

Abstract
Despite tremendous advances over the last 15 years in identifying vulnerable atherosclerotic plaques, the incidence of death and disability caused by such lesions still remains the number one health threat in developed countries. Therefore, new systemic or focal therapies aimed at decreasing the overall burden of disease, and a change to a more benign phenotype, are needed. Because cell death is a prominent feature of advanced atherosclerotic plaques with a major impact on plaque destabilization, an increasing number of compounds targeting the apoptotic or autophagic machinery in atherosclerosis are being explored, predominantly at the preclinical level. This review will provide an overview of these compounds, with a focus on both inhibition and stimulation of cell death, to prevent acute coronary syndromes and sudden cardiac death.
Keywords: atherosclerosis, cell death, apoptosis, necrosis, autophagy
Introduction
Atherosclerosis is a vascular disease marked by atheromatous plaques in the intima of medium- and large-size arteries, and is the leading cause of death among adults in the industrialized world (Lusis, 2000). In addition to causing ischaemia by obstructing blood flow in arteries that supply the heart, brain, kidneys and lower extremities; atherosclerotic plaques may rupture, followed by the formation of thrombi. Occasionally, these thrombi may cause myocardial infarction, stroke or gangrene of the legs. A well-developed plaque consists of a fibrous cap overlying a lipid mass, called the necrotic core (Lusis, 2000). The fibrous cap is composed primarily of smooth muscle cells (SMCs) and a relatively dense extracellular matrix made of collagen, elastin and proteoglycans. It also contains macrophages, T lymphocytes and foam cells, especially beneath and to the side of the cap (Lusis, 2000). The necrotic centre of an advanced plaque contains a mixture of lipids, mainly cholesterol and cholesterol esters, as well as cellular debris. In the last two decades, major advances in cardiovascular research have established a fundamental role for inflammation in all stages of atherosclerosis, and provided strong support for the hypothesis that atherosclerosis is an inflammatory disorder (Galkina and Ley, 2009). Once formed, destabilization of an atherosclerotic plaque may be initiated, either by external factors such as increased blood pressure and shear stress (Slager et al., 2005), or by factors within the atherosclerotic plaque, in particular inflammation and the induction of cell death (Halvorsen et al., 2008). Current therapies for stabilizing the vulnerable plaque are based on drugs with lipid-lowering potential. Despite their value in long-term therapy, these drugs are unsuitable for the acute management of rupture-prone plaques (Madjid et al., 2004), suggesting that novel therapies as coadjutants to the standard protocol are needed. Interestingly, an increasing number of compounds targeting cell death molecules are being explored at the preclinical and clinical level for the treatment of many human disorders, in particular cancer (Green and Kroemer, 2005; Hotchkiss et al., 2009). Some drug targets elucidated in studies of cell death have already led to Food and Drug Administration-approved pharmacological agents, although many more are in clinical trials (see ClinicalTrials.gov for details). Because cell death is an essential physiological process occurring daily to remove damaged or unwanted cells, this evolution is challenging. In this review, we will focus on the pharmacological modulation of cell death in atherosclerosis and whether this approach may contribute to the beneficial effects of currently applied plaque-stabilizing therapies.
Cell death in atherosclerosis
Apoptosis is a major event in atherosclerosis, with increasing frequencies (up to 1–2%) as the plaque develops (Mallat and Tedgui, 2000). Although the process has been described for merely all cell types in the plaque, macrophages comprise up to 50% of apoptotic cells in advanced lesions (Lutgens et al., 1999). Besides apoptosis, a growing body of evidence suggests that autophagy and necrotic death is stimulated in advanced atherosclerotic plaques (Crisby et al., 1997; Martinet and De Meyer, 2009). The consequences of cell death in atherosclerotic plaques depend on the type of death, but also on the stage of the plaque and the cell type that is involved (Mallat and Tedgui, 2000; Tabas, 2005; Gautier et al., 2009). Endothelial cell death, for example, may play a pivotal role in plaque progression as injured endothelial cells attract phagocytes and produce cytokines and growth factors that act on adjacent SMCs to promote their growth (Mallat and Tedgui, 2000). Damaged endothelial cells also become procoagulant and promote the formation of thrombi.
Because SMCs are responsible for the synthesis of interstitial collagen fibres, cell death of SMCs will lead to a decrease of collagen in the plaque, which subsequently leads to plaque destabilization and rupture (Clarke et al., 2006). Moreover, apoptotic SMCs in advanced plaques are often not scavenged and could be an important source of calcifying matrix vesicles (Clarke et al., 2008). If dying or apoptotic SMCs are not swiftly phagocytosed, secondary necrosis of apoptotic SMCs results in the leakage of interleukin (IL) 1-α (Clarke et al., 2010). This cytokine acts on the surrounding viable SMCs inducing them to release IL-6 and monocyte chemotactic protein (MCP)-1, thereby perpetuating a chronic inflammatory state (Clarke et al., 2010). Furthermore, SMCs undergoing apoptosis increase the thrombogenicity of the plaque as they have the same potency to generate thrombin as platelets due to the exposure of phosphatidylserine on the surface early in the process of apoptosis (Mallat and Tedgui, 2000). Overall, loss of SMCs is detrimental for plaque stability and increases the risk of thrombosis.
Macrophages are responsible for the collagen breakdown and loss of SMCs in the plaque, thus contributing to the thinning of the fibrous cap. In this regard, one may conclude that death of macrophages results in plaque stabilization. However, recent evidence suggests that the consequences of macrophage death largely depend on their phagocytic capacity (Tabas, 2005). In early lesions, in which phagocytosis is highly efficient, increased cell death of macrophages limits lesion cellularity and progression without an increase in necrosis or inflammation. In advanced plaques, clearance of apoptotic bodies is defective (Schrijvers et al., 2005) so that macrophage cell death may result in secondary necrosis, enlargement of the necrotic core and enhanced inflammation (Tabas, 2005; Gautier et al., 2009) (Figure 1).
Figure 1.
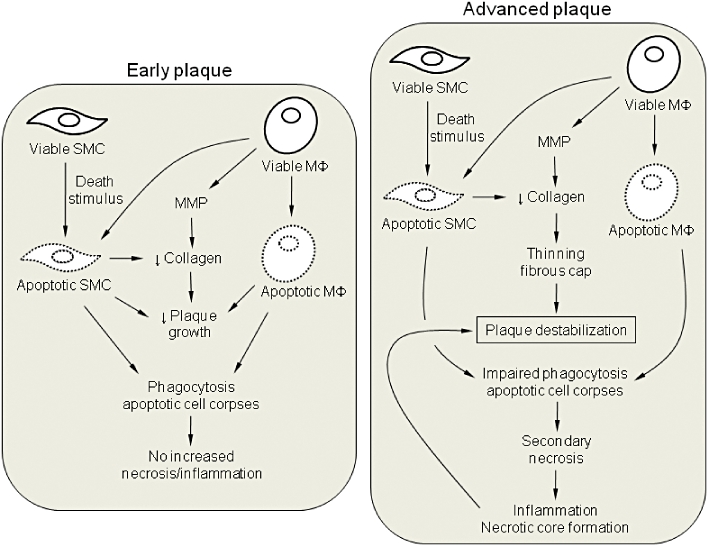
Schematic diagram of the consequences of apoptotic cell death in atherosclerosis. In early plaques, smooth muscle cells (SMCs) and macrophages (MΦ) undergo apoptosis and are then rapidly and safely cleared by neighbouring phagocytes. The net effect is a reduction in lesion cellularity and plaque growth. In advanced lesions, phagocytic clearance of apoptotic cells is not efficient so that secondary necrosis may occur, leading to inflammation, necrotic core formation and further plaque destabilization. Importantly, in both early and advanced plaques, death of SMCs may lead to a reduction in collagen synthesis, which in turn, leads to thinning of the fibrous cap, particularly in the more advanced stages of atherosclerosis. Loss of collagen is enhanced by the release of matrix metalloproteinases (MMPs).
Pharmacological inhibition of cell death in atherosclerosis (Table 1)
Table 1.
