

Abstract
For a future structure-activity relationship (SAR) study, a library of desketoraloxifene analogues has been prepared by parallel synthesis using iodocyclization and subsequent palladium-catalyzed coupling reactions. Points of desketoraloxifene diversification involve the two phenolic hydroxyl groups and the aliphatic amine side chain. This approach affords oxygen-bearing 3-iodobenzo[b]thiophenes 4 in excellent yields, which are easily further elaborated using a two-step approach involving Suzuki-Miyaura and Mitsunobu coupling reactions to give multimethoxy-substituted desketoraloxifene analogues 6. Various hydroxyl-substituted desketoraloxifene analogues 7 were subsequently generated by demethylation with BBr3.
Keywords: parallel synthesis, desketoraloxifene, iodocyclization, benzo[b]thiophene, selective estrogen receptor modulator (SERM), palladium coupling
INTRODUCTION
Early cancer drug discovery efforts focused on the design of small molecule nonsteroidal estrogen receptor (ER) ligands with antagonist properties against breast and other reproductive tissues.1 The estrogen receptors alpha and beta (ERα and ERβ) are members of a large family of nuclear receptors that regulate gene transcription in response to small molecule binding.2 Due to the validated therapeutic importance of these receptors in diseases, such as osteoporosis and breast cancer, a number of drugs have been developed that target these estrogen receptors.3
Some of the more important estrogen antagonist structures cited in the literature are summarized in Figure 1. Tamoxifen (I)4 is a well-established estrogen antagonist. Traditionally the design of modulators has involved the preparation of triarylethylene analogues of this parent structure. 4-Hydroxytamoxifen (II)5 is an effective antiestrogen for estrogen receptor positive breast tissue. However, hydroxytamoxifen was subsequently discovered to have undesirable estrogenic properties on the endometrium. Several additional selective estrogen receptor modulators (SERMs), including the benzoxepin scaffold (III),6 the 2-phenylspiroindene scaffold (IV),7 ERA-923 (V),6b, 8 nafoxidine (VI),9 and trioxifene (IV)6b, 10 are presently in late stages of clinical trials. Most approaches to SERMs have involved modifications of the nonsteroidal antagonists tamoxifen (I) and raloxifene (VIII). Although the current SERMs have clear advantages over conventional hormone replacement therapy (HRT), they retain some of the disadvantages as well. Clearly, an “ideal SERM” has not yet emerged.
Figure 1.

Chemical structures of representative synthetic SERMs with A and B rings corresponding to tamoxifen (I) and raloxifene (VIII).
Because more potent and safer chemotherapeutic agents are needed, due to the potential side effects of tamoxifen (I), considerable attention has been paid to the development of less toxic SERMs.11 Benzothiophene derivatives, specifically those with oxygen-bearing substituents at the C-2, C-5 and/or C-6 positions are biologically important compounds. Many of these are known to be medicinally and physiologically active substances. Raloxifene (VIII) is a SERM), which is currently under clinical evaluation for the prevention and treatment of postmenopausal osteoporosis.4b, 12 Another benzothiophene SERM, arzoxifene (IX), is a highly effective agent for the prevention of mammary cancer induced in the rat by the carcinogen nitrosomethylurea and is significantly more potent than raloxifene in this regard.11, 13 Desmethylarzoxifene (DMA) (X), with a 4′-OH group, is an active metabolite of arzoxifene (IX), which has been observed in highly variable steady-state plasma concentrations.11, 13–14
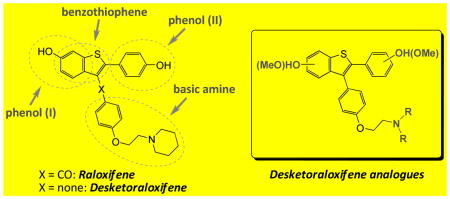
Interestingly, removal of the ketone moiety in raloxifene results in a benzothiophene analogue SERM, desketoraloxifene (Figure 1) (XI), which is more planar and conformationally more similar to 4-hydroxytamoxifen (II). Desketoraloxifene (XI) has been found to be a much stronger activator of the Activator Protein-1 (AP-1) site by ERα than ERβ, and mimics 4-hydroxytamoxifen (II) more than raloxifene (VIII).6b, 12b, 15
The benzo[b]thiophene SERMs VIII–XI have four important structural features, the benzothiophene aromatic ring, two phenolic hydroxyl groups and the basic aliphatic amine side chain, which are primarily responsible for their biological activity (Figure 2).15 Any new methodology suitable for the investigation of structure activity relationships (SAR) of benzothiophene-based SERMs14, 16 must take into account those four key structural features and be aware that many SERMs in clinical use and clinical development are also highly susceptible to oxidative metabolism by electrophilic, redox active quinoids simply because they are based on polyaromatic phenol scaffolds.17
Figure 2.

Structure of Benzo[b]thiophene SERMs VIII–XI and the key points of diversification introduced in analogues.
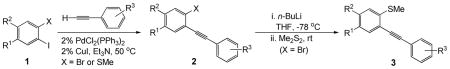
In general, benzo[b]thiophenes are of interest because of their frequent appearance in nature and wide range of biological and physiological effects.18 We have recently shown that the electrophilic cyclization of 2-(1-alkynyl)thioanisoles readily prepared by Sonogashira chemistry provides a very mild, high yielding synthesis of benzothiophenes bearing a bromine, iodine, sulfur or selenium group in the 3 position (Scheme 2).19
Scheme 2.

Synthesis of 2,3-Disubstituted Benzo[b]thiophenes by Electrophilic Cyclization
This basic strategy appeared particularly useful for the synthesis of desketoraloxifene analogs 6. In this series, we proposed to initially change the substituents at the C-2, C-3, C-5, and C-6 positions of the benzothiophene ring system. This decision was based on the structure of desketoraloxifene (XI), which has a para-substituted phenol at the 2-position, a basic aliphatic amine-containing chain at the 3-position, and an hydroxyl group at the 6-position of the benzothiophene ring system. Herein, we demonstrate the efficient preparation of oxygen-functionalized 3-iodobenzo[b]thiophenes 4 by electrophilic cyclization using I2 and their further elaboration to desketoraloxifene 7 analogues by solution-phase parallel synthesis.
