

Abstract
SIBLING (Small Integrin-Binding Ligand N-linked Glycoproteins) family is the major group of noncollagenous proteins in bone and dentin. These extremely acidic and highly phosphorylated extracellular proteins play critical roles in the formation of collagenous mineralized tissues. While the lack of individual SIBLINGs causes significant mineralization defects in vivo, none of them led to a complete cessation of mineralization suggesting that these proteins have overlapping functions. To assess whether different SIBLINGs regulate biomineralization in a similar manner, and how phosphorylation impacts their activity, we studied the effects of two SIBLINGs, dentin matrix protein 1 (DMP1) and dentin phosphophoryn (DPP), on mineral morphology and organization in vitro. Our results demonstrate distinct differences in the effects of these proteins on mineralization. We show that phosphorylation has a profound effect on the regulation of mineralization by both proteins. Specifically, both phosphorylated proteins facilitated organized mineralization of collagen fibrils and phosphorylated DMP1 induced formation of organized mineral bundles in the absence of collagen. In summary, these results indicate that the primary structure and phosphorylation uniquely determine functions of individual SIBLINGs in regulation of mineral morphology and organization.
Introduction
Mineralized collagenous tissues such as bone and dentin are nanostructured composites, with mechanical properties uniquely adapted to their function. These functional properties of bone tissues are determined by an intricate hierarchical organization spanning atomic to macroscopic scales.1, 2 A basic building block of bone tissue is the mineralized collagen fibril, within which plate-like mineral particles are organized in parallel arrays with their c-axes co-aligned with the fibril axis.2 Besides the mineralized collagen fibrils, organized arrays of mineral crystals were also observed outside of the fibrils. 3, 4 Fibrillar collagen type I, is the major component of the organic matrix of bone tissues. Noncollagenous proteins (NCPs), which comprise less than 10% of this matrix, play essential roles in the regulation of mineralization 5, 6 as well as in the mechanical integrity of bone.3, 7 Small Integrin Binding Ligand N-linked Glycoproteins or SIBLINGs is the major group of NCPs.5, 8 SIBLING proteins share a number of common characteristics. Specifically, they contain a large fraction of aspartic and glutamic acids, which can comprise up to a third of their sequence. In addition, these proteins contain numerous serines, up to 90% of which can be phosphorylated.9 Consequently, native SIBLING proteins are extremely acidic,10 and adopt a random, open conformation in solution in their monomeric form. 8, 11 This open conformation provides a structural freedom for interaction, both with other macromolecules and with surfaces such as cell membranes and mineral crystals.6, 8, 11-13 This freedom of interaction is believed to be the structural basis of the multifunctionality of SIBLING proteins.8
In this manuscript, we focus on two major SIBLING proteins: Dentin Matrix Protein 1 (DMP1) and Dentin Phosphophoryn (DPP). DMP1 was first cloned from dentin 14 and was subsequently found in bone,15, 16 where it is primarily expressed by osteocytes.17 DMP1 is a multifunctional protein: it plays a role in biomineralization events in bone and dentin 18-20 the regulation of phosphate homeostasis 21, and in the differentiation of odonto- and osteoblasts.22, 23 The DMP1 amino acid sequence contains Ser (22%), Glu (15%) and Asp (13%), with a calculated pI=4.15 in its nonphosphorylated form, which can be much lower in vivo where roughly half of all its serines are phosphorylated 14. DMP1 adopts extended, largely unstructured conformation, and in the presence of calcium, it undergoes self-assembly into filaments.13 It has been shown to bind to N-telopeptide sequences of collagen and to affect collagen fibrogenesis.24 In gelatin gel assays DMP1 can, depending on its level of phosphorylation, alternatively regulate calcium phosphate nucleation or inhibit mineralization. It is also known to affect crystal size in a concentration dependent manner.25. Furthermore, in vitro studies show that DMP1 supramolecular assemblies can induce crystal nucleation and regulate the phase transition from amorphous calcium phosphate (ACP) to carbonated apatite.6, 13, 26
DPP is another SIBLING protein, particularly rich in aspartic acid and serine. Its sequence chiefly comprises Ser-Asp-Asp repeat motifs, with up to 90% of all serines phosphorylated.9 DPP is a proteolytic cleavage product of a larger pro-protein, so called dentin sialophosphoprotein (DSPP). DPP is the most abundant noncollagenous protein in dentin, and plays a critical role in its mineralization.27, 28 Like DMP1, DPP adopts extended and largely unstructured conformations in solution.29 In the presence of Ca2+ DPP tends to form a compact globular structure and assemble into filaments 30, 31. With a specific affinity for collagen molecules, DPP localizes to the boundary between the gap and overlap regions of collagen fibrils.32, 33 Collectively, studies on DPP's specific role in mineralization suggest that its effect varies significantly depending on its concentration, its degree of phosphorylation, and its conformational state (i.e. either surface bound or in solution).30, 34-36
Although it is clear that SIBLING proteins are key players in biomineralization, few studies have focused specifically on identifying the individual roles of these proteins. In order to achieve a deeper understanding of the functional specificity of individual SIBLINGS, we have carried out a comparative study of the direct effects of DPP and DMP1 on mineral morphology, organization and phase alone or in the presence of collagen fibrils. Our findings provide novel insight into the mechanisms by which acidic noncollagenous proteins regulate the formation of mineralized collagenous tissues which can lead to the development of novel bio-inspired strategies for biomaterials aimed at hard tissue repair.
Materials and Methods
Generation of Recombinant DMP1 (rDMP1) and Recombinant DPP
Mouse DMP1 or DPP cDNAs were inserted into the pGEX vector following digestion from pcDNA3 vector (DMP1 clone was a generous gift from Dr. J. Feng (Baylor College of Dentistry, Dallas, TX)). The DMP1 or DPP-pGEX was then transformed into the bacterial host BL21. Cells were cultured in LB+Amp media overnight at 37°C. Protein expression was induced by 0.4 mM IPTG for 2-6 hrs. The bacterial lysate was cleared by centrifugation and applied directly to Glutathione Sepharose 4B (Amersham). After washing with PBS, GST-bound protein was eluted with thrombin. Thrombin was removed from eluates with p-aminobenzamindine immobilized on Sepharose 4 Fast Flow matrix (Amersham). The purified protein was electrophoresed on a polyacrylamide gel to verify the molecular mass, and subjected to western blot analysis.
DMP1 or DPP Adenovirus Transfection and Purification from Cultured Mammalian Cells
DMP1 or DPP adenovirus was added to MC3T3-E1 cells which were grown in serum free media for three days. The media was then collected, immediately frozen at -80C and then lyophilized. The sample was resuspended in 6M urea in 20mM Tris-HCl. The cells and ECM were lysed in 4M Guanidine-HCl pH 7.4, in the presence of a protease inhibitor cocktail (C# 118361450011) 1 tablet / 50ml of medium) and 10mM NaF for several hours. Guanidine buffer was exchanged with 6M urea in 20mM Tris-HCl using Amicon ultracentrifuge filter units (Millipore C#UFC901024). The samples collected from the media or the cells/matrix were purified by FPLC, using an anion exchange column – HiTrap Q HP ((Cat. no. 17-1154-01) from GE Healthcare). The proteins were eluted from the column by increase salt concentration, using an elution buffer containing 6M urea in 20mM Tris-HCl+ 0.8M NaCl (PH7.2) at a flow rate 0.4ml/min for 80min. The quality of the protein preparation was assessed using SDS PAGE and Western Blot technique, using antibodies generously provided by Dr. J. Feng (Baylor College of Dentistry, Dallas, TX) 37(Supplementary Figure 1). The phosphorylation of the proteins was assessed using phosphoprotein specific ProQ stain 37(Supplementary Figure 1).
Preparation of Purified Collagen Solution
An acidic solution of type I collagen was obtained from rat tail tendon. The tendons were collected from 4-8-week-old rats and washed in protease inhibitor buffer. The tendons were then briefly rinsed in deionized water, homogenized, and solubilized in 2mM HCl (pH 2.8). The collagen solution was centrifuged at 25000g to remove aggregates. Collagen was then purified by several cycles of selective salt precipitation followed by acid dissolution as reported elsewhere 38-40. The final concentration of the collagen stock solution was ∼ 10 mg/ml.