Compounds used to stabilize atherosclerotic plaques via modulation of cell death
| Compound | Remarks | Reference |
|---|---|---|
| Pharmacological inhibition of cell death | ||
| Caspase inhibitors | ||
| Pancaspase inhibitors (e.g. z-VAD-fmk) | Inhibits apoptosis, but may induce autophagy and necrotic cell death; macrophages treated with z-VAD-fmk secrete pro-inflammatory cytokines | (Martinet et al., 2006; Sarai et al., 2007) |
| Antioxidants | ||
| Antioxidant vitamins (e.g. vitamin C and E) | Prevent oxidative injury; In vivo effectiveness is questionable; Analogues targeted at the mitochondria might be more effective | (Madamanchi et al., 2005; Victor et al., 2009) |
| Polyphenols (e.g. resveratrol) | May act as pro-apoptotic and anti-apoptotic agents, depending on their concentration | (Das and Das, 2007) |
| ER stress inhibitors | ||
| Chemical chaperones (e.g. PBA, TUDCA) | Alleviates lipid-induced ER stress and suppresses lesion development | (Erbay et al., 2009; Dong et al., 2010) |
| Antioxidants (e.g. N-acetylcysteine, Tempol) | Reduces oxidant stress-mediated ER stress | (Tabas, 2010) |
| Cytokine blocking agents | ||
| Anti-TNFα (e.g. infliximab) | Inhibits the pro-inflammatory effects of TNFα; Treatment may be associated with serious side effects, including worsening of atherosclerosis | (Di Micco et al., 2009; Murdaca et al., 2009) |
| Interleukin-1 receptor antagonist | Inhibits IL-1 actions by binding to the type 1 IL-1 receptor | (Elhage et al., 1998; Crossman et al., 2008) |
| Lipid lowering drugs | ||
| HMG-CoA reductase inhibitors (statins) | Hydrophilic statins are anti-apoptotic, although hydrophobic statins promote apoptosis | (Katsiki et al., 2010) |
| Efferocytosis stimulating drugs | ||
| Thiazolidinediones (e.g. pioglitazone) | Selectively improve phagocytosis of apoptotic cells, at least in vitro, but enhance macrophage apoptosis in atherosclerotic plaques | (Thorp et al., 2007) |
| Fish oil | Increases ω-3 fatty acids, which are thought to be important factors contributing to efficient phagocytosis of apoptotic cells | (Li et al., 2009) |
| Pharmacological stimulation of cell death | ||
| Pro-apoptotic drugs | ||
| TRAIL | Induces selective macrophage apoptosis; stimulates smooth muscle cell migration; CD14+ monocytes are resistant to TRAIL-induced apoptosis | (Secchiero et al., 2006) |
| Protein synthesis inhibitors | ||
| mTOR inhibitors (e.g. everolimus) | Induces selective macrophage autophagy | (Verheye et al., 2007) |
| Cycloheximide, anisomycin | Induces selective macrophage apoptosis | (Croons et al., 2007, 2009) |
| NO donors | ||
| Molsidomine | Induces selective macrophage apoptosis, possibly through induction of ER stress | (De Meyer et al., 2003; Martinet et al., 2007) |
| Liposomes with cytotoxic drugs | ||
| Clodronate-containing liposomes | Intracellular delivery of cytotoxic drugs via phagocytosis; selective induction of macrophage apoptosis; nonphagocytic cells are resistant to the treatment | (van Rooijen and Hendrikx, 2010) |
ER, endoplasmic reticulum; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; mTOR, mammalian target of rapamycin; NO, nitric oxide; PBA, 4-phenyl butyric acid; TRAIL, TNF-related apoptosis inducing ligand; TUDCA, tauroursodeoxycholic acid.
Pharmacological inhibition of apoptosis by caspase inhibitors (Figure 2A)
Figure 2.
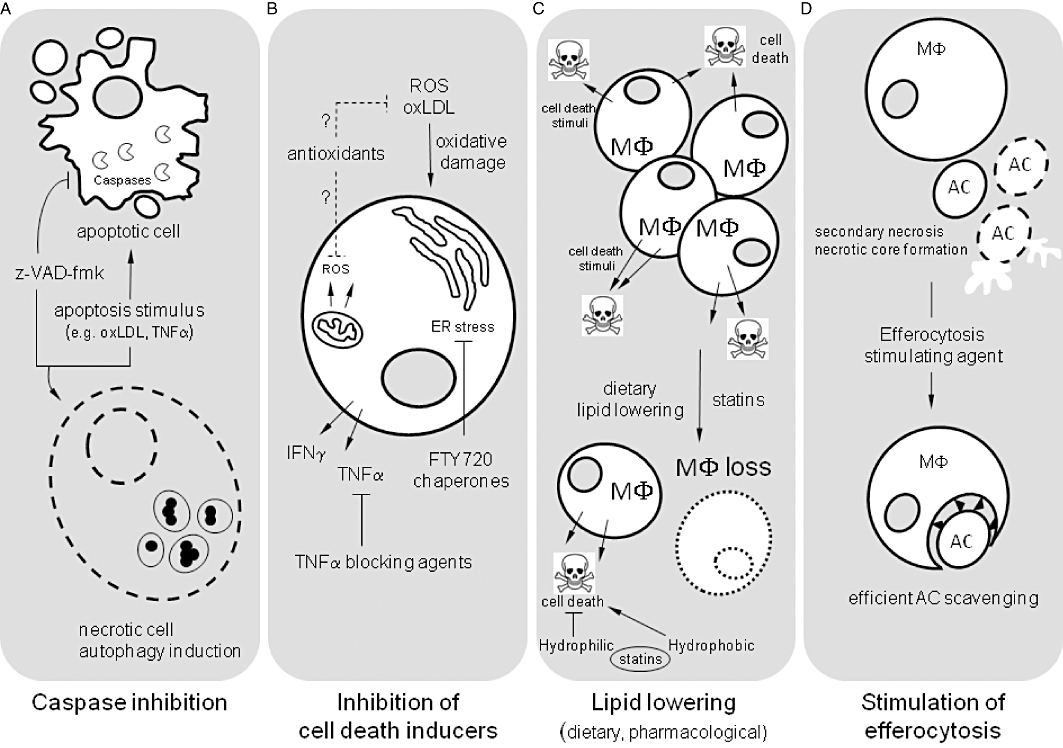
Overview of potential mechanisms that inhibit cell death in atherosclerosis. (A) Caspases are key effector molecules during apoptosis. Synthetic peptide inhibitors such as z-VAD-fmk are efficient in blocking apoptotic cell death in atherosclerotic plaques. However, inhibition of the classical caspase-dependent apoptotic pathway by z-VAD-fmk may induce autophagy and necrotic cell death. (B) A number of proapoptotic factors have been identified that contribute to apoptosis in atherosclerotic plaques. Examples include oxidative damage (through formation of ROS and oxLDL), endoplasmic reticulum (ER) stress and the release of pro-inflammatory cytokines such as TNFα. These apoptotic stimuli can be tackled by antioxidants, chemical chaperones or TNFα blocking agents respectively. (C) Apoptosis in atherosclerotic plaques is associated with the progressive accumulation of macrophages (MΦ) which may express a variety of cell death stimuli. Dietary lipid lowering leads to a substantial loss of macrophages. Also statins help, even though hydrophilic and hydrophobic statins may have differential effects on cell death. (D) Advanced atherosclerosis is characterized by defective clearance of apoptotic cells (AC). Several compounds stimulate AC scavenging and may prevent secondary necrosis and necrotic core formation. LDL, low-density lipoprotein; oxLDL, oxidized low density lipoprotein; ROS, reactive oxygen species.
Caspases were probably one of the first obvious therapeutic targets for modulating apoptosis, and they remain the most viable approach to blocking apoptotic cell death. Indeed, a substantial decrease in macrophage apoptosis was observed in plaques of cholesterol-fed rabbits that were infused intravenously with the broad caspase inhibitor z-VAD-fmk, the executioner caspase-3-specific inhibitor DEVD-cho or inhibitors of caspase-1 and −9 (YVAD-cho and z-LEHD-fmk, respectively) (Sarai et al., 2007). Moreover, local delivery of z-VAD-fmk during balloon injury inhibits SMC apoptosis and reduces subsequent neointimal hyperplasia (Beohar et al., 2004). However, in the context of caspase inhibitors, cells often do not die by apoptosis but succumb to a delayed form of cell death that can have an apoptosis-like morphology or manifest as autophagic or necrotic death (Green and Kroemer, 2005). Indeed, cell culture experiments have demonstrated that broad caspase inhibitors such as z-VAD-fmk can induce autophagy and necrotic cell death in macrophages (Yu et al., 2004; Martinet et al., 2006). One theory that may explain z-VAD-fmk-induced macrophage death is that inhibition of caspase-8 by z-VAD-fmk prevents cleavage of its substrate receptor-interacting protein 1 (RIP1) (Yu et al., 2004). Large amounts of uncleaved RIP1 stimulate autophagy-related genes so that autophagy is initiated. Autophagy can lead to the degradation of catalase (Yu et al., 2006), a key enzyme in the degradation of hydrogen peroxide. The resulting accumulation of reactive oxygen species (ROS) in the cell leads to membrane peroxidation, loss of membrane integrity and eventually necrotic cell death. Although this explanation seems plausible, it must be taken into account that z-VAD-fmk is not a specific inhibitor of caspases so that other interpretations for the induction of macrophage death should be considered. Chloro/fluoromethyl ketone peptide inhibitors such as z-VAD-fmk can also potently bind and inhibit cysteine proteases such as calpains (Knoblach et al., 2004) and cathepsins (Schotte et al., 1999), as well as cytosolic peptide:N-glycanase (PNGase) (Misaghi et al., 2006), an enzyme involved in deglycosylation of unfolded glycoproteins. Inhibition of PNGase may induce endoplasmic reticulum (ER) stress which in the presence of z-VAD-fmk may result in macrophage necrosis. Importantly, z-VAD-fmk-treated macrophages over-express and secrete several chemokines and cytokines, including tumour necrosis factor alpha (TNFα) (Martinet et al., 2006). The combination of z-VAD-fmk and TNFα, but not TNFα alone, induces SMC necrosis (Martinet et al., 2006), suggesting that z-VAD-fmk is detrimental and not beneficial for atherosclerotic plaque stability. Increasing knowledge of the molecular mechanisms associated with neurodegenerative, autoimmune and inflammatory pathologies suggests that novel anti-caspase therapies targeting caspase-1 could prove to be among the safer clinical interventions, as caspase-1 is responsible for maturation of the pro-inflammatory cytokines IL-1β and IL-18, and not involved in the apoptotic caspase cascade. Several caspase-1 inhibitors such as VX-740 (pralnacasan) and VX-765 have shown promising results in clinical trials for rheumatoid arthritis and osteoarthritis (Cornelis et al., 2007). Because activated caspase-1 has been observed in vulnerable plaques (Kolodgie et al., 2000) and caspase-1 inhibitors affect apoptosis in such lesions (Sarai et al., 2007), therapeutic modulation of caspase-1 activity is likely to offer significant health benefits for patients with severe atherosclerosis. One drawback of anti-caspase-1 therapy is that downstream targets of caspase-1 are of key importance in host defence against infection so that local inhibition will be essential to minimize side effects. Also of note, it has been postulated that caspase activity is not necessarily a lethal event in macrophages, and may be required for normal physiological functions such as macrophage differentiation (Nhan et al., 2005). It is presently unclear whether inhibition of such functions has a positive or negative effect on the structure and stability of the plaque.