RESULTS AND DISCUSSION
Using our previously developed benzothiophene methodology, we envisioned an efficient strategy that would lead to a library of methoxy- and hydroxy-substituted desketoraloxifene analogues 6/7 with multiple points of diversity present in the benzothiophene SERM desketoraloxifene analogues. Our basic strategy for generating a large number of such analogues is outlined in Scheme 1. Retrosynthetically, we planned to utilize the oxygen-bearing 3-iodobenzo[b]thiophene derivatives 4 as key intermediates that can be efficiently prepared using our alkyne iodocyclization chemistry.
Scheme 1.

Retrosynthetic Route to Fully Substituted Desketoraloxifene Analogues
The requisite precursors 2/3, bearing appropriate oxygen substituents and an alkyne moiety, can be easily prepared by palladium/copper-catalyzed Sonogashira coupling, according to a reported method (1.0 equiv of 1, 1.1 equiv of terminal alkyne, 2 mol % of PdCl2(PPh3)2, 2 mol % of CuI, and Et3N as the solvent at 50 °C for 5–8 h).19b As can be seen from the results reported in Table 1, using the sequence of reactions shown, involving the Sonogashira coupling of compounds 1, and subsequent lithiation of compounds 2{5–15}, followed by methylthiolation with dimethyl disulfide, afforded the corresponding sulfide products 3{1–11} in good to excellent yields.
Table 1.
Sequential Preparation of Alkynes 2{1–15} and 3{1–11} from Aryl Halides 1
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | X | alkyne 2 | yield (%)a | alkyne 3 | yield (%)a |
| 1 | H | H | H | SMe | 2{1} | 88 | ||
| 2 | H | H | 4-MeO | SMe | 2{2} | 88 | ||
| 3 | H | H | 3-MeO | SMe | 2{3} | 77 | ||
| 4 | H | H | 2-MeO | SMe | 2{4} | 79 | ||
| 5 | MeO | H | 4-MeO | Br | 2{5} | 94 | 3{1} | 86 |
| 6 | MeO | H | 3-MeO | Br | 2{6} | 91 | 3{2} | 93 |
| 7 | MeO | H | 2-MeO | Br | 2{7} | 87 | 3{3} | 89 |
| 8 | MeO | H | 3,5-(MeO)2 | Br | 2{8} | 83 | 3{4} | 83 |
| 9 | H | MeO | 4-MeO | Br | 2{9} | 73 | 3{5} | 81 |
| 10 | H | MeO | 2-MeO | Br | 2{10} | 77 | 3{6} | 90 |
| 11 | MeO | MeO | 4-MeO | Br | 2{11} | 92 | 3{7} | 87 |
| 12 | MeO | MeO | 3-MeO | Br | 2{12} | 79 | 3{8} | 63 |
| 13 | MeO | MeO | 2-MeO | Br | 2{13} | 71 | 3{9} | 91 |
| 14 | OCH2O | 4-MeO | Br | 2{14} | 84b | 3{10} | 73 | |
| 15 | OCH2O | 2-MeO | Br | 2{15} | 83b | 3{11} | 63 | |
Isolated yields after column chromatography.
Our first goal was the efficient preparation of a variety of oxygen-bearing 3-iodobenzothiophenes 4. Those 3-iodobenzo[b]thiophenes 4 have been smoothly prepared in excellent yields by electrophilic cyclization of the corresponding methylthio-containing alkynes 2{1–4} and 3{1–11} using I2 in CH2Cl2 at room temperature for 30 min (Scheme 3 and Figure 3). The chemoselectivity of this reaction is also quite interesting. In examples where MeS and MeO groups are both present ortho to the alkyne, only the desired 3-iodobenzo[b]thiophenes 4 were produced rapidly in high yields (Scheme 4, Figure 3; 4{4,7,10,13,15}).20 None of the possible 3-iodobenzofuran products were observed. In fact, most of the crude 3-iodobenzo[b]thiophenes 4 were of sufficient purity (>95%) for immediate further use based on their clean 1H NMR spectra. All of the reactions were monitored by thin layer chromatography and the products purified by column chromatography (see the Supporting Information for the experimental details).
Scheme 3.

Synthesis of Oxygen-Bearing 3-Iodobenzo[b]thiophenes 4 from 2{1–4}/3 by Iodocyclization
Figure 3.

Synthesis of the oxygen-bearing 3-iodobenzo[b]thiophenes 4{1–15}
Scheme 4.

Competition between MeO- and MeS Group
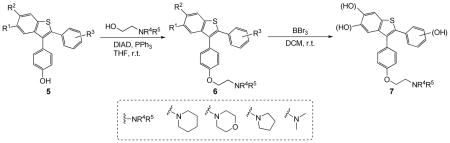
The 3-iodobenzo[b]thiophenes 4, having oxygen substituents at the C-5 and/or C-6 benzothiophene positions, are promising desketoraloxifene analogs (6/7). These 3-iodobenzo[b]thiophenes 4 are easily elaborated using a two-step approach involving Suzuki-Miyaura and subsequent Mitsunobu coupling reactions to give desketoraloxifene analogs 6. Thus, the palladium-catalyzed Suzuki-Miyaura coupling of 3-iodobenzo[b]thiophenes 4 with the tetrahydropyranyl (THP) ether-protected boronic acid p-THPOC6H4B(OH)2, followed by aqueous HCl deprotection, afforded the desired phenolic products 5 in good yields (Scheme 5, see the Supporting Information).19b Unfortunately, we could not obtain the desired compound 5 when we used 4-hydroxyphenylboronic acid directly.
Scheme 5.

Suzuki-Miyaura Coupling to Form Phenolic Benzothiophenes 5
For second-generation diversity, various amine-coupled SERM precursors have been produced by reaction of the phenolic benzothiophenes 5 with four different alkylaminoethanol moieties, specifically 1-(2-hydroxyethyl)piperidine, 1-(2-hydroxyethyl)morpholine, 1-(2-hydroxyethyl)pyrrolidine and 2-(dimethylamino)ethanol under Mitsunobu reaction conditions21 using Ph3P and diethyl azodicarboxylate (DEAD) for 24–36 h at room temperature to afford multimethoxy-substituted desketoraloxifene analogues 6 in good yields. The desketoraloxifene analogues 6 allow a wide variety of diversity to be incorporated into the final products. The methoxy-substituted desketoraloxifene analogues 6 have been demethylated using BBr316f to provide the hydroxy-substituted desketoraloxifene analogues 7. These processes have been performed in parallel on approximately a 40–50 mg scale, starting from the methoxy-substituted desketoraloxifene analogues 6. Each coupling reaction was worked up by washing with saturated aqueous sodium bicarbonate, water, and brine, and then the crude products were extracted with 5% methanol in chloroform. Concentration of the organic layer delivered each targeted compound in a modest yield and good purity. Overall, only nine compounds (products 7{10}, 7{11}, 7{16}, 7{20}, 7{34}, 7{35}, 7{36}, 7{38} and 7{39}) failed to afford the anticipated desketoraloxifene analogues by preparative HPLC, primarily because of poor solubility. All of the crude products 7 were isolated by either column chromatography or preparative HPLC. The results of the synthesis of the desketoraloxifene analog library are summarized in Table 2.