Stock Solutions
High purity CaCl2·2H2O, NaCl and NaOH were obtained from Sigma-Aldrich, Na2HPO4·2H2O from Fluka, and KH2PO4 was obtained from Spectrum Chemicals. A 10× PBS buffer with 10.58mM KH2PO4, 29.66mM Na2HPO4·2H2O and 1551.72mM NaCl was prepared in deionized distilled water (DDW) with a resistivity of 18.2MΩ/cm. The pH of this buffer solution was adjusted to 7.2 (pH 7.8 at 1X dilution) using NaOH. Stock solutions of CaCl2 (6.68mM) Na2HPO4·2H2O (4mM) and NaOH (5mM) were prepared using DDW. A 10X PBS buffer with 100 mM sodium phosphate and 1550 mM NaCl was purchased from Fluka.
Assembly of Collagen Fibrils
To achieve effective removal of cross-linked collagen and to obtain collagen fibrils with larger diameter, additional steps were taken prior to the assembly of collagen fibrils on TEM grids. Nine ml of acidic collagen solution and 1ml of 10X, 4mM PBS solution were mixed together to obtain 0.1-0.2% collagen solution in 1X, 4mM PBS. This solution was incubated in a closed vial at 37°C for 3 hours. The incubation resulted in a gel of self-assembled collagen fibrils. The resultant gel was stored at 4°C for one week upon which the gel resolved into a clear solution. This solution was centrifuged at 25,000g at 4°C for 30 minutes. The supernatant was collected and stored at 4°C. To obtain a thin layer of collagen fibrils, a 20μL droplet of this mixed solution was placed on an inert polyethylene substrate in a humidity chamber, and a carbon coated Ni grid (EMS, HatfField, PA) was placed on top of the droplet. The humidity chamber was sealed and the sample was incubated at 37°C for 3h. After incubation, the grid was quickly washed with DDW, blotted against a filter paper and air-dried. Prior to the mineralization experiments we have analyzed the quality of collagen self-assembly in the samples positively stained with uranyl acetate as described elsewhere 38, 41 (Supplementary Figure 2).
Mineralization Experiments without Collagen
Mineralization experiments were conducted as previously described.38 Briefly, for the control mineralization experiments, ×3.4 PBS was prepared. Five μl of ×3.4 PBS was mixed with 5μl of DDW, 5μl of 4mM Na2HPO4 and 5μl of 6.8mM CaCl2 stock solutions to achieve final concentrations of 1.67mM CaCl2, 9.5 mM phosphate and 127 NaCl at pH 7.7. We chose pH 7.7, which is slightly higher than physiological pH 7.4 based on our earlier studies and preliminary experiments. We have found that the kinetics of the reactions and their reproducibility were optimal at this pH. Droplets of mineralization solution were placed in a humidity chamber and carbon coated Ni grids ((#400) EMS, Hatfield, PA) were placed on the top of the droplets, sealed and incubated for 16 hours. After the incubation, the TEM grids were rinsed with DDW, blotted against a filter paper and air dried. The experiments with the proteins were performed in the same way; however 5μL of aqueous protein solution, of different concentrations, was added to the mineralization medium instead of DDW, to establish final protein concentrations of 250, 125, 62.5 and 31.25μg/ml.
Collagen Mineralization in the Presence of Phosphorylated and Non-phosphorylated DPP and DMP1
Collagen mineralization has been performed on a TEM grid coated with pre-assembled collagen fibrils, as described in the section above. The final concentration of phosphorylated and non-phosphorylated DPP and DMP1 added to the mineralization solution was 125 μg/ml. The grids were incubated for 16 hours in the humidity chamber at 37°C.
Transmission Electron Microscopy (TEM) and Selected Area Electron Diffraction (SAED) Analysis
TEM and SAED studies were conducted using JEOL 1210 operated at 100 kV and JEOL 2000EX operated at 200 kV. The micrographs were recorded using an AMT CCD camera (AMT, Danvers, MA). An aluminum-coated TEM grid (EMS Hatfield, PA) was used as a standard for the calibration of SAED patterns for d-spacing calculations. The micrographs were analyzed using ImageJ 1.38× image processing software (Bethesda, MD). The statistical analysis was performed using MS Excel.
Results
In the absence of proteins, plate shaped apatitic crystals form
In the control mineralization experiments carried out using bare carbon-coated grid in the absence of proteins (DPP and DMP1), plate-like particles were observed (Figure 1; Table 1). The electron diffraction analysis and FTIR spectroscopy of the samples indicated the presence of carbonated apatite (Figure 1, inset, Supplementary Figure 3).
Figure 1.
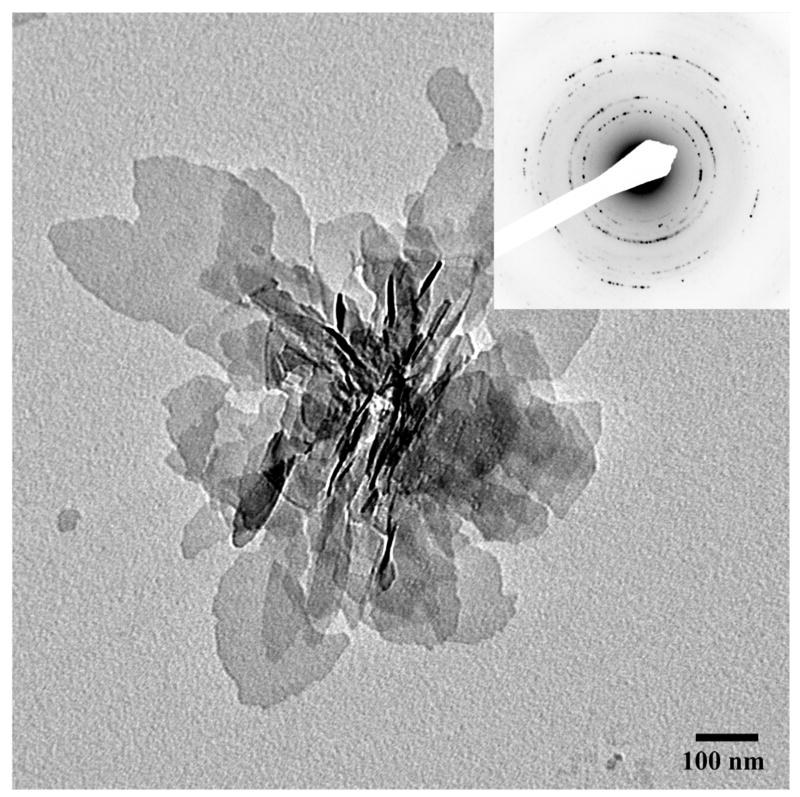
TEM micrograph and the corresponding diffraction pattern of randomly oriented plate-like apatitic crystals formed in the control experiment without proteins. The inset represents electron diffraction analysis. 86×86mm (300 × 300 DPI)
Table 1.
Dimensions of mineral particles obtained in mineralization with DMP1 and p-DMP1 after 16 hours of incubation at 37°C. The average values and standard deviations were obtained from at least 50 individual crystals per group.
| Protein | Concentration (μg/ml) | Thickness (nm) | Length (nm) | Width (nm) | Aspect ratio | Crystal phase |
|---|---|---|---|---|---|---|
| DMP1 | 250 | 3 ± 0.3* | 66± 20 | 34 ± 7 | 1.92 | apatitic phase |
| 125 | 3 ± 0.3 | 76 ± 16 | 39± 12 | 1.94 | apatitic phase | |
| 62.5 | 3 ± 0.3 | 80 ± 18 | 44 ± 9 | 1.80 | apatitic phase | |
| 31.25 | 3± 0.3 | 117±43 | 67±20 | 1.74 | apatitic phase | |
| p-DMP1 | 250 | 3 ± 0.4 | 74± 23 | 39 ± 9 | 1.92 | apatitic phase |
| 125 | 3 ± 0.4 | 51 ± 14 | 29± 5 | 1.76 | apatitic phase | |
| 62.5 | 3 ± 0.4 | 55± 17 | 34 ± 9 | 1.61 | apatitic phase | |
| 31.25 | 3 ± 0.3 | 38± 13 | 23 ± 4 | 1.68 | apatitic phase | |
| control | 0 | 3 ± 0.3 | 144 ± 53 | 80 ± 27 | 1.79 | apatitic phase |
Standard deviation
Non-phosphorylated and phosphorylated DMP1 have profoundly different effects on mineral organization
TEM analysis of the reaction products in the presence of nonphosphorylated DMP1 (DMP1) at concentrations varying from 250 to 31.25μg/mL, revealed numerous plate-shaped particles randomly distributed throughout the grid (Figure 2). The diffraction patterns of the samples were identical to those of the control and indicated the presence of a crystalline apatitic phase. These crystallites were significantly shorter in these experiments than in the control, however their thickness was similar to those of the control (Table 1). Variations in the DMP1 concentration had only a marginal effect on the particle dimensions, although a small but statistically significant decrease in crystal size was observed at higher protein concentrations (Table 1) (p<0.005). This decrease in size can be due to a mild inhibition of crystal growth by DMP1; alternatively DMP1 can act as a nucleator, increasing a number of crystalline nuclei per volume, hence leading to decrease in crystal size. These results indicate that DMP1 regulates the size of the crystals, while this effect seems to be only moderately influenced by the protein concentration. In the presence of phosphorylated DMP1 (p-DMP1) at 31.25 to 250μg/mL concentration range, plate-like apatitic crystals formed. As in experiments with nonphosphorylated DMP1 the crystals were significantly smaller than those of the control (p<0.001) (Figure 3, Table 1). Varying the p-DMP1 concentration, however, did have a greater effect on the crystal size than in the experiments with nonphosphorylated DMP1 (Table 1). Quite surprisingly, the direction of the change was opposite to that in the experiments with nonphosphrorylated DMP1. Namely, the size of crystals formed at the highest p-DMP1 concentration (250μg/mL) was two times larger than at the lowest concentration (31.25μg/mL) (Table 1).