Pharmacological inhibition of cell death inducers in atherosclerosis (Figure 2B)
Based on cell culture models that attempt to model specific conditions in atherosclerotic plaques, a number of factors or mechanisms such as oxidative or ER stress and the production of pro-inflammatory cytokines have been proposed that contribute to the induction of cell death (Mallat and Tedgui, 2000; Tabas, 2010). It should be noted, however, that at least some of the death inducers are different in early versus late lesions, and that cell death in atherosclerosis is almost certainly multifactorial. This complexity suggests that pharmacological inhibition of cell death through manipulation of its inducers may not be easy to accomplish.
Oxidative stress
Plaque formation is associated with the production of ROS which are currently assumed to induce oxidative damage and cell death. One of the earliest and best-studied oxidative events in atherosclerosis is the oxidative modification of low-density lipoprotein (LDL). A large body of evidence suggests that oxidized LDL (oxLDL) as well as its main oxides (7-ketocholesterol and 7β-hydroxycholesterol) exhibit cytotoxic effects to vascular cells leading to both apoptosis, autophagy and necrotic death (Mallat and Tedgui, 2000; Martinet and De Meyer, 2009; Vejux and Lizard, 2009). The type of death that is induced largely depends on the oxidation degree, the exposure time and the concentration of oxLDL. Mildly oxidized LDL is usually internalized and degraded through a receptor-mediated pathway, which mainly involves the lectin-like endothelial oxLDL receptor-1 (LOX-1) (Kataoka et al., 2001; Chen et al., 2004). Expression of LOX-1 can be induced by its ligand oxLDL and leads to stimulation of the proapoptotic factor Bax, decreased expression of the antiapoptotic proteins Bcl-2 and c-IAP-1, and activation of the intrinsic pathway of apoptosis that involves the release of cytochrome C, activation of caspase-9 and −3, but not caspase-8 (Kataoka et al., 2001; Chen et al., 2004). Uptake of oxLDL through LOX-1 also induces ROS production, reduces nitric oxide (NO), activates NF-κB and thereby up-regulates expression of MCP-1 and metalloproteinases. In addition, some groups report enhanced expression of Fas ligand (FasL), down-regulation of FADD-like interleukin-1 beta-converting enzyme inhibitory protein (FLIP) and activation of mitogen-activated protein and Jun kinases (Sata and Walsh, 1998; Napoli et al., 2000; Lee and Chau, 2001), indicating that oxLDL-induced apoptosis is very complex. Highly oxidized LDL exhibits dramatic cytotoxic effects on vascular cells, leading to both apoptosis and necrosis (Martinet and Kockx, 2001). Autophagy is induced after oxLDL-mediated oxidative injury to facilitate the removal of damaged organelles and to prevent the accumulation of oxidatively modified proteins (Nowicki et al., 2007).
Although the intake of antioxidant vitamins such as vitamin C and E seems recommendable to prevent oxidative injury and the induction of cell death, many antioxidant supplementation studies did not show any effect. Several reasons may account for antioxidant ineffectiveness including the administration of a nonoptimal dose and/or type of antioxidant, the complexity of redox reactions in vivo and the potential for a paradoxical increase in oxidant generation by antioxidants themselves (Madamanchi et al., 2005). Nonetheless, there is at least one study showing a correlation between antioxidant administration and decreased apoptosis in rabbit atherosclerotic plaques (Li et al., 2004). Because mitochondria are the most important biological source of ROS, antioxidants targeted at the mitochondria (e.g. mitoquinone, mitovitamin E) are currently appealing novel agents to attenuate oxidative stress and cell death in atherosclerosis (Victor et al., 2009). These compounds concentrate on the matrix-facing surface of the inner mitochondrial membrane and therefore, protect directly against mitochondrial oxidative damage evoked by the respiratory chain. However, large clinical trials are needed to evaluate the effectiveness of mitochondrially targeted antioxidants in more detail.
Besides vitamins, polyphenols have clear antioxidant activities, at least in vitro. However, contradictory results have been reported with regard to modulation of cell apoptosis. In fact, polyphenols can act as pro-apoptotic or anti-apoptotic agents depending on their concentration. For example resveratrol, one of the most recognized and widely studied polyphenols, inhibits apoptotic cell death at very low concentrations, but when used in higher doses, it facilitates apoptotic cell death (Das and Das, 2007). Therefore, additional studies are needed to better elucidate the mechanisms of action and the real in vivo effectiveness of polyphenols in order to propose them as potential candidate drugs in atherosclerosis treatment.
Finally, it is noteworthy that high-density lipoproteins (HDL) act directly on endothelial cell viability by enhancing their resistance against oxLDL-induced apoptosis (Suc et al., 1997). Partly as a result of this finding, there has been an increasing interest in HDL-raising strategies to prevent atherotrombotic events.
ER stress
An important trigger of macrophage death in atherosclerosis is the progressive accumulation of free cholesterol in the ER membrane (Tabas et al., 2009), which is normally cholesterol-poor and highly fluid. Probably by altering the function of integral ER membrane proteins, this event induces the ER stress signal transduction pathway, known as the unfolded protein response. This process occurs at all stages of plaque development and enhances cell survival, particularly in early lesions (Zhou et al., 2005; Tabas, 2010). However, prolonged or severe ER stress can result in CCAAT/enhancer-binding protein homologous protein (CHOP)-induced apoptosis. DeVries-Seimon et al. (2005) demonstrated that p38 MAP kinase (MAPK) is necessary for free cholesterol-mediated CHOP induction and apoptosis. Yet, two additional signalling pathways must cooperate with p38-CHOP (DeVries-Seimon et al., 2005). One involves the type A scavenger receptor, the other pathway requires JNK. Recently, it was demonstrated that the synthetic sphingosine analogue FTY720, currently in clinical trials as an immunological drug, is able to decrease cholesterol toxicity in macrophages by reducing the delivery of scavenged lipoprotein cholesterol to the ER (Blom et al., 2010). Moreover, FTY720 stimulates the production of 27-hydroxycholesterol, an endogenous ligand of the liver X receptor (LXR), leading to LXR-induced up-regulation of ATP-binding cassette transporter A1 proteins and increased efflux of cholesterol (Blom et al., 2010).
Apart from high levels of free cholesterol, pathological ER stress during plaque development can be triggered by excess intracellular concentrations of saturated fatty acids (FAs). Indeed, Erbay et al. (2009) recently demonstrated that the cytosolic lipid-binding protein aP2 promotes ER stress through binding of saturated fats. Administration of the chemical chaperone 4-phenyl butyric acid (PBA) for 2 weeks in mice inhibits aP2, alleviates toxic lipid-induced ER stress, and suppresses apoptotic cell death as well as plaque formation (Erbay et al., 2009). Inhibition of aP2 increases expression of LXR-a, which subsequently stimulates the expression of the lipogenic protein stearoyl-Coenzyme A desaturase-1 (SCD-1). Elevated SCD-1 activity converts toxic-saturated fats into monounsaturated fats for further metabolism. Comparable to PBA, also oral administration of the chemical chaperone tauroursodeoxycholic acid significantly inhibits both ER stress and lesion development in LDL receptor-deficient mice (Dong et al., 2010).