Table 2.
Desketoraloxifene Analog Librarya
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| product 6/7 | R1 | R2 | R3 | NR4R5 | ion HRMS | HRMS (calcd) | HRMS (found) | purity (%)e | yield (%)f |
| 6{1} | H | H | H | piperidino | [M+H]+ | 413.1813 | 414.1894 | 98 | 83 |
| 6{2} | H | H | H | morpholino | [M+H]+ | 415.1606 | 416.1682 | >99 | 78 |
| 6{3} | H | H | H | pyrrolidino | [M+H]+ | 399.1657 | 400.1732 | 92 | 81h |
| 6{4} | H | H | H | NMe2 | [M+H]+ | 373.1500 | 374.1576 | 98 | 79 |
|
| |||||||||
| 6{5} | H | H | 4-MeO | piperidino | [M+H]+ | 443.1919 | 444.1987 | 98 | 83 |
| 7{1}b | H | H | 4-OH | piperidino | [M+H]+ | 429.1762 | 430.1835 | 98 | 27g |
| 6{6} | H | H | 4-MeO | morpholino | [M+H]+ | 445.1712 | 446.1788 | >99 | 73 |
| 7{2}b | H | H | 4-OH | morpholino | [M+H]+ | 431.1555 | 432.1636 | 75 | 23g |
| 6{7} | H | H | 4-MeO | pyrrolidino | [M]+ | 429.1762 | 429.1765 | 85 | |
| 7{3}b | H | H | 4-OH | pyrrolidino | [M+H]+ | 415.1606 | 416.1681 | 74 | 46g |
| 6{8} | H | H | 4-MeO | NMe2 | [M+H]+ | 403.1606 | 404.1673 | 98 | 81 |
| 7{4}b | H | H | 4-OH | NMe2 | [M+H]+ | 389.1449 | 390.1528 | 70 | 56 |
|
| |||||||||
| 6{9} | H | H | 3-MeO | piperidino | [M]+ | 443.1919 | 443.1916 | 78 | |
| 7{5}b | H | H | 3-OH | piperidino | [M+H]+ | 429.1763 | 430.1843 | >99 | 53g |
| 6{10} | H | H | 3-MeO | morpholino | 73 | ||||
| 7{6}b | H | H | 3-OH | morpholino | [M+H]+ | 431.1555 | 432.1636 | >99 | 47 |
| 6{11} | H | H | 3-MeO | pyrrolidino | 76 | ||||
| 7{7}b | H | H | 3-OH | pyrrolidino | [M+H]+ | 415.1606 | 416.1682 | 98 | 35g |
| 6{12} | H | H | 3-MeO | NMe2 | 69 | ||||
| 7{8}b | H | H | 3-OH | NMe2 | [M+H]+ | 389.1450 | 390.1525 | >99 | 51g |
|
| |||||||||
| 6{13} | H | H | 2-MeO | piperidino | [M]+ | 443.1919 | 443.1917 | 76 | |
| 7{9}b | H | H | 2-OH | piperidino | [M+H]+ | 429.1763 | 430.1841 | 92 | 17g |
| 6{14} | H | H | 2-MeO | morpholino | 78 | ||||
| 7{10}b | H | H | 2-OH | morpholino | ndi | ||||
| 6{15} | H | H | 2-MeO | pyrrolidino | 67 | ||||
| 7{11}b | H | H | 2-OH | pyrrolidino | ndi | ||||
| 6{16} | H | H | 2-MeO | NMe2 | 71 | ||||
| 7{12}b | H | H | 2-OH | NMe2 | [M+H]+ | 389.1450 | 390.1525 | >99 | 12g |
|
| |||||||||
| 6{17} | MeO | H | 4-MeO | piperidino | [M+H]+ | 473.2025 | 474.2050 | 99 | 87 |
| 7{13}c | OH | H | 4-OH | piperidino | [M+H]+ | 445.1712 | 446.1787 | 99 | 38g |
| 6{18} | MeO | H | 4-MeO | morpholino | 74h | ||||
| 7{14}c | OH | H | 4-OH | morpholino | [M+H]+ | 447.1504 | 448.1579 | 87 | 56g |
| 6{19} | MeO | H | 4-MeO | pyrrolidino | [M]+ | 429.1762 | 429.1768 | 81 | |
| 7{15}c | OH | H | 4-OH | pyrrolidino | [M+H]+ | 431.1555 | 432.1638 | 86 | 16g |
| 6{20} | MeO | H | 4-MeO | NMe2 | 81 | ||||
| 7{16}c | OH | H | 4-OH | NMe2 | ndi | ||||
|
| |||||||||
| 6{21} | MeO | H | 3-MeO | piperidino | [M]+ | 473.2025 | 473.2031 | 86 | |
| 7{17}c | OH | H | 3-OH | piperidino | [M+H]+ | 445.1712 | 446.1790 | 94 | 48g |
| 6{22} | MeO | H | 3-MeO | morpholino | [M+H]+ | 475.1817 | 476.1890 | 97 | 83h |
| 6{23} | MeO | H | 3-MeO | pyrrolidino | 71 | ||||
| 6{24} | MeO | H | 3-MeO | NMe2 | [M+H]+ | 433.1712 | 434.1790 | 96 | 82 |
| 7{18}c | OH | H | 3-OH | NMe2 | [M+H]+ | 405.1399 | 406.1475 | 89 | 17g |
|
| |||||||||
| 6{25} | MeO | H | 2-MeO | piperidino | [M+H]+ | 473.2025 | 474.2095 | 99 | 87 |
| 7{19}c | OH | H | 2-OH | piperidino | [M+H]+ | 445.1712 | 446.1787 | 95 | 52 |
| 6{26} | MeO | H | 2-MeO | morpholino | 81h | ||||
| 6{27} | MeO | H | 2-MeO | pyrrolidino | [M+H]+ | 459.1868 | 460.1946 | 98 | 83 |
| 7{20}c | OH | H | 2-OH | pyrrolidino | ndi | ||||
| 6{28} | MeO | H | 2-MeO | NMe2 | [M]+ | 433.1712 | 433.1720 | 83 | |
| 7{21}c | OH | H | 2-OH | NMe2 | [M+H]+ | 405.1399 | 406.1475 | 93 | 42g |
