

Abstract
Cardiac repolarization abnormalities can be caused by a wide range of cardiac and non-cardiac compounds and may lead to the development of life-threatening Torsades de Pointes (TdP) ventricular arrhythmias. Drug-induced Torsades de Pointes is associated with unexpected and unexplained sudden cardiac deaths resulting in the withdrawal of several compounds in the past. To better understand the mechanism of such unexpected sudden cardiac deaths, the concept of repolarization reserve has recently emerged. According to this concept, pharmacological, congenital or acquired impairment of one type of transmembrane ion channel does not necessarily result in excessive repolarization changes because other repolarizing currents can take over and compensate. In this review, the major factors contributing to repolarization reserve are discussed in the context of their clinical significance in physiological and pathophysiological conditions including drug administration, genetic defects, heart failure, diabetes mellitus, gender, renal failure, hypokalaemia, hypothyroidism and athletes' sudden deaths. In addition, pharmacological support of repolarization reserve as a possible therapeutic option is discussed. Some methods for the quantitative estimation of repolarization reserve are also recommended. It is concluded that repolarization reserve should be considered by safety pharmacologists to better understand, predict and prevent previously unexplained drug-induced sudden cardiac deaths.
Keywords: adverse drug reactions; cardiovascular pharmacology; K-channels; repolarization, repolarization reserve; safety pharmacology; sudden cardiac death; Torsades de Pointes
Introduction
It is well known that certain antiarrhythmic drugs induce life-threatening Torsades de Pointes (TdP) arrhythmia in 3–5% of patients. This was not considered particularly surprising because the drugs in question were expected to affect cardiac transmembrane ion channels and the electrophysiological properties of the heart as part of their therapeutic effect. Recently, however, there has been increasing concern about TdP arrhythmia and sudden death caused by non-cardiac drugs. The incidence of TdP induced by non-cardiac drugs is usually low (less than 1:10 000 or 1:100 000). However, several of them including astemizole, cisapride, grepafloxacin, terfenadine, terolidine have been withdrawn from the market due to their torsadogenic properties preventing their further therapeutic application in patients not susceptible to TdP and leading to the loss of several billion dollars in development costs and increasing uncertainty for future drug development programmes. Therefore, the need for minimizing the proarrhythmic risk of novel developmental drugs is urgent and represents a significant challenge (Pugsley et al., 2008; Farkas and Nattel, 2010).
In the background of these arrhythmias and sudden deaths, variable repolarization abnormalities are suspected due to unrecognized drug adverse effects, possibly in combination with other factors including genetic mutations and polymorphisms, electrophysiological remodelling, and changes in serum ion concentrations. Although unexpected and unexplained sudden deaths related to TdP have low incidence, they often occur at a young age or in individuals without previous medical history. Because our understanding of the underlying mechanisms is still poor, effective prevention remains elusive. In many cases, the degree of repolarization defect is so subtle that warning signs cannot be detected by common and widely used diagnostic tools such as ECG or routine preclinical drug safety methods. It is therefore almost impossible to identify the population at highest risk. During the last decade the concept of repolarization reserve has emerged (Roden, 1998; 2006; 2008;) and has been demonstrated experimentally (Iost et al., 1999; Varróet al., 2000; Lengyel et al., 2001; Jost et al., 2005). This concept seems to be a useful approach for safety pharmacologists to better understand, predict and possibly prevent unexpected drug related, and previously unexplained, sudden cardiac deaths.
Definitions and background
The term of repolarization reserve was first coined by Roden (Roden, 1998) in an editorial in 1998:
The greater problem is predicting the development of torsade de pointes in the absence of high doses or plasma concentrations. We have developed the concept of “repolarization reserve” to address this issue. We postulate that there exist in the normal ventricle and conducing system mechanisms to effect orderly and rapid repolarization that runs essentially no risk of setting up re-entrant circuits or of generating EADs. Indeed, the normal function of the delayed rectifier currents IKr and IKs is a major contributor to such stable repolarization, or a large repolarization reserve. Identified risk factors for torsades de pointes reduce this repolarization reserve, making it more likely that the further added stress of, for, e.g. an IKr blocking drug or a subtle genetic defect, is sufficient to precipitate torsades de pointes in individual patients.
At the time, experimental evidence was not available to support this concept; instead, it represented a way of thinking about cardiac repolarization based on general principles. Even so, the impact of this editorial was immediate and substantial. It is now an important principle in understanding unexpected proarrhythmic complications associated with genetic disorders of ion channels, certain diseases and drug effects. The concept has also served to provide a theoretical background for safety pharmacology related to drug-induced QT prolongation.
During the 10 years after the concept of repolarization reserve was first proposed, it has evolved yielding the following definition (Roden, 2008):
The concept of “repolarization reserve”, the idea is that the complexity of repolarization includes some redundancy. As a consequence, loss of 1 component (such as IKr) ordinarily will not lead to failure of repolarization (i.e. marked QT prolongation); as a corollary, individuals with subclinical lesions in other components of the system, say IKs or calcium current, may display no QT change until IKr block is superimposed.
At around the same time of the original proposal, specific inhibitors (chromanol 293B, L-735,821 and HMR-1556) of the slow delayed rectifier potassium current (IKs) became available (Busch et al., 1996; Salata et al., 1996; Gögelein et al., 2000), and both researchers and pharmaceutical industry considered IKs block to have beneficial class III antiarrhythmic effects causing less or negligible proarrhythmic complications compared with rapid delayed rectifier potassium current (IKr) inhibition (Hondeghem, 1992; Busch et al., 1994; Nair and Grant, 1997). Several drug companies therefore launched projects to develop novel and specific IKs inhibitors. Surprisingly, however, in our experiments as shown on Figure 1, IKs block did not elicit any measurable effects on repolarization in either canine papillary muscle or Purkinje fibre preparations (Iost et al., 1999; Varróet al., 2000). These observations contradicted some published studies (Bosch et al., 1998; Cordeiro et al., 1998), as well as the general belief that decrease in a major and important potassium current should substantially delay cardiac repolarization. However, when action potential duration (APD) was lengthened by pharmacological means, IKs inhibition further lengthened repolarization (Figure 1C). Based on these results, we hypothesized that the slowly activating and relatively small IKs represented only a negative feedback mechanism limiting excessive repolarization lengthening. In particular, we stated that:
Figure 1.
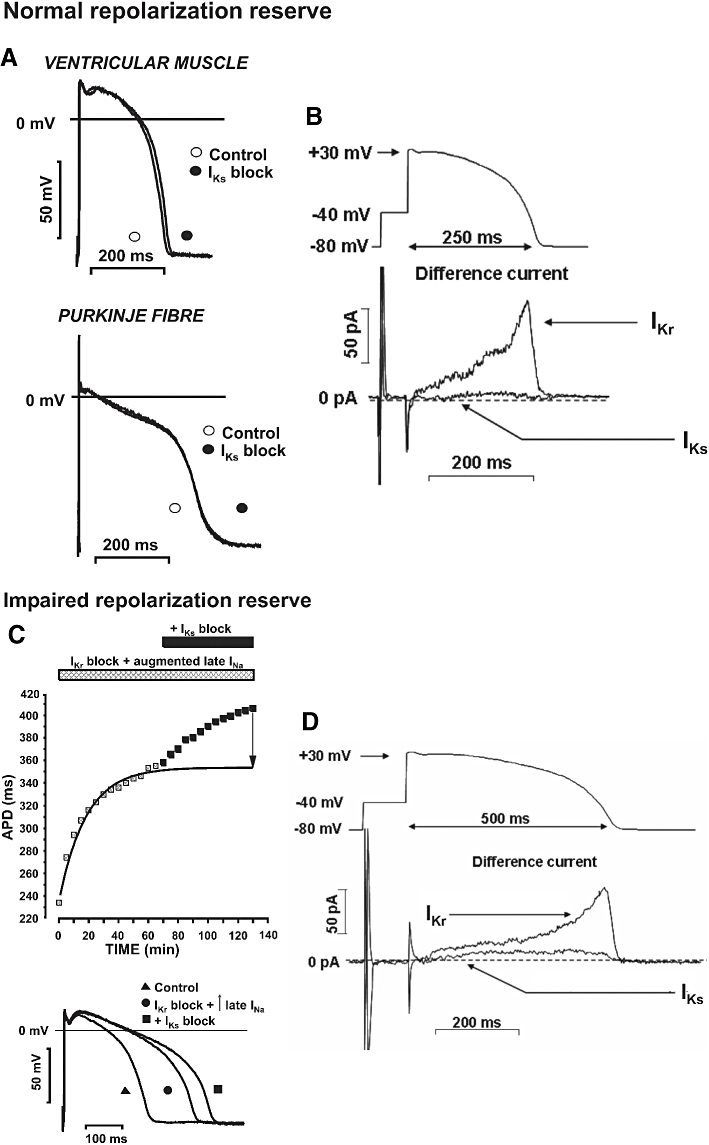
Experimental evidence of repolarization reserve. (A) The lack of marked prolongation of the action potential after IKs block (by L-735,821) on dog right ventricular papillary muscle and Purkinje fibres at 1 Hz stimulation frequency. (B) The magnitude of IKr and IKs after normal action potential-like voltage clamp pulses. Short voltage clamp pulses corresponding to ‘normal action potential’ elicit relatively large IKr (E-4031 sensitive current) but small IKs (L-735,821 sensitive current). (C) Effect of IKs block on dog ventricular action potential duration (APD) previously prolonged by application of E-4031 (IKr block) and veratrine (augmented late INa). Note that in preparations where repolarization was lengthened, inhibition of IKs resulted in significant action potential prolongation. (D) Increasing the duration of the voltage clamp pulses beyond ‘normal’ did not change the density of IKr but increased that of IKs. From Varróet al. (2000), with permission.
Although IKs may have little role in normal action potential repolarization, it probably plays a vital role when cardiac APD is abnormally lengthened by other means (e.g. by reductions in IKr or Ik1 or increases in INa or ICa). As such, pharmacological block of IKs might be expected to have severe detrimental consequences when this protective mechanism is eliminated. For example, if repolarization is excessively lengthened due to drug-induced IKr block, hypokalaemia, genetic abnormality, or bradycardia, the subsequent increase in APD would favour IKs activation and provide a negative feedback mechanism to limit further APD lengthening.
The principle of repolarization reserve was thereby directly and experimentally demonstrated (Iost et al., 1999; Varróet al., 2000), and in the conclusion of this paper the concept was described without the term ‘repolarization reserve’.
Major currents influencing repolarization reserve
Cardiac muscle repolarization is governed by the simultaneous and balanced activities (activation and inactivation) of several inward and outward currents flowing through different ion channels or electrogenic ion pumps (Figure 2). Although this review focuses on ventricular tissue and it is well known that there are significant differences in the density of these channels and exchangers between various regions of the heart (Nerbonne and Kass, 2005; Gaborit et al., 2007), the concept of repolarization reserve can also be applied to these areas.
Figure 2.
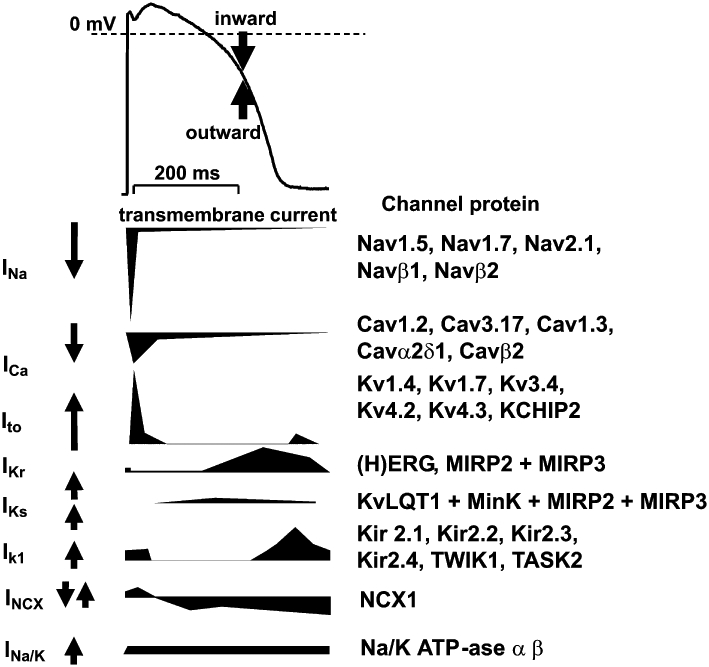
Ventricular action potential (top) and schematic, illustrative representation (not proportional to actual current densities) of the most important underlying transmembrane ionic currents and electrogenic pumps (bottom). Channel proteins, responsible for mediating the respective current, are indicated on the right. Upward and downward deflections and arrows denote outward and inward currents respectively.
Inward sodium current (INa)
The large fast sodium current is inactivated within a few milliseconds but the sodium current does not vanish completely during the plateau phase. Over the limited range of membrane potentials where activation and inactivation overlap, a relatively small steady-state sodium current can be recorded known as the ‘Na+– window current’ (Attwell et al., 1979). In addition, a small fraction of the sodium current slowly inactivates during the plateau phase (Carmeliet, 1987). This so-called persistent sodium current functionally opposes repolarizing potassium currents and delays repolarization. It is augmented in a number of situations, for example, by administration of veratrine and Anemonia sulcata toxin (ATX II), in long QT syndrome 3 (LQT3) syndrome and heart failure (Schwartz et al., 1995), further opposing the function of outward currents. In case there are compensatory ionic movements for this current, the net result is diminished repolarization reserve; however, when the increase of this inward current is substantial, prolongation of repolarization may occur.
L-type inward calcium current (ICa,L)
The influence of inward calcium currents on repolarization is more complex than that of INa and is still not completely understood. The calcium current, like INa, exhibits both slowly inactivating and window components but in addition, inactivation of the current is dependent on the free cytosolic calcium concentration ([Ca2+]i). Because [Ca2+]i changes dynamically during the action potential, and is regulated by many factors (Bers, 2001) including sarco/endoplasmic reticulum CA2+ ATPase, phospholamban, ryanodine receptor, calmodulin and protein kinase A (PKA) and C (PKC), the impact of inward calcium currents on the repolarization is difficult to predict accurately; and it represents one of the most problematic targets for mathematical modelling of cardiac action potentials. Moreover, if inward currents (both INa and ICa) are augmented, the plateau voltage is shifted towards more positive values that may result in enhanced activation of outward potassium currents that, depending on the actual balance of currents, may shorten repolarization.