Finally, it is noteworthy that homocysteine as well as oxidative stress evoked by oxLDL or ROS may stimulate ER stress in vascular cells (Malhotra and Kaufman, 2007; Sanson et al., 2009; Tabas, 2010). Antioxidants such as N-acetylcysteine and Tempol, as well as up-regulation of ER-resident chaperones, including the anti-apoptotic ER-associated oxygen-regulated protein 150, prevent ER stress and promote cell survival (Sanson et al., 2009; Tabas, 2010). Hypothetically, administration of molecular or chemical chaperones which facilitate proper protein folding and stability could help to prevent ROS and/or oxLDL-induced ER stress, but thorough in vivo evidence for this effect in atherosclerosis is lacking.
Pro-inflammatory cytokines
Several pro-inflammatory cytokines that contribute to atherogenesis can have profound effects on cell death. Interferon gamma (IFNγ), for example, is a pro-inflammatory cytokine produced by T cells, NK cells and macrophages. It is highly expressed in atherosclerotic lesions and has been shown to induce apoptosis in endothelial cells, SMCs and macrophages (Kleemann et al., 2008). Several pro-apoptotic proteins including TNF-related apoptosis inducing ligand (TRAIL), Fas and TNF receptor 1 were suggested to be responsible for IFNγ-induced cell death (Inagaki et al., 2002; Li et al., 2002; Stefanescu et al., 2008). Next to IFNγ, IL-1β in plaques has important pro-inflammatory and pro-apoptotic effects, in particular on endothelial cells and SMCs, and is strongly induced in monocytes by direct contact with stimulated T lymphocytes (Kirii et al., 2003). Also, TNFα is highly expressed by activated macrophages and other immune cells in atherosclerotic lesions (Kleemann et al., 2008). TNFα promotes macrophage-induced apoptosis of SMCs by cooperative interactions with NO and FasL/Fas (Boyle et al., 2003), particularly when combined with other pro-inflammatory cytokines such as IFNγ and IL-1β (Geng et al., 1996).
Given the substantial role of IFNγ, IL-1β and TNFα in cell death of plaque SMCs, therapeutically targeting these cytokines could enhance the stability of atherosclerotic plaques. Indeed, plaques from mice injected with a plasmid encoding a soluble INFγ receptor construct had a more stable phenotype with reduced inflammatory cells and increased collagen and SMC content (Koga et al., 2007). IL-1 receptor antagonist (IL-1ra), the only known naturally occurring cytokine antagonist that inhibits IL-1 actions by binding to the type 1 IL-1 receptor, is available for the treatment of a number of inflammatory conditions as a recombinant, non-glycosylated protein (Anakinra). Because IL-1ra inhibits the formation of intimal fatty streaks in mouse models of atherosclerosis (Elhage et al., 1998), clinical trials are now ongoing to evaluate the effect of IL-1ra on acute coronary syndromes (Crossman et al., 2008). Furthermore, anti-TNFα monoclonal antibody therapy (e.g. infliximab) is now licensed for use in severe chronic inflammation of the gut and joints, even though serious side effects, such as tuberculosis, infections, sepsis and even death have been reported with the use of TNF-blocking agents (Murdaca et al., 2009). In this regard, it is noteworthy that patients with rheumatoid arthritis that were treated with infliximab show significant worsening of atherosclerosis (Di Micco et al., 2009), suggesting that this approach might not be ideal to tackle cell death in atherosclerosis.
Pharmacological inhibition of apoptosis by lipid lowering (Figure 2C)
Plaques lose lipid-laden macrophages after a prolonged period of dietary lipid lowering (Martinet et al., 2009). This plaque-stabilizing effect is not related to the induction of macrophage apoptosis, but probably a consequence of multiple factors such as decreased macrophage replication, impaired monocyte recruitment and emigration of macrophage foam cells from the plaque (Llodra et al., 2004; Martinet et al., 2009). In addition to loss of macrophages, several beneficial effects of dietary lipid lowering on plaque stability and clinical outcome have been described, including a substantial reduction in apoptotic cell death (Martinet et al., 2009). Decreased apoptosis in plaques after dietary lipid lowering is most likely attributed to less inflammation.
Lipid-lowering therapy with 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, better known as statins, is probably one of the most important pharmacological strategies to treat patients with vulnerable plaques. Statins block the conversion of HMG-CoA to mevalonate with consecutive attenuation of the biosynthesis of cholesterol. This effect is associated with a reduction (up to 63%) in LDL cholesterol (Knopp, 1999). Furthermore, statins exert pleiotropic effects, including the ability to modulate cell death in the vascular wall (Katsiki et al., 2010), which may occur independent of their cholesterol-lowering properties. In general, lipophilic statins (atorvastatin, simvastatin, lovastatin, fluvastatin, pitavastatin) have been shown to render SMCs and endothelial cells susceptible to apoptosis or even induce apoptosis in these cells at high doses (Katsiki et al., 2010). On the contrary, hydrophilic statins (pravastatin or rosuvastatin) have been reported to suppress apoptosis in both human and animal plaques (Katsiki et al., 2010). A possible explanation for this discrepancy is that hydrophilic statins are highly hepatoselective, although the lipophilic ones are much more widely taken up by a broad range of tissues and cells via passive diffusion. Statin-induced apoptosis is most likely linked to the reduced synthesis of important intermediates, such as the isoprenoids farnesyl pyrophosphate and geranylgeranyl pyrophosphate, involved in the post-translational prenylation of several proteins that regulate a variety of cellular processes (Katsiki et al., 2010). Inhibition of apoptosis by hydrophilic statins is poorly understood, even though rosuvastatin may protect endothelial cells from death by phosphorylating Akt and endothelial NO synthase (Enomoto et al., 2009). Because statins also exert beneficial pleiotropic effects on the vasculature, the physiological significance of statin-induced apoptosis of vascular cells is controversial and remains to be determined. Indeed, despite their different effects on vascular cell apoptosis, both lipophilic and hydrophilic statins are efficient LDL-lowering substances that decrease the occurrence of cardiovascular disease so that the current clinical guidelines recommend the use of statins, without specification of lipophilic or hydrophilic characteristics.
Pharmacological inhibition of necrotic death by stimulating efferocytosis (Figure 2D)
The phagocytic clearance of apoptotic cells by macrophages, a process also known as efferocytosis, is severly impaired in advanced lesions and leads to plaque necrosis (Schrijvers et al., 2005). Besides expansion of the necrotic core, the accumulation of uncleared apoptotic cells has a number of consequences that promote plaque destabilization such as enhanced inflammation, rapid matrix breakdown and an increased level of thrombogenicity (Tabas, 2005). Glucocorticoids are known to stimulate phagocytic clearance of apoptotic cells, but the serious adverse effects of glucocorticoid therapy render this approach impractical. Thiazolidinediones (TZDs), which affect cells through activation of the nuclear receptor peroxisome proliferator-activated receptor (PPAR)-γ and through other ‘off-target’ mechanisms, selectively improve phagocytosis of apoptotic cells in vitro, but at the same time enhance macrophage apoptosis in advanced atherosclerotic lesions of nondiabetic LDL receptor-null mice (Thorp et al., 2007). Consequently, the net in vivo effect of these drugs in this atherosclerosis model is increased plaque necrosis and subsequent plaque destabilization. In addition, data from several sources have established that TZDs are associated with oedema, weight gain and heart failure in humans. Nonetheless, TZDs have become a well-established component of treatment for type 2 diabetes, and recent evidence suggests that there is no cardiovascular risk, at least not for pioglitazone (Erdmann et al., 2009). In obesity and type 2 diabetes, the defect in efferocytosis of macrophages seems to be related to increased concentrations of saturated FAs and/or decreased concentrations of the ω-3 FA eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) (Li et al., 2009). This change in membrane lipid composition may lead to defective phosphatidylinositol 3-kinase activation and failure to generate phosphatidylinositol 3-phosphate in the macrophage phagocytic membrane. Interestingly, feeding ob/ob mice a fish oil diet increases ω-3 FA without appreciable changes in the content of saturated FA, and leads to a reversal of the defect in efferocytosis and a reduction in plaque size (Li et al., 2009). These findings suggest that the level of ω-3 FA in the macrophage membrane is an important factor contributing to efficient efferocytosis. Furthermore, because oxLDL or oxLDL antibodies interfere with phagocytosis of apoptotic cells, it is tempting to speculate that antioxidants can improve clearance of dying cells. However, as suggested above, clinical trials in humans with antioxidants showed only limited success in preventing coronary artery disease, although it is possible that more encouraging results will be obtained in the future through the use of drugs that are targeted to specific oxidation reactions in atherosclerosis. It should also be noted that certain antioxidants may not promote but inhibit recognition of apoptotic cells by phagocytes by inhibiting oxidation of externalized phosphatidylserine (Tyurina et al., 2004). Examples of other drugs that might promote phagocytic clearance of apoptotic cells are the cholesterol-lowering agent lovastatin (Morimoto et al., 2006), the macrolide antibiotic azithromycin (Hodge et al., 2006) and members of the lipoxin family (Godson et al., 2000). Future studies are needed to determine whether these drugs may provide the basis for a novel therapeutic strategy to prevent the progression of advanced atherosclerotic plaques via modulation of phagocytosis. Indeed, it has been proposed that abundant phagocytosis of apoptotic cells might be associated with the production of ROS and tissue injury which has prompted the search for attenuation mechanisms of phagocytosis (de Almeida and Linden, 2005).