|
| |||||||||
| 6{29} | MeO | H | 3,5-(MeO)2 | piperidino | [M]+ | 503.2130 | 503.2132 | 77 | |
| 7{22}d | OH | H | 3,5-(OH)2 | piperidino | [M+H]+ | 461.1661 | 462.1737 | 33 | 7g |
| 6{30} | MeO | H | 3,5-(MeO)2 | morpholino | 78 | ||||
| 6{31} | MeO | H | 3,5-(MeO)2 | pyrrolidino | 73 | ||||
| 6{32} | MeO | H | 3,5-(MeO)2 | NMe2 | 73 | ||||
| 7{23}d | OH | H | 3,5-(OH)2 | NMe2 | [M+H]+ | 421.1348 | 422.1362 | 82 | 21g |
|
| |||||||||
| 6{33} | H | MeO | 4-MeO | piperidino | [M+H]+ | 473.2025 | 474.2105 | 98 | 83 |
| 7{24}c | H | OH | 4-OH | piperidino | [M+H]+ | 445.1712 | 446.1793 | 97 | 78 |
| 6{34} | H | MeO | 4-MeO | morpholino | 78 | ||||
| 7{25}c | H | OH | 4-OH | morpholino | [M+H]+ | 447.1504 | 448.1585 | 55 | 43g |
| 6{35} | H | MeO | 4-MeO | pyrrolidino | 75 | ||||
| 7{26}c | H | OH | 4-OH | pyrrolidino | [M+H]+ | 431.1555 | 432.1633 | 13 | 38g |
| 6{36} | H | MeO | 4-MeO | NMe2 | 76 | ||||
| 7{27}c | H | OH | 4-OH | NMe2 | [M+H]+ | 405.1399 | 406.1471 | 30 | 47 |
|
| |||||||||
| 6{37} | H | MeO | 2-MeO | piperidino | [M]+ | 473.2025 | 473.2019 | 78 | |
| 7{28}c | H | OH | 2-OH | piperidino | [M+H]+ | 445.1712 | 446.1914 | 97 | 37g |
| 6{38} | H | MeO | 2-MeO | morpholino | 77 | ||||
| 7{29} | H | OH | 2-OH | morpholino | [M+H]+ | 447.1504 | 448.1712 | 97 | 49g |
| 6{39} | H | MeO | 2-MeO | pyrrolidino | 81 | ||||
| 7{30}c | H | OH | 2-OH | pyrrolidino | [M+H]+ | 431.1555 | 432.1703 | 92 | 35g |
| 6{40} | H | MeO | 2-MeO | NMe2 | 82 | ||||
| 7{31}c | H | OH | 2-OH | NMe2 | [M+H]+ | 405.1399 | 406.1538 | 99 | 31g |
|
| |||||||||
| 6{41} | MeO | MeO | 4-MeO | piperidino | [M+H]+ | 503.2130 | 504.2146 | 95 | 76 |
| 7{32}d | OH | OH | 4-OH | piperidino | [M+H]+ | 461.1661 | 462.1732 | >99 | 12g |
| 6{42} | MeO | MeO | 4-MeO | morpholino | 75h | ||||
| 7{33}d | OH | OH | 4-OH | morpholino | [M+H]+ | 463.1453 | 464.1471 | 97 | 53g |
| 6{43} | MeO | MeO | 4-MeO | pyrrolidino | [M]+ | 489.1974 | 489.1981 | 79 | |
| 7{34}d | OH | OH | 4-OH | pyrrolidino | ndi | ||||
| 6{44} | MeO | MeO | 4-MeO | NMe2 | 69 | ||||
| 7{35}d | OH | OH | 4-OH | NMe2 | ndi | ||||
|
| |||||||||
| 6{45} | MeO | MeO | 3-MeO | piperidino | [M]+ | 503.2130 | 503.2134 | 79 | |
| 7{36}d | OH | OH | 3-OH | piperidino | ndi | ||||
| 6{46} | MeO | MeO | 3-MeO | morpholino | 81h | ||||
| 6{47} | MeO | MeO | 3-MeO | pyrrolidino | [M]+ | 489.1974 | 489.1982 | 73 | |
| 6{48} | MeO | MeO | 3-MeO | NMe2 | [M]+ | 463.1817 | 463.1826 | 72 | |
|
| |||||||||
| 6{49} | MeO | MeO | 2-MeO | piperidino | [M+H]+ | 503.2130 | 504.2209 | 99 | 77 |
| 6{50} | MeO | MeO | 2-MeO | morpholino | 72 | ||||
| 7{37}d | OH | OH | 2-OH | morpholino | [M+H]+ | 463.1453 | 464.1527 | >99 | 17g |
| 6{51} | MeO | MeO | 2-MeO | pyrrolidino | 68 | ||||
| 7{38}d | OH | OH | 2-OH | pyrrolidino | ndi | ||||
| 6{52} | MeO | MeO | 2-MeO | NMe2 | 71 | ||||
| 7{39}d | OH | OH | 2-OH | NMe2 | ndi | ||||
|
| |||||||||
| 6{53} | OCH2O | 4-MeO | piperidino | [M]+ | 487.1817 | 487.1817 | 81 | ||
| 6{54} | OCH2O | 4-MeO | morpholino | [M+H]+ | 489.1610 | 490.1692 | >99 | 77h | |
| 6{55} | OCH2O | 4-MeO | pyrrolidino | [M+H]+ | 473.1661 | 474.1754 | 98 | 69 | |
| 6{56} | OCH2O | 4-MeO | NMe2 | 79 | |||||
|
| |||||||||
| 6{57} | OCH2O | 2-MeO | piperidino | [M+H]+ | 487.1817 | 488.1896 | 93 | 76 | |
| 6{58} | OCH2O | 2-MeO | morpholino | 63h | |||||
| 6{59} | OCH2O | 2-MeO | pyrrolidino | 69h | |||||
| 6{60} | OCH2O | 2-MeO | NMe2 | [M]+ | 447.1504 | 447.1512 | 76 | ||
Reagents and conditions: i. Mitsunobu Coupling: 5 (0.2 mmol), alkylaminoethanol (1.5 equiv), DIAD (1.5 equiv), PPh3 (2.0 equiv), THF (2.0 mL), rt, 24–36 h. ii. Demethylation: 6 (0.1mmol), BBr3, CH2Cl2 (1.0 mL), rt, N2, 3 h.
