

Abstract
We report, for the first time, that certain N-acetylthiourea derivatives serve as highly potent and isozyme selective activators for the recombinant form of human histone deacetylase-8 in the assay system containing Fluor-de-Lys as a fluorescent substrate. The experimental data reveals that such activating feature is manifested via decrease in the Km value of the enzyme’s substrate and increase in the catalytic turnover rate of the enzyme
Keywords: Enzyme Activation, Epigenetic Regulation, Histone Deacetylase, Isozyme selectivity, Thiourea Derivative
Of different modes of epigenetic regulation of gene expression, acetylation and deacetylation of histore core proteins have gained considerable interest in the recent years. This has been particularly important from the point of view of developing therapeutic agents for the treatment of various human diseases.1 The acetylation of the lysine groups of histone is accomplished by the action of histone acyltransferase (HAT), and their deacetylation reaction is catalyzed by various isoforms of histone deacetylases (HDACs). The dynamic (catalytic) actions of both these enzymes dictate the extent of acetylated form of histone within the chromatin structure.2 Since the acetylation eliminates the positive charges from the histone’s lysine residues, their electrostatic interactions with negatively charged phosphate groups of DNA is impaired, resulting in loosening of the nucleosomal assembly and subsequently facilitating the transcriptional activation of various genes. The reversal of the overall process (i.e., the suppression of the gene expression) is manifested due to the diminution in the extent of acetyl functionality within the histone core, which “compacts” the nucleosomal assembly due to favorable electrostatic interaction between the positively charged histone and negatively charged phosphate groups of DNA. In contrast to this simplistic model, it has been observed that the transcriptional activation and suppression of genes is not only controlled by the extent of histone acetylation/deacetylation but also on the type of histone molecules which are acetylated.3 Since the extent and/or the type of histone acetylation within the nucleosome is maintained by the catalytic efficiencies as well as selectivity of HAT and HDAC isozymes, it is possible to control the expression of various genes simply by modulating the activities of these enzymes by using isozyme selective activators and inhibitors. With realization that small molecular weight HDAC inhibitors have anticancer effect, there has been a growing interest (by major Pharmaceutical and Biotech companies) in designing novel inhibitors against the above enzyme not only for the treatment of cancers but various other diseases.4,5 Presently, two HDAC inhibitors, namely SAHA and romidepsin, have been approved by FDA for the treatment of cutaneous lymphoma, and there are about a dozen of other inhibitors which are at various stages of clinical trials.6,7
To date, 14 different types of HDACs are known to be involved in promoting deacetylase reaction of different histones as well as selected non-histone proteins.8 Based on the phylogenetic relationship, HDACs have been grouped in four different classes: (1) Class I (HDAC 1, 2, 3, and 8), (2) Class II (HDAC-4, -5, -6, -7, -9 and -10), (3) Class III (Sirtuins), and (4) class IV (HDAC 11). Of these, whereas Class I, II and IV HDACs catalyze metal dependent deacetylation reaction, Class III HDACs/Sirtuins utilize NAD+ as the coenzyme and its ADP-ribose moiety serves as the acetyl group acceptor during the catalytic cycle.9
Of these, except for Class III HDACs (Sirtuins), there has been limited interest in delineating the roles of HDAC activators in gene expression, leading to the modulation of different physiological/pathological processes.10 The activation of selected isoforms of HDACs has been found to be desirable in treating chronic obstructive pulmonary disease (COPD), which is caused by increased expressions of pro-inflammatory factors due to reduced HDAC activity, and theophyllin has been found to relieve the symptoms of COPD by indirectly activating HDACs (especially HDAC-2).11,12 Aside from the histone mediated regulation at the transcriptional level, circumstantial evidence suggest the potential role of activation of specific isoforms of HDACs in promoting physiologically desirable metabolic processes.13 Hence, not only HDAC inhibitors but also the small molecular weight HDAC-activators are expected to find applications in human health and diseases. However, to the best of our knowledge, except for a US patent application13, there has been no report (in any scientific journal) of small molecular weight compounds serving as the direct activators of metallo-HDACs (i.e., HDAC class I, II, and IV).