Pharmacological stimulation of cell death in atherosclerosis
Macrophages play a central role in atherosclerotic plaque destabilization via production of cytotoxic amounts of ROS, release of pro-inflammatory cytokines, synthesis of matrix-degrading enzymes and expression of membrane-bound FasL, all of which stimulate SMC death (Mallat and Tedgui, 2000). Traditional anti-inflammatory agents such as glucocorticoids and cyclophosphamide have serious side effects, making them almost certainly inappropriate for atherosclerotic macrophage clearance. Fortunately, resolvins and protectins, two new groups of chemical mediators endogenously generated from the fatty acids EPA and DHA, may stabilize atherosclerotic plaques through pro-resolution and counter-modulating inflammation (Chen et al., 2008), albeit further studies are necessary. A promising alternative approach would be to induce cell death in macrophages via pharmacological therapy. Although this method requires specificity so that only macrophages would die, monocytes and macrophages are known to be more susceptible to a variety of apoptosis stimuli as compared with SMCs and endothelial cells (Zeini et al., 2007). Indeed, several compounds have recently been identified that selectively clear macrophages by induction of cell death in atherosclerotic plaques, as outlined below.
Selective macrophage death via therapeutics directly stimulating the apoptotic pathways
Because the apoptotic machinery is present in all cell types, apoptosis-inducing therapeutics that can be used in the clinic are extremely scarce. Nevertheless, impressive advances have been made to trigger apoptosis safely in vivo, particularly in the field of cancer. Even though initiation of apoptosis will not be a panacea for all of humanity's ailments, at least one promising therapy in cancer therapy – namely recombinant TRAIL – may be useful to treat vascular disease (Figure 3, [1]). TRAIL is a type II transmembrane protein belonging to the TNF family of ligands, and occurs as a trimer at the cell surface. It can be proteolytically cleaved at its C-terminus to form a soluble ligand. Serum levels of soluble TRAIL are significantly decreased in patients affected by or predisposed to coronary artery disease (Schoppet et al., 2006), suggesting that circulating TRAIL may be involved in the pathophysiology of cardiovascular disease. Systemic administration of recombinant TRAIL in diabetic ApoE−/− mice results in apoptosis of macrophages that have infiltrated in the plaque and attenuates the development of atherosclerotic lesions (Secchiero et al., 2006). Interestingly, TRAIL treatment does not decrease but rather shows a tendency to increase the number of plaque SMCs by stimulating SMC migration (Secchiero et al., 2006). Moreover, circulating CD14+ monocytes are resistant to TRAIL-induced apoptosis (Secchiero et al., 2006). Therefore, TRAIL must be considered a promising therapeutic agent, not only for anti-tumour therapy, but also for its anti-macrophage and anti-atherosclerotic activity.
Figure 3.
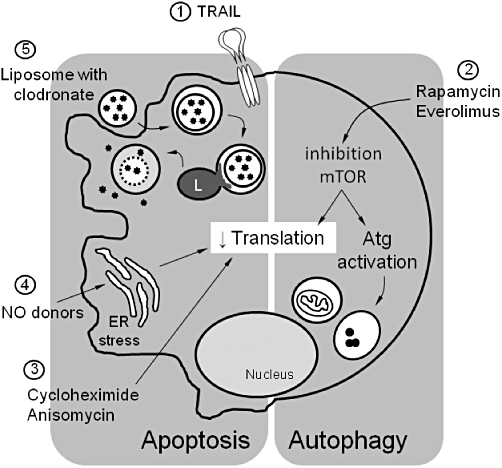
Overview of different strategies that induce selective macrophage death in atherosclerotic plaques. (1) TRAIL is expressed as a type II transmembrane protein, but its extracellular domain can be proteolytically cleaved from the cell surface and acts as a soluble cytokine interacting with transmembrane receptors belonging to the TNF-receptor family. Systemic soluble TRAIL selectively induces apoptosis of macrophages in atherosclerotic plaques. (2) Rapamycin and its structural analogue everolimus inhibit mammalian target of rapamycin (mTOR). Inhibition of mTOR downregulates translation and activates autophagy-related (Atg) genes. These effects may selectively induce autophagosome formation and autophagic cell death in macrophages of atherosclerotic plaques. (3) Inhibition of translation by the protein synthesis inhibitors cycloheximide or anisomycin selectively stimulates macrophage apoptosis in atherosclerotic plaques. (4) Treatment of atherosclerotic plaques with nitric oxide (NO) donors such as molsidomine depletes macrophages via apoptosis, potentially via induction of ER stress and subsequent inhibition of protein translation. (5) Liposomes encapsulating clodronate (or other lethal drugs) are ingested by macrophages via endocytosis. After fusion with lysosomes (L), the phospholipid bilayers of the liposomes are disrupted under the influence of lysosomal phospholipases. Clodronate is released into the cytosol and triggers apoptotic cell death. ER, endoplasmic reticulum; TRAIL, TNF-related apoptosis inducing ligand.
Selective macrophage death via protein synthesis inhibitors
Mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that acts as a sensor for energy, growth factors and nutrients. Inhibition of mTOR activity by rapamycin (or its analogues everolimus, zotarolimus or biolimus) leads to the inhibition of protein translation and stimulates death of plaque macrophages via autophagy without affecting the viability of SMCs (Figure 3, [2]) (Verheye et al., 2007). The mechanism underlying the selective death of macrophages by everolimus is poorly understood. It has been proposed, based on their oxygen consumption and high-level expression of markers for DNA synthesis and/or repair, that plaque macrophages are metabolically highly active and thus, more dependent on protein synthesis than SMCs for their survival (Verheye et al., 2007). In addition, inhibition of translation in SMCs by rapamycin induces a modulation towards a differentiated, quiescent, contractile phenotype, which may render SMCs relatively insensitive to cell death mediated by mTOR inhibition (Martin et al., 2004).
Because inhibition of protein translation seems to be a major trigger that drives selective induction of macrophage death (Verheye et al., 2007), plaques from cholesterol-fed rabbits have been treated with several protein synthesis inhibitors. Local administration of cycloheximide, a protein synthesis inhibitor that binds on 80S ribosomes (thereby blocking the activity of peptidyl transferase), induces macrophage death in rabbit plaques without influencing the viability and reactivity of the SMCs or the endothelium; however, in contrast to everolimus, apoptosis and not autophagy is induced (Figure 3, [3]) (Croons et al., 2007). Cycloheximide is widely used in cell death research, but the detailed mechanism of cycloheximide-induced apoptosis is unclear. A few possibilities have been suggested, including activation of ER stress (Ito et al., 2006) and over-expression of the pro-apoptotic genes c-myc, c-fos, c-jun and p53 (Alessenko et al., 1997). Similar to cycloheximide, treatment of plaques with anisomycin triggers selective macrophage apoptosis via a signalling pathway that requires p38 MAPK (Croons et al., 2009).
Selective macrophage death via NO donors
Because NO promotes vasodilation and inhibits several key processes in vascular disease, such as platelet aggregation and interaction of leukocytes with the vessel wall, NO donors are effective in the prevention and/or treatment of coronary artery disease. Recently, it was demonstrated that exogenous in vivo NO treatment using nitratethiol, nitrosothiol or nitroglycerine protects SMCs against apoptosis and drives cells to quiescence through up-regulation of p53 and increased levels of Bcl-2/Bax (Duran et al., 2009). Moreover, endothelial cell apoptosis was inhibited in rabbit lesions if the animals received L-arginine, an essential substance for nitric oxide synthesis (Nematbakhsh et al., 2008). Interestingly, treatment of plaques in cholesterol-fed rabbits with the NO donor molsidomine leads to the formation of a large subendothelial macrophage-free layer solely consisting of SMCs and extracellular matrix (De Meyer et al., 2003). Two mechanisms may explain selective loss of macrophages in molsidomine-treated plaques. First, NO donors can decrease the expression of adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) in endothelial cells so that molsidomine attenuates macrophage influx in the vessel wall. Because VCAM-1 expression was not affected in plaques from molsidomine-treated rabbits (De Meyer et al., 2003), this explanation seems unlikely. Second, NO stimulates selective macrophage death or sensitizes macrophages to undergo cell death. This theory is more plausible, as NO is able to trigger selective macrophage apoptosis via induction of ER stress (Martinet et al., 2007). Of note, in an early phase of ER stress, translational attenuation occurs to reduce the load of potentially misfolded proteins on the ER. Accordingly, macrophage death by NO may add further evidence that inhibition of protein synthesis leads to selective depletion of macrophages in atherosclerotic plaques (Figure 3, [4]). Furthermore, it should be noted that macrophages which acquired resistance to the apoptotic effects of endogenously generated NO by inducible NO synthase (iNOS) paradoxically become hypersensitive to cell death induced by exogenously added NO donors (Mohr et al., 1998). Given that human plaques contain many macrophages that over-express iNOS (Cromheeke et al., 1999), administration of an NO donor may preferentially eliminate the activated (iNOS-positive) macrophages, thereby favouring features of atherosclerotic plaque stability.
