;89:715–725. doi: 10.1016/s0092-8674(00)80254-8. [DOI] [PubMed] [Google Scholar]
- Schlenstedt G, Smirnova E, Deane R, Solsbacher J, Kutay U, Gorlich D, Ponstingl H, Bischoff FR. Yrb4p, a yeast ran-GTP-binding protein involved in import of ribosomal protein L25 into the nucleus. EMBO J. 1997;16:6237–6249. doi: 10.1093/emboj/16.20.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KR, Smith RL, O’Shea EK. Phosphate-regulated inactivation of the kinase PHO80-PHO85 by the CDK inhibitor PHO81. Science. 1994;266:122–126. doi: 10.1126/science.7939631. [DOI] [PubMed] [Google Scholar]
- Seedorf M, Silver PA. Importin/karyopherin protein family members required for mRNA export from the nucleus. Proc Natl Acad Sci. 1997;94:8590–8595. doi: 10.1073/pnas.94.16.8590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sidorova JM, Mikesell GE, Breeden LL. Cell cycle-regulated phosphorylation of Swi6 controls its nuclear localization. Mol Biol Cell. 1995;6:1641–1658. doi: 10.1091/mbc.6.12.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- Toone WM, Kuge S, Samuels M, Morgan BA, Toda T, Jones N. Regulation of the fission yeast transcription factor pap1 by oxidative stress: Requirement for the nuclear export factor crm1 (Exportin) and the stress-activated MAP kinase Sty1/Spc1. Genes & Dev. 1998;12:1453–1463. doi: 10.1101/gad.12.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis K. Importins and exportins: How to get in and out of the nucleus. Trends Biochem Sci. 1998;23:157–196. doi: 10.1016/s0968-0004(98)01204-3. [DOI] [PubMed] [Google Scholar]
- Weis K, Mattaj IW, Lamond AI. Identification of hSRP1 alpha as a functional receptor for nuclear localization sequences. Science. 1995;268:1049–1053. doi: 10.1126/science.7754385. [DOI] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Williams SC, Angerer ND, Johnson PF. C/EBP proteins contain nuclear localization signals imbedded in their basic regions. Gene Expr. 1997;6:371–385. [PMC free article] [PubMed] [Google Scholar]
- Xiao CY, Hubner S, Elliot RM, Caon A, Jans DA. A consensus cAMP-dependent protein kinase (PK-A) site in place of the CcN motif casein kinase II site simian virus 40 large T-antigen confers PK-A-mediated regulation of nuclear import. J Biol Chem. 1996;271:6451–6457. doi: 10.1074/jbc.271.11.6451. [DOI] [PubMed] [Google Scholar]
- Xiao Z, McGrew JT, Schroeder AJ, Fitzgerald-Hayes M. CSE1 and CSE2, two new genes required for accurate mitotic chromosome segregation in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:4691–4702. doi: 10.1128/mcb.13.8.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]