In pursuit of investigating the structural-functional and inhibitory features of the recombinant form of human HDAC-8, we have been synthesizing a variety of structurally diverse small molecular weight compounds, which would interact at the active site pocket of the enzyme to unravel their influence on the enzyme catalysis. With precedent of selected benzamide and thiourea derivatives serving as the inhibitors of different class of HDACs,14 we synthesized N(phenylcarbothiol)benzamide (TM-2-51) using benzoylchloride and aniline as the precursors as outlined in Scheme 1.
Scheme 1.

Synthetic scheme of TM-2-51
We evaluated the effectiveness of TM-2-51 (Scheme 1) on the HDAC-8 catalyzed reaction using Fluor-de-Lys® as the commercial fluorogenic substrate as described previously.15 To our surprise, we observed that the initial rate of the HDAC-8 catalyzed reaction was significantly enhanced in the presence of TM-2-51, suggesting that the latter compound served as an “activator” rather than inhibitor of HDAC-8. This preliminary finding prompted us to synthesize differently substituted N-acetylthiourea derivatives (see the supporting information) and evaluated their efficacies (at 10 μM; the concentration of compounds customarily used in our lab for initial screening of inhibitors/activators against HDAC-8 as well as other pathogenic enzymes (Table 1).
Table 1.
Fold-activation of HDAC-8 by 10 μM concentration of selected N-acetylthiourea derivatives
| Entry | Structure | Compound ID | Fold-Activationa |
|---|---|---|---|
| 1 |
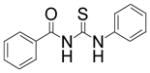
|
TM-2-51 | 12 |
| 2 |
|
TM-2-87 | 2 |
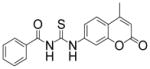
| 3 |
|
TM-2-88 | 8.4 |
| 4 |
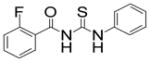
|
TM-2-138 | 3.5 |
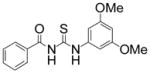
| 5 |
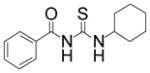
|
TM-2-97 | 5.6 |
| 6 |
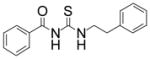
|
TM-2-105 | 2.0 |
| 7 |

|
TM-2-90 | 3.3 |
| 8 |
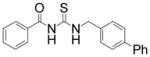
|
TM-2-101 | 1.1 |
| 9 |
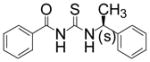
|
TM-2-104 | 4.5 |
| 10 |

|
TM-2-130 | 1.5 |
| 11 |
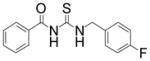
|
TM-2-131 | 3.0 |
| 12 |
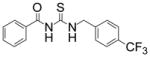
|
TM-2-126 | 1.2 |
| 13 |
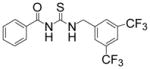
|
TM-2-125 | 1.1 |
| 14 |
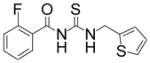
|
TM-2-107 | 3.0 |
| 15 |
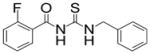
|
TM-2-139 | 1.0 |
Defined as the ratio of the initial rate of the HDAC-8 catalyzed reaction in the absence and presence and of 10 μM activator
A casual perusal of the data of Table 1 reveals that most of N-acetylthioureas, with a few exceptions, serve as activators of HDAC-8. Depending on the structure of the activator, the activity of HDAC-8 is increased up to 12-fold by TM-2-51 (at 10 μM concentration) and there appears to be a strong structure-activity relationship among N-acetylthiourea derivatives. As long as one of thiourea’s nitrogen forms the amide linkage with benzoic acid and the other nitrogen is directly attached to a six member aromatic (TM-2-51, TM-2-88) or cyclic (TM-2-97) ring, the activator exhibits maximal potency. Several factors, e.g. the presence of methylene group(s) between thiourea nitrogen and aromatic ring (TM-2-105, TM-2-90, TM-2-101, TM-2-125, TM-2-107, TM-2-139), the presence of fluoro group(s) at either of the aromatic rings (TM-2-138, TM-2-126, TM-2-125, TM-2-139) and/or bulky substituents (TM-2-87 and TM-2-101) all appear to impair the activation potencies of N-acetylthioureas.