Selective macrophage death via clodronate-containing liposomes
Selective death of macrophages can be accomplished by taking advantage of their phagocytic function. One of the best studied approaches is liposome-mediated intracellular delivery of cytotoxic drugs such as clodronate (Figure 3, [5]) (van Rooijen and Hendrikx, 2010). Liposomes are ingested by macrophages via endocytosis, forming endosomes which then fuse with lysosomes. Lysosomal phospholipases disrupt the phospholipid bilayers of the liposomes and the drug is intracellularly released. Released clodronate from dying macrophages or free clodronate from leakage of the liposomes do not enter nonphagocytic cells (van Rooijen and Hendrikx, 2010), making this approach specific for phagocytic cells of the mononuclear phagocyte system. In animal models of restenosis, systemic administration of clodronate-containing liposomes reduces macrophage numbers in the arterial lesion, SMC proliferation and MMP-2 activity (Danenberg et al., 2002). However, also the number of circulating blood monocytes decreases as a consequence of the systemic treatment. Local administration of clodronate-containing liposomes through intra-articular injection selectively depletes macrophages in patients with rheumatoid arthritis (Barrera et al., 2000). Local application of liposomes to the atherosclerotic lesion could therefore represent a promising strategy for selective depletion of macrophages in atherosclerotic plaques.
Concluding remarks
Pharmacological agents such as statins may target plaque destabilization, and have been shown to reduce the incidence of acute coronary syndromes. Yet, cardiovascular disease resulting from atherosclerosis and thrombosis remains a major cause of death and disability among adults in Western countries. Because cell death is an important trigger of plaque rupture, an increasing number of compounds targeting the apoptotic or autophagic machinery in atherosclerosis are being explored, predominantly at the preclinical level. Inhibition of cell death or its inducers is, of course, an interesting approach from a theoretical point of view, but further studies will be needed to determine whether compounds such as caspase inhibitors or antioxidants will find their way into routine clinical use for the treatment of patients with vulnerable plaques. Not only the efficacy and absence of non-specific effects are crucial for selection of the most appropriate drugs, early studies in mice with an apoptosis defect in macrophages suggested that macrophage apoptosis is a critical self-defence mechanism in suppressing atherogenesis (Liu et al., 2005). In this light, inhibition of apoptosis would not be a valuable approach for the prevention and treatment of atherosclerosis. Recent studies, however, brought more insight into this complex matter and indicate that macrophage apoptosis does not have a discernable effect on inflammation or plaque size, unless there is an additional defect in phagocytosis of the apoptotic corpses (Clarke and Bennett, 2009). In that case, secondary necrosis could occur which may result in local and systemic inflammation, and increased plaque size. Therefore, if selective induction of macrophage death would be ensued as an alternative option to stabilize vulnerable plaques instead of cell death inhibition, combined therapy with efferocytosis-stimulating drugs would be recommendable. In line with these findings, a growing body of evidence suggests that enhancing cell death in early plaques would be beneficial while being detrimental during the late stages of atherosclerosis (Tabas, 2005; Gautier et al., 2009). Accordingly, therapeutic strategies should be conceived to selectively promote cell death in early lesional macrophages and/or to selectively prevent cell death in advanced lesions, which is obviously not self-evident. Indeed, it is presently unknown how pro-apoptotic drugs can be targeted specifically to early lesions. Given that macrophages in advanced lesions probably die by mechanisms that are different from those in early lesions, one may try to target cell death pathways (e.g. accumulation of cytotoxic levels of free cholesterol) that only occur after lesions have progressed to advanced stages. Importantly, strategies aimed at selectively decreasing macrophage numbers by inducing cell death look only promising if depletion of peripheral blood monocytes is avoided. Local drug delivery via coated stents can decrease or avoid unwanted systemic effects, but also have some drawbacks. First, stents have relatively fast release rates (hours to months) of the coated drug so that it will only be a matter of time before monocytes will reinfiltrate the plaque. Additional systemic therapy with, for example, statins or NO-donors may be required to prevent this process. Second, drug-eluting stents show delayed arterial healing which appears to be responsible for late in-stent thrombosis. Hence, drug-eluting stent technology should be optimized to balance the benefits against the risks of late in-stent thrombosis.
Acknowledgments
This work was supported by the Fund for Scientific Research-Flanders and the University of Antwerp. We dedicate this paper to Em Prof Dr Norbert Buyssens, who passed away on February 11, 2011. We have always admired him for his erudition and are grateful for his scientific advice and the many fruitful discussions that stimulated our research in the past 20 years.
Glossary
Abbreviations
- CHOP
C/EBP homologous protein
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- ER
endoplasmic reticulum
- FA
fatty acids
- FasL
Fas ligand
- FLIP
FLICE inhibitory protein
- HDL
high-density lipoprotein
- IL-1ra
interleukin-1 receptor antagonist
- iNOS
inducible nitric oxide synthase
- LDL
low-density lipoprotein
- LOX-1
lectin-like oxidized low density lipoprotein receptor-1
- LXR
liver X receptor
- MCP-1
monocyte chemotactic protein-1
- mTOR
mammalian target of rapamycin
- NO
nitric oxide
- oxLDL
oxidized low-density lipoprotein
- PBA
4-phenyl butyric acid
- RIP1
receptor-interacting protein 1
- ROS
reactive oxygen species
- SCD-1
stearoyl-Coenzyme A desaturase-1
- SMC
smooth muscle cell
- TUDCA
tauroursodeoxycholic acid
- TZD
thiazolidinedione
- UPR
unfolded protein response
- VCAM-1
vascular cell adhesion molecule-1
Conflict of interest
The authors have nothing to declare.