Rapid delayed rectifier outward potassium current (IKr)
IKr activates rapidly on membrane depolarizations (Gintant, 1996) that are more positive than −30 mV (τ∼ 40 ms at +30 mV) but its inactivation precedes depolarization-dependent activation (Spector et al., 1996). As a consequence, IKr channels are largely closed during the plateau phase and only open when membrane potential returns to around 0 mV. Recovery from inactivation is very rapid, and as membrane potential slowly changes towards negative values these channels open again and then slowly deactivate (close), resulting in the transient nature of IKr. Contrary to what one might expect from the Nernst equation, increased extracellular K+ concentration is known to enhance, while decreased [K+]o acutely reduces IKr (Sanguinetti and Jurkiewicz, 1992; Yang et al., 1997). Interestingly, the surface density of IKr in rabbit hearts and human ether-a-go-go-related gene (HERG) channel in human cell lines have been recently found to be under direct control of extracellular K+ concentration, with [K+]o reduction resulting in accelerated internalization and degradation of the channel (Guo et al., 2009). These mechanisms may play important roles in changes in repolarization reserve following electrolyte disturbances. Due to its peculiar gating properties, that is, its very fast inactivation and recovery from inactivation, an apparent inward rectification (also see Ik1 later) occurs. IKr may play a positive feedback role contributing to repolarization lengthening when repolarization is already prolonged (Virág et al., 2009) due to other factors such as LQT3 or bradycardia. In this setting shown on Figure 3, with longer action potentials there is less IKr (and also less Ik1, see later) developing at isochronal points further weakening the force of repolarization in a complex voltage and time dependent process (Virág et al., 2009). Thus, repolarization becomes more vulnerable when depolarizing factors [INa, ICa,L, Na+-Ca2+ exchanger (NCX)] are augmented or repolarizing factors (Ik1, IKs, Na/K) are reduced. Even a small decrease in IKr may lead therefore to substantial prolongation of APD at slow rates or may have an additive effect with other interventions that lengthen repolarization. IKr is widely recognized as one of the most important outward currents controlling repolarization. A marked decrease or inhibition of IKr results in substantial repolarization lengthening in many species, including human (Varróet al., 2000; Lengyel et al., 2001; Jost et al., 2005). When the decrease in IKr is only fractional, the APD is not necessarily prolonged due to possible compensatory functions by other outward currents. However, further impairment of the current may have detrimental consequences such as excessive repolarization lengthening leading to TdP arrhythmia. This is the most likely reason why relatively weak IKr inhibition by certain non-cardiac drugs provokes arrhythmias related to repolarization abnormalities. Thus, IKr is not only a robust outward current determining the course of apparent repolarization but itself is clearly the most important contributor to repolarization reserve.
Figure 3.
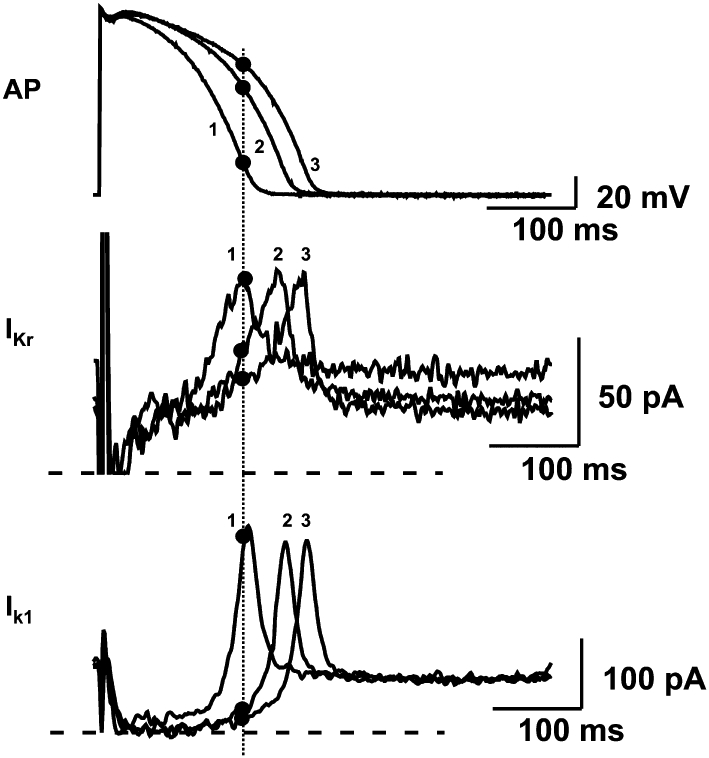
When action potential duration has been prolonged previously by other means in dog papillary muscle (top panel), the positive feedback mechanism by which reduction of IKr (middle panel) and Ik1 (lower panel) further augments repolarization delay. Increased duration and thereby decreased rate of repolarization decreases the contribution of IKr and Ik1 currents to the repolarization process during the time course of the action potential, in turn producing a further slowing of repolarization. Numbers denote three recordings with different action potential durations. Dotted line identifies isochronal points on these recordings. IKr and Ik1 were measured from isolated ventricular myocytes subjected to action potential waveforms used as command signals in voltage-clamp conditions. AP, action potentials. Modified from Virág et al. (2009), with permission.
Slow delayed rectifier outward potassium current (IKs)
IKs, which flows through KvLQT1 + MinK + MIRP protein constituted channels, activates slowly (500–1000 ms) during the plateau phase and deactivates rapidly (100–200 ms) at negative membrane potentials (for a recent review, see Jost et al., 2007). Its relatively small amplitude between 0 mV and +20 mV and slow activation kinetics, mean that there is relatively little current activating during the normal course of the action potential (Varróet al., 2000; Jost et al., 2005) and consequently little influence on repolarization (Varróet al., 2000; Jost et al., 2005). However, when action potential is lengthened and plateau voltage is shifted to more positive voltage, IKs (Han et al., 2001) ‘creates an available reserve of channels that are ready to open on demand’ (Carmeliet, 2006), thereby forming a transmembrane current that has the main function of participating in repolarization reserve (Figure 1D), and opposing excessive APD lengthening (Varróet al., 2000; Jost et al., 2005). IKs is activated by PKC and by sympathetic stimulation via PKA (Yazawa and Kameyama, 1990; Walsh and Kass, 1991). The latter has important consequences, because increases in the amplitude of IKs and shifts in its activation voltage towards negative potentials enhance IKs current density during elevated sympathetic tone. Moreover, sympathetic stimulation enhances ICa,L that in turn shifts the plateau towards more positive voltage and lengthening repolarization. These changes further activate IKs which, as a negative feedback limits the effect of the enhancement of ICa,L (Han et al., 2001; Volders et al., 2003). It becomes evident therefore that impairment of IKs due to loss of function mutation (LQT1) or IKs blocking drugs in the presence of elevated sympathetic tone, leaves the repolarization lengthening caused by augmented ICa,L unopposed and substantial prolongation of APD can develop. Thus, IKs does not necessarily contribute to repolarization in normal settings and at low sympathetic tone. However, under certain conditions (e.g. enhanced sympathetic activation), this current provides a protective mechanism that creates a repolarization reserve counterbalancing repolarization lengthening only when necessary (Johnson et al., 2010). It should be noted that IKs has exhibited transmural expression differences (Gintant, 1995) that, with the transmural and regional expression heterogeneities other currents may also contribute to enhanced heterogeneity of repolarization and, possibly, regional differences in repolarization reserve. In canine wedge preparations, Akar et al. (2002) have clearly demonstrated a direct link between such transmural repolarization heterogeneity and the development of TdP.
Inward rectifier potassium current (Ik1)
Ik1 flows through a mixture of Kir 2.1, 2.2, 2.3 showing strong inward rectification (for a recent review, see Anumonwo and Lopatin, 2010). Consequently, Ik1 current is decreasing close to zero at membrane potentials more positive than −30 mV and reopening very rapidly when membrane potentials reach more negative values. The inward rectification is due to channel block by Mg2+ and polyamines from intracellular sites on depolarization (Matsuda et al., 1987). When the positive membrane potential is changing to the negative direction, the Mg2+ and polyamine-induced channel block is relieved and Ik1 increases. This process forms a negative feedback during repolarization making it a regenerative process. This phenomenon resembles that mentioned in connection with IKr, but the potential range of Ik1 is more negative than that of IKr. Ik1 plays a similar role to IKr in the self-limiting repolarization process after APD lengthening (Figure 3) following bradycardia (Virág et al., 2009). These channels are fully open at rest (i.e. during diastole) so that Ik1 opposes any kind of depolarization due to enhanced pacemaker activity and Ca2+ overload-related delayed afterdepolarizations (DAD). This feature represents a special form of repolarization reserve. Consequently, the impairment of Ik1 may have a proarrhythmic potential by not opposing depolarization that can thereby reach the threshold for propagation of extrasystoles. In addition, smaller Ik1 also increases duration of repolarization and its heterogeneity. The density of Ik1 shows considerable variations among species being relatively weak in human ventricle (Jost et al., 2008) but strong in dog, rabbit and guinea pig (Varróet al., 1993). A possible result in humans is that a moderate Ik1 defect or block is not reflected in significant repolarization lengthening. However, the inhibition of Ik1, the current being a part of the repolarization reserve, together with the malfunction of other ion channels, may lead to excessive lengthening of the APD. Indeed, loss of function mutations of Kir 2.1 channels in LQT7 (Andersen–Tawil syndrome), although substantially decreasing Ik1, do not result in marked QTc lengthening yet greatly enhance proarrhythmic risks for these patients (Zhang et al., 2005). An important role for Ik1 in repolarization reserve has also been suggested using mathematical modelling where reduction of Ik1 in addition to IKr inhibition resulted in early afterdepolarizations (EAD) (Ishihara et al., 2009). Based on this analysis, it is likely that Ik1 in an important contributor to repolarization reserve both in diastole and during the final repolarization phase.
Transient outward potassium current (Ito)
Ito consists of the rapidly (Ito,f) and slowly recovering (Ito,s) components that are carried by channels with distinct pore-forming subunits, Kv4.3/Kv4.2 for Ito,f, and Kv1.4 for Ito,s, respectively, and show considerable species variations (recently reviewed by Niwa and Nerbonne, 2010). Ito has not been recorded in guinea pig. In human (Gaborit et al., 2007) and dog (Fülöp et al., 2006), Kv4.3 is considered to provide the α subunit, while in rabbit Kv1.4 seems to be the dominant protein (Wang et al., 1999). In general, Ito is a large and rapidly activating (τ∼ 1–2 ms) and inactivating (5–10 ms) outward current at membrane potentials more positive than −20 mV (Patel and Cambell, 2005). Therefore, Ito contributes to the initial phase 1 repolarization and its influence on the APD is believed to be limited. It must be emphasized that Ito can alter the amplitude of the plateau and thereby indirectly change the activation, inactivation and deactivation of other transmembrane currents. In addition, Ito (Kv4.3 current) has a second, slower phase of inactivation (τ∼ 15–30 ms) which may be responsible for a reasonable amplitude during the plateau, and its activation and inactivation may overlap producing a similar type of window current during the final repolarization as described for ICa or INa. Thus, one can suspect that although changes in Ito current may not lead to marked changes in repolarization; however, as part of the repolarization reserve together with other currents, Ito may also have an impact on repolarization. Because Ito has been shown to vary between subendocardial, subepicardial and midmyocardial layers (Litovsky and Antzelevitch, 1988; Antzelevitch and Fish, 2001) it may contribute to spatial repolarization heterogeneity as well.
Sodium – potassium pump current (INa/K)
The INa/K is due to the activity of the cardiac Na/K pump that hydrolyses adenosine triphosphate (ATP) and is stimulated by increased intracellular cAMP concentrations. This electrogenic ion transport mechanism is mainly responsible for maintaining resting membrane potential and pumps out three Na+ from the cell in exchange for the inward movement of two K+. In the range of membrane potentials corresponding to the action potential and diastole, the pump current is outward (De Weer et al., 1988) and therefore contributes to the repolarization reserve during both systole and diastole. The magnitude of this current depends primarily on intracellular [Na+] which explains why INa/K is so sensitive to frequency of stimulation. In certain pathological settings, including heart failure, Na/K pump expression is altered and INa/K also shows a characteristic transmural distribution (Gao et al., 2005).
Na+/Ca2+ exchange current (INCX)
In physiological conditions, the sarcolemmal NCX is one of the main mechanisms that are responsible for restoring physiological low concentrations of intracellular Ca2+ in diastole (Blaustein and Lederer, 1999). Because three Na+ are transported in exchange for one Ca2+ per cycle, the transport is electrogenic and critically depends on membrane potential and intracellular [Ca2+], both changing dynamically during the action potential. It is difficult therefore to estimate accurately the current magnitude produced by NCX, and efforts to this end are also hampered by the lack of sufficiently specific inhibitors. At the beginning of the action potential when intracellular [Ca2+] is low and membrane potential is positive, the NCX current is outward; on the other hand, during the later plateau phase, in early and late repolarization and also during diastole, it is inward. When calcium overload is present, NCX can carry a depolarizing current that may provoke both DAD and EAD contributing to arrhythmogenesis (Bers et al., 2002). NCX also exhibits differences in transmural expression (Xiong et al., 2005) and it is known that NCX is up-regulated in heart failure (Schillinger et al., 2000). Therefore, this trigger mechanism is more likely to induce arrhythmias in case repolarization reserve is decreased, as it is the case in heart failure.
Alterations in calcium cycling
The function and/or density of several ion channels have been shown to be modulated by [Ca2+]i. Calmodulin, a ubiquitous Ca2+-sensing protein regulates human cardiac sodium channels (Tan et al., 2002), and INa density was found to be acutely modulated by [Ca2+]i (Casini et al., 2009). Ito is regulated by Ca2+ and calmodulin-dependent protein kinase II (Tessier et al., 1999; Sergeant et al., 2005) and Ito is down-regulated following rapid ventricular pacing in a dog model of tachycardiomyopathy via Ca2+/calmodulin-dependent protein kinase II and calcineurin/nuclear factor of activated T-cells systems (Xiao et al., 2008a). Importantly, calmodulin has been shown to be necessary for both correct channel assembly and gating of IKs (Ghosh et al., 2006; Shamgar et al., 2006) and Ca2+ was found to modulate the delayed rectifier potassium current in guinea pig (Tohse et al., 1987) and to inhibit HERG (Schönherr et al., 2000). As these currents play important roles in repolarization, it is reasonable to assume that disease (e.g. heart failure) or drug-induced impairment of calcium cycling will have significant effects on repolarization which might be important for a better understanding of the molecular mechanisms responsible for repolarization reserve. In this regard, the activation of calmodulin-dependent protein kinase II in heart failure and cardiac hypertrophy has been shown to exert direct or indirect effects on the expression and functional properties of Ito and Ik1 (recently reviewed by Nerbonne, 2011). Also, preclinical measures of repolarization reserve, particularly mechanisms underlying beat-to-beat variability of repolarization are probably at least in part related to alterations of Ca2+ cycling in the chronic atrioventricular block dog model (Oros et al., 2008). Furthermore, cardiac repolarization alternans is mechanistically related to changes in Ca2+ cycling (Pruvot et al., 2004).