Given that TM-2-51 serves as the most potent activator of HDAC-8 at 10 μM concentration, it was of interest to determine its apparent binding affinity for the enzyme and its maximal activation potency at saturating concentration. Toward this goal, we measured the initial rate of the HDAC-8 catalyzed reaction in the presence of increasing concentration of the activator. Figure 1 shows the increase in the fold activation of the enzyme (given by the ratio of the enzyme catalyzed rate in the presence, v, and absence, v0, of the activator) as a function of the activator concentration. The solid smooth line is the best fit of the data for the hyperbolic dependence of v/v0 on the activator concentration for an apparent activation constant (Ka) of 12.4 ± 1.2 μM and the maximum fold activation as being equal to 26.8 ± 1.1. Hence, at saturating concentration of TM-2-51, HDAC-8 catalyzed reaction is enhanced by about 27 fold.
Figure 1.

Fold activation of HDAC8 catalyzed reaction is dependent on activator concentration with apparent activation constant of 12.4 ± 1.2 μM
To further probe the kinetic mechanism of activation of the HDAC-8 catalyzed reaction by TM-2-51, we determined the Km value of the enzyme’s substrate and kcat of HDAC-8 in the absence and presence of saturating concentration of the activator. As apparent from the data of Table 2, the activator decreased the Km value of the substrate by about 3-fold and it increased the kcat value of the enzyme by 5-fold. Hence, the specificity constant (kcat/Km value) of the enzyme is enhanced by about 16 fold in the presence of saturating concentration of the activator. It should be noted that the latter value is somewhat lower than that obtained from the data of Figure 1. We believe the origin of this discrepancy lies in the mechanistic complexity of the enzyme activation at different concentrations of the substrate. We are currently investigating this feature and we will report our finding subsequently.
Table 2.
Steady-state kinetic parameters of HDAC-8 catalyzed reaction in the absence and presence of TM-2-51
| Condition | Km (μM) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|
| Control (No activator) | 744 | 0.007 | 9.4 |
| + Activator | 230 | 0.036 | 150 |
To ascertain whether TM-2-51 as well as other N-acetylthioureas of Table-1 functions as selective activator for HDAC-8, or they serve as non-specific “pan” activators for other HDAC isozymes, we determined the initial rates of the selected HDAC (viz., HDAC-1, HDAC-2, HDAC-3, HDAC-4, HDAC-6, HDAC-10 and HDAC-11) catalyzed reaction in the presence of 10 and 100 μM concentrations of different activators. We observed that none of the N-acetylthioureas of Table-1 activated other HDAC isozymes at either of the above concentrations (see the supporting information). In fact, we observed that at 100 μM concentration, some of the N-acetylthioureas inhibited several HDACs by 4 to 15%, and the compound TM-2-107 (at 100 μM concentration) inhibited the HDAC-10 catalyzed reaction by 47%. Such weak inhibitory feature of some of thiourea derivatives is similar to that observed with sirtuins.14 In view of these results, we propose that depending on the structural variants, some of N-acetylthioureas serve as the highly potent and isozyme selective activators for the recombinant form of HDAC-8 particularly in the Fluoro-de-Lys Assay system.
The question arose as to how some of N-acetylthiourea derivatives selectively activated HDAC-8 but not other HDAC isozymes, and the activation was manifested via increasing the binding affinity of the enzyme’s substrate (i.e., decrease in the Km value) as well as increase in the catalytic turnover rate (kcat value) of the enzyme. Clearly, the activators stabilize both the ground as well as the transition state (albeit by slightly different magnitudes) of the HDAC-8 catalyzed reaction. This can be achieved either by binding of activators within or in the vicinity of the enzyme’s active site pocket leading to the stabilization of enzyme bound substrate as well as facilitating the deacetylation reaction, or due to their binding at some putative allosteric site and eliciting their influence via long range conformational changes in the enzyme structure. However, to further ascertain whether the activators indeed interacted with HDAC-8, we performed isothermal microcalorimetric studies for the binding of TM2-51 to the enzyme. The experimental data revealed that the binding affinity of TM2-51 to HDAC-8 was similar to the activation constant (see Fig. 1) of the compound, suggesting that the observed activation phenomenon was not due to some unforeseen kinetic complexity (data not shown).