2.0 Equiv of BBr3 used.
4.0 Equiv of BBr3 used.
6.0 Equiv of BBr3 used.
UV purity determined at 214 nm after preparative HPLC.
Isolated yields after column chromatography. All isolated products were characterized by 1H and13C NMR spectroscopy (see the Supporting Information).
Isolated yield after preparative HPLC.
An inseparable mixture was obtained.
The final product was not purified, because of poor solubility.
Desketoraloxifene (XI) itself is an extremely useful compound for biological screening. The dimethoxy-substituted desketoraloxifene analog 6{33} was readily prepared from phenolic benzothiophene 5{9} using 1-(2-hydroxyethyl)piperidine under Mitsunobu coupling conditions. Compound 6{33} was then readily converted by demethylation using BBr3 to desketoraloxifene (XI) in 78% yield (Scheme 6).
Scheme 6.

Demethylation to Form Desketoraloxifene (XI)
In conclusion, a total synthesis of desketoraloxifene (XI) and numerous analogues 6/7 have been accomplished from simple alkynes bearing electron-rich aromatic rings by electrophilic cyclization using I2. An efficient synthesis of the key oxygen-bearing intermediate 3-iodobenzo[b]thiophenes 4 has been successfully carried out in good to excellent yields by iodocyclization using I2. For the synthesis of benzothiophene SERMs, the desketoraloxifene analogues 6/7 have been prepared starting from various oxygen-bearing 3-iodobenzo[b]thiophenes 4 by a two-step approach involving sequential Suzuki-Miyaura and Mitsunobu couplings. The benzothiophene SERM desketoraloxifene analog 6/7 library is presently being evaluated against various biological screens by the National Institutes of Health Molecular Library Screening Center Network. We believe that this approach to oxygen-bearing 3-iodobenzo[b]thiophenes 4 should readily afford many other functionalized desketoraloxifene analogues 6 using known chemistry and parallel synthesis strategies.
Experimental Section
General Procedure for the Regioselective Sonogashira Reaction to Form Compounds 2
To a solution of dihalobenzene (1) (10.0 mmol), 2 mol % PdCl2(PPh3)2 and 2 mol % CuI in Et3N (20 mL), the terminal alkyne (11.0 mmol) was added. The reaction mixture was stirred vigorous at 50 °C for 5–8 h under an Ar atmosphere. The resulting mixture was diluted with EtOAc (2 × 200 mL). The separated organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using ethyl acetate/hexanes as the eluent to afford the corresponding products 2.
4-Bromo-3-[(4-methoxyphenyl)ethynyl]anisole [2{5}]
The product was obtained as a yellow oil (94% yield): 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.80 (s, 3H), 6.71 (dd, J = 3.1, 8.9 Hz, 1H), 6.87 (d, J = 8.9 Hz, 2H), 7.05 (d, J = 3.1 Hz, 1H), 7.44 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 55.5, 55.7, 87.1, 94.0, 114.2 (×2), 115.0, 116.2, 116.3, 117.6, 126.3, 133.1, 133.4 (×2), 158.6, 160.1.; HRMS calcd for C16H13BrO2 [M+], 316.0099, found 316.0094.
General Procedure for Methylthiolation to Form Compounds 3
Bromoalkyne 2 (8.0 mmol) was dissolved in dry THF (80 mL) under an Ar atmosphere and cooled to −78 °C for 0.5 h. Then, n-BuLi (2.0 M solution in cyclohexane, 12.0 mmol) was added dropwise to the stirred solution. After the addition was complete, the reaction solution was stirred for an additional 1 h at −78 °C. Dimethyl disulfide (9.6 mmol) was then added and the reaction mixture was stirred further at this temperature before being allowed to warm to room temperature for 2 h under an Ar atmosphere. The resulting mixture was diluted with EtOAc (2 × 160 mL). The separated organic layer was washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using ethyl acetate/hexanes as the eluent to afford the corresponding products 3.
4-Methoxy-2-[(4-methoxyphenyl)ethynyl]thioanisole [3{1}]
The product was obtained as a colorless oil (86% yield): 1H NMR (400 MHz, CDCl3) δ 2.46 (s, 3H), 3.76 (s, 3H), 3.76 (s, 3H), 6.83 (dd, J = 2.8, 8.7 Hz, 1H), 6.86 (d, J = 9.0 Hz, 2H), 7.04 (d, J = 2.8 Hz, 1H), 7.14 (d, J = 8.7 Hz, 1H), 7.51 (d, J = 9.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 16.5, 55.4, 55.5, 86.1, 95.4, 114.1 (×2), 115.2, 115.5, 117.1, 123.9, 127.8, 131.9, 133.2 (×2), 157.3, 159.9; HRMS calcd for C17H16O2S [M+], 284.0871, found 284.0873.
General Procedure for Iodocyclization Using I2 to Form Compounds 4
To a solution of 5.0 mmol of the alkyne 10 and 20 mL of CH2Cl2 was added gradually 1.2 equiv of I2 dissolved in 30 mL of CH2Cl2. The reaction mixture was allowed to stir at room temperature for up to 10 min. The reaction was monitored by TLC to establish completion. The remaining I2 was removed by washing with satd aq Na2S2O3. The mixture was then extracted by EtOAc (2 × 100 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by flash chromatography using EtOAc/hexanes as the eluent to afford the corresponding products 4.
3-Iodo-5-methoxy-2-(4-methoxyphenyl)benzo[B]thiophene [4{5}]
The product was obtained as a pale yellow solid (94% yield): mp 114–115 °C (uncorrected); 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 3.90 (s, 3H), 6.95–7.00 (m, 3H), 7.24 (d, J = 2.4 Hz, 1H), 7.58–7.60 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 55.5, 55.8, 78.8, 108.4, 114.0 (×2), 115.7, 123.0, 127.1, 131.1 (×2), 131.3, 143.2, 143.5, 158.6, 160.2; HRMS calcd for C16H13IO2S[M+], 395.9681, found 395.9684.