Figure 2.
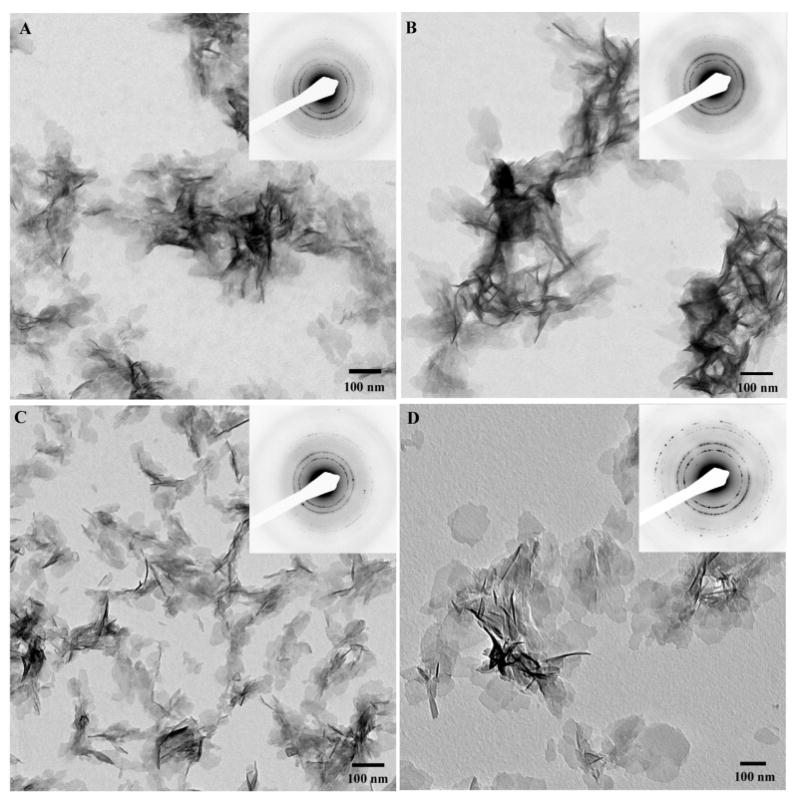
TEM micrographs and corresponding diffraction patterns of crystallites formed in the presence of recombinant nonphosphorylated DMP1 at concentrations of 250μg/mL (A), 125μg/mL (B), 62.5μg/mL (C), and 31.25μg/mL (D). Note that the size of the crystallites decreased with increasing protein concentration. 173×173mm (300 × 300 DPI)
Figure 3.
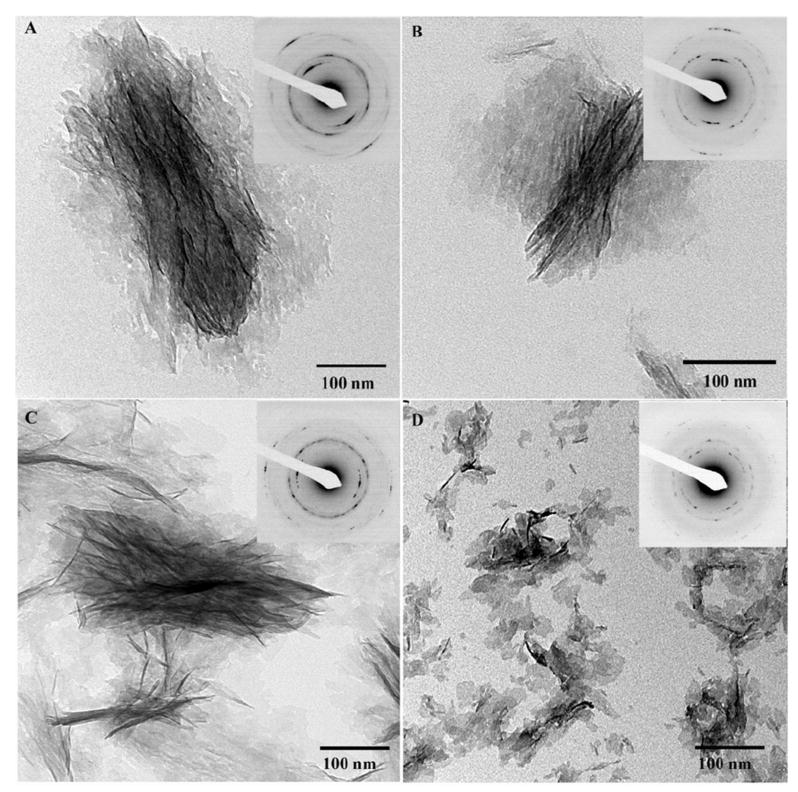
TEM micrographs and corresponding diffraction patterns of mineralization products formed in the presence of phosphorylated DMP1 at concentrations of 250μg/mL (A), 125μg/mL (B), 62.5μg/mL (C), and 31.25μg/mL (D). Note that the organization of mineral particles increases with the protein concentration. 722×713mm (72 × 72 DPI)
Phosphorylation of DMP1 had a profound concentration-dependent effect on the structural organization of mineral crystallites. In mineralization experiments carried out in the presence of 31.25μg/mL p-DMP1, randomly oriented individual plate-like crystals were observed throughout the grid (Figure 3D), similar to crystals formed in the presence of nonphosphorylated DMP1. The electron diffraction and HRTEM lattice analysis have confirmed the apatitic nature of these crystals (Figure 3 and Supplementary Figure 4). Increase in the protein concentration lead to a gradual increase in the level of organization of the crystallites. At concentrations of 62.5μg/mL and 125μg/mL the crystals were loosely organized into bundles of varying size and shape (Figure 3B, C) while at the highest p-DMP1 concentration of 250μg/mL, the mineral particles were packed tightly into 579 ± 104nm long and 231 ± 50nm wide bundles (Figure 3A). Electron diffraction analysis revealed that in these bundles, the crystals were arranged with their c-axes co-aligned, with increasing level of alignment directly correlating with the increase in the protein concentration. Specifically, the angular spread of 002 reflections was 53°±20° for 62.5μg/mL p-DMP1, 44°±14° for 125μg/mL p-DMP1 and 38°±6° for 250mg/mL p-DMP1 concentration, indicating that the alignment of the crystallites in the bundles improved with an increase in the protein concentration (Figure 3).