To determine the putative binding site(s) of activators on the enzyme’s surface, we performed modeling studies by docking TM2-51 to the known crystal structure of HDAC-8-substrate complex via AutoDock Vina18 (as described in the supplementary information). A blind docking approach (i.e., without specifying the putative binding regions on the structural coordinates of the enzyme) revealed that most of the activator molecules clustered in the vicinity of the active site pocket of HDAC-8 (see Fig. S1 of supplementary information), and one of aromatic rings of the activator was stacked between Phe306 and the coumarin moiety of the substrate. Such stacking is believed to increase the binding affinity of the substrate in the presence of the activator (as observed experimentally; Table 2). Since Tyr306 (the corresponding amino acid present in the native enzyme) has been known to be involved in polarization of the carbonyl group of the acetylated lysine moiety of the substrate and in stabilization of the oxyanion intermediate during catalysis,19 it is conjectured that the enzyme bound activator would increase the kcat value of the enzyme (as also observed experimentally; see Table 2). Aside from our docking results, the structural data of HDAC-8 suggests that the enzyme’s surface in the vicinity of the active site is extremely malleable which facilitates the binding of a variety of ligands.19 Hence, it is not surprising that TM-2-51 as well as its derivatives selectively activates HDAC-8 (via binding to the enzyme site) as compared to other HDAC isozymes (see Table S1 of supporting information).
However, we realize that our demonstration of N-acetylthiourea-mediated activation of HDAC-8 primarily relies on the employment of Fluor-de-Lys as the fluorogenic substrate, and thus one can argue that the observed activation is due to potential interaction between the fluorescent moiety of the substrate and the aromatic rings of the activators as observed in the case of sirtuin-1.16–17 In case of sirtuin-1, it has been demonstrated that the putative activators bind to the enzyme only in the presence of fluorogenic substrates17,20 and the activation is manifested via lowering the Km value of the substrate. This is in marked contrast to our observation that N-acetylthioureas directly bind to HDAC-8 (in the absence of substrate or any other ligand) and their binding results in the decrease in the Km value of the substrate as well as increase in the kcat value of the enzyme. These coupled with the fact that the overall activation profile (at sub-saturating substrate concentrations) conforms to the marked co-operativity (our unpublished results) prompt us to surmise that N-acetylthiourea-mediated activation of HDAC-8 is real, and it is not an artifact of the employment of Fluor-de-Lys as the fluorogenic substrate. We are currently evaluating the mode of binding our activators to HDAC-8 and their overall mechanism of activation, and we will report these findings subsequently.
Supplementary Material
Acknowledgments
This research was supported by the NIH grants CA113746 and CA132034 to DKS, and the NIH COBRE grant NCRR-P20-RR15566 to GC.
Footnotes
Synthetic details of different activators, experimental protocol for the activation of the enzyme, and docking of an activator to the enzyme site are provided as the supplementary information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Papait R, Monti E, Bonapace IM. Curr Opin Drug Discov Devel. 2009;12:264–275. [PubMed] [Google Scholar]
- 2.Kouzarides T. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Shahbazian MD, Grunstein M. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 4.Marson CM. Anticancer Agents Med Chem. 2009;9:661–692. doi: 10.2174/187152009788679976. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, de Zoeten EF, Greene MI, Hancock WW. Nat Rev Drug Discov. 2009;8:969–981. doi: 10.1038/nrd3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campas-Moya C. Drugs Today. 2009;45:787–795. doi: 10.1358/dot.2009.45.11.1437052. [DOI] [PubMed] [Google Scholar]
- 7.Ma X, Ezzeldin HH, Diasio RB. Drugs. 2009;69:1911–1934. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Holbert MA, Marmorstein R. Curr Opin Struct Biol. 2005;15:673–680. doi: 10.1016/j.sbi.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Michan S, Sinclair D. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 11.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 12.Ito K, Lim S, Caramori G, Cosio B, Chung KF, Adcock IM, Barnes PJ. Proc Natl Acad Sci USA. 2002;99:8921–8926. doi: 10.1073/pnas.132556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai LH, Haggarty SJ, Kim D. US2010/0075926 A1. US Patent Application Publication. 2010 March 25;
- 14.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJR, Pass G, Woods J, Lane DP, Westwood NJ. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh RK, Mandal T, Balasubramanian N, Cook G, Srivastava DK. Anal Biochem. 2011;408:309–315. doi: 10.1016/j.ab.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dittenhafer-Reed KE, Feldman JL, Denu JM. Chembiochem. 2011;12:281–289. doi: 10.1002/cbic.201000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trott O, Olson AJ. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. J Biol Chem. 285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.