References
- Alessenko AV, Boikov PY, Filippova GN, Khrenov AV, Loginov AS, Makarieva ED. Mechanisms of cycloheximide-induced apoptosis in liver cells. FEBS Lett. 1997;416:113–116. doi: 10.1016/s0014-5793(97)01161-7. [DOI] [PubMed] [Google Scholar]
- de Almeida CJ, Linden R. Phagocytosis of apoptotic cells: a matter of balance. Cell Mol Life Sci. 2005;62:1532–1546. doi: 10.1007/s00018-005-4511-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera P, Blom A, van Lent PL, van Bloois L, Beijnen JH, van Rooijen N, et al. Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis Rheum. 2000;43:1951–1959. doi: 10.1002/1529-0131(200009)43:9<1951::AID-ANR5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Beohar N, Flaherty JD, Davidson CJ, Maynard RC, Robbins JD, Shah AP, et al. Antirestenotic effects of a locally delivered caspase inhibitor in a balloon injury model. Circulation. 2004;109:108–113. doi: 10.1161/01.CIR.0000105724.30980.CD. [DOI] [PubMed] [Google Scholar]
- Blom T, Back N, Mutka AL, Bittman R, Li Z, de Lera A, et al. FTY720 stimulates 27-hydroxycholesterol production and confers atheroprotective effects in human primary macrophages. Circ Res. 2010;106:720–729. doi: 10.1161/CIRCRESAHA.109.204396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle JJ, Weissberg PL, Bennett MR. Tumor necrosis factor-alpha promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler Thromb Vasc Biol. 2003;23:1553–1558. doi: 10.1161/01.ATV.0000086961.44581.B7. [DOI] [PubMed] [Google Scholar]
- Chen J, Mehta JL, Haider N, Zhang X, Narula J, Li D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res. 2004;94:370–376. doi: 10.1161/01.RES.0000113782.07824.BE. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang J, Nie R, Zhou S. Endogenous pro-resolving and anti-inflammatory lipid mediators: the new hope of atherosclerotic diseases. Med Hypotheses. 2008;71:237–240. doi: 10.1016/j.mehy.2008.03.026. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Bennett MR. Cause or consequence: what does macrophage apoptosis do in atherosclerosis? Arterioscler Thromb Vasc Biol. 2009;29:153–155. doi: 10.1161/ATVBAHA.108.179903. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529–1538. doi: 10.1161/CIRCRESAHA.108.175976. [DOI] [PubMed] [Google Scholar]
- Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res. 2010;106:363–372. doi: 10.1161/CIRCRESAHA.109.208389. [DOI] [PubMed] [Google Scholar]
- Cornelis S, Kersse K, Festjens N, Lamkanfi M, Vandenabeele P. Inflammatory caspases: targets for novel therapies. Curr Pharm Des. 2007;13:367–385. doi: 10.2174/138161207780163006. [DOI] [PubMed] [Google Scholar]
- Crisby M, Kallin B, Thyberg J, Zhivotovsky B, Orrenius S, Kostulas V, et al. Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis. 1997;130:17–27. doi: 10.1016/s0021-9150(96)06037-6. [DOI] [PubMed] [Google Scholar]
- Cromheeke KM, Kockx MM, De Meyer GRY, Bosmans JM, Bult H, Beelaerts WJ, et al. Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovasc Res. 1999;43:744–754. doi: 10.1016/s0008-6363(99)00148-0. [DOI] [PubMed] [Google Scholar]
- Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GRY. Selective clearance of macrophages in atherosclerotic plaques by the protein synthesis inhibitor cycloheximide. J Pharmacol Exp Ther. 2007;320:986–993. doi: 10.1124/jpet.106.113944. [DOI] [PubMed] [Google Scholar]
- Croons V, Martinet W, Herman AG, Timmermans JP, De Meyer GRY. The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen-activated protein kinase. J Pharmacol Exp Ther. 2009;329:856–864. doi: 10.1124/jpet.108.149948. [DOI] [PubMed] [Google Scholar]
- Crossman DC, Morton AC, Gunn JP, Greenwood JP, Hall AS, Fox KA, et al. Investigation of the effect of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-HEART Study) Trials. 2008;9:8. doi: 10.1186/1745-6215-9-8. DOI: 10.1186/1745-6215-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danenberg HD, Fishbein I, Gao J, Monkkonen J, Reich R, Gati I, et al. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106:599–605. doi: 10.1161/01.cir.0000023532.98469.48. [DOI] [PubMed] [Google Scholar]
- Das S, Das DK. Resveratrol: a therapeutic promise for cardiovascular diseases. Recent Pat Cardiovasc Drug Discov. 2007;2:133–138. doi: 10.2174/157489007780832560. [DOI] [PubMed] [Google Scholar]
- De Meyer GRY, Kockx MM, Knaapen MW, Martinet W, De Cleen DM, Bult H, et al. Nitric oxide donor molsidomine favors features of atherosclerotic plaque stability during cholesterol lowering in rabbits. J Cardiovasc Pharmacol. 2003;41:970–978. doi: 10.1097/00005344-200306000-00021. [DOI] [PubMed] [Google Scholar]
- DeVries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, et al. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco P, Ferrazzi P, Libre L, Mendolicchio L, Quaglia I, De Marco M, et al. Intima-media thickness evolution after treatment with infliximab in patients with rheumatoid arthritis. Int J Gen Med. 2009;2:141–144. doi: 10.2147/ijgm.s5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran X, Vilahur G, Badimon L. Exogenous in vivo NO-donor treatment preserves p53 levels and protects vascular cells from apoptosis. Atherosclerosis. 2009;205:101–106. doi: 10.1016/j.atherosclerosis.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242–244. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- Enomoto S, Sata M, Fukuda D, Nakamura K, Nagai R. Rosuvastatin prevents endothelial cell death and reduces atherosclerotic lesion formation in ApoE-deficient mice. Biomed Pharmacother. 2009;63:19–26. doi: 10.1016/j.biopha.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–1391. doi: 10.1038/nm.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann E, Charbonnel B, Wilcox R. Thiazolidinediones and cardiovascular risk – a question of balance. Curr Cardiol Rev. 2009;5:155–165. doi: 10.2174/157340309788970333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Huby T, Witztum JL, Ouzilleau B, Miller ER, Saint-Charles F, et al. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation. 2009;119:1795–1804. doi: 10.1161/CIRCULATIONAHA.108.806158. [DOI] [PubMed] [Google Scholar]
- Geng YJ, Wu Q, Muszynski M, Hansson GK, Libby P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin-1 beta. Arterioscler Thromb Vasc Biol. 1996;16:19–27. doi: 10.1161/01.atv.16.1.19. [DOI] [PubMed] [Google Scholar]
- Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115:2610–2617. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Oie E, et al. Atherosclerotic plaque stability – what determines the fate of a plaque? Prog Cardiovasc Dis. 2008;51:183–194. doi: 10.1016/j.pcad.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Hodge S, Hodge G, Brozyna S, Jersmann H, Holmes M, Reynolds PN. Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur Respir J. 2006;28:486–495. doi: 10.1183/09031936.06.00001506. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki Y, Yamagishi S, Amano S, Okamoto T, Koga K, Makita Z. Interferon-gamma-induced apoptosis and activation of THP-1 macrophages. Life Sci. 2002;71:2499–2508. doi: 10.1016/s0024-3205(02)02042-8. [DOI] [PubMed] [Google Scholar]
- Ito K, Kiyosawa N, Kumagai K, Manabe S, Matsunuma N, Yamoto T. Molecular mechanism investigation of cycloheximide-induced hepatocyte apoptosis in rat livers by morphological and microarray analysis. Toxicology. 2006;219:175–186. doi: 10.1016/j.tox.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Kume N, Miyamoto S, Minami M, Morimoto M, Hayashida K, et al. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:955–960. doi: 10.1161/01.atv.21.6.955. [DOI] [PubMed] [Google Scholar]
- Katsiki N, Tziomalos K, Chatzizisis Y, Elisaf M, Hatzitolios AI. Effect of HMG-CoA reductase inhibitors on vascular cell apoptosis: beneficial or detrimental? Atherosclerosis. 2010;211:9–14. doi: 10.1016/j.atherosclerosis.2009.12.028. [DOI] [PubMed] [Google Scholar]
- Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblach SM, Alroy DA, Nikolaeva M, Cernak I, Stoica BA, Faden AI. Caspase inhibitor z-DEVD-fmk attenuates calpain and necrotic cell death in vitro and after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24:1119–1132. doi: 10.1097/01.WCB.0000138664.17682.32. [DOI] [PubMed] [Google Scholar]
- Knopp RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341:498–511. doi: 10.1056/NEJM199908123410707. [DOI] [PubMed] [Google Scholar]
- Koga M, Kai H, Yasukawa H, Yamamoto T, Kawai Y, Kato S, et al. Inhibition of progression and stabilization of plaques by postnatal interferon-gamma function blocking in ApoE-knockout mice. Circ Res. 2007;101:348–356. doi: 10.1161/CIRCRESAHA.106.147256. [DOI] [PubMed] [Google Scholar]
- Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, Hui-Liang Y, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol. 2000;157:1259–1268. doi: 10.1016/S0002-9440(10)64641-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Chau L. Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2001;280:C709–C718. doi: 10.1152/ajpcell.2001.280.3.C709. [DOI] [PubMed] [Google Scholar]