Phosphorylated and non-phosphorylated DPP have distinctly different effects on mineral formation
When mineralization reaction was carried out in the presence of DPP and p-DPP, a very different effect on mineral morphology and crystallization behavior was observed. In experiments with 250μg/mL of nonphosphorylated DPP, spherical particles 70.3 nm in diameter formed (Figure 4A) (Table 2). Detailed analysis of these particles at high magnification suggests that they are clusters of much smaller units, less than 10nm in diameter. Electron diffraction analysis revealed a diffused ring pattern indicative of amorphous calcium phosphate (ACP) (Figure 4A, inset). These particles often formed larger assemblies of different shapes, i.e. long chains, doughnuts and networks. At a DPP concentration of 125μg/mL, similar particles of a slightly smaller diameter were observed (Figure 4B, Table 2). In addition, bundles of thin plate-like particles associated with the spherical particles were present. Electron diffraction analysis indicated that these plate-like particles were also amorphous (Figure 4B, inset). At lower DPP concentrations of 62.5μg/mL and 31.25μg/mL, only thin plate-like mineral particles formed (Figure 4C, D). The average particle size increased two-fold with a decrease in concentration of DPP from 62.5μg/mL to 31.25μg/mL (Table 2). Electron diffraction studies of these samples indicated the presence of a crystalline apatitic mineral phase similar to that of the control (Figure 4C and D, insets). Overall, these results indicate that nonphosphorylated DPP alone can stabilize ACP at high concentrations and has a negative dose dependent effect on the mineral crystal size at lower concentrations.
Figure 4.
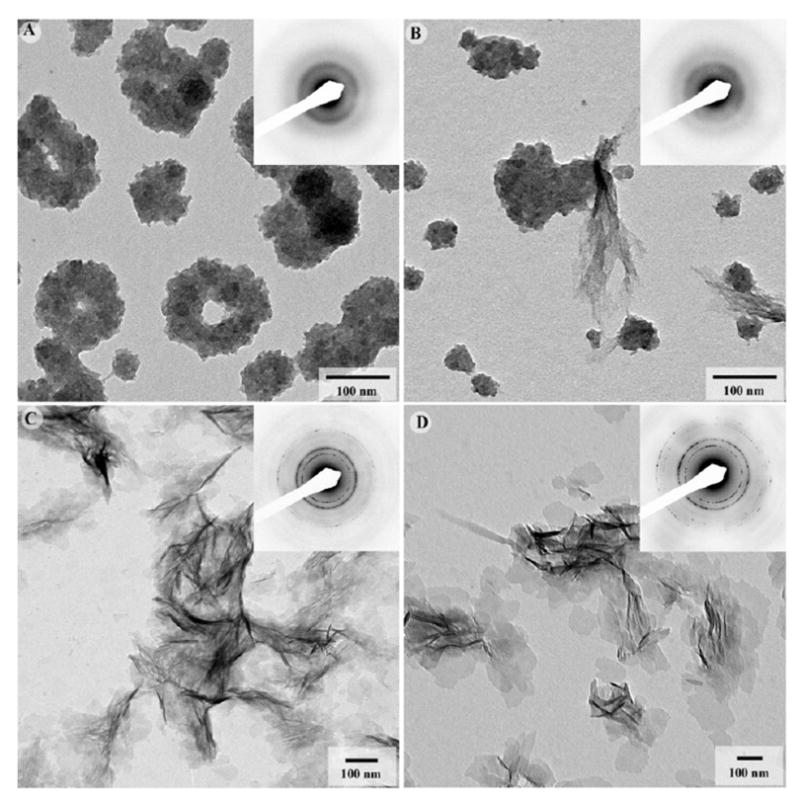
TEM micrographs and corresponding diffraction patterns of mineralization products formed in the presence of nonphosphorylated recombinant DPP at concentrations of 250μg/mL (A), 125μg/mL (B), 62.5μg/mL (C), and 31.25μg/mL (D). At higher concentrations aggregates of ACP particles formed, while at lower concentrations randomly oriented plate-shaped apatitic crystallites were observed. 173×173mm (300 × 300 DPI)
Table 2.
Dimensions of mineral particles obtained in mineralization with DPP and p-DPP after 16 hours of incubation at 37°C. The average values and standard deviations were obtained from at least 50 individual crystals per group.
| Protein | Concentration (μg/ml) | Thickness / diameter* (nm) | Length (nm) | Width (nm) | Aspect ratio | Crystal phase |
|---|---|---|---|---|---|---|
| DPP | 250 | 70 ± 18** | - | - | - | ACP |
| 125 | 60 ± 19 | - | - | - | ACP | |
| 62.5 | 3 ± 0.2 | 60± 19 | 36 ± 8 | 1.64 | Apatitic phase | |
| 31.25 | 3 ± 0.3 | 71± 23 | 41 ± 12 | 1.73 | Apatitic phase | |
| p-DPP | 250 | 4 ± 1 | - | - | - | ACP |
| 125 | 3 ± 0.3 | 28 ± 7 | - | ACP/poorly crystalline apatite | ||
| 62.5 | 3 ± 0.3 | 66 ± 17 | 52 ± 15 | 1.28 | apatitic phase | |
| 31.25 | 3 ± 0.3 | 71 ± 23 | 41 ± 12 | 1.73 | apatitic phase | |
| control | 0 | 3 ± 0.3 | 144± 53 | 80 ± 27 | 1.79 | apatitic phase |
In the experiments with 125 and 250μg/ml of DPP and 250μg/ml p-DPP spherical particles formed
Standard deviation
In the mineralization experiments with 250μg/mL of pDPP, a very small number of nanoparticles 4.1 ± 1.2nm in diameter were seen scattered throughout the grid (Figure 5A), strongly suggesting that the phosphorylated molecule inhibits mineralization. Electron diffraction from these particles resulted in a weak diffuse ring pattern indicative of ACP. At 125μg/mL of p-DPP, the amount of material on the grid was still quite low. A few tiny needle shaped particles were observed in this sample (Figure 5B; Table 2). The diffraction pattern from this sample contained a very weak 002 ring and a broad 211/112/300 composite ring (Figure 5B inset), suggesting that the mineral in this sample was poorly crystalline. At 62.5μg/mL of p-DPP, plate-like mineral particles randomly distributed throughout the grid were observed (Figure 5C; Table 2). The diffraction patterns revealed intense sharp rings, indicating the presence of apatitic crystalline phase. HRTEM analysis of the mineral crystals also confirmed the apatitic nature of the crystals (Supplementary Figure 5). The mineral particles formed at 31.25μg/mL of p-DPP were similar to those observed at 62.5μg/mL (Figure 5D).
Figure 5.
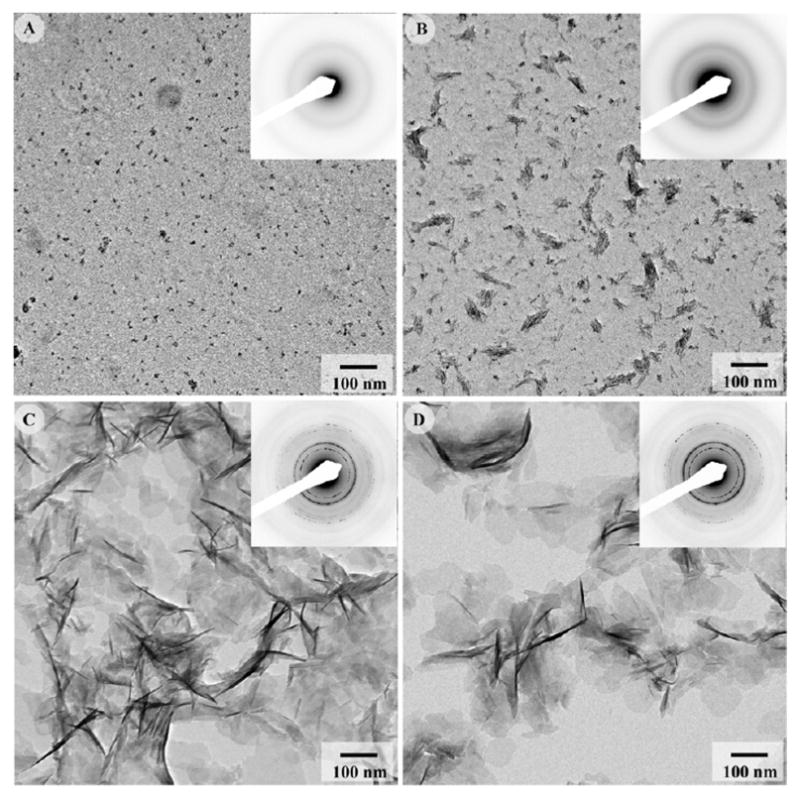
TEM micrographs and corresponding diffraction patterns of mineralization products formed in the presence of phosphorylated DPP at concentrations of 250μg/mL (A), 125μg/mL (B), 62.5μg/mL (C), and 31.25μg/mL (D). At higher concentrations, phosphorylated DPP inhibited mineral formation, while at lower concentrations its affect was similar to that of nonphosphorylated DPP. 171×170mm (300 × 300 DPI)
Phosphorylated DMP1and DPP facilitate the formation of highly organized mineralized collagen fibrils
It has been previously shown that the collagen fibrils alone, do not affect morphology, distribution and organization of mineral. 38, 42, 43
We have conducted our collagen mineralization studies at protein concentrations of 125 μg/mL. This concentration has been chosen based on our earlier studies 38, 41 and preliminary experiments with phosphorylated DPP. Specifically, at higher concentrations this protein strongly inhibited the mineralization reaction, while at lower concentrations, the driving force toward precipitation was too strong, diminishing the effect of protein and leading to the uncontrolled mineral precipitation.