General Procedure for Suzuki-Miyaura Coupling to Form Compounds 5
To a solution of 3-iodobenzo[b]thiophene 4 (1.0 mmol) and 5 mol % Pd(PPh3)4 in toluene (10 mL) was added K2CO3 (2.5 mmol) under an Ar atmosphere. To the resulting mixture was added p-(THPO)C6H4B(OH)2 (1.5 mmol) dissolved in ethanol (2 mL) and water (0.5 mL) and the reaction mixture heated to 80 °C for 6–8 h with vigorous stirring. After concentration of the solvent under reduced pressure, 10% aq HCl was added to the crude product in THF (0.1 M conc.) at room temperature and stirred for 1 h. The mixture was then extracted by EtOAc (2 × 20 mL), and the aqueous phase was also extracted with EtOAc or CH2Cl2. The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by flash chromatography using EtOAc/hexanes as the eluent to afford the corresponding product 5.
3-(4-Hydroxyphenyl)-5-methoxy-2-(4-methoxyphenyl)benzo[B]thiophene [5{5}]
The product was obtained as a pale yellow oil (89% yield): 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 3H), 3.78 (s, 3H), 5.12 (br s, 1H), 6.78 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 6.96–7.03 (m, 2H), 7.20 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 55.5, 55.8, 105.8,114.0 (×2), 114.3, 115.9 (×2), 122.9, 127.1, 128.3, 130.8 (×2), 131.1, 131.85 (×2), 131.89, 140.7, 142.4, 155.0, 157.8, 159.2; HRMS calcd for C22H18O3S [M+], 362.0977, found 362.0983.
General Procedure for the Mitsunobu Reaction to Form Compounds 6
To a solution of 5 (0.2 mmol), triphenylphosphine (PPh3) (0.4 mmol), and alkylaminoethanol (0.3 mmol) in anhydrous THF (2 mL) was added diisopropylazodicarboxylate (DIAD) (0.3 mmol) with stirring at 0–5 °C. The resulting solution was stirred at room temperature for 24–32 h (monitored by TLC until completion) and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using methanol/ethyl acetate/hexanes as the eluent to afford the corresponding products 6.
5-Methoxy-2-(4-methoxyphenyl)-3-{4-[2-(1-piperidinyl)ethoxy]phenyl}benzo[B]thiophene [6{17}]
The product was obtained as a pale yellow oil (89% yield): 1H NMR (400 MHz, CDCl3) δ 1.41–1.50 (m, 2H), 1.59–1.66 (m, 4H), 2.50–2.58 (m, 4H), 2.81 (t, J = 6.0 Hz, 2H), 3.779 (s, 3H), 3.780 (s, 3H), 4.15 (t, J = 6.0 Hz, 2H), 6.78 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 8.8 Hz, 2H), 6.95–7.03 (m, 2H), 7.20–7.27 (m, 4H), 7.70 (d, J = 8.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 24.4, 26.2 (×2), 55.3 (×2), 55.4, 55.8, 58.3, 66.1, 105.7, 114.0 (×2), 114.4, 115.0 (×2), 122.9, 127.1, 128.2, 130.8 (×2), 131.0, 131.6 (×2), 132.0, 140.6, 142.4, 157.8, 158.2, 159.2; HRMS calcd for C29H32NO3S [M+H+], 474.2103, found 474.2050.
General Procedure for Demethylation to Compounds 7
To a solution of 6 (0.10 mmol) in anhydrous CH2Cl2 (2 mL) cooled in an ice water bath under N2 was added BBr3 (1.0 M solution in CH2Cl2; 4.0 equiv) while stirring. The solution turned orange in color. This solution was stirred for 3 h after slowly warming to room temperature. The reaction was quenched with satd aq NaHCO3 (2 × 2 mL) and the product was extracted with 5% CH3OH/CHCl3 (3 × 5 mL). The combined organic layers were dried over anhydrous MgSO4 and concentrated under a vacuum to yield the crude product, which was purified by column chromatography using 5–10% CH3OH/CHCl3 as the eluent to provide the desketoraloxifene analogues 7.
Desketoraloxifene [7{24}, XI]
The product was obtained as a white solid (68% yield): 1H NMR (400 MHz, DMSO-d6) δ 1.34–1.43 (m, 2H), 1.48–1.57 (m, 4H), 2.50–2.53 (m, 4H), 2.70–2.76 (m, 2H), 4.10 (t, J = 5.7 Hz, 2H), 6.67 (d, J = 8.7 Hz, 2H), 6.84 (dd, J = 2.2, 8.7 Hz, 1H), 6.99 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.17 (d, J = 8.7 Hz, 2H), 7.23 (d, J = 8.7 Hz, 1H), 7.28 (d, J = 2.2 Hz, 1H), 9.62 (s, 1H), 9.65 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 23.7, 25.3 (×2), 54.3 (×2), 57.2, 65.3, 107.0, 114.6, 114.7 (×2), 115.3 (×2), 123.2, 124.6, 127.4, 130.1 (×2), 130.7, 131.0 (×2), 133.5, 134.8, 138.8, 155.1, 156.9, 157.6; HRMS calcd for C27H27NO3S [M+H+], 446.1790, found 446.1793.
Supplementary Material
Acknowledgments
We thank Johnson Matthey, Inc. and Kawaken Fine Chemicals Co. Ltd. for donations of palladium catalysts, and Frontier Scientific and Synthonix for donations of boronic acids.
Funding Sources
Financial support of this work was provided by the National Institute of General Medical Sciences (GM070620 and GM079593) and the National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (GM069663).