Mechanisms of arrhythmias in the setting of impaired repolarization reserve
Transmembrane potassium channel expression patterns show transmural and regional differences, probably creating only relatively small repolarization heterogeneity; however, these differences can be greatly enhanced by impaired repolarization reserve.
The hypothetical mechanism of arrhythmia development due to increased repolarization heterogeneity is shown on Figure 4. In normal circumstances, conduction in the heart is fast (1–2 m·s−1) and the duration of myocardial cell action potential is long (200–300 ms). Thus, these cells are resistant to early stimulation because they are in a refractory state. The length of refractoriness is characterized by the effective refractory period. Importantly, in the normal setting the difference between the APD and consequently the effective refractory periods of adjacent cells is very small, creating only small repolarization heterogeneity. Fast conduction and relatively homogenous repolarization prevents the circular re-entry of excitation and arrhythmia will not develop. However, when the duration of repolarization and therefore the refractory period are prolonged in a heterogenous fashion, that is, the intrinsic transmural/regional repolarization heterogeneities are further augmented by repolarization reserve impairment in cells/regions in a different degree, an arrhythmia substrate is created. As a consequence, an extrasystole generated after a normal sinus stimulus can propagate in the direction of cells with shorter APD but its propagation is blocked in the direction of cells with longer APD (Figure 4). Thus, the extra stimulus can travel, usually with reduced conduction velocity, back in a complicated path towards its site of origin and in other directions where excitability is regained, leading to the generation of TdP or even ventricular fibrillation. Ventricular fibrillation does not revert to sinus rhythm spontaneously in humans and leads to sudden cardiac death without intervention in a few minutes. It is important to note that two independent factors are needed for the arrhythmia described above, and illustrated in Figure 4, to develop. Heterogeneity of repolarization following prolongation of repolarization is itself not sufficient for arrhythmia development, establishing only the possibility of an arrhythmia (‘substrate’). In order to induce arrhythmia, an extrasystole in the vulnerable period is needed (‘trigger’) that can travel the re-entry paths created by the heterogeneous repolarization. The timing of this trigger extrasystole is critical, because before the vulnerable period its conduction is blocked and after the vulnerable period it does not lead to tachycardia or fibrillation, only to a harmless single extrasystole. Increased repolarization heterogeneity leads to longer vulnerability periods and with more frequent extrasystoles, the chances become greater for the generation of serious arrhythmias. In an elegant study, Akar et al. (2002) have experimentally demonstrated the development of TdP arrhythmia in dogs based on mechanisms described above (Figure 5). In these experiments, increased transmural repolarization heterogeneity within relatively short transmural distances was observed and the development of TdP was dependent on both bradycardia and administration of the IKr blocker d-sotalol in canine wedge preparations (Figure 5). The steep gradients in the duration of repolarization appear to be mechanistically related to the block and re-entry as shown on Figure 6. These studies also illustrate that additional hits on repolarization during bradycardia, particularly in midmyocardial cells, where decreased IKs expression has been shown (Liu and Antzelevitch, 1995) can precipitate TdP. In this model, TdP arrhythmia was induced by applying an extra stimulus that travelled a re-entry path around a region with delayed repolarization (Figure 6), thus, decreased repolarization reserve facilitates arrhythmias at both the substrate and trigger level by increasing repolarization heterogeneity and by decreasing repolarization force during diastole which consequently helps subthreshold depolarizations to reach threshold and to produce propagating extrasystoles.
Figure 4.
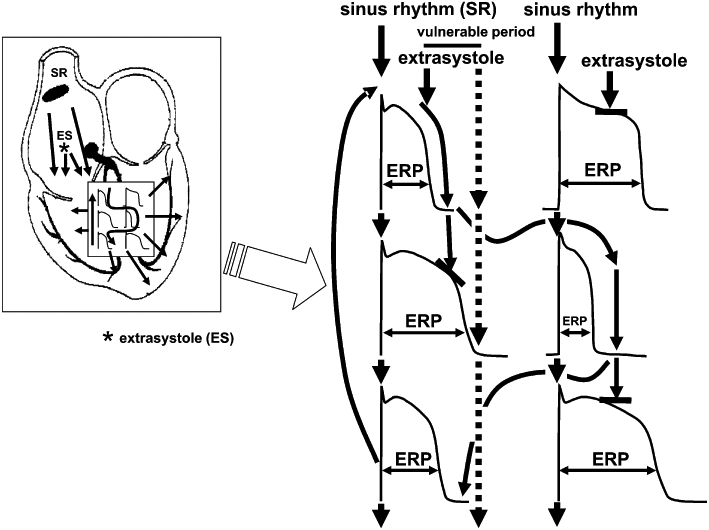
The simplified illustration of the mechanism of arrhythmia development due to lengthening of repolarization and decreased repolarization reserve leading to enhanced spatial repolarization heterogeneity. When the duration of repolarization and therefore the refractory period are prolonged in a heterogenous fashion, that is, the intrinsic transmural/regional repolarization heterogeneities are further augmented by repolarization reserve impairment in cells/regions in a different degree, an arrhythmia substrate is created. An extra stimulus in the vulnerable period then can travel back in a complicated path to its site of origin due to enhanced differences in refractoriness of myocardial cells leading to serious ventricular arrhythmia. Dotted arrows show an extra stimulus occurring after the vulnerable period propagating on the physiological path not leading to serious arrhythmia. ERP, effective refractory period.
Figure 5.
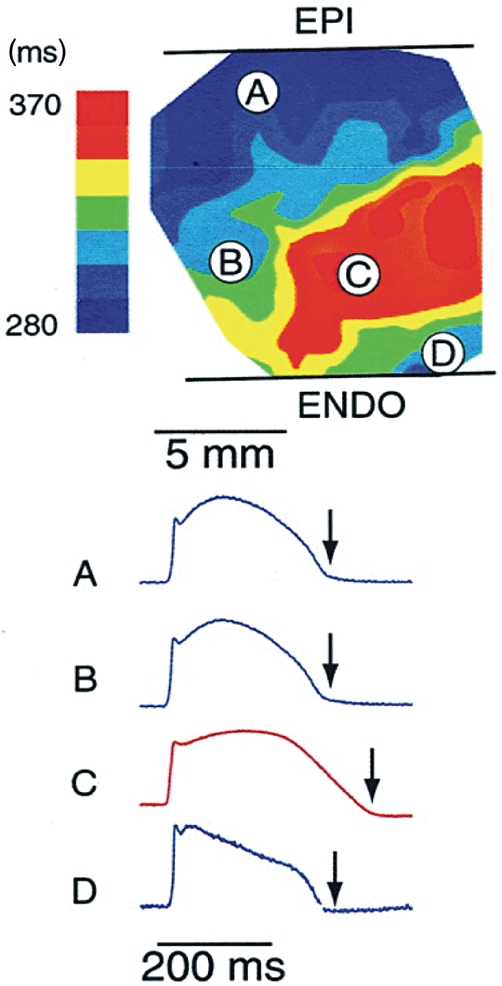
Experimental demonstration of spatial differences in duration of repolarization. Upper panel shows transmural heterogeneity of action potential durations using color codes in bradycardia and d-sotalol administration mimicking decreased repolarization reserve in the canine wedge long QT syndrome 2 model. Optical action potentials are shown from selected transmural sites (A–D) on lower panel. Arrows denote the significant differences in the action potential duration within relatively short transmural distances. EPI, epicardium; ENDO, endocardium. Adapted from Akar et al. (2002), with permission.
Figure 6.
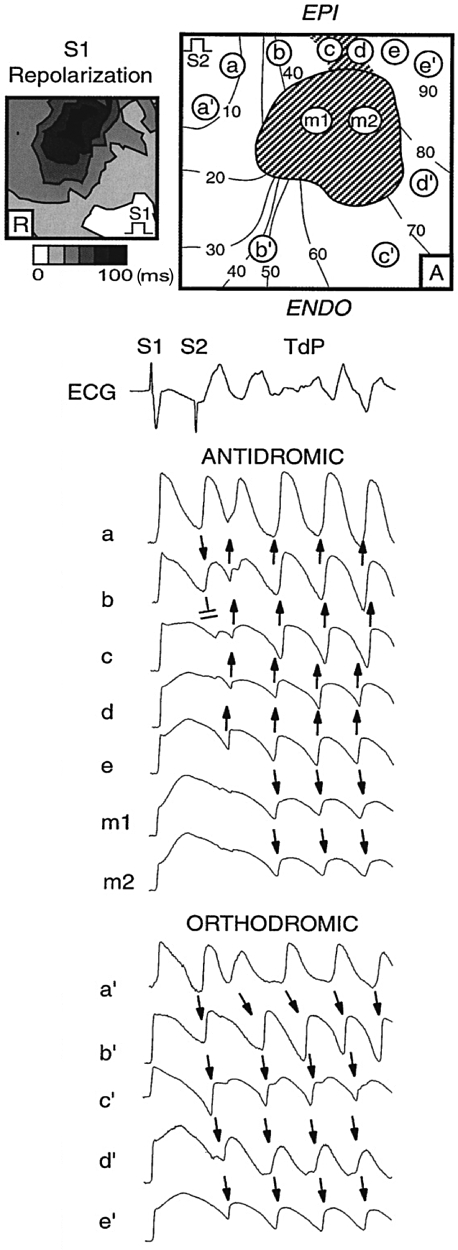
Experimental demonstration of the development of Torsades de Pointes provoked by a premature stimulus (S2) in the canine wedge model during bradycardia and selective IKr block by d-sotalol. Spatial action potential duration heterogeneity created a region with delayed repolarization (c to d and m1, m2) around which the premature stimulus could propagate only in the orthodromic direction (a' to e') but not in the antidromic direction (a to e and m1, m2). EPI, epicardium; ENDO, endocardium; TdP, Torsades de Pointes. Adapted from Akar et al. (2002), with permission.
In addition, when repolarizing force is weakened (Ik1, IKr, IKs), it would further enhance both normal and abnormal triggered activity by counterbalancing the depolarizing influence of EAD or DAD or If to a lesser degree.
Clinical significance of reduced repolarization reserve
It is known that decreased repolarization reserve occurs in pathological settings and in certain special circumstances. Therefore, it should be possible to recognize diminished repolarization reserve. However, such situations are usually complex, and attenuated repolarization reserve can be recognized only retrospectively, sometimes only after a fatal outcome. Some patients with a QTc interval within the normal range may respond to drug treatment with excessive lengthening of repolarization and arrhythmias, occasionally leading to sudden cardiac death. In other patients these drugs do not even prolong cardiac APD or only lengthen it moderately. In accordance with these observations, Kääb et al. (2003) reported that patients who experienced TdP arrhythmia with QT prolonging drugs developed more QTc lengthening after i.v. sotalol, an IKr blocking drug, than those of the control group consisting of patients without history of TdP. An interesting observation of this study showed that in both groups the baseline QTc was normal and did not differ between the two groups (Figure 7). It can be assumed that the striking differences in response to the administered drug were due to differences between the two groups in the repolarization reserve.
Figure 7.
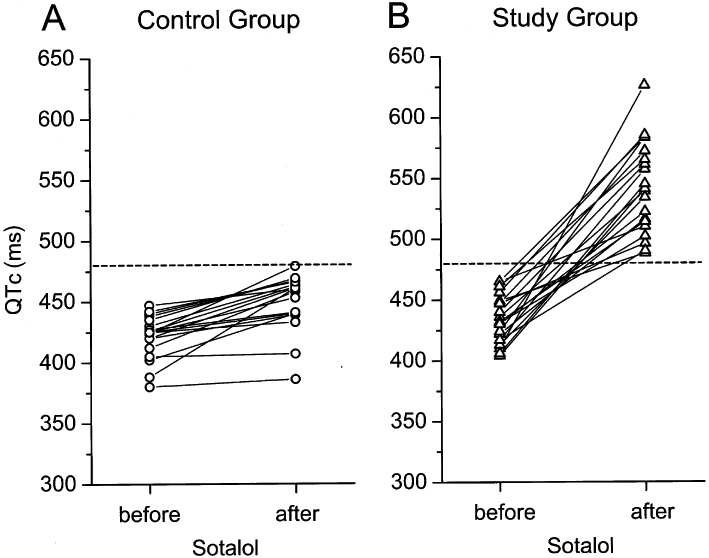
Individual QTc intervals in control individuals and in patients with suspected acquired long QT syndrome (study group) before and after intravenous administration of sotalol. The dotted line indicates a cut-off value of 480 ms that distinguished best between the study population and the control group. From Kääb et al. (2003), with permission.
Drugs and dietary constituents
Drugs that lengthen ventricular repolarization, measured on the ECG as QTc prolongation, are well known to be susceptible to potentially fatal arrhythmia, such as TdP and ventricular fibrillation (Kannankeril and Roden, 2007). Although drug-induced lethal arrhythmia related to QT prolongation is rare, its existence is well established. In the case of class III antiarrhythmics, the therapeutic mechanism includes manifest prolongation of repolarization by inhibition of one or more types of cardiac potassium channels. The proarrhythmic risk with these drugs is therefore obvious and as such it can be expected and monitored. To deal with the problem posed by non-cardiac drugs that are usually prescribed by non-cardiologists represents a significantly more difficult task. These drugs often do not cause manifest, or at least easily recognizable, cardiac repolarization lengthening because repolarization reserve compensates for relatively weak potassium channel inhibition. Alternatively, relatively weak potassium channel inhibition by drugs can be a source of decreased repolarization reserve which manifests as repolarization prolongation and arrhythmia in cases where additional factors such as genetic mutation or disease-induced electrophysiological remodelling alone, do not present as clinical symptoms. Moreover, dietary ingredients and supplements may have similar effects. For example, grapefruit juice has been reported to elicit moderate and marginally noticeable QT prolongation (Figure 8) in normal subjects (Zitron et al., 2005) which was attributed to the HERG channel inhibitory property of some of its flavonoid ingredients (Scholz et al., 2005; Zitron et al., 2005). Pink grapefruit juice ingestion has also been found to increase QT variability index in a small set of patients with dilated and hypertensive cardiomyopathies (Piccirillo et al., 2008). In addition, it is well known that grapefruit juice strongly inhibits P-450 which plays an important role of the metabolism of several drugs including those that block cardiac potassium channels and impair repolarization, a mechanism that further contributes to decreased repolarization reserve and enhanced proarrhythmic risk. Importantly, many old drugs, food ingredients, additives and preservatives have never been properly studied in terms of potassium channel pharmacology. Therefore, our current knowledge in this respect may represent only the tip of the iceberg. One must also take into consideration the possible individual variation in the strength of repolarization reserve; thus, evaluation of the effect of drugs on cardiac repolarization by conventional statistical approaches for entire populations can be misleading. One or two consistent positive cases in the study population deserve particular attention even without statistical significance in the incidence of drug-related events between study groups.