- Li JH, Kluger MS, Madge LA, Zheng L, Bothwell AL, Pober JS. Interferon-gamma augments CD95(APO-1/Fas) and pro-caspase-8 expression and sensitizes human vascular endothelial cells to CD95-mediated apoptosis. Am J Pathol. 2002;161:1485–1495. doi: 10.1016/s0002-9440(10)64424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Sun Y, Liang CP, Thorp EB, Han S, Jehle AW, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res. 2009;105:1072–1082. doi: 10.1161/CIRCRESAHA.109.199570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hellsten A, Jacobsson LS, Blomqvist HM, Olsson AG, Yuan XM. Alpha-tocopherol and astaxanthin decrease macrophage infiltration, apoptosis and vulnerability in atheroma of hyperlipidaemic rabbits. J Mol Cell Cardiol. 2004;37:969–978. doi: 10.1016/j.yjmcc.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutgens E, de Muinck ED, Kitslaar PJ, Tordoir JH, Wellens HJ, Daemen MJ. Biphasic pattern of cell turnover characterizes the progression from fatty streaks to ruptured human atherosclerotic plaques. Cardiovasc Res. 1999;41:473–479. doi: 10.1016/s0008-6363(98)00311-3. [DOI] [PubMed] [Google Scholar]
- Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- Madjid M, Zarrabi A, Litovsky S, Willerson JT, Casscells W. Finding vulnerable atherosclerotic plaques: is it worth the effort? Arterioscler Thromb Vasc Biol. 2004;24:1775–1782. doi: 10.1161/01.ATV.0000142373.72662.20. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130:947–962. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KA, Rzucidlo EM, Merenick BL, Fingar DC, Brown DJ, Wagner RJ, et al. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286:C507–C517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- Martinet W, De Meyer GRY. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104:304–317. doi: 10.1161/CIRCRESAHA.108.188318. [DOI] [PubMed] [Google Scholar]
- Martinet W, Kockx MM. Apoptosis in atherosclerosis: focus on oxidized lipids and inflammation. Curr Opin Lipidol. 2001;12:535–541. doi: 10.1097/00041433-200110000-00009. [DOI] [PubMed] [Google Scholar]
- Martinet W, De Meyer GRY, Timmermans JP, Herman AG, Kockx MM. Macrophages but not smooth muscle cells undergo benzyloxycarbonyl-Val-Ala-DL-Asp(O-Methyl)-fluoromethylketone-induced nonapoptotic cell death depending on receptor-interacting protein 1 expression: implications for the stabilization of macrophage-rich atherosclerotic plaques. J Pharmacol Exp Ther. 2006;317:1356–1364. doi: 10.1124/jpet.106.102970. [DOI] [PubMed] [Google Scholar]
- Martinet W, Croons V, Timmermans JP, Herman AG, De Meyer GRY. Nitric oxide selectively depletes macrophages in atherosclerotic plaques via induction of endoplasmic reticulum stress. Br J Pharmacol. 2007;152:493–500. doi: 10.1038/sj.bjp.0707426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, Croons V, Herman AG, De Meyer GRY. Apoptosis does not mediate macrophage depletion in rabbit atherosclerotic plaques after dietary lipid lowering. Ann N Y Acad Sci. 2009;1171:365–371. doi: 10.1111/j.1749-6632.2009.04685.x. [DOI] [PubMed] [Google Scholar]
- Misaghi S, Korbel GA, Kessler B, Spooner E, Ploegh HL. z-VAD-fmk inhibits peptide:N-glycanase and may result in ER stress. Cell Death Differ. 2006;13:163–165. doi: 10.1038/sj.cdd.4401716. [DOI] [PubMed] [Google Scholar]
- Mohr S, McCormick TS, Lapetina EG. Macrophages resistant to endogenously generated nitric oxide-mediated apoptosis are hypersensitive to exogenously added nitric oxide donors: dichotomous apoptotic response independent of caspase 3 and reversal by the mitogen-activated protein kinase kinase (MEK) inhibitor PD 098059. Proc Natl Acad Sci U S A. 1998;95:5045–5050. doi: 10.1073/pnas.95.9.5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, et al. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol. 2006;176:7657–7665. doi: 10.4049/jimmunol.176.12.7657. [DOI] [PubMed] [Google Scholar]
- Murdaca G, Colombo BM, Puppo F. Anti-TNF-alpha inhibitors: a new therapeutic approach for inflammatory immune-mediated diseases: an update upon efficacy and adverse events. Int J Immunopathol Pharmacol. 2009;22:557–565. doi: 10.1177/039463200902200301. [DOI] [PubMed] [Google Scholar]
- Napoli C, Quehenberger O, De Nigris F, Abete P, Glass CK, Palinski W. Mildly oxidized low density lipoprotein activates multiple apoptotic signaling pathways in human coronary cells. FASEB J. 2000;14:1996–2007. doi: 10.1096/fj.99-0986com. [DOI] [PubMed] [Google Scholar]
- Nematbakhsh M, Haghjooyjavanmard S, Mahmoodi F, Monajemi AR. The prevention of endothelial dysfunction through endothelial cell apoptosis inhibition in a hypercholesterolemic rabbit model: the effect of L-arginine supplementation. Lipids Health Dis. 2008;7:27. doi: 10.1186/1476-511X-7-27. DOI: 10.1186/1476-511X-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nhan TQ, Liles WC, Schwartz SM. Role of caspases in death and survival of the plaque macrophage. Arterioscler Thromb Vasc Biol. 2005;25:895–903. doi: 10.1161/01.ATV.0000159519.07181.33. [DOI] [PubMed] [Google Scholar]
- Nowicki M, Zabirnyk O, Duerrschmidt N, Borlak J, Spanel-Borowski K. No upregulation of lectin-like oxidized low-density lipoprotein receptor-1 in serum-deprived EA.hy926 endothelial cells under oxLDL exposure, but increase in autophagy. Eur J Cell Biol. 2007;86:605–616. doi: 10.1016/j.ejcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- van Rooijen N, Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol. 2010;605:189–203. doi: 10.1007/978-1-60327-360-2_13. [DOI] [PubMed] [Google Scholar]
- Sanson M, Auge N, Vindis C, Muller C, Bando Y, Thiers JC, et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ Res. 2009;104:328–336. doi: 10.1161/CIRCRESAHA.108.183749. [DOI] [PubMed] [Google Scholar]
- Sarai M, Hartung D, Petrov A, Zhou J, Narula N, Hofstra L, et al. Broad and specific caspase inhibitor-induced acute repression of apoptosis in atherosclerotic lesions evaluated by radiolabeled annexin A5 imaging. J Am Coll Cardiol. 2007;50:2305–2312. doi: 10.1016/j.jacc.2007.08.044. [DOI] [PubMed] [Google Scholar]
- Sata M, Walsh K. Endothelial cell apoptosis induced by oxidized LDL is associated with the down-regulation of the cellular caspase inhibitor FLIP. J Biol Chem. 1998;273:33103–33106. doi: 10.1074/jbc.273.50.33103. [DOI] [PubMed] [Google Scholar]
- Schoppet M, Sattler AM, Schaefer JR, Hofbauer LC. Osteoprotegerin (OPG) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) levels in atherosclerosis. Atherosclerosis. 2006;184:446–447. doi: 10.1016/j.atherosclerosis.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Schotte P, Declercq W, Van Huffel S, Vandenabeele P, Beyaert R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999;442:117–121. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GRY, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- Secchiero P, Candido R, Corallini F, Zacchigna S, Toffoli B, Rimondi E, et al. Systemic tumor necrosis factor-related apoptosis-inducing ligand delivery shows antiatherosclerotic activity in apolipoprotein E-null diabetic mice. Circulation. 2006;114:1522–1530. doi: 10.1161/CIRCULATIONAHA.106.643841. [DOI] [PubMed] [Google Scholar]
- Slager CJ, Wentzel JJ, Gijsen FJ, Thury A, van der Wal AC, Schaar JA, et al. The role of shear stress in the destabilization of vulnerable plaques and related therapeutic implications. Nat Clin Pract Cardiovasc Med. 2005;2:456–464. doi: 10.1038/ncpcardio0298. [DOI] [PubMed] [Google Scholar]
- Stefanescu R, Bassett D, Modarresi R, Santiago F, Fakruddin M, Laurence J. Synergistic interactions between interferon-gamma and TRAIL modulate c-FLIP in endothelial cells, mediating their lineage-specific sensitivity to thrombotic thrombocytopenic purpura plasma-associated apoptosis. Blood. 2008;112:340–349. doi: 10.1182/blood-2007-10-119552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suc I, Escargueil-Blanc I, Troly M, Salvayre R, Negre-Salvayre A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc Biol. 1997;17:2158–2166. doi: 10.1161/01.atv.17.10.2158. [DOI] [PubMed] [Google Scholar]
- Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Seimon T, Timmins J, Li G, Lim W. Macrophage apoptosis in advanced atherosclerosis. Ann N Y Acad Sci. 2009;1173(Suppl 1):E40–E45. doi: 10.1111/j.1749-6632.2009.04957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E, Kuriakose G, Shah YM, Gonzalez FJ, Tabas I. Pioglitazone increases macrophage apoptosis and plaque necrosis in advanced atherosclerotic lesions of nondiabetic low-density lipoprotein receptor-null mice. Circulation. 2007;116:2182–2190. doi: 10.1161/CIRCULATIONAHA.107.698852. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Serinkan FB, Tyurin VA, Kini V, Yalowich JC, Schroit AJ, et al. Lipid antioxidant, etoposide, inhibits phosphatidylserine externalization and macrophage clearance of apoptotic cells by preventing phosphatidylserine oxidation. J Biol Chem. 2004;279:6056–6064. doi: 10.1074/jbc.M309929200. [DOI] [PubMed] [Google Scholar]
- Vejux A, Lizard G. Cytotoxic effects of oxysterols associated with human diseases: induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Mol Aspects Med. 2009;30:153–170. doi: 10.1016/j.mam.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, Timmermans JP, et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol. 2007;49:706–715. doi: 10.1016/j.jacc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Victor VM, Apostolova N, Herance R, Hernandez-Mijares A, Rocha M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: mitochondria-targeted antioxidants as potential therapy. Curr Med Chem. 2009;16:4654–4667. doi: 10.2174/092986709789878265. [DOI] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeini M, Lopez-Fontal R, Traves PG, Benito G, Hortelano S. Differential sensitivity to apoptosis among the cells that contribute to the atherosclerotic disease. Biochem Biophys Res Commun. 2007;363:444–450. doi: 10.1016/j.bbrc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]