In the presence of 125μg/ml of DMP1 and collagen fibrils, mineral particles identical in shape and size to the crystallites formed in the presence of 125μg/ml of DMP1 alone were observed (Figure 6) (Table 2). These particles were randomly distributed throughout the grid, although the mineral density around the collagen fibrils appeared to be slightly higher when compared to that on the support film. The crystallites associated with collagen were randomly oriented, with no particular organization of their crystallographic axes.
Figure 6.
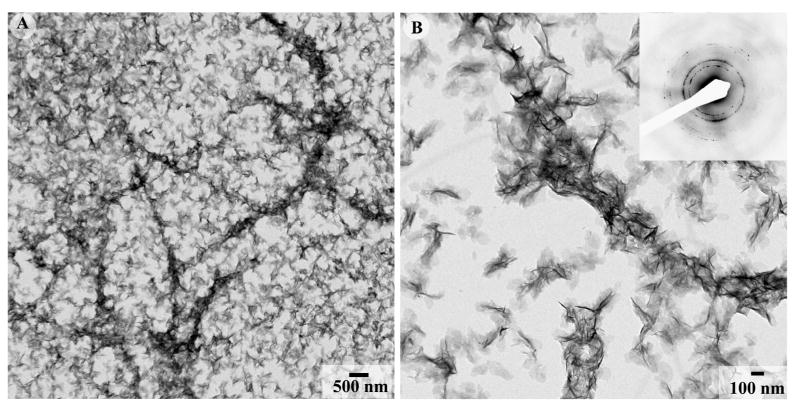
TEM micrographs of apatitic crystallites formed in the presence of 125μg/ml DMP1 and reconstituted collagen fibrils at low (A) and intermediate magnification (B) with the inset containing the corresponding diffraction pattern. Note that the crystals are randomly oriented throughout the grid. 173×86mm (300 × 300 DPI)
Under identical reaction conditions, mineralization experiments with 125μg/ml of p-DMP1 produced remarkably different results. TEM analysis revealed arrays of ribbon-like mineral particles deposited within the collagen fibrils (Figure 7; Table 1). These ribbon-like mineral particles were preferentially oriented along the fibril axis. Electron diffraction studies of the mineralized fibrils confirmed the crystalline nature of the mineral. Furthermore, they indicate that the c-axes of the crystals were co-aligned with the long axes of the fibrils, with 002 reflections forming distinctive arcs, with an angular spread of ∼19±7° (N=6) (Figure 7C, inset). This structural organization is the hallmark of mineralized collagen fibrils of bone and dentin 44. Typically, the mineralized regions of the collagen fibrils observed in this experiment were more than two times wider than the non-mineralized portions of the same collagen fibril (Figure 7; Supplementary Figures 6 and 7, supplementary movie), suggesting that mineral was not exclusively localized within the collagen fibrils and that the organized mineral deposition on the fibrils surface also takes place. Besides the mineralized collagen fibrils, bundles of organized crystallites similar to those formed in the mineralization experiments with 125μg/ml p-DMP1 without collagen were also observed (Figure 7A, B).
Figure 7.
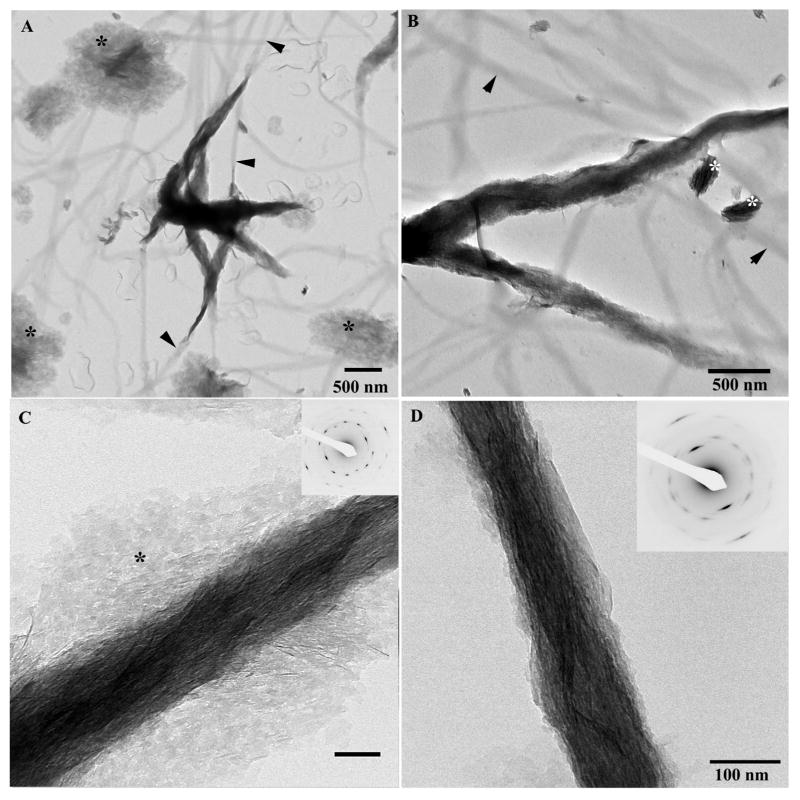
TEM micrographs of collagen fibrils mineralized in the presence of 125μg/ml phosphorylated DMP1 at low (A) intermediate (B) magnifications and higher magnifications (C) mineralized fibril, with a large portion of crystallites localized extrafibrillarly (asterisks). Inset represents the diffraction pattern taken from the field of the micrograph; note that the reflections are highly organized indicating an extremely high level of alignment of extra- and intrafibrillar crystallites. (D) micrograph of the mineralized fibril at intermediate magnification. Inset in (D) contains the corresponding diffraction pattern. Arrowheads indicate nonmineralized portions of collagen fibrils; note that these non-mineralized fibrils are thinner than the mineralized portions. Asterisks indicate organized bundles of mineral particles. 173×173mm (300 × 300 DPI)
Phosphorylated DPP induces highly organized intrafibrillar collagen mineralization
When the mineralization was conducted in the presence of reconstituted collagen fibrils and 125μg/ml DPP, spherical aggregates similar to those observed without collagen formed throughout the grid (Figure 8). The electron diffraction analysis of this sample indicates the presence of ACP (Figure 8B, inset).
Figure 8.

TEM micrographs of collagen fibrils mineralized in the presence of 125μg/ml nonphosphorylated recombinant DPP at low (A) and intermediate magnification (B). In some areas ACP aggregates exhibit higher density in the vicinity of the collagen fibrils. 173×86mm (300 × 300 DPI)
In contrast mineralization of collagen fibrils in the presence of 125μg/ml of p-DPP resulted in stacks of ribbon-like mineral particles deposited within the collagen fibrils (Figure 9). In these experiments the mineralization was primarily associated with mineralized collagen fibrils with very little mineral deposition outside of the fibrils. Analysis of the diffraction patterns from isolated mineralized fibrils revealed that these mineral particles were fully crystalline and organized with their c-axes aligned along the collagen fibril axis (Figure 9A,B inset). The average angular spread of 002 reflections arcs was ∼34±8° (N=6), indicating a lower degree of alignment than in the collagen fibrils mineralized in the presence of DMP1. Unlike the mineralized fibrils formed in the presence of p-DMP1, the mineralized regions of the collagen fibrils in these experiments were of the same width as non-mineralized fibrils, implying an intrafibrillar nature of the mineral (Figure 9).
Figure 9.