Figure 8.
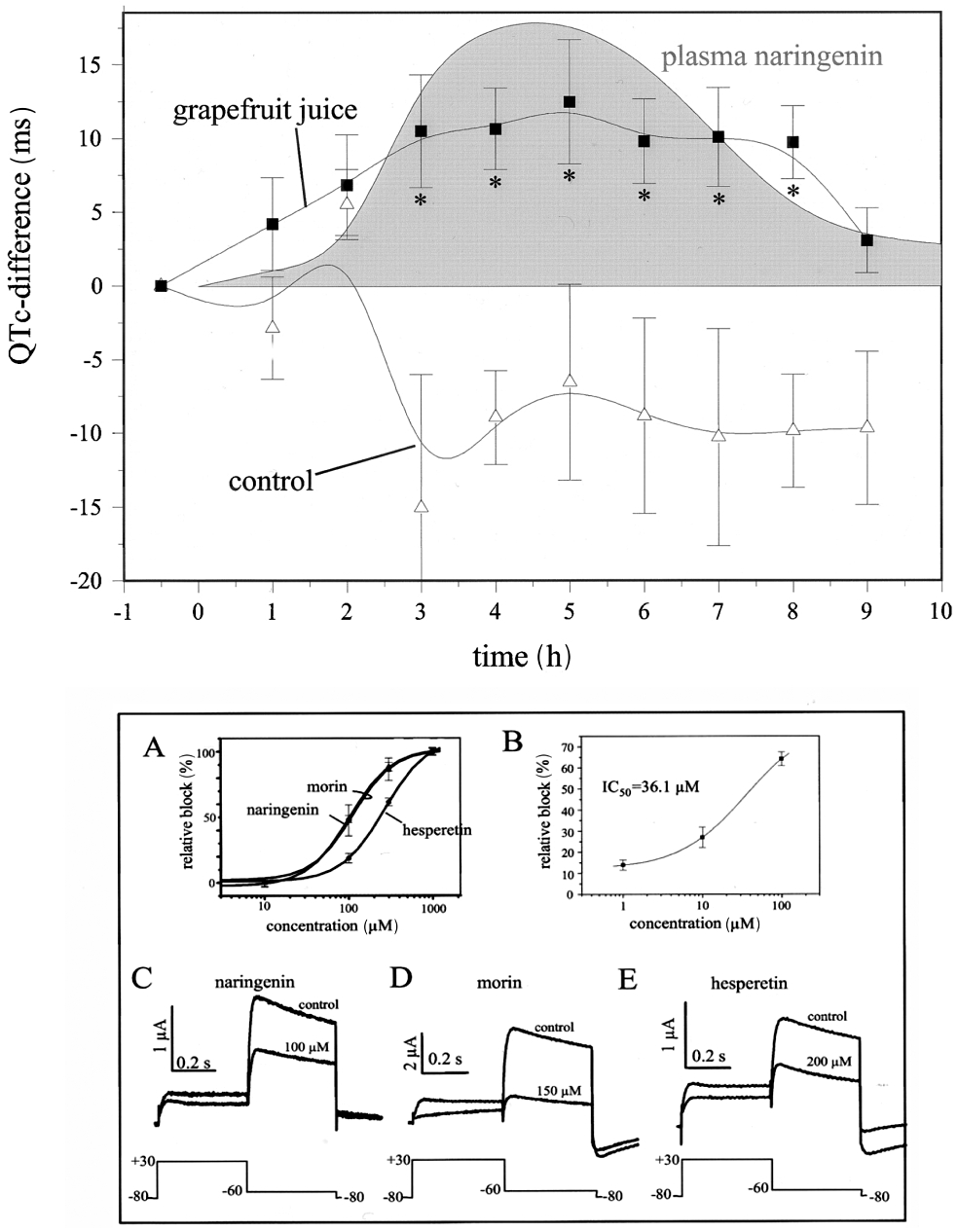
Prolongation of repolarization following grapefruit juice ingestion in healthy volunteers (upper panel) and inhibition of the human ether-a-go-go-related gene current by grapefruit juice flavonoids naringenin, morin and hesperetin in Xenopus oocytes (lower panel). From Zitron et al. (2005), with permission.
Genetic components
A recent study from Boulet et al. (2006) described a loss of function mutation in the KCNQ1 gene encoding the pore-forming subunit of IKs channel in a 40-year-old woman. This subject had experienced TdP arrhythmia and syncope with a QTc interval of 430 ms (well within the normal range), indicating that a genetic loss of IKs function alone does not necessarily prolong repolarization. However, in case other unfavourable conditions set in (e.g. hypokalaemia, drug effects, possible down-regulation of potassium channels), the impairment of repolarization reserve by this mutation can diminish the necessary protection, making this patient more susceptible to arrhythmia than those with normal IKs channels.
This case highlights the importance of ‘silent’, ‘subclinical’ or ‘concealed’ long QT syndrome (Varró and Papp, 2006) where repolarization processes are not apparently affected as measured by conventional diagnostic tools, but where the repolarization reserve is impaired. These individuals may be more susceptible to drug-induced arrhythmias than those with normal genetic backgrounds.
Another very interesting case illustrating decreased repolarization reserve due to congenital factors involved a 31-year-old female patient with known asymptomatic LQT syndrome (Hermans et al., 2003). The patient was also diabetic and was admitted to hospital with polydipsia where she developed convulsive syncopes due to TdP arrhythmic epidoses and exhibited an excessively long QTc interval of 580 ms on the ECG. She drank large amounts of grapefruit juice and tonic water because of her polydipsia before admission. Following discontinuation of these drinks, the patient's QTc interval decreased to 450 ms and the TdP episodes disappeared. In this patient, ventricular repolarization reserve was markedly impaired by congenital LQT syndrome and autonomic neuropathy due to diabetes mellitus; however, she had asymptomatic LQT. Significant QTc prolongation with concomitant TdP development most likely occurred as a result of multiple additional hits on ventricular repolarization: ingestion of HERG blocking flavonoids in grapefruit juice (naringin, naringenin, hesperetin, morin, etc.) and quinine in tonic water.
Ion channel polymorphisms represent a particularly interesting chapter in investigations on repolarization reserve, because they can cause mild ion channel dysfunction, however, might have relatively high incidence in the general population and were also reported to be associated with drug-induced arrhythmias (Sesti et al., 2000; Roden, 2004; Darbar and Roden, 2006; McBride et al., 2009). These polymorphisms highlight individual differences in repolarization reserve and therefore in drug-induced proarrhythmia susceptibility in the population.
Chronic heart failure
Heart failure is still a devastating disease with a 5 year life expectancy worse than cancer, with approximately half of heart failure patients dying due to sudden cardiac death caused by lethal arrhythmias and not due to end-stage pump failure (Packer, 1985; Kjekshus, 1990). It has been shown that both electrophysiological and structural remodelling occurs during the progression of heart failure. This includes the substantial degree of reduction of the repolarization reserve, due to down-regulation of different types of potassium channels. The results obtained so far regarding the nature of potassium channel down-regulation are conflicting, depending on the form of the disease (hypertrophic vs. dilated), experimental model (pacing vs. valve destruction) and the species studied (rat, rabbit, dog). In general, it has been reported that Ik1, IKs, Ito and IKr channels were down-regulated (Kääb et al., 1998; Näbauer and Kääb, 1998; Li et al., 2002). The persistent or slowly inactivating sodium current is also increased in heart failure (Valdivia et al., 2005). This latter change in the presence of reduced repolarization reserve results in APD lengthening and increased repolarization heterogeneity, a substrate for arrhythmias. The up-regulation of NCX, pacemaker current (If) and malfunction of the ryanodine receptor proteins (Stillitano et al., 2008; Tateishi et al., 2009) enhance ectopic activity because transient depolarizations are not opposed properly during diastole due to impaired repolarization reserve which later also contributes to the resting potential. These also increase arrhythmia risks by trigger mechanisms.
Diabetes mellitus
Repolarization abnormalities, including mild prolongation of the QT interval have been observed in patients with diabetes mellitus (Giunti et al., 2007); thus, both type 1 and type 2 diabetes mellitus are associated with increased risk for sudden cardiac death (Gill et al., 2008), which is not attributable to general pathophysiological changes, such as atherosclerosis, hyperlipidaemia or hypertension, commonly observed in diabetes. The mechanisms responsible for repolarization abnormalities in long term chronic diabetes are not well explored. The reduction of Ito, IKs and IKr in different diabetic animal models (Shimoni et al., 1998; Lengyel et al., 2007b) of short-term type 1 diabetes suggests that decreased density of these currents may contribute to reduced repolarization reserve in diabetes mellitus. In accordance with these experimental data, increased beat-to-beat QT variability was also recently reported (Lengyel et al., 2008) in patients with type 1 diabetes mellitus.
In addition to the repolarization abnormalities caused by endocrine diseases, compounds used for the treatment of these pathologies can also adversely influence cardiac ventricular repolarization resulting in QTc prolongation and impairment of repolarization reserve (Gonzalez et al., 2010).
Gender
It is well known that women are at a significantly greater risk than men for developing TdP arrhythmia in response to drugs that prolong cardiac repolarization. This is likely to be related to the fact that women have longer QTc and reduced repolarization reserve compared with men (James et al., 2007).
The basis for the gender differences in risk of TdP and QTc is not clear. This topic has been under intensive investigation currently (Rodriguez et al., 2001; James and Hancox, 2003; Kurokawa et al., 2008; Ridley et al., 2008) and gender-related differences in cardiac electrophysiology were recently reviewed in detail by Jonsson et al. (2010). It can be assumed that gender differences are related to the influence of sex hormones on cardiac repolarization and the underlying transmembrane currents. This notion is supported by findings showing at birth QTc intervals are very similar in males and females (Stramba-Badiale et al., 1995) and during puberty, the QTc intervals are shorten in men (Rautaharju et al., 1992).
Evidence suggests that oestrogen reduces IKr and prolongs repolarization (Kurokawa et al., 2008) while testosterone increases IKr (Ridley et al., 2008). Sex hormones can also alter the interactions of drugs with these potassium channels (James and Hancox, 2003; Kurokawa et al., 2008). A smaller Ito and a larger ICa,L were measured in female cardiomyocytes (Verkerk et al., 2005); however, these results were obtained using cells isolated from failing hearts.
Interestingly, when studying IKr block-induced QTc prolongation in women at different stages of the menstrual cycle, drug-induced QTc prolongation was greatest during menses and at the ovulatory stage of the cycle, compared with the luteal phase (Figure 10), despite the fact that in pre-drug conditions the QTc did not differ in the cycle (Rodriguez et al., 2001). This observation suggests that progesterone may be protective by opposing repolarization lengthening caused by IKr inhibition. In a recent study, it was shown that testosterone activated HERG channels via androgen receptors (Ridley et al., 2008). This finding further emphasizes the possibility of gender-based adverse consequences of drug-receptor interactions.
Figure 10.
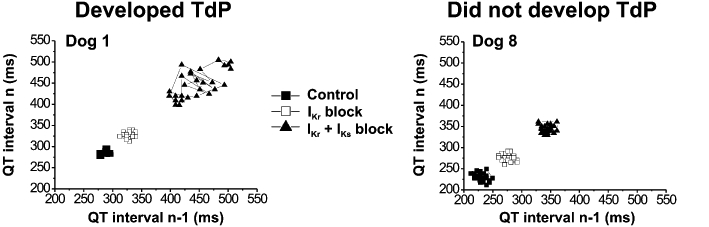
The development of Torsades de Pointes (TdP) arrhythmia correlated better with the increase in short-term QT variability than with QTc changes in dogs. Two representative individual Poincaré plots show increased short-term variability of QT following combined IKr (by dofetilide) and IKs (by HMR-1556) block in a dog that later developed TdP (left panel) compared with a dog where TdP did not occur (right panel). From Lengyel et al. (2007a), with permission.
A very recent review has concluded that testosterone mediates the increased repolarization reserve in men by decreasing ICa,L and enhancing IK and therefore testosterone plays a significant role in the reduced susceptibility to drug-induced ventricular arrhythmias in males (Jonsson et al., 2010).
Renal failure
It is known that patients with chronic renal failure are at increased risk for ventricular arrhythmia and sudden death (Johansson et al., 2004). In chronic renal failure, QT dispersion (Lőrincz et al., 1999) and the QT variability index are increased compared with normal subjects. These results suggest the presence of impairment of repolarization reserve. Chronic renal failure patients are prone to electrolyte derangements both due to the disease and medications used for its treatment. Importantly, rapid electrolyte shifts may also alter repolarization (Severi et al., 2008) and significantly contribute to the high, arrhythmia-related cardiac mortality rates seen in dialysis patients (Herzog et al., 2008). However, it must be also noted that chronic renal failure patients are usually co-morbid patients, that is, they have additional diseases such as diabetes, coronary artery sclerosis, hypertension, cardiac hypertrophy and are on multiple medications (for a recent review, see Schmid et al., 2010). All these factors may affect repolarization, making it difficult to determine the extent renal dysfunction contributes to alterations in repolarization reserve.