Footnotes
Supporting Information. Synthetic methods, spectral assignments and copies of 1H and 13C NMR spectra for all previously unreported starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lerner LJ, Holthaus FJ, Jr, Thompson CR. A Nonsteroidal Estrogen Antagonist, 1-[p-(2-Diethylaminoethoxy)phenyl]-1-phenyl-2-(p-methoxyphenyl)ethanol. Endocrinology. 1958;63:295–318. doi: 10.1210/endo-63-3-295. [DOI] [PubMed] [Google Scholar]
- 2.Weatherman RV, Fletterick RJ, Scanlan TS. Nuclear-Receptor Ligands and Ligand-binding Domains. Annu Rev Biochem. 1999;68:559–581. doi: 10.1146/annurev.biochem.68.1.559. [DOI] [PubMed] [Google Scholar]
- 3.Gustafsson JA. Therapeutic Potential of Selective Estrogen Receptor Modulators. Curr Opin Chem Biol. 1998;2:508–511. doi: 10.1016/s1367-5931(98)80127-0. [DOI] [PubMed] [Google Scholar]
- 4.(a) Harper MJ, Walpole AL. Contrasting Endocrine Activities of cis and trans Isomers in a Series of Substituted Triphenylethylenes. Nature. 1966;212:87. doi: 10.1038/212087a0. [DOI] [PubMed] [Google Scholar]; (b) Jordan VC, Phelps E, Lindgren JU. Effects of anti-Estrogens on Bone in Castrated and Intact Female Rats. Breast Cancer Res Tr. 1987;10:31–35. doi: 10.1007/BF01806132. [DOI] [PubMed] [Google Scholar]; (c) Jordan VC. Tamoxifen: A Most Unlikely Pioneering Medicine. Nat Rev Drug Discovery. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]; (d) Itami K, Kamei T, Yoshida J. Diversity-Oriented Synthesis of Tamoxifen-Type Tetrasubstituted Olefins. J Am Chem Soc. 2003;125:14670–14671. doi: 10.1021/ja037566i. [DOI] [PubMed] [Google Scholar]
- 5.Killackey MA, Hakes TB, Pierce VK. Endometrial Adenocarcinoma in Breast Cancer Patients Receiving Antiestrogens. Cancer Treat Rep. 1985;69:237–238. [PubMed] [Google Scholar]
- 6.(a) Lloyd DG, Hughes RB, Zisterer DM, Williams DC, Fattorusso C, Catalanotti B, Campiani G, Meegan MJ. Benzoxepin-Derived Estrogen Receptor Modulators: A Novel Molecular Scaffold for the Estrogen Receptor. J Med Chem. 2004;47:5612–5615. doi: 10.1021/jm0495834. [DOI] [PubMed] [Google Scholar]; (b) Carta G, Knox AJS, Lloyd DG. Unbiasing Scoring Functions: A New Normalization and Rescoring Strategy. J Chem Inf Model. 2007;47:1564–1571. doi: 10.1021/ci600471m. [DOI] [PubMed] [Google Scholar]
- 7.Blizzard TA, Morgan JD, Mosley RT, Birzin ET, Frisch K, Rohrer SP, Hammond ML. 2-Phenylspiroindenes: A Novel Class of Selective Estrogen Receptor Modulators (SERMs) Bioorg Med Chem Lett. 2003;13:479–483. doi: 10.1016/s0960-894x(02)00985-x. [DOI] [PubMed] [Google Scholar]
- 8.(a) Greenberger LM, Annable T, Collins KI, Komm BS, Lyttle CR, Miller CP, Satyaswaroop PG, Zhang Y, Frost P. A New Antiestrogen, 2-(4-Hydroxy-phenyl)-3-methyl-1-[4-(2-piperidin-1-yl-ethoxy)-benzyl]-1H-indol-5-ol Hydrochloride (ERA-923), Inhibits the Growth of Tamoxifen-sensitive and -resistant Tumors and is Devoid of Uterotropic Effects in Mice and Rats. Clinical Cancer Research. 2001;7:3166–3177. [PubMed] [Google Scholar]; (b) Miller CP, Collini MD, Tran BD, Harris HA, Kharode YP, Marzolf JT, Moran RA, Henderson RA, Bender RHW, Unwalla RJ, Greenberger LM, Yardley JP, Abou-Gharbia MA, Lyttle CR, Komm BS. Design, Synthesis, and Preclinical Characterization of Novel, Highly Selective Indole Estrogens. J Med Chem. 2001;44:1654–1657. doi: 10.1021/jm010086m. [DOI] [PubMed] [Google Scholar]
- 9.(a) Steinbaum FL, De Jager R, Krakoff I. Clinical Trial of Nafoxidine in Advanced Breast Cancer. Medical and Pediatric Oncology. 1978;4:123–126. doi: 10.1002/mpo.2950040207. [DOI] [PubMed] [Google Scholar]; (b) Castaner J, Thorpe P. Nafoxidine. Drugs Future. 1978;3:211–215. [Google Scholar]; (c) Legha SS, Slavik M, Carter SK. Nafoxidine - An Antiestrogen for the Treatment of Breast Cancer. Cancer. 1976;38:1535–1541. doi: 10.1002/1097-0142(197610)38:4<1535::aid-cncr2820380415>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]; (d) Laios I, Journe F, Laurent G, Nonclercq D, Toillon RA, Seo HS, Leclercq G. Mechanisms Governing the Accumulation of Estrogen Receptor Alpha in MCF-7 Breast Cancer Cells Treated with Hydroxytamoxifen and Related Antiestrogens. J Steroid Biochem Mol Biol. 2004;87:207–221. doi: 10.1016/j.jsbmb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Neubauer BL, McNulty AM, Chedid M, Chen K, Goode RL, Johnson MA, Jones CD, Krishnan V, Lynch R, Osborne HE, Graff JR. The Selective Estrogen Receptor Modulator Trioxifene (LY133314) Inhibits Metastasis and Extends Survival in the PAIII Rat Prostatic Carcinoma Model. Cancer Res. 2003;63:6056–6062. [PubMed] [Google Scholar]
- 11.Liu H, Liu J, van Breemen RB, Thatcher GRJ, Bolton JL. Bioactivation of the SERM Desmethylated Arzoxifene to Quinoids: 4′-Fluoro Substitution Prevents Quinoid Formation. Chem Res Toxicol. 2005;18:162–173. doi: 10.1021/tx049776u. [DOI] [PubMed] [Google Scholar]
- 12.(a) Delmas PD, Bjarnason NH, Mitlak BH, Ravoux AC, Shah AS, Huster WJ, Draper M, Christiansen C. Effects of Raloxifene on Bone Mineral Density, Serum Cholesterol Concentrations, and Uterine Endometrium in Postmenopausal Women. N Engl J Med. 1997;337:1641–1647. doi: 10.1056/NEJM199712043372301. [DOI] [PubMed] [Google Scholar]; (b) Weatherman RV, Carroll DC, Scanlan TS. Activity of a Tamoxifen-Raloxifene Hybrid Ligand for Estrogen Receptors at an AP-1 Site. Bioorg Med Chem Lett. 2001;11:3129–3131. doi: 10.1016/s0960-894x(01)00646-1. [DOI] [PubMed] [Google Scholar]