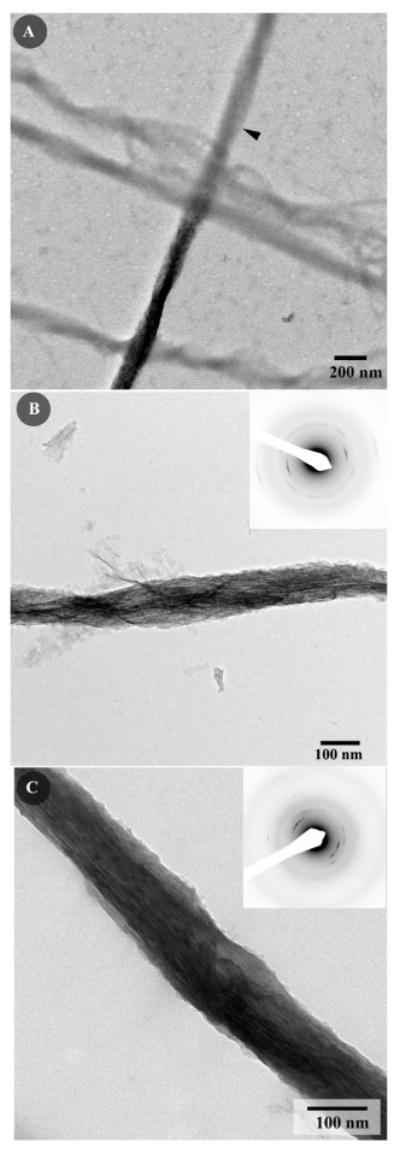
TEM micrographs of collagen fibrils mineralized in the presence of 125μg/ml phosphorylated DPP at intermediate magnification (A); note that that the mineralized and nonmineralized portions of the fibril are similar in diameter (arrowhead indicates the nonmineralized portion of the fibril). (B;C) intermediate magnification micrographs of the mineralized fibrils, the insets contain the corresponding diffraction patterns. Note that the crystallites are oriented with their c-axes aligned parallel to the long axis of the collagen fibril; however unlike the fibrils mineralized in the presence of phosphorylated DMP1 the mineral is located primarily intrafibrillarly. 86×260mm (300 × 300 DPI)
Discussion
Overall, the results of our mineralization experiments demonstrate that two SIBLING proteins DMP1 and DPP regulate phase, morphology and organization of calcium phosphate mineral in a different manner. Furthermore, phosphorylation of these proteins leads to remarkable changes in their functional properties, which is especially evident in their effect on collagen mineralization.
The results of our experiments and the published data 38, 45, 46 strongly suggest that the charge density of the proteins plays a significant role in the regulation of calcium phosphate mineral phase and morphology. Accordingly, the molecule with lowest charge density, in our case nonphosphorylated DMP1, induced a significant reduction of crystal size compared to the control, however it did not inhibit mineral formation or stabilize metastable mineral phases. At the same time, nonphosphorylated DPP with its higher negative charge density had a strong influence over mineral particle size and morphology, and stabilized ACP at higher concentrations. Finally, p-DPP, which has the highest negative charge density among the macromolecules studied, prevented mineral precipitation at the highest concentrations. At lower concentrations, it strongly influenced the size and shape of the crystallites. (Table 3) This effect of charge density on mineralization can be explained by the higher calcium binding capacity of macromolecules with higher negative charge. Specifically, calcium chelation by soluble macromolecules can reduce the concentration of free Ca2+ in solution, effectively decreasing the driving forces of mineral precipitation and at high enough protein concentrations, arresting this process. At lower concentrations, negatively charged proteins can bind to the mineral particles' surfaces and either decrease the rate of crystal growth, or slow down the transformation of transient amorphous mineral.
Table 3.
Summary of the Mineralization Results. L- length, W- width, T- thickness; * Size varies in a concentration-dependent manner; ** dimension as % of control
| Mineralization Experiment | Mineral Phase | Mineral Shape and Size | Mineral Organization |
|---|---|---|---|
| No proteins (control) | Apatitic | Plates L=143 nm W= 80 nm T= 3 nm |
Random |
| DMP1 alone | Apatitic | Plates* L= 66-116 nm (46-81 %)** W= 34-67 nm (42-96%) T= 3 nm (100%) |
Random |
| DPP alone | ACP at high concentrations | Spheres ∼ 60-70 nm | Large spherical aggregates |
| Apatitic at lower concentrations | Plates* L= 59-114 nm (41-79%) W= 36-61 nm (45-76 %) T= 3 nm (100%) |
Random | |
| p-DMP1 alone | Apatitic | Plates* L= 74-39 nm (51-27%) W= 39-22 nm (48-27%) T= 3 nm |
At higher concentrations, crystallites form parallel arrays with their c-axes coaligned |
| p-DPP alone | Inhibits mineral formation at higher concentrations | Nanodots ∼4 nm | Random |
| Apatitic at lower concentrations | Plates L= 60-70 nm (41-48%) W= 40-51 nm (50-63 %) T= 3 nm |
Random | |
| DMP1 with collagen fibrils | Apatitic | Plates L= 66 nm (46%) W= 34 nm (42%) T= 3 nm (100%) |
Random |
| DPP with collagen fibrils | ACP | Spheres ∼60-70 nm | Large spherical aggregates |
| p-DMP1 with collagen fibrils | Apatitic | Plates | Mineralized collagen fibrils with parallel crystalline arrays coaligned with the fibril axis, the mineral is located both intra- and extrafibrillarly |
| p-DPP with collagen fibrils | Apatitic | Plates | Mineralized collagen fibrils with parallel crystalline arrays co-aligned with the fibril axis, the mineral is located intrafibrillarly |
At the same time simple variations in charge density cannot explain a number of our observations. For example, the major difference between phosphorylated and nonphosphorylated DMP1 was not its effect on the mineral phase formed or the size of the crystals but the ability of the phosphorylated molecule to induce templated nucleation of mineral particles. It has been recently shown that Ca2+ causes conformational changes and self-assembly of DMP1 6, 30 and these assemblies can organize forming mineral particles 6, 13. These earlier studies have been conducted using recombinant DMP1 or synthetic peptides lacking phosphates. In contrast, in our experiments recombinant nonphosphorylated DMP1 did not have any effect on the organization of the mineral crystals which might be due to the fact that the protein was in solution and not immobilized on a surface, as reported by He et al.6 Parallel arrays of plate-shaped crystals is the hallmark mineral arrangement in bone and dentin, and usually is associated with mineralized collagen fibrils. At the same time a number of studies have reported the presence of highly organized mineral particles outside of the collagen fibrils in bone 4, 47, 48. These extrafibrillar crystals can comprise up to 70% of total mineral in the tissue according to some reports 49. It has been proposed that these extrafibrillar mineral particles interact with the acidic macromolecules in the extrafibrillar space, contributing to the bone mechanical performance 3, 50. Furthermore, in some tissues, such as highly mineralized peritubular dentin, which lacks collagen, well organized crystalline arrays were observed 51. Importantly, recent immunolabeling studies demonstrate specific localization of DMP1 to dentin tubules 52, 53 and peritubular dentin 54. These published studies together with the data presented here strongly suggest that phosphorylated DMP1 might play a role in the formation of organized crystalline arrays not associated with collagen fibrils (Table 3).