Body temperature abnormalities
Fever can indirectly interfere with repolarization reserve which may have important clinical implications (Burashnikov et al., 2008). Elevation of body temperature by 1–3°C enhances both inward and outward currents. Therefore, pharmacological inhibition, loss of function mutations or down-regulation of potassium currents may shift the balance of currents towards depolarization and thereby unexpected repolarization lengthening may occur when body temperature increases. On the other hand, hypothermia has also been shown to profoundly affect cardiac repolarization by prolonging APD in guinea pig hearts (Duker et al., 1987) and papillary muscle (Bjørnstad et al., 1993) and by promoting monophasic APD heterogeneities in open chest dogs (Kuo et al., 1983). In a recent study using canine left ventricular wedge preparations Piktel et al. (2011) found that therapeutic hypothermia (cooling to 32°C) increased dispersion of repolarization to a lesser degree and was associated with decreased arrhythmia susceptibility compared with severe hypothermia (cooling to 26°C) and subsequent rewarming.
Hypokalaemia
Hypokalaemia is a common factor that can decrease repolarization reserve. Low extracellular potassium increases IKs and Ik1 according to the Nernst equation but paradoxically reduces IKr and prolongs QT interval (Sanguinetti and Jurkiewicz, 1992; Yang et al., 1997). Furthermore, lowering of extracellular potassium was reported to increase drug-induced IKr block (Yang and Roden, 1996), although this latter observation has recently been questioned (Limberis et al., 2006).
Hypothyroidism
Hypothyroidism decreases (i.e. down-regulates) a number of different potassium currents (Bosch et al., 1999; Le Bouter et al., 2003) and pacemaker channels (Le Bouter et al., 2003), resulting in cardiac repolarization lengthening (Galetta et al., 2008) and bradycardia (Galetta et al., 2008). Even in its subclinical form, it decreases repolarization reserve as reflected by an increased QTc dispersion and prolonged QTc (Galetta et al., 2008). The reduction of repolarization reserve induced by hypothyroidism is also associated with an increased incidence of TdP arrhythmias (Fredlund and Olsson, 1983).
Increased sympathetic tone
As mentioned earlier, increased sympathetic tone enhances IKs, ICa and possibly IKr (Volders et al., 2003; Lengyel et al., 2004). In this situation, pharmacological inhibition, loss of function mutation and down-regulation of IKs and possibly IKr all tend to leave the enhanced depolarizing effect of augmented ICa unopposed, producing repolarization lengthening. In this regard, increased instability of repolarization was demonstrated in vivo in dogs following intense β-adrenergic receptor stimulation followed by ventricular extrasystoles and TdP (Gallacher et al., 2007). A very recent in vitro study by Johnson et al. (2010) has shown that β-adrenergic stimulation of IKs rescued dog left ventricular myocytes from excessive increase in beat-to-beat variability of their APD following IKr blockade and augmented late INa (impaired repolarization reserve). Also, β-adrenergic stimulation elicited largely increased repolarization variability in these cells when IKs was pharmacologically inhibited. As both IKs and IKr exhibit transmural and apico-basal heterogeneity (Antzelevitch, 2007), dispersion of repolarization can also be substantially increased in these circumstances providing further substrate for arrhythmia.
In addition, elevated sympathetic tone enhances If and spontaneous Ca2+ release from sarcoplasmic reticulum favouring the triggering of arrhythmias. In accordance with this, in LQT1 (IKs loss of function mutation) and LQT2 (IKr loss of function mutation), but not in LQT3 (INa gain function mutation), sympathetic tone is well recognized as a precipitating factor for arrhythmias.
Competitive athletes
Top athletes represent a special population where repolarization reserve may play an important role (Varró and Baczkó, 2010). Competitive sports activities and training induces physiological adaptation in the cardiovascular system leading to the development of athlete's heart that is characterized by reversible cardiac hyperthrophy, where down-regulation of certain potassium channels can be expected (Hart, 2003). In top competitive athletes, significant bradycardia develops that is another feature of athlete's heart and is attributed to increased vagal activity. Vagally mediated bradycardia has been identified as a key factor in the induction of TdP arrhythmias in an in vivo rabbit proarrhythmia model (Farkas et al., 2008).
In competitive athletes, mild changes in repolarization reserve alone probably do not represent a significant risk for arrhythmia development, but the effects of otherwise mild factors such as different medications, bradycardia, certain ingredients of dietary supplements, early and undiagnosed cardiomyopathy and doping agents (steroids or amphetamines) on repolarizing currents can superimpose and result in repolarization abnormalities that can occasionally lead to sudden cardiac death (Maron, 2007). Indeed, in spite of the fact that the preconditioning effect of regular exercise has been shown to protect against arrhythmias (Szekeres et al., 1993) and tissue damage (Das and Das, 2008), top athletes have two to four times higher mortality due to sudden cardiac death compared with age and sex matched controls not involved in sports activities (Corrado et al., 2007).
Repolarization reserve: a dynamically changing factor
Recent research indicates that repolarization reserve is not a static feature of cardiac tissue but a dynamically changing one. After 24 h incubation with dofetilide, a potent IKr blocker, continuously paced dog ventricular myocytes elicited shorter APD and blunted dofetilide response (Xiao et al., 2008b). Patch-clamp measurements revealed enhancement of IKs in cultured myocytes incubated with dofetilide compared with IKs in control cells. Accordingly, the KvLQT1 and MinK protein levels were increased. Interestingly, the mRNA for both KvLQT1 and MinK did not change, suggesting regulatory events at the posttranslational level. In the same experiments, the expression of microRNA 133a and 133b (miR-133a and miR-133b) were diminished in cells previously incubated with dofetilide. In another earlier study, it was shown that muscle specific miR-133 depressed KvLQT1 protein expression without changing KvLQT1 mRNA (Luo et al., 2007). Based on these results, it was proposed that sustained reduction of IKr leads to compensatory up-regulation of IKs consistent with a feedback control of ion channel expression. This newly recognized modulation of repolarization reserve might happen via changes in certain miRNA levels working at the translational level. In fact, enhanced miR-133 expression was associated with a decreased density of ERG protein and IKr in diabetic rabbit hearts (Xiao et al., 2009). These results are not without controversy and further studies are needed to confirm them; even so, they suggest that repolarization reserve might be regulated and altered by disease and chronic drug treatment and this should be taken into account in developing future therapies.
Measurement of repolarization reserve
Despite its potential importance, the estimation of cardiac repolarization reserve of individual patients is not a common procedure in clinical practice. One may argue that it would be more appropriate to test individuals for their status of repolarization reserve and apply drugs accordingly, rather than restrict drug development if the compound is suspected to cause HERG inhibition or QTc lengthening. It is very likely that patients with normal repolarization reserve would not respond to drugs with mild-to-moderate K+ channel inhibitory properties with arrhythmias and sudden cardiac death. However, patients with diminished repolarization reserve may do so. Therefore, reliably predictive non-invasive tests in clinical settings and easy to use animal models are sorely needed. In this regard, it is clear that the pharmaceutical industry needs novel and reliable models that are able to go beyond the assessment of QT interval alterations caused by developmental drugs, thus enabling the industry to predict the proarrhythmic side effects of compounds under development (Pugsley et al., 2009).
Due to the complex nature of repolarization reserve, its accurate measurement in a quantitative manner is difficult. In experimental settings, repolarization reserve based on theoretical considerations can be estimated by drugs which increase depolarization force, as would be produced by veratridine or the toxin ATX II. In these cases, the degree of repolarization prolongation produced would be expected to be opposed by the sum of repolarizing potassium currents. If limited APD prolongation is observed, then a strong repolarization reserve is predicted. Conversely, substantial APD lengthening argues for weak repolarization reserve. For pragmatic reasons, it is more appropriate to test HERG/IKr channel inhibition, and analyse the evoked repolarization lengthening. This is based on the common experience that most drugs reported to cause TdP arrhythmias inhibit IKr and the repolarization reserve in these cases would include repolarizing currents largely other than IKr. Drug testing for safety pharmacology purposes, should also be performed using preparations in which repolarization reserve had been previously attenuated by ion channel remodelling (Volders et al., 1999) or by pharmacological means (Figure 9) (Lengyel et al., 2004; 2007a;). Special techniques based on action potential configurations have also been described to assess proarrhythmic drug effects based on repolarization instability (Hondeghem and Carlsson, 2001) at the preclinical level.
Figure 9.
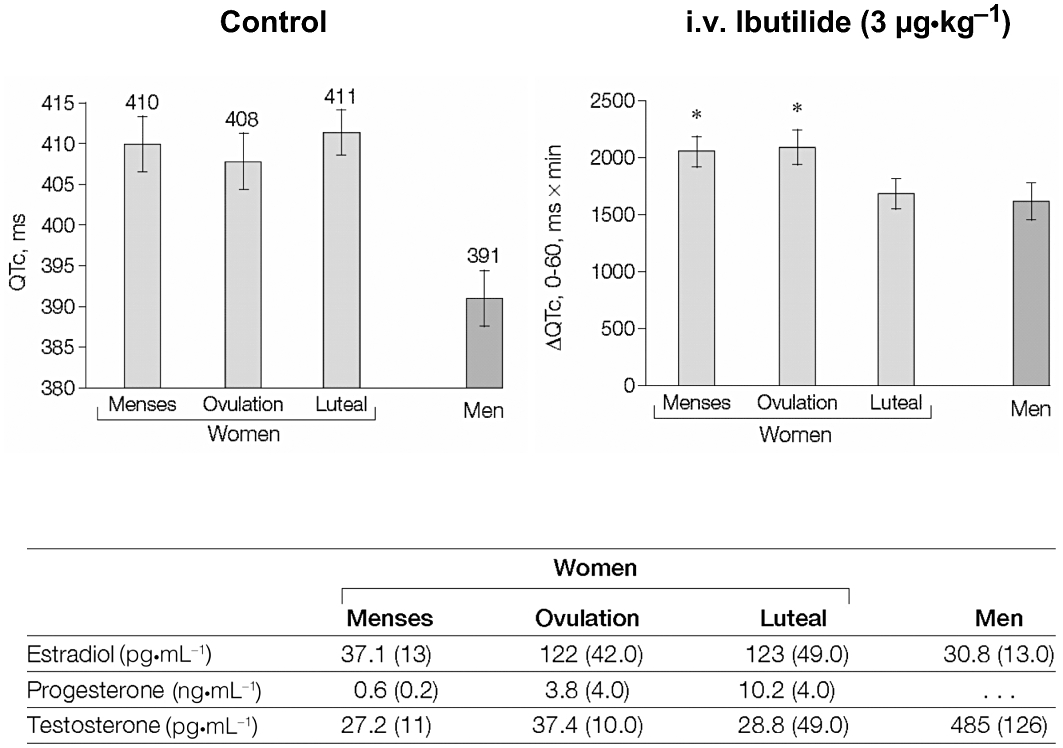
Possible influence of hormonal changes during the menstrual cycle on repolarization reserve. Left panel shows baseline QTc intervals in women during the different phases of the menstrual cycle and in men. Right panel shows mean change in QTc interval area under the curve during the first hour following ibutilide (IKr blocker) infusion. Lower panel table shows sex hormone levels in study subjects, with low levels of estradiol and progesterone during menses, a peak of estradiol during ovulation, and the highest values of progesterone during the luteal phase. Men had the lowest estradiol serum levels, and their testosterone level was 15 times higher than in women. Table data are presented as mean (SD). These data suggest that progesterone may oppose IKr block-induced repolarization lengthening in women. From Rodriguez et al. (2001), with permission.
Differences in the QT intervals of consecutive heart beats, measured as the variability index (Thomsen et al., 2004) or short-term beat-to-beat QT variability (Berger, 2003), have been suggested as a measure of proarrhythmic risk related to changes in repolarization reserve. The collected results are encouraging. These studies (Thomsen et al., 2004; Lengyel et al., 2007a) suggest that the development of TdP arrhythmia in dogs and rabbits correlated better with short-term QT variability than with QTc changes in general (Figure 10). In this context, the short-term temporal beat-to-beat variability of the QT interval has been found to reflect latent repolarization disorders and increased susceptibility to sudden cardiac death in patients with nonischaemic heart failure with no marked prolongation of the QT interval (Hinterseer et al., 2010). Also, short-term variability of the QT interval was found to be increased in humans with congenital LQT syndromes (Hinterseer et al., 2009).
Very recently, a novel parameter, the so-called ‘absolute’ variability has also been suggested to show good correlation between repolarization instability and development of TdP arrhythmias in an in vivo rabbit proarrhythmia model (Farkas et al., 2010). Further studies are needed to validate the utility of this parameter using compounds that had been withdrawn from the market due to their torsadogenic side effects.
At the clinical level, as data shown on Figure 8 suggests, simple QTc measurements from ECG recordings are not sufficient to measure repolarization reserve. Applying other ECG parameters such as QTc dispersion, Tpeak to Tend and T-wave morphology analysis, also give uncertain results. Provocative pharmacological tests have been suggested (Kilborn et al., 2000) but these may raise ethical concerns. In this study, intravenous application of a short acting IKr blocking class III antiarrhythmic drug, ibutilide, lengthened QTc and the magnitude of this effect had a good Gaussian distribution (Figure 11). Furthermore, the long QTc edge of the curve (indicated by squares), and the subjects far out of the range (indicated by a circle), may identify patients with decreased repolarization reserve and increased risk for proarrhythmia following drug treatment (Kilborn et al., 2000).
Figure 11.
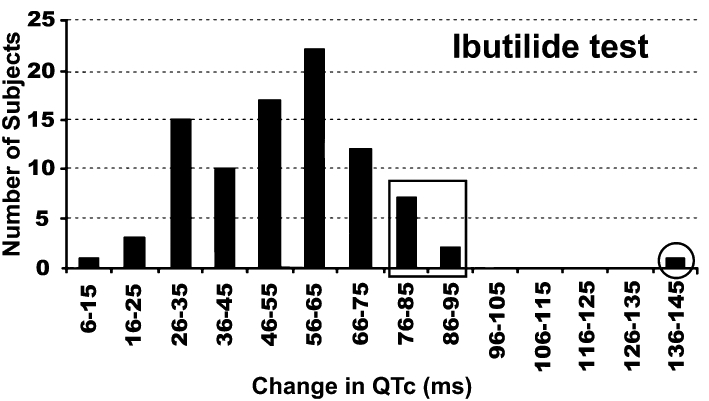
Possible clinical manifestation of reduced repolarization reserve. Distribution of QTc prolongation in patients following ibutilide (IKr blocker) administration. Patients who had QTc prolongation far out of range may be individuals with decreased repolarization reserve and increased proarrhythmic risk (indicated by circle). From Kilborn et al. (2000), with permission.