- 13.Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, Sporn MB. Arzoxifene, A New Selective Estrogen Receptor Modulator for Chemoprevention of Experimental Breast Cancer. Cancer Res. 2001;61:8412–8415. [PubMed] [Google Scholar]
- 14.Qin Z, Kastrati I, Chandrasena REP, Liu H, Yao P, Petukhov PA, Bolton JL, Thatcher GRJ. Design and Synthesis of Benzothiophene SERMs with Modulated Oxidative Activity and Receptor Affinity. J Med Chem. 2007;50:2682–2692. doi: 10.1021/jm070079j. [DOI] [PubMed] [Google Scholar]
- 15.Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, Matsumoto K, Palkowitz AD, Sato M, Termine JD, Winter MA, Yang NN, Dodge JA. Molecular Determinants of Tissue Selectivity in Estrogen Receptor Modulators. Proc Natl Acad Sci USA. 1997;94:14105–14110. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Richardson TI, Frank SA, Wang M, Clarke CA, Jones SA, Ying B-P, Kohlman DT, Wallace OB, Shepherd TA, Dally RD, Palkowitz AD, Geiser AG, Bryant HU, Henck JW, Cohen IR, Rudmann DG, McCann DJ, Coutant DE, Oldham SW, Hummel CW, Fong KC, Hinklin R, Lewis GHTADJ. Structure-Activity Relationships of SERMs Optimized for Uterine Antagonism and Ovarian Safety. Bioorg Med Chem Lett. 2007;17:3544–3549. doi: 10.1016/j.bmcl.2007.04.044. [DOI] [PubMed] [Google Scholar]; (b) Palkowitz AD, Glasebrook AL, Thrasher KJ, Hauser KL, Short LL, Phillips DL, Muehl BS, Sato M, Shetler PK, Cullinan GJ, Pell TR, Bryant HU. Discovery and Synthesis of [6-Hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene: A Novel, Highly Potent, Selective Estrogen Receptor Modulator. J Med Chem. 1997;40:1407–1416. doi: 10.1021/jm970167b. [DOI] [PubMed] [Google Scholar]; (c) Grese TA, Cho S, Finley DA, Godfrey AG, Jones CD, Lugar CWI, Martin MJ, Matsumoto K, Pennington LD, Winter MA, Adrian MD, Cole HW, Magee DE, Phillips DL, Rowley ER, Short LL, Glasebrook AL, Bryant HU. Structure-Activity Relationships of Selective Estrogen Receptor Modulators: Modifications to the 2-Arylbenzothiophene Core of Raloxifene. J Med Chem. 1997;40:146–167. doi: 10.1021/jm9606352. [DOI] [PubMed] [Google Scholar]; (d) Yu L, Liu H, Li W, Zhang F, Luckie C, van Breemen RB, Thatcher GRJ, Bolton JL. Oxidation of Raloxifene to Quinoids: Potential Toxic Pathways via a Diquinone Methide and o-Quinones. Chem Res Toxicol. 2004;17:879–888. doi: 10.1021/tx0342722. [DOI] [PubMed] [Google Scholar]; (e) Grese TA, Pennington LD, Sluka JP, Adrian MD, Cole HW, Fuson TR, Magee DE, Phillips DL, Rowley ER, Shetler PK, Short LL, Venugopalan M, Yang NN, Sato M, Glasebrook AL, Bryant HU. Synthesis and Pharmacology of Conformationally Restricted Raloxifene Analogues: Highly Potent Selective Estrogen Receptor Modulators. J Med Chem. 1998;41:1272–1283. doi: 10.1021/jm970688z. [DOI] [PubMed] [Google Scholar]; (f) Schmid CR, Sluka JP, Duke KM. Nucleophilic Aromatic Substitution on 3-Aroyl-2-arylbenzothiophenes. Rapid Access to Raloxifene and Other Selective Estrogen Receptor Modulators. Tetrahedron Lett. 1999;40:675–678. [Google Scholar]
- 17.(a) Macgregor JI, Jordan VC. Basic Guide to the Mechanisms of Antiestrogen Action. Pharmacol Rev. 1998;50:151–196. [PubMed] [Google Scholar]; (b) Bolton JL, Yu L, Thatcher GRJ. Quinoids Formed from Estrogens and Antiestrogens. Methods Enzymol. 2004;378:110–123. doi: 10.1016/S0076-6879(04)78006-4. [DOI] [PubMed] [Google Scholar]
- 18.(a) Bianchini C, Meli A. Thiophene, Benzo[b]thiophene and Dibenzo[b,d]thiophene as Precursors to Highly Conjugated Organosulfur Compounds. Synlett. 1997:643–649. [Google Scholar]; (b) Irie M, Uchida K. Synthesis and Properties of Photochromic Diarylethenes with Heterocyclic Aryl Groups. Bull Chem Soc Jpn. 1998;71:985–996. [Google Scholar]; (c) Zhang TY, O’Toole J, Proctor CS. Recent Advances in the Synthesis and Applications of Benzo[b]thiophenes. Sulfur Rep. 1999;22:1–47. [Google Scholar]; (d) Pelkey ET. Five-Membered Ring Systems: Thiophenes & Se, Te Analogs. Prog Heterocycl Chem. 2003;15:116–139. [Google Scholar]
- 19.(a) Yue D, Larock RC. Synthesis of 2,3-Disubstituted Benzo[b]thiophenes via Palladium-Catalyzed Coupling and Electrophilic Cyclization of Terminal Acetylenes. J Org Chem. 2002;67:1905–1909. doi: 10.1021/jo011016q. [DOI] [PubMed] [Google Scholar]; (b) Cho CH, Neuenswander B, Lushington GH, Larock RC. Solution-Phase Parallel Synthesis of a Multi-substituted Benzo[b]thiophene Library. J Comb Chem. 2009;11:900–906. doi: 10.1021/cc9000604. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cho CH, Neuenswander B, Larock RC. Diverse Methyl Sulfone-Containing Benzo[b]thiophene Library via Iodocyclization and Palladium-Catalyzed Coupling. J Comb Chem. 2010;12:278–285. doi: 10.1021/cc900172u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cho CH, Jung DI, Larock RC. A New Approach to Desketoraloxifene Analogs from Oxygen-Bearing 3-Iodobenzo[b]thiophenes Prepared via Iodocyclization. Tetrahedron Lett. 2010;51:6485–6488. doi: 10.1016/j.tetlet.2010.09.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehta S, Waldo JP, Larock RC. Competition Studies in Alkyne Electrophilic Cyclization Reactions. J Org Chem. 2009;74:1141–1147. doi: 10.1021/jo802196r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitsunobu O, Yamada Y. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull Chem Soc Japan. 1967;40:2380–2382. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.