Probably, the most remarkable finding of the current study is the observation of organized mineralization of collagen fibrils in the presence of phosphorylated DMP1 and DPP. While nonphosphorylated molecules did not affect collagen mineralization, both P-DMP1 and P-DPP lead to the formation of highly organized mineralized collagen fibrils, similar to mineralized fibrils in bone and dentin (Table 3). The formation of well-organized mineralized fibrils has been previously achieved using polyaspartic acid and fetuin 38, 55, 56. It has been proposed that the organized mineralization of collagen occurs via inhibition of crystallization outside of the collagen fibrils and infiltration of stabilized mineral prenucleation clusters into collagen fibrils, followed by their intrafibrillar crystallization, guided by collagen molecules. Our data suggest that collagen mineralization in the presence of p-DPP likely occurs via similar mechanism. Specifically, as in the case of polyaspartic acid 38, phosphorylated DPP is a strong inhibitor of mineralization and in the experiments in the presence of this protein the mineralization occurs in association with collagen fibrils, primarily intrafibrillarly. In contrast a significant amount, up to 50%, of mineral not associated with the fibrils was revealed in the experiments in the presence of p-DMP1, indicating that this protein does not have a strong inhibitory effect as polyaspartic acid or p-DPP. Furthermore, the crystals not associated with collagen fibrils were organized into bundles, identical to those found in mineralization experiments with p-DMP1 without collagen. Secondly, the mineralized and nonmineralized portions of collagen fibrils varied only slightly in thickness in the experiments with p-DPP, while in the presence of DMP1 the mineralized portions of collagen fibrils were in some places more than 2 times wider than their nonmineralized portions. These differences suggest a different mechanism of collagen mineralization in the presence of DMP1. We propose that in the case of DMP1-mineral complexes initially organize on the surfaces of collagen fibrils (extrafibrillar), and the mineralization progresses intrafibrillarly at a later stage. The alignment of the crystals these collagen fibrils mineralized in the presence of p-DMP1 was much higher than in the crystalline bundles formed with p-DMP1 alone or in the collagen fibrils mineralized in the presence of p-DPP, suggesting that supramolecular interactions between collagen fibrils and p-DMP1 assemblies might enhance alignment of the crystallites. It has been shown that DMP1 binds to collagen fibrils 24 and that DMP1 can form supramolecular assemblies under mineralizing conditions 13, 54, supporting this hypothesis; nevertheless, additional studies are needed to elucidate the interactions between collagen fibrils and DMP1 during the mineralization process. Interestingly, in our recent study collagen mineralization in the presence of amelogenin, occurred via the latter mechanism 41, providing further support to our hypothesis. Amelogenin is the major protein of forming dental enamel, which lacks collagen. However, amelogenin is also found in the mantle dentin layer 57, 58 at the dentin-enamel boundary. Amelogenin in mantle dentin can play a very important role in maintaining the continuity between dentin and enamel tissues, by forming supramolecular complexes with collagen and guiding the transition between dentin and enamel mineral 41. Similarly to DMP1 amelogenin is able to organize forming mineral particles in parallel arrays in the absence of collagen. It also can bind to collagen fibrils and its assemblies organize on the surface of the collagen fibrils along the fibril axis. It has been shown that initially the amelogenin-mineral complexes align on the surface of collagen fibrils, followed by the propagation of the mineral intrafibirillarly, providing the continuity between extra and intrafibrillar mineral 41. There are therefore many parallels between the mineralization processes in the presence of DMP1 and amelogenin. Overall, the data presented here and the results of earlier studies, strongly suggest that there might be more than one mechanism by which noncollagenous proteins regulate formation of mineralized collagen fibrils with similar structural organization.
Conclusions
In conclusion, the results of this in vitro study show that although SIBLING proteins share a number of similarities, such as high negative charge, lack of structural organization in solution and high degree of phosphorylation their effects on calcium phosphate mineralization are distinct. We also demonstrate that the control over organized collagen mineralization by SIBLING proteins requires their phosphorylation. Furthermore our data suggest that the formation of mineralized collagen fibrils with similar structural organization might occur via different modes of mineralization. Overall, these data suggest that the variations in concentrations of individual SIBLING proteins and their phosphorylation levels can lead to differences in the mineral phase formed, degree of mineralization and mineral organization at the the nano- and microscopic scales. Such variations can provide the basis for structural and functional microheterogeniety of the mineralized tissues. We believe that these findings will lead to a better understanding of biomineralization processes in bone and dentin tissues, as well as inspire design strategies for the synthesis of novel nanostructured composite materials.
Supplementary Material
Supplementary figure 1 contains the characterization of DMP1 produced in the MC3T3;
Supplementary Figure 2 contains TEM micrographs of reconstituted collagen fibrils used in the study;
Supplementary Figure 3 contains FTIR spectrum of carbonated apatite produced in the control;
Supplementary Figure 4 contains HRTEM micrographs and corresponding power spectra of mineral formed in the presence of phosphorylated DMP1;
Supplementary Figure 5 contains HRTEM micrographs and corresponding power spectra of mineral formed in the presence of phosphorylated DPP;
Supplementary Figure 6 contains additional images of the collagen fibrils mineralized in the presence of phosphorylated DMP1;
Supplementary Figure 7 contains still images from the tomographic reconstruction in Supplementary Movie 1
Supplementary Movie 1 contains a tomographic reconstruction of a reconstituted collagen fibril mineralized in the presence of phosphorylated DMP1.
Acknowledgments
The study is supported by NIH grants DE016703 to EB and R01 DE016123 to CS. The TEM studies were conducted at the Center for Biologic Imaging, University of Pittsburgh School of Medicine. We acknowledge the scientific editorial contributions of Leslie Bannon.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fratzl P, Weinkamer R. Progr Mat Sci. 2007;52(8):1263–1334. [Google Scholar]
- 2.Weiner S, Wagner HD. Annu Rev Mater Sci. 1998;28:271–298. [Google Scholar]
- 3.Fantner GE, Hassenkam T, Kindt JH, Weaver JC, Birkedal H, Pechenik L, Cutroni JA, Cidade GAG, Stucky GD, Morse DE, Hansma PK. Nat Mater. 2005;4(8):612–616. doi: 10.1038/nmat1428. [DOI] [PubMed] [Google Scholar]
- 4.Landis WJ, Hodgens KJ, Song MJ, Arena J, Kiyonaga S, Marko M, Owen C, McEwen BF. J Struct Biol. 1996;117(1):24–35. doi: 10.1006/jsbi.1996.0066. [DOI] [PubMed] [Google Scholar]
- 5.George A, Veis A. Chem Rev. 2008;108(11):4670–4693. doi: 10.1021/cr0782729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He G, Dahl T, Veis A, George A. Nat Mater. 2003;2(8):552–558. doi: 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- 7.Adams J, Fantner GE, Fisher LW, Hansma PK. Nanotechnology. 2008;19(38) doi: 10.1088/0957-4484/19/38/384008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher LW, Torchia DA, Fohr B, Young MF, Fedarko NS. Biochem Biophys Res Commun. 2001;280(2):460–465. doi: 10.1006/bbrc.2000.4146. [DOI] [PubMed] [Google Scholar]
- 9.Stetler-Stevenson WG, Veis A. Biochemistry. 1983;22(18):4326–4335. doi: 10.1021/bi00287a025. [DOI] [PubMed] [Google Scholar]
- 10.Weiner S, Addadi L. J Mater Chem. 1997;7(5):689–702. [Google Scholar]
- 11.Evans JS. Curr Opin Coll Int Sci. 2003;8(1):48–54. [Google Scholar]
- 12.Burke EM, Guo Y, Colon L, Rahima M, Veis A, Nancollas GH. Colloids Surf B. 2000;17(1):49–57. [Google Scholar]