Lengyel et al. (2004) showed that eliprodil, a new investigational neuroprotective drug, did not change cardiac repolarization significantly in normal rabbit hearts, but lengthened it substantially accompanied by TdP episodes in preparations where repolarization reserve was previously reduced by inhibition of Ik1 (Figure 12).
Figure 12.
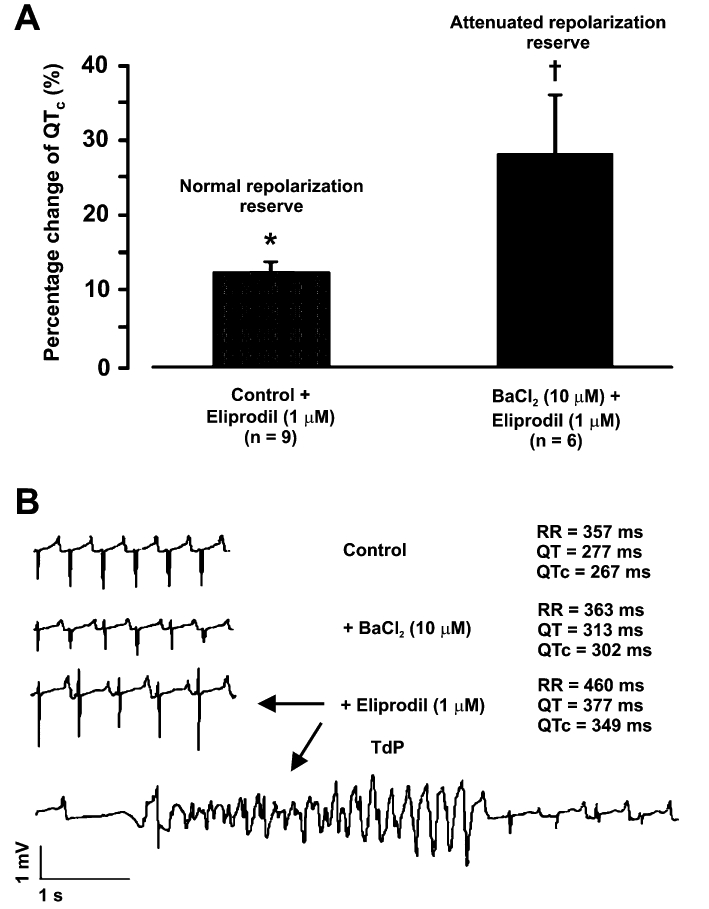
Repolarization (QTc) lengthening effect of eliprodil, a new investigational neuroprotective compound, in Langendorff-perfused rabbit hearts with normal and attenuated repolarization reserve (upper panel). Eliprodil never induced arrhythmias in normal hearts but often caused Torsades de Pointes (TdP) arrhythmias in preparations where repolarization reserve was reduced via Ik1 block by BaCl2 (lower panel). From Lengyel et al. (2004), with permission.
Possible pharmacological enhancement of repolarization reserve
As mentioned previously, decreasing repolarization reserve by drugs can have detrimental consequences by increasing the risk of proarrhythmia. As such, decreasing repolarization reserve represents an important adverse side effect of drug treatment. In addition, it must be emphasized that drugs which block multiple potassium channels without decreasing inward currents such as INa or ICa are expected to have higher proarrhythmic risk than selective potassium channel inhibitors because they tend to further depress repolarization reserve (Kodama et al., 1999; Biliczki et al., 2002). Therapeutically, it would be useful to enhance repolarization reserve and thereby attenuate the proarrhythmic risk caused by repolarization abnormalities.
Inhibition of inward sodium and calcium currents may represent pharmacological interventions that may have indirect beneficial effects on repolarization reserve. Amiodarone, a drug that inhibits both INa and ICa, is one of the best examples illustrating beneficial actions on repolarization. Amiodarone lengthens repolarization substantially but its proarrhythmic effect is much less than that of class III antiarrhythmics which lengthen APD by inhibiting outward potassium currents only. One may speculate that for these purposes, relatively weak INa and ICa block would be sufficient, because persistent (or window) inward currents seem more sensitive to drug-induced inhibition than the corresponding peak currents (Kodama et al., 1999). In addition, if peak inward currents are substantially affected, impairment of impulse conduction both at the ventricular and atrioventricular levels would be expected with concomitant side effects of proarrhythmia and decreased force of contraction. These effects may have detrimental consequences, especially in diseased hearts.
Another option is to enhance repolarization reserve by increasing outward potassium currents. The best candidate in this case is IKs, because stimulating this current should not abbreviate repolarization markedly when APD is in the normal range; however, as a negative feedback mechanism, enhanced IKs would limit excessive proarrhythmic repolarization lengthening. In this regard, the benzodiazepine derivative L-364,373 enhanced IKs in guinea pig and rabbit myocytes (Salata et al., 1998; Xu et al., 2002); however, this effect was not confirmed in dog (Magyar et al., 2006). Further studies are needed on the effect and potency of L-364,373 before firm conclusions can be drawn. In a recent study, the pharmacological activation of IKs by the benzodiazepine R-L3 resulted in enhanced repolarizing capacity and counteracted pharmacologically reduced IKr in isolated guinea pig cardiomyocytes (Nissen et al., 2009).
Recent data indicates that IKr can be augmented by NS-3623 and NS-1643, novel compounds that shorten ventricular action potentials in different preparations (Hansen et al., 2006a,b; 2007; Diness et al., 2008). NS-3623 was also shown to elicit post-repolarization refractoriness as an additional antiarrhythmic mechanism (Hansen et al., 2007).
Repolarization reserve could also be enhanced by opening ATP-sensitive potassium channels (KATP). There are a number of compounds that activate KATP (Lathrop et al., 1990; Carlsson et al., 1992; Jahangir and Terzic, 2005), but these are not sufficiently selective for the cardiac sarcolemmal KATP channels. Therefore, unwanted effects (e.g. hypotension and altered glucose homeostasis) resulting from activation of KATP channels expressed in other tissues can be expected following their application. However, pinacidil and nicorandil have been shown to suppress abnormal automaticity, triggered activity and TdP in a guinea-pig LQT1 model (Yang et al., 2004). It should be noted that excessive cardiac sarcolemmal KATP activation would shorten action potential in the myocardium promoting the development of re-entry arrhythmias (Janse and Wit, 1989).
Conclusions
Repolarization reserve is a relatively new concept that is helpful in explaining the development of arrhythmias based on repolarization abnormalities and drug-induced proarrhythmic adverse effects. According to this concept, inhibition or functional impairment of one transmembrane ion channel does not necessarily result in excessive repolarization changes because other currents can compensate. Impaired repolarization reserve, which is common in certain pathological settings and following drug treatment, is not easily detected by conventional in vivo or in vitro electrophysiological measurements. In some circumstances, however, reduction in repolarization reserve leads to marked repolarization lengthening and is proarrhythmic. Better predictive methods are needed to identify the conditions in which preventable arrhythmias due to impaired repolarization reserve occur. Another important future goal is to avoid discrediting otherwise useful novel compounds solely on their minor IKr (HERG) blocking properties. Instead, the repolarization reserve status of individual patients could be assessed when they have a specific need for a drug with known effects on cardiac repolarization and thus unwanted drug-induced arrhythmias could be limited in these individuals as, for example, is the case with patients with a specific requirement for penicillin therapy who have presumed penicillin allergy.
Acknowledgments
This work was supported by grants from the Hungarian Scientific Research Fund (OTKA CNK 77855), National Research and Development Programmes (NKTH Cardio08), János Bolyai Research Scholarship (I. B.), National Development Agency (TÁMOP-4.2.2-08/1-2008-0013 and TÁMOP-4.2.1/B-09/1/KONV-2010-0005) and European Community (EU FP7 grant ICT-2008-224381, preDICT).
Glossary
Abbreviations
- APD
action potential duration
- ATP
adenosine triphosphate
- ATX II
Anemonia sulcata toxin
- cAMP
3′-5′-cyclic adenosine monophosphate
- DAD
delayed afterdepolarization
- EAD
early afterdepolarization
- ERP
effective refractory period
- ICa,L
L-type inward calcium current
- IKr
rapid delayed rectifier potassium current
- IKs
slow delayed rectifier potassium current
- INa
inward sodium current
- Ito
transient outward potassium current
- LQT1
long QT syndrome 1
- LQT2
long QT syndrome 2
- LQT3
long QT syndrome 3
- LQT7
long QT syndrome 7
- LQTS
long QT syndromes
- miRNA
micro ribonucleic acid
- NCX
Na+-Ca2+ exchanger
- PKA
protein kinase A
- PKC
protein kinase C
- TdP
Torsades de Pointes
Conflict of interest
The authors declare that they have no conflicts of interest related to this work. Both authors have made written contributions to this manuscript, and have approved the final version.
References
- Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace. 2007;9(Suppl 4):iv4–i15. doi: 10.1093/europace/eum166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Fish J. Electrical heterogeneity within the ventricular wall. Basic Res Cardiol. 2001;96:517–527. doi: 10.1007/s003950170002. [DOI] [PubMed] [Google Scholar]
- Anumonwo JM, Lopatin AN. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010;48:45–54. doi: 10.1016/j.yjmcc.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Cohen I, Eisner D, Ohba M, Ojeda C. The steady state TTX-sensitive (‘window’) sodium current in cardiac Purkinje fibres. Pflug Arch Eur J Physiol. 1979;379:137–142. doi: 10.1007/BF00586939. [DOI] [PubMed] [Google Scholar]
- Berger RD. QT variability. J Electrocardiol. 2003;36:83–87. doi: 10.1016/j.jelectrocard.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd edn. Dordrecht, Boston, London: Kluwer Academic Publishers; 2001. [Google Scholar]
- Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl 1):I36–I42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- Biliczki P, Virág L, Iost N, Papp JG, Varró A. Interaction of different potassium channels in cardiac repolarization in dog ventricular preparations: role of repolarization reserve. Br J Pharmacol. 2002;137:361–368. doi: 10.1038/sj.bjp.0704881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørnstad H, Tande PM, Lathrop DA, Refsum H. Effects of temperature on cycle length dependent changes and restitution of action potential duration in guinea pig ventricular muscle. Cardiovasc Res. 1993;27:946–950. doi: 10.1093/cvr/27.6.946. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Gaspo R, Busch AE, Lang HJ, Li GR, Nattel S. Effects of the chromanol 293B, a selective blocker of the slow component of the delayed rectifier K+ current, on repolarisation in human and guinea pig ventricular myocytes. Cardiovasc Res. 1998;38:441–450. doi: 10.1016/s0008-6363(98)00021-2. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Wang Z, Li GR, Nattel S. Electrophysiological mechanisms by which hypothyroidism delays repolarization in guinea pig hearts. Am J Physiol. 1999;277:H211–H220. doi: 10.1152/ajpheart.1999.277.1.H211. [DOI] [PubMed] [Google Scholar]
- Boulet IR, Raes AL, Ottschytsch N, Snyders DJ. Functional effects of a KCNQ1 mutation associated with the long QT. Cardiovasc Res. 2006;70:466–474. doi: 10.1016/j.cardiores.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Burashnikov A, Shimizu W, Antzelevitch C. Fever accentuates transmural dispersion of repolarization and facilities development of early afterdepolarizations and torsade de pointes under long-QT conditions. Circ Arrhythm Electrophysiol. 2008;1:202–208. doi: 10.1161/CIRCEP.107.691931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Malloy K, Groh WJ, Varnum MD, Adelman JP, Maylie J. The novel class III antiarrhythmics NE-10064 and NE-10133 inhibit IsK channels expressed in Xenopus oocytes and IKs in guinea pig cardiac myocytes. Biochem Biophys Res Commun. 1994;15:265–270. doi: 10.1006/bbrc.1994.1922. [DOI] [PubMed] [Google Scholar]
- Busch AE, Suessbrich H, Waldegger S, Sailer E, Greger R, Lang H, et al. Inhibition of IKs in guinea pig cardiac myocytes and guinea pig IsK channels by the chromanol 293B. Pflug Arch Eur J Physiol. 1996;432:1094–1096. doi: 10.1007/s004240050240. [DOI] [PubMed] [Google Scholar]
- Carlsson L, Abrahamsson C, Drews L, Duker G. Antiarrhythmic effects of potassium channel openers in rhythm abnormalities related to delayed repolarization. Circulation. 1992;85:1627–1629. doi: 10.1161/01.cir.85.4.1491. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Slow inactivation of the sodium current in rabbit cardiac Purkinje fibres. Pflug Arch Eur. J Physiol. 1987;408:18–26. doi: 10.1007/BF00581835. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Repolarization reserve in cardial cells. J Med Biol Eng. 2006;26:97–105. [Google Scholar]
- Casini S, Verkerk AO, van Borren MM, van Ginneken AC, Veldkamp MW, de Bakker JM, et al. Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc Res. 2009;81:72–81. doi: 10.1093/cvr/cvn274. [DOI] [PubMed] [Google Scholar]
- Cordeiro JM, Spitzer KW, Giles WR. Repolarizing K+ currents in rabbit heart Purkinje cells. J Physiol. 1998;508:811–823. doi: 10.1111/j.1469-7793.1998.811bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado D, Michieli P, Basso C, Schiavon M, Thiene G. How to screen athletes for cardiovascular diseases. Cardiol Clin. 2007;25:391–397. doi: 10.1016/j.ccl.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Darbar D, Roden DM. Pharmacogenetics of antiarrhythmic therapy. Expert Opin Pharmacother. 2006;7:1583–1590. doi: 10.1517/14656566.7.12.1583. [DOI] [PubMed] [Google Scholar]
- Das M, Das DK. Molecular mechanism of preconditioning. IUBMB Life. 2008;60:199–203. doi: 10.1002/iub.31. [DOI] [PubMed] [Google Scholar]
- De Weer P, Gadsby DC, Rakowski RF. Voltage dependence of the Na-K pump. Annu Rev Physiol. 1988;50:225–241. doi: 10.1146/annurev.ph.50.030188.001301. [DOI] [PubMed] [Google Scholar]
- Diness TG, Yeh YH, Qi XY, Chartier D, Tsuji Y, Hansen RS, et al. Antiarrhythmic properties of a rapid delayed-rectifier current activator in rabbit models of acquired long QT syndrome. Cardiovasc Res. 2008;79:61–69. doi: 10.1093/cvr/cvn075. [DOI] [PubMed] [Google Scholar]
- Duker G, Sjöquist PO, Johansson BW. Monophasic action potentials during induced hypothermia in hedgehog and guinea pig hearts. Am J Physiol. 1987;253:H1083–H1088. doi: 10.1152/ajpheart.1987.253.5.H1083. [DOI] [PubMed] [Google Scholar]
- Farkas A, Dempster J, Coker SJ. Importance of vagally mediated bradycardia for the induction of torsade de pointes in an in vivo model. Br J Pharmacol. 2008;154:958–970. doi: 10.1038/bjp.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas AS, Nattel S. Minimizing repolarization-related proarrhythmic risk in drug development and clinical practice. Drugs. 2010;70:573–603. doi: 10.2165/11535230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Farkas AS, Rudas L, Makra P, Csík N, Leprán I, Forster T, et al. Biomarkers and endogenous determinants of dofetilide-induced Torsades de Pointes in alpha(1)-adrenoceptor-stimulated, anaesthetized rabbits. Br J Pharmacol. 2010;161:1477–1495. doi: 10.1111/j.1476-5381.2010.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredlund BO, Olsson SB. Long QT interval and ventricular tachycardia of ‘torsade de pointe’ type in hypothyroidism. Acta Med Scand. 1983;213:231–235. doi: 10.1111/j.0954-6820.1983.tb03724.x. [DOI] [PubMed] [Google Scholar]
- Fülöp L, Bányász T, Szabó G, Tóth G, Biró T, Lőrincz I, et al. Effects of sex hormones on ECG parameters and expression of cardiac ion channels in dogs. Acta Physiol. 2006;188:163–171. doi: 10.1111/j.1748-1716.2006.01618.x. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Le Bouter S, Szuts V, Varró A, Escande D, Nattel S, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582:675–693. doi: 10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galetta F, Franzoni F, Fallahi P, Tocchini L, Braccini L, Santoro G, et al. Changes in heart rate variability and QT dispersion in patients with overt hypothyroidism. Eur J Endocrinol. 2008;158:85–90. doi: 10.1530/EJE-07-0357. [DOI] [PubMed] [Google Scholar]
- Gallacher DJ, Van de Water A, van der Linde H, Hermans AN, Lu HR, Towart R, et al. In vivo mechanisms precipitating torsades de pointes in a canine model of drug- induced long-QT1 syndrome. Cardiovasc Res. 2007;76:247–256. doi: 10.1016/j.cardiores.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Gao J, Wang W, Cohen IS, Mathias RT. Transmural gradients in Na/K pump activity and [Na+] in canine. Biophys J. 2005;89:1700–1709. doi: 10.1529/biophysj.105.062406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Nunziato DA, Pitt GS. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ Res. 2006;98:1048–1054. doi: 10.1161/01.RES.0000218863.44140.f2. [DOI] [PubMed] [Google Scholar]
- Gill GV, Woodward A, Casson IF, Weston PJ. Cardiac arrhythmia and noctural hypoglycaemia in type 1 diabetes – the ‘dead in bed’ syndrome revisited. Diabetologia. 2008;52:42–45. doi: 10.1007/s00125-008-1177-7. [DOI] [PubMed] [Google Scholar]
- Gintant GA. Regional differences in IK density in canine left ventricle: role of IKs in electrical heterogeneity. Am J Physiol. 1995;268:H604–H613. doi: 10.1152/ajpheart.1995.268.2.H604. [DOI] [PubMed] [Google Scholar]
- Gintant GA. Two components of delayed rectifier current in canine atrium and ventricle: does IKs play a role in the reverse rate dependence of class III agents? Circ Res. 1996;78:26–37. doi: 10.1161/01.res.78.1.26. [DOI] [PubMed] [Google Scholar]
- Giunti S, Bruno G, Lillaz E, Gruden G, Lolli V, Chaturvedi N, et al. Incidence and risk factors of prolonged QTc interval in type 1 diabetes: the EURODIAB Prospective Complications Study. Diabetes Care. 2007;30:2057–2063. doi: 10.2337/dc07-0063. [DOI] [PubMed] [Google Scholar]
- Gögelein H, Bruggemann A, Gerlach U, Brendel J, Busch AE. Inhibition of IKs channels by HMR 1556. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:480–488. doi: 10.1007/s002100000284. [DOI] [PubMed] [Google Scholar]
- Gonzalez CD, de Sereday M, Sinay I, Santoro S. Endocrine therapies and QTc prolongation. Curr Drug Saf. 2010;5:79–84. doi: 10.2174/157488610789869157. [DOI] [PubMed] [Google Scholar]
- Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, et al. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–2757. doi: 10.1172/JCI39027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Wang Z, Nattel S. Slow delayed rectifier current and repolarization in canine cardiac Purkinje cells. Am J Physiol. 2001;280:H1075–H1080. doi: 10.1152/ajpheart.2001.280.3.H1075. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Diness TG, Christ T, Demnitz J, Ravens U, Olesen SP, et al. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) Mol Pharmacol. 2006a;69:266–277. doi: 10.1124/mol.105.015859. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Diness TG, Christ T, Wettwer E, Ravens U, Olesen SP, et al. Biophysical characterization of the new human ether-a-go-go-related gene channel opener NS3623 [N-(4-bromo-2-(1H-tetrazol-5-yl)-phenyl)-N'-(3′-trifluoromethylphenyl)urea] Mol Pharmacol. 2006b;70:1319–1329. doi: 10.1124/mol.106.026492. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Olesen SP, Grunnet M. Pharmacological activation of rapid delayed rectifier potassium current suppresses bradycardia-induced triggered activity in the isolated guinea pig heart. J Pharmacol Exp Ther. 2007;321:996–1002. doi: 10.1124/jpet.106.118448. [DOI] [PubMed] [Google Scholar]
- Hart G. Exercise-induced cardiac hypertrophy: a substrate for sudden death in athletes? Exp Physiol. 2003;88:639–644. doi: 10.1113/eph8802619. [DOI] [PubMed] [Google Scholar]
- Hermans K, Stockman D, Van den Branden F. Grapefruit and tonic: a deadly combination in a patient with the long QT syndrome. Am J Med. 2003;114:511–512. doi: 10.1016/s0002-9343(03)00071-8. [DOI] [PubMed] [Google Scholar]
- Herzog CA, Mangrum JM, Passman R. Sudden cardiac death and dialysis patients. Semin Dial. 2008;21:300–307. doi: 10.1111/j.1525-139X.2008.00455.x. [DOI] [PubMed] [Google Scholar]
- Hinterseer M, Beckmann BM, Thomsen MB, Pfeufer A, Pozza RD, Loeff M, et al. Relation of increased short-term variability of QT interval to congenital long-QT syndrome. Am J Cardiol. 2009;103:1244–1248. doi: 10.1016/j.amjcard.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Hinterseer M, Beckmann BM, Thomsen MB, Pfeufer A, Ulbrich M, Sinner MF, et al. Usefulness of short-term variability of QT intervals as a predictor for electrical remodeling and proarrhythmia in patients with nonischemic heart failure. Am J Cardiol. 2010;106:216–220. doi: 10.1016/j.amjcard.2010.02.033. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM. Development of class III antiarrhythmic agents. J Cardiovasc Pharmacol. 1992;20:S17–S22. [PubMed] [Google Scholar]
- Hondeghem LM, Carlsson L. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Iost N, Virág L, Opincariu M, Szécsi J, Varró A, JGy P. Does IKs play an important role in the repolarization in normal human ventricular muscle? Circulation. 1999;100(Suppl 1):I–495. [Google Scholar]
- Ishihara K, Sarai N, Asakura K, Noma A, Matsuoka S. Role of Mg2+ block of the inward rectifier K+ current in cardiac repolarization reserve: A quantitative simulation. J Mol Cell Cardiol. 2009;47:76–84. doi: 10.1016/j.yjmcc.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Jahangir A, Terzic A. KATP channel therapeutics at the bedside. J Mol Cell Cardiol. 2005;39:99–112. doi: 10.1016/j.yjmcc.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AF, Choisy SCM, Hancox JC. Recent advances in understanding sex differences in cardiac repolarization. Prog Biophys Mol Biol. 2007;94:265–319. doi: 10.1016/j.pbiomolbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- James AF, Hancox JC. Sex, drugs and arrhythmia: are gender differences in risk of torsades de pointes simply a matter of testosterone? Cardiovasc Res. 2003;57:1–4. doi: 10.1016/s0008-6363(02)00740-x. [DOI] [PubMed] [Google Scholar]
- Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049–1169. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- Johansson M, Gao SA, Friberg P, Annerstedt M, Bergström G, Carlström J, et al. Elevated temporal QT variability index in patients with chronic renal failure. Clin Sci. 2004;107:583–588. doi: 10.1042/CS20040122. [DOI] [PubMed] [Google Scholar]
- Johnson DM, Heijman J, Pollard CE, Valentin JP, Crijns HJ, Abi-Gerges N, et al. IKs restricts excessive beat-to-beat variability of repolarization during beta-adrenergic receptor stimulation. J Mol Cell Cardiol. 2010a;48:122–130. doi: 10.1016/j.yjmcc.2009.08.033. [DOI] [PubMed] [Google Scholar]
- Jonsson MKB, Vos MA, Duker G, Demolombe S, van Veen TAB. Gender disparity in cardiac electrophysiology: Implications for cardiac safety pharmacology. Pharmacol Ther. 2010;127:9–18. doi: 10.1016/j.pharmthera.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Jost N, Papp JG, Varró A. Slow delayed rectifier potassium current (IKs) and the repolarization reserve. Ann Noninvas Electrocardiol. 2007;12:64–78. doi: 10.1111/j.1542-474X.2007.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N, Varró A, Szuts V, Kovacs PP, Seprenyi G, Biliczki P, et al. Molecular basis of repolarization reserve differences between dogs and man. Circulation. 2008;118(Suppl):342. [Google Scholar]
- Jost N, Virág L, Bitay M, Takács J, Lengyel C, Biliczki P, et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- Kääb S, Dixon J, Duc J, Ashen D, Näbauer M, Beuckelmann DJ, et al. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
- Kääb S, Hinterseer M, Näbauer M, Steinbeck G. Sotalol testing unmasks altered repolarization in patients with suspected acquired long-QT-syndrome – a case-control pilot study using i.v. sotalol. Eur Heart J. 2003;24:649–657. doi: 10.1016/s0195-668x(02)00806-0. [DOI] [PubMed] [Google Scholar]
- Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol. 2007;22:39–43. doi: 10.1097/HCO.0b013e32801129eb. [DOI] [PubMed] [Google Scholar]
- Kilborn MJ, Liu XK, Rodriguez I, Barbey JT, Woosley RL, Kilborn MJ. Determination of QT response phenotype with low-dose ibutilide. Circulation. 2000;102:II–673. [Google Scholar]
- Kjekshus J. Arrhythmias and mortality in congestive heart failure. Am J Cardiol. 1990;65:421–481. doi: 10.1016/0002-9149(90)90125-k. [DOI] [PubMed] [Google Scholar]
- Kodama I, Kamiya K, Toyama J. Amiodarone: ionic and cellular mechanisms of action of the most promising class III agent. Am J Cardiol. 1999;84:20R–28R. doi: 10.1016/s0002-9149(99)00698-0. [DOI] [PubMed] [Google Scholar]
- Kuo CS, Munakata K, Reddy CP, Surawicz B. Characteristics and possible mechanism of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation. 1983;67:1356–1367. doi: 10.1161/01.cir.67.6.1356. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Tamagawa M, Harada N, Honda S, Bai CX, Bai CX, et al. Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization. J Physiol. 2008;586:2961–2973. doi: 10.1113/jphysiol.2007.150367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop DA, Nánási P, Varró A. In vitro cardiac models of dog Purkinje fibre triggered and spontaneous electrical activity: effects of nicorandil. Br J Pharmacol. 1990;99:119–123. doi: 10.1111/j.1476-5381.1990.tb14664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bouter S, Demolombe S, Chambellan A, Bellocq C, Aimond F, Toumaniantz G, et al. Microarray analysis reveals complex remodelling of cardiac ion channel expression with altered thyroid status: relation to cellular and integrated electrophysiology. Circ Res. 2003;92:234–242. doi: 10.1161/01.res.0000053185.75505.8e. [DOI] [PubMed] [Google Scholar]
- Lengyel C, Iost N, Virág L, Varró A, Lathrop DA, Papp JG. Pharmacological block of the slow component of the outward delayed rectifier current (IKs) fails to lengthen rabbit ventricular muscle QTc and action potential duration. Br J Pharmacol. 2001;132:101–110. doi: 10.1038/sj.bjp.0703777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel C, Horváth C, Biliczki P, Dézsi P, Németh M, Iost N, et al. Effect of a neuroprotective drug, eliprodil on cardiac repolarization: importance of the decreased repolarisation reserve in the development of proarrythmic risk. Br J Pharmacol. 2004;143:152–158. doi: 10.1038/sj.bjp.0705901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel C, Varró A, Tábori K, Papp JG, Baczkó I. Combined pharmacological block of IKr and IKs increases short-term QT interval variability and provokes torsades de pointes. Br J Pharmacol. 2007a;151:941–951. doi: 10.1038/sj.bjp.0707297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel C, Virág L, Bíró T, Jost N, Magyar J, Biliczki P, et al. Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res. 2007b;73:512–520. doi: 10.1016/j.cardiores.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Lengyel C, Orosz A, Takács R, Várkonyi TT, Baczkó I, Wittmann T, et al. Short-term QT variability in patients with long-standing type 1 diabetes mellitus. Diabetologia. 2008;51(Suppl 1):S556. [Google Scholar]
- Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- Limberis JT, Su Z, Cox BF, Gintant GA, Martin RL. Altering extracellular potassium concentration does not modulate drug block of human ether-a-go-go-related gene (hERG) channels. Clin Exp Pharmacol Physiol. 2006;33:1059–1065. doi: 10.1111/j.1440-1681.2006.04487.x. [DOI] [PubMed] [Google Scholar]
- Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ Res. 1988;62:116–126. doi: 10.1161/01.res.62.1.116. [DOI] [PubMed] [Google Scholar]
- Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res. 1995;76:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- Lőrincz I, Mátyus J, Zilahi Z, Kun C, Karányi Z, Kakuk G. QT dispersion in patients with end-stage renal failure and during hemodialysis. J Am Soc Nephrol. 1999;10:1297–1302. doi: 10.1681/ASN.V1061297. [DOI] [PubMed] [Google Scholar]
- Luo X, Xiao J, Lin H, Li B, Lu Y, Yang B, et al. Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J Cell Physiol. 2007;212:358–367. doi: 10.1002/jcp.21030. [DOI] [PubMed] [Google Scholar]
- Magyar J, Horváth B, Bányász T, Szentandrássy N, Birinyi P, Varró A, et al. L-364,373 fails to activate the slow delayed rectifier K+ current in canine ventricular cardiomyocytes. Naunyn-Sch Arch Pharmacol. 2006;373:85–90. doi: 10.1007/s00210-006-0047-4. [DOI] [PubMed] [Google Scholar]