- 13.He G, Gajjeraman S, Schultz D, Cookson D, Qin C, Butler WT, Hao J, George A. Biochemistry. 2005;44(49):16140–16148. doi: 10.1021/bi051045l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George A, Sabsay B, Simonian PA, Veis A. J Biol Chem. 1993;268(17):12624–12630. [PubMed] [Google Scholar]
- 15.Hirst KL, IbarakiOconnor K, Young MF, Dixon MJ. J Dent Res. 1997;76(3):754–760. doi: 10.1177/00220345970760030701. [DOI] [PubMed] [Google Scholar]
- 16.MacDougall M, Gu TT, Luan XG, Simmons D, Chen JK. J Bone Miner Res. 1998;13(3):422–431. doi: 10.1359/jbmr.1998.13.3.422. [DOI] [PubMed] [Google Scholar]
- 17.Toyosawa S, Shintani S, Fujiwara T, Ooshima T, Sato A, Ijuhin N, Komori T. J Bone Miner Res. 2001;16(11):2017–2026. doi: 10.1359/jbmr.2001.16.11.2017. [DOI] [PubMed] [Google Scholar]
- 18.Ling YF, Rios HF, Myers ER, Lu YB, Feng JQ, Boskey AL. J Bone Miner Res. 2005;20(12):2169–2177. doi: 10.1359/JBMR.050815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu YB, Ye L, Yu SB, Zhang SB, Xie YX, McKee MD, Li YC, Kong J, Eick JD, Dallas SL, Feng JQ. Dev Biol. 2007;303(1):191–201. doi: 10.1016/j.ydbio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng JQ, Huang H, Lu Y, Ye L, Xie Y, Tsutsui TW, Kunieda T, Castranio T, Scott G, Bonewald LB, Mishina Y. J Dent Res. 2003;82(10):776–780. doi: 10.1177/154405910308201003. [DOI] [PubMed] [Google Scholar]
- 21.Feng JQ, Ward LM, Liu SG, Lu YB, Xie YX, Yuan BZ, Yu XJ, Rauch F, Davis SI, Zhang SB, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Nat Genet. 2006;38(11):1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almushayt A, Narayanan K, Zaki AE, George A. Gene Ther. 2006;13(7):611–620. doi: 10.1038/sj.gt.3302687. [DOI] [PubMed] [Google Scholar]
- 23.Narayanan K, Srinivas R, Ramachandran A, Hao JJ, Quinn B, George A. Proc Natl Acad Sci U S A. 2001;98(8):4516–4521. doi: 10.1073/pnas.081075198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He G, George A. J Biol Chem. 2004;279(12):11649–11656. doi: 10.1074/jbc.M309296200. [DOI] [PubMed] [Google Scholar]
- 25.Tartaix PH, Doulaverakis M, George A, Fisher LW, Butler WT, Qin C, Salih E, Tan M, Fujimoto Y, Spevak L, Boskey AL. J Biol Chem. 2004;279(18):18115–18120. doi: 10.1074/jbc.M314114200. [DOI] [PubMed] [Google Scholar]
- 26.Weiner S, Addadi L. Trends Biochem Sci. 1991;16(7):252–256. doi: 10.1016/0968-0004(91)90098-g. [DOI] [PubMed] [Google Scholar]
- 27.Sreenath T, Thyagarajan T, Hall B, Longenecker G, D'Souza R, Hong S, Wright JT, MacDougall M, Sauk J, Kulkarni AB. J Biol Chem. 2003;278(27):24874–24880. doi: 10.1074/jbc.M303908200. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki S, Sreenath T, Haruyama N, Honeycutt C, Terse A, Cho A, Kohler T, Müller R, Goldberg M, Kulkarni AB. Matrix Biol. 2009;28(4):221–229. doi: 10.1016/j.matbio.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans JS, Chan SI. Biopolymers. 1994;34(4):507–527. doi: 10.1002/bip.360340407. [DOI] [PubMed] [Google Scholar]
- 30.He G, Ramachandran A, Dahl T, George S, Schultz D, Cookson D, Veis A, George A. J Biol Chem. 2005;280(39):33109–33114. doi: 10.1074/jbc.M500159200. [DOI] [PubMed] [Google Scholar]
- 31.Li JH, Olton D, Lee D, Kumta PN, Sfeir C. Cells Tissues Organs. 2009;189(1-4):252–255. doi: 10.1159/000158571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beniash E, Traub W, Veis A, Weiner S. J Struct Biol. 2000;132(3):212–225. doi: 10.1006/jsbi.2000.4320. [DOI] [PubMed] [Google Scholar]
- 33.Dahl T, Sabsay B, Veis A. J Struct Biol. 1998;123(2):162–168. doi: 10.1006/jsbi.1998.4025. [DOI] [PubMed] [Google Scholar]
- 34.Boskey AL, Maresca M, Doty SB, Sabsay B, Veis A. Bone Miner. 1990;11(1):55–65. doi: 10.1016/0169-6009(90)90015-8. [DOI] [PubMed] [Google Scholar]
- 35.Hunter GK, Hauschka PV, Poole AR, Rosenberg LC, Goldberg HA. Biochem J. 1996;317(1):59–64. doi: 10.1042/bj3170059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silverman L, Boskey AL. Calcif Tissue Int. 2004;75(6):494–501. doi: 10.1007/s00223-004-0019-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sfeir C, Lee D, Li J, Zhang X, Boskey AL, Kumta PN. J Biol Chem. 2011;286(23):20228–20238. doi: 10.1074/jbc.M110.209528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deshpande AS, Beniash E. Cryst Growth Des. 2008;8(8):3084–3090. doi: 10.1021/cg800252f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuznetsova N, Leikin S. Biophys J. 1999;76(1):A319–A319. [Google Scholar]
- 40.Miller EJ, Rhodes RK. Methods Enzymol. 1982;82:33–64. doi: 10.1016/0076-6879(82)82059-4. [DOI] [PubMed] [Google Scholar]
- 41.Deshpande AS, Fang PA, Simmer JP, Margolis HC, Beniash E. J Biol Chem. 2010;285(25):19277–19287. doi: 10.1074/jbc.M109.079939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunter GK, Poitras MS, Underhill TM, Grynpas MD, Goldberg HA. J Biomed Mater Res. 2001;55(4):496–502. doi: 10.1002/1097-4636(20010615)55:4<496::aid-jbm1042>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Saito T, Arsenault AL, Yamauchi M, Kuboki Y, Crenshaw MA. Bone. 1997;21(4):305–311. doi: 10.1016/s8756-3282(97)00149-x. [DOI] [PubMed] [Google Scholar]
- 44.Beniash E. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2011;3(1):47–69. doi: 10.1002/wnan.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diegmueller JJ, Cheng XG, Akkus O. Cryst Growth Des. 2009;9(12):5220–5226. [Google Scholar]
- 46.Ofir PBY, Govrin-Lippman R, Garti N, Furedi-Milhofer H. Cryst Growth Des. 2004;4(1):177–183. [Google Scholar]
- 47.Hassenkam T, Fantner GE, Cutroni JA, Weaver JC, Morse DE, Hansma PK. Bone. 2004;35(1):4–10. doi: 10.1016/j.bone.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 48.Prostak KS, Lees S. Calcif Tissue Int. 1996;59(6):474–479. doi: 10.1007/BF00369213. [DOI] [PubMed] [Google Scholar]
- 49.Bonar LC, Lees S, Mook HA. J Mol Biol. 1985;181(2):265–270. doi: 10.1016/0022-2836(85)90090-7. [DOI] [PubMed] [Google Scholar]
- 50.Gupta HS, Fratzl P, Kerschnitzki M, Benecke G, Wagermaier W, Kirchner HOK. J Royal Soc Interface. 2007;4(13):277–282. doi: 10.1098/rsif.2006.0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiner S, Veis A, Beniash E, Arad T, Dillon JW, Sabsay B, Siddiqui F. J Struct Biol. 1999;126(1):27–41. doi: 10.1006/jsbi.1999.4096. [DOI] [PubMed] [Google Scholar]
- 52.Maciejewska I, Cowan C, Svoboda K, Butler WT, D'Souza R, Qin C. J Histochem Cytochem. 2009;57(2):155–166. doi: 10.1369/jhc.2008.952630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orsini G, Ruggeri A, Mazzoni A, Nato F, Falconi M, Putignano A, Di Lenarda R, Nanci A, Breschi L. Eur J Histochem. 2008;52(4):215–220. doi: 10.4081/1219. [DOI] [PubMed] [Google Scholar]
- 54.Beniash E, Deshpande AS, Fang PA, Lieb NS, Zhang X, Sfeir CS. J Struct Biol. 2011;174(1):100–106. doi: 10.1016/j.jsb.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nudelman F, Pieterse K, George A, Bomans PHH, Friedrich H, Brylka LJ, Hilbers PAJ, de With G, Sommerdijk N. Nat Mater. 2010;9(12):1004–1009. doi: 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olszta MJ, Cheng XG, Jee SS, Kumar R, Kim YY, Kaufman MJ, Douglas EP, Gower LB. Mater Sci Eng, R. 2007;58(3-5):77–116. [Google Scholar]
- 57.McKee MD, Nanci A. Microsc Res Tech. 1995;31(1):44–62. doi: 10.1002/jemt.1070310105. [DOI] [PubMed] [Google Scholar]
- 58.Nanci A, Zalzal S, Lavoie P, Kunikata M, Chen WY, Krebsbach PH, Yamada Y, Hammarstrom L, Simmer JP, Fincham AG, Snead ML, Smith CE. J Histochem Cytochem. 1998;46(8):911–934. doi: 10.1177/002215549804600806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1 contains the characterization of DMP1 produced in the MC3T3;
Supplementary Figure 2 contains TEM micrographs of reconstituted collagen fibrils used in the study;
Supplementary Figure 3 contains FTIR spectrum of carbonated apatite produced in the control;
Supplementary Figure 4 contains HRTEM micrographs and corresponding power spectra of mineral formed in the presence of phosphorylated DMP1;
Supplementary Figure 5 contains HRTEM micrographs and corresponding power spectra of mineral formed in the presence of phosphorylated DPP;
Supplementary Figure 6 contains additional images of the collagen fibrils mineralized in the presence of phosphorylated DMP1;
Supplementary Figure 7 contains still images from the tomographic reconstruction in Supplementary Movie 1
Supplementary Movie 1 contains a tomographic reconstruction of a reconstituted collagen fibril mineralized in the presence of phosphorylated DMP1.