- Maron BJ. Hypertropic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399–414. doi: 10.1016/j.ccl.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through the inwardly rectifying K+ channel and blocking by internal Mg2. Nature. 1987;325:156–158. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- McBride BF, Yang T, Roden DM. Influence of the G2677T/C3435T haplotype of MDR1 on P-glycoprotein trafficking and ibutilide-induced block of HERG. Pharmacogenomics J. 2009;9:194–201. doi: 10.1038/tpj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näbauer M, Kääb S. Potassium channel down-regulation in heart failure. Cardiovasc Res. 1998;37:324–334. doi: 10.1016/s0008-6363(97)00274-5. [DOI] [PubMed] [Google Scholar]
- Nair LA, Grant AO. Emerging class III antiarrhythmic agents: mechanism of action and proarrhythmic potential. Cardiovasc Drugs Ther. 1997;11:149–167. doi: 10.1023/a:1007784814823. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM. Repolarizing Cardiac Potassium Channels: Multiple Sites and Mechanisms for CaMKII-Mediated Regulation. Heart Rhythm. 2011 doi: 10.1016/j.hrthm.2011.01.018. Published online 2011 Jan 10. doi: 10.1016/j.hrthm.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Nissen JD, Diness JG, Diness TG, Hansen RS, Grunnet M, Jespersen T. Pharmacologically induced long QT type 2 can be rescued by activation of IKs with benzodiazepine R-L3 in isolated guinea pig cardiomyocytes. J Cardiovasc Pharmacol. 2009;54:169–177. doi: 10.1097/FJC.0b013e3181af6db3. [DOI] [PubMed] [Google Scholar]
- Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oros A, Beekman JD, Vos MA. The canine model with chronic, complete atrio-ventricular block. Pharmacol Ther. 2008;119:168–178. doi: 10.1016/j.pharmthera.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Packer M. Sudden unexpected death in patients with congestive heart failure: a second frontier. Circulation. 1985;72:681–685. doi: 10.1161/01.cir.72.4.681. [DOI] [PubMed] [Google Scholar]
- Patel SP, Cambell DL. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo G, Magrì D, Matera S, Magnanti M, Pasquazzi E, Schifano E, et al. Effects of pink grapefruit juice on QT variability in patients with dilated or hypertensive cardiomyopathy and in healthy subjects. Transl Res. 2008;151:267–272. doi: 10.1016/j.trsl.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Piktel JS, Jeyaraj D, Said TH, Rosenbaum DS, Wilson LD. Enhanced dispersion of repolarization explains increased arrhythmogenesis in severe vs. therapeutic hypothermia. Circ Arrhythm Electrophysiol. 2011;4:79–86. doi: 10.1161/CIRCEP.110.958355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- Pugsley MK, Authier S, Curtis MJ. Principles of safety pharmacology. Br J Pharmacol. 2008;154:1382–1399. doi: 10.1038/bjp.2008.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley MK, Authier S, Towart R, Gallacher DJ, Curtis MJ. Beyond the safety assessment of drug-mediated changes in the QT interval … what's next? J Pharmacol Toxicol Methods. 2009;60:24–27. doi: 10.1016/j.vascn.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Rautaharju PM, Zhou S, Wong S, Calhoun H, Berenson GS, Prineas R, et al. Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol. 1992;8:690–695. [PubMed] [Google Scholar]
- Ridley JM, Shuba YM, James AF, Hancox JC. Modulation by testosterone of an endogenous hERG potassium channel current. J Physiol Pharmacol. 2008;59:395–407. [PubMed] [Google Scholar]
- Roden DM. Taking the idio out of idiosyncratic-predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- Roden DM. Human genomics and its impact on arrhythmias. Trends Cardiovasc Med. 2004;14:112–116. doi: 10.1016/j.tcm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- Roden DM. Repolarization reserve. A moving target. Circulation. 2008;118:981–982. doi: 10.1161/CIRCULATIONAHA.108.798918. [DOI] [PubMed] [Google Scholar]
- Rodriguez I, Kilborn MJ, Liu X-K, Pezzullo JC, Woosley RL. Drug-induced QT prolongation in woman during the menstrual cycle. J Am Med Assoc. 2001;285:1322–1326. doi: 10.1001/jama.285.10.1322. [DOI] [PubMed] [Google Scholar]
- Salata JJ, Jurkiewicz NK, Sanguinetti MC, Siegl PK, Claremon DC, Remy DC, et al. The novel class III antiarrhythmic agent, L-735,821 is a potent and selective blocker of IKs in guinea pig ventricular myocytes (abstract) Circulation. 1996;94:I–529. [Google Scholar]
- Salata JJ, Jurkiewicz NK, Wang J, Evans BE, Orme HT, Sanguinetti MC. A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol Pharmacol. 1998;54:220–230. doi: 10.1124/mol.54.1.220. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Role of external Ca2+ and K+ in gating of cardiac delayed rectifier K+ currents. Pflugers Arch. 1992;420:180–186. doi: 10.1007/BF00374988. [DOI] [PubMed] [Google Scholar]
- Schillinger W, Janssen PM, Emami S, Henderson SA, Ross RS, Teucher N, et al. Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na+-Ca 2+ exchanger. Circ Res. 2000;87:581–587. doi: 10.1161/01.res.87.7.581. [DOI] [PubMed] [Google Scholar]
- Schmid H, Schiffl H, Lederer SR. Pharmacotherapy of end-stage renal disease. Expert Opin Pharmacother. 2010;11:597–613. doi: 10.1517/14656560903544494. [DOI] [PubMed] [Google Scholar]
- Scholz EP, Zitron E, Kiesecker C, Lück S, Thomas D, Kathöfer S, et al. Inhibition of cardiac HERG channels by grapefruit flavonoid naringenin: implications for the influence of dietary compounds on cardiac repolarisation. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:516–525. doi: 10.1007/s00210-005-1069-z. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Löber K, Heinemann SH. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin. EMBO J. 2000;19:3263–3271. doi: 10.1093/emboj/19.13.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- Sergeant GP, Ohya S, Reihill JA, Perrino BA, Amberg GC, Imaizumi Y, et al. Regulation of Kv4.3 currents by Ca2+/calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol. 2005;288:C304–C313. doi: 10.1152/ajpcell.00293.2004. [DOI] [PubMed] [Google Scholar]
- Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, et al. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci U S A. 2000;97:10613–10618. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severi S, Grandi E, Pes C, Badiali F, Grandi F, Santoro A. Calcium and potassium changes during haemodialysis alter ventricular repolarization duration: in vivo and in silico analysis. Nephrol Dial Transplant. 2008;23:1378–1386. doi: 10.1093/ndt/gfm765. [DOI] [PubMed] [Google Scholar]
- Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, et al. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ Res. 2006;98:1055–1063. doi: 10.1161/01.RES.0000218979.40770.69. [DOI] [PubMed] [Google Scholar]
- Shimoni Y, Ewart HS, Severson D. Type I and II models of diabetes produce different modifications of K+ currents in rat heart: role of insulin. J Physiol. 1998;507:485–496. doi: 10.1111/j.1469-7793.1998.485bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillitano F, Lonardo G, Zicha S, Varró A, Cerbai E, Mugelli A. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Stramba-Badiale M, Spagnolo D, Bosi G, Schwartz PJ. On behalf of the misnes investigators. Are gender differences in QTc present at birth? Am J Cardiol. 1995;75:1278–1281. [PubMed] [Google Scholar]
- Szekeres L, JGy P, Szilvássy Z, Udvary E, Végh A. Moderate stress by cardiac pacing may induce both short term and long term cardioprotection. Cardiovasc Res. 1993;27:593–596. doi: 10.1093/cvr/27.4.593. [DOI] [PubMed] [Google Scholar]
- Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- Tateishi H, Yano M, Tateishi H, Yano M, Mochizuki M, Suetomi T, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81:536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier S, Karczewski P, Krause EG, Pansard Y, Acar C, Lang-Lazdunski M, et al. Regulation of the transient outward K+ current by Ca2+/calmodulin-dependent protein kinases II in human atrial myocytes. Circ Res. 1999;85:810–819. doi: 10.1161/01.res.85.9.810. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, et al. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004;110:2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- Tohse N, Kameyama M, Irisawa H. Intracellular Ca2+ and protein kinase C modulate K+ current in guinea pig heart cells. Am J Physiol. 1987;253:H1321–H1324. doi: 10.1152/ajpheart.1987.253.5.H1321. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Varró A, Baczkó I. Possible mechanisms of sudden cardiac death in top athletes: a basic cardiac electrophysiological point of view. Pflug Arch Eur. J Physiol. 2010;460:31–40. doi: 10.1007/s00424-010-0798-0. [DOI] [PubMed] [Google Scholar]
- Varró A, Baláti B, Iost N, Takács J, Virág L, Lathrop DA, et al. The role of IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol. 2000;523:67–81. doi: 10.1111/j.1469-7793.2000.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varró A, Lathrop DA, Hester SB, Nánási PP, JGy P. Ionic currents and action potentials in rabbit, rat, and guinea pig ventricular myocytes. Basic Res Cardiol. 1993;88:93–102. doi: 10.1007/BF00798257. [DOI] [PubMed] [Google Scholar]
- Varró A, Papp JG. Low penetrance, subclinical congenital LQTS: Concealed LQTS or silent LQTS? Cardiovasc Res. 2006;70:404–406. doi: 10.1016/j.cardiores.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Verkerk AO, Wilders R, Veldkamp MW, de Geringel W, Kirkels JH, Tan HL. Gender disparities in cardiac cellular electrophysiology and arrhythmia susceptibility in human failing ventricular myocytes. Int Heart J. 2005;46:1105–1118. doi: 10.1536/ihj.46.1105. [DOI] [PubMed] [Google Scholar]
- Virág L, Acsai K, Hála O, Bitay M, Bogáts G, Papp JGy VA. Self augmentation of the repolarization lengthening is related to the shape of the action potential: the role of the intrinsic properties of IKr and Ik1 and its implication to reverse rate dependency. Br J Pharmacol. 2009;156:1076–1084. doi: 10.1111/j.1476-5381.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volders PG, Sipido KR, Vos MA, Spätjens RL, Leunissen JD, Carmeliet E, et al. Downregulation of delayed rectifier K+ currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation. 1999;100:2455–2461. doi: 10.1161/01.cir.100.24.2455. [DOI] [PubMed] [Google Scholar]
- Volders PGA, Stengl M, van Opstal JM, Gerlach U, Spätjens RL, Beekman JDM, et al. Probiting the contribution of IKs to canine ventricular repolarization. Key role for β-adrenergic receptor stimulation. Circulation. 2003;107:2753–2760. doi: 10.1161/01.CIR.0000068344.54010.B3. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Distinct voltage-dependent regulation of a heart-delayed IK by protein kinases A and C. Am J Physiol. 1991;261:C1081–C1090. doi: 10.1152/ajpcell.1991.261.6.C1081. [DOI] [PubMed] [Google Scholar]
- Wang Z, Feng J, Shi H, Pond A, Nerbonne JM, Nattel S. Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circ Res. 1999;84:551–561. doi: 10.1161/01.res.84.5.551. [DOI] [PubMed] [Google Scholar]
- Xiao L, Coutu P, Villeneuve LR, Tadevosyan A, Maguy A, Le Bouter S, et al. Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res. 2008a;103:733–742. doi: 10.1161/CIRCRESAHA.108.171157. [DOI] [PubMed] [Google Scholar]
- Xiao L, Xiao J, Luo X, Lin H, Wang Z, Nattel S. Feedback remodelling of cardiac potassium current expression. A novel potential mechanism for control of repolarization reserve. Circulation. 2008b;118:983–992. doi: 10.1161/CIRCULATIONAHA.107.758672. [DOI] [PubMed] [Google Scholar]
- Xiao J, Luo X, Lin H, Zhang Y, Lu Y, Wang N, et al. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem. 2009;282:12363–12367. doi: 10.1074/jbc.C700015200. [DOI] [PubMed] [Google Scholar]
- Xiong W, Tian Y, DiSilvestre D, Tomaselli GF. Transmural heterogeneity of Na+-Ca2+ exchange: evidence for differential expression in normal and failing hearts. Circ Res. 2005;97:207–209. doi: 10.1161/01.RES.0000175935.08283.27. [DOI] [PubMed] [Google Scholar]
- Xu X, Salata JJ, Wang J, Wu Y, Yan GX, Liu T, et al. Increasing IKs corrects abnormal repolarization in rabbit models of acquired LQT2 and ventricular hypertrophy. Am J Physiol. 2002;283:H664–H670. doi: 10.1152/ajpheart.00076.2002. [DOI] [PubMed] [Google Scholar]
- Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:207–411. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]
- Yang T, Snyders DJ, Roden DM. Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circ Res. 1997;80:782–789. doi: 10.1161/01.res.80.6.782. [DOI] [PubMed] [Google Scholar]
- Yang Z, Shi G, Li C, Wang H, Liu K, Liu Y. Electrophysiologic effects of nicorandil on the guinea pig long QT1 syndrome model. J Cardiovasc Electrophysiol. 2004;15:815–820. doi: 10.1046/j.1540-8167.2004.03632.x. [DOI] [PubMed] [Google Scholar]
- Yazawa K, Kameyama M. Mechanism of receptor-mediated modulation of the delayed outward potassium current in guinea-pig ventricular myocytes. J Physiol. 1990;421:135–150. doi: 10.1113/jphysiol.1990.sp017937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, et al. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation. 2005;111:2720–2726. doi: 10.1161/CIRCULATIONAHA.104.472498. [DOI] [PubMed] [Google Scholar]
- Zitron E, Scholz E, Owen RW, Lück S, Kiesecker C, Thomas D, et al. QTc prolongation by grapefruit juice and its potential pharmacological basis: HERG channel blockade by flavonoids. Circulation. 2005;111:835–838. doi: 10.1161/01.CIR.0000155617.54749.09. [DOI] [PubMed] [Google